

ABSTRACT
Hypercalcemia is defined as a serum calcium concentration that is greater than two standard deviations above the normal mean, which in children may vary with age and sex, reflecting changes in the normal physiology at each developmental stage. Hypercalcemic disorders in children may present with hypotonia, poor feeding, vomiting, constipation, abdominal pain, lethargy, polyuria, dehydration, failure to thrive, and seizures. In severe cases renal failure, pancreatitis and reduced consciousness may also occur and older children and adolescents may present with psychiatric symptoms. The causes of hypercalcemia in children can be classified as parathyroid hormone (PTH)‐dependent or PTH‐independent, and may be congenital or acquired. PTH‐independent hypercalcemia, ie, hypercalcemia associated with a suppressed PTH, is commoner in children than PTH‐dependent hypercalcemia. Acquired causes of PTH‐independent hypercalcemia in children include hypervitaminosis; granulomatous disorders, and endocrinopathies. Congenital syndromes associated with PTH‐independent hypercalcemia include idiopathic infantile hypercalcemia (IIH), William's syndrome, and inborn errors of metabolism. PTH‐dependent hypercalcemia is usually caused by parathyroid tumors, which may give rise to primary hyperparathyroidism (PHPT) or tertiary hyperparathyroidism, which usually arises in association with chronic renal failure and in the treatment of hypophosphatemic rickets. Acquired causes of PTH‐dependent hypercalcemia in neonates include maternal hypocalcemia and extracorporeal membrane oxygenation. PHPT usually occurs as an isolated nonsyndromic and nonhereditary endocrinopathy, but may also occur as a hereditary hypercalcemic disorder such as familial hypocalciuric hypercalcemia, neonatal severe primary hyperparathyroidism, and familial isolated primary hyperparathyroidism, and less commonly, as part of inherited complex syndromic disorders such as multiple endocrine neoplasia (MEN). Advances in identifying the genetic causes have resulted in increased understanding of the underlying biological pathways and improvements in diagnosis. The management of symptomatic hypercalcemia includes interventions such as fluids, antiresorptive medications, and parathyroid surgery. This article presents a clinical, biochemical, and genetic approach to investigating the causes of pediatric hypercalcemia. © 2017 The Authors. Journal of Bone and Mineral Research Published by Wiley Periodicals Inc.
Keywords: NEONATES, PARATHYROID HORMONE, VITAMIN D, SYNDROMES, GENETICS
Introduction
Hypercalcemia in children is less common than in adults, but it nevertheless is more likely to be of clinical significance.1 The differential diagnosis of hypercalcemia in children (Table 1) and adults is similar, but there are marked differences in the frequencies with which they occur. Thus, congenital causes are more frequent in children than acquired causes, such as malignancy, which are more common in adults (Table 1).1 The causes of the hypercalcemia also depend on the age of the child, with congenital anomalies being more common in neonates, and with adolescents being affected by conditions typically seen in adults. Establishing the causes of hypercalcemia in a child may be challenging; this article reviews these etiologies and proposes a clinical algorithm to facilitate their diagnosis.
Table 1.
Causes of Hypercalcemia in Children
| PTH‐dependent hypercalcemia | PTH‐independent hypercalcemia | |
|---|---|---|
| Genetic | FHH1‐3; nsPHPT; NSHPT; FIHP; MEN 1, 2, 3 and 4; HPT‐JT | IIH; Williams syndrome; Down syndrome; hypophosphatasia; Jansen's disease; inborn errors of metabolism (eg, CLD, Bartter syndrome, blue diaper syndrome, sucrose‐isomaltase deficiency, primary oxalosis, IMAGe syndrome) |
| Acquired | Tertiary hyperparathyroidism due to chronic renal failure or treatment for hypophosphatemic rickets; gestational maternal hypocalcemia | Hypervitaminosis D and A; malignancies causing osteolysis (eg, ALL, AML), or secreting PTHrP (eg, lymphoma, medulloblastoma, rhabdomyosarcoma, hepatoblastoma, or hepatocellular carcinoma), or secreting 1,25(OH)2D3 (eg, lymphoma or ovarian dysgerminoma); drugs (eg, thiazides), chemotherapy including 13‐cis‐retinoic acid; milk‐alkali syndrome; granulomatous disease (eg, subcutaneous fat necrosis of the newborn, tuberculosis, sarcoidosis, HIV immune reconstitution syndrome, cat scratch fever, histoplasmosis, coccidiomycosis, leprosy); endocrinopathies (eg, thyrotoxicosis, congenital hypothyroidism, Addison's disease, pheochromocytoma); distal renal tubular acidosis. Multicystic dysplastic kidney disease and renal dysplasia; chronic inflammatory disorders (eg, Crohn's disease); infection (eg, disseminated CMV); immobilization; nutritional and phosphate depletion in preterm neonates |
Causes more likely to be contributing to neonatal or infantile hypercalcemia are shown in italics.
PHPT = primary hyperparathyroidism; FHH1/FHH2/FHH3 = familial hypocalciuric hypercalcemia types 1, 2, and 3; nsPHPT = nonsyndromic primary hyperparathyroidism; NSHPT = neonatal severe primary hyperparathyroidism; FIHP = familial isolated hyperparathyroidism; MEN1/MEN2/MEN3/MEN4 = multiple endocrine neoplasia types 1, 2, 3, and 4; HPT‐JT = hyperparathyroid jaw‐tumor syndrome; IIH = idiopathic infantile hypercalcemia; CLD = congenital lactase deficiency; IMAGe syndrome = syndrome characterized by intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies; ALL = acute lymphoblastic leukemia; AML = acute myeloid leukemia; 1,25(OH)2D3 = 1,25‐dihydroxyvitamin D3; HIV = human immunodeficiency virus; CMV = cytomegalovirus.
Definition and Presentation of Hypercalcemia
Hypercalcemia is defined as a serum calcium concentration that is greater than two standard deviations above the normal mean, which in adults is usually an ionized calcium above ∼1.32 mmol/L (normal range, 1.16 to 1.32 mmol/L) (Table 2) and a total serum calcium, which comprises 55% to 60% ionized calcium plus 40% to 45% protein‐bound (mainly to albumin) calcium, of ∼2.60 mmol/L (normal range, 2.20 to 2.60 mmol/L, 8.5 to 10.5 mg/dL). It is important to distinguish true hypercalcemia from an increased total calcium level secondary to an increase in protein binding because the two conditions may also overlap. For example, hypercalcemia can cause severe dehydration that in turn may result in hyperalbuminemia, resulting in a concurrent increase in calcium binding. The total calcium adjusted for albumin is calculated by the following formulas: adjusted calcium = total calcium – albumin + 4.0, where calcium is in mg/100 mL and albumin in g/100 mL2; or adjusted calcium = total calcium + ({40 – albumin} × 0.02), where calcium is in mmol/L and albumin in g/L (equivalent to 0.02 mmol/L calcium for every 1 g/L albumin 40 g/L). Moreover, ionized and total serum calcium concentrations may vary with age and sex, reflecting changes in the normal physiology at each developmental stage; reference ranges have been established for the different age groups (Table 2). Generally, ionized and total serum calcium concentrations are higher in preterm and full‐term neonates, where ionized calcium is above the 95% reference limits for adults from about the third day until at least 2 weeks postpartum.3
Table 2.
Age‐Specific Reference Intervals for Serum Total and Ionized Calcium Concentrations
| Total calcium a | Ionized calcium b | |||
|---|---|---|---|---|
| Reference | mg/dL | mmol/L | mg/dL | mmol/L |
| Cord blood | 8.2–11.2 | 2.05–2.80 | 5.20–6.40 b | 1.30–1.60 b |
| Neonate (24 hours) | NR | NR | 4.40–5.44 b | 1.10–1.36 b |
| Neonate (5 days) | NR | NR | 4.88–5.92 b | 1.22–1.38 b |
| Birth to 90days | 8.0–11.3 a | 2.00–2.80 a | NR | NR |
| 91–180 days | 8.9–11.2 a | 2.20–2.80 a | NR | NR |
| 181–364 days | 9.0–11.3 a | 2.30–2.80 a | NR | NR |
| 1–3 years | 8.9–11.1 a | 2.20–2.80 a | 4.80–5.52 b | 1.20–1.38 b |
| 4–11 years | 8.7–10.7 a | 2.20–2.70 a | 4.80–5.52 b | 1.20–1.38 b |
| 12–18 years | 8.5–10.7 a | 2.10–2.70 a | 4.80–5.52 b | 1.20–1.38 b |
| >19 years | 8.5–10.5 a | 2.20–2.60 a | 4.64–5.28 b | 1.16–1.32 b |
NR = not reported.
Serum total calcium measured in vitamin D–replete children and young adults, excluding those from renal, endocrine, and critical care unit; thus, these individuals likely had a plasma albumin in the normal range (with serum 25(OH)D3 concentrations of 30 to 80 mg/dL or 75 to 200 nmol/L) and adapted from Roizen JD, Shah V, Levine MA, Carlow DC (Determination of reference intervals for serum total calcium in the vitamin D‐replete pediatric population. J Clin Endocrinol Metab. 2013;98(12):E1946–50).
Cord blood calcium concentrations and serum ionized calcium concentrations adapted from Alan Wu (Tietz Clinical Guide to Laboratory Tests. 4th ed. Philadelphia: Saunders; 2006).
The presentation of hypercalcemia in children may range from an incidental asymptomatic biochemical finding to symptoms of hypotonia, poor feeding, vomiting, constipation, abdominal pain, lethargy, failure to thrive, polyuria, dehydration, and seizures.4, 5 In severe cases, renal failure, pancreatitis, and reduced consciousness may also occur, and older children and adolescents may present with psychiatric symptoms.6 Clinical history and examination may provide diagnostic clues and guide further investigations. The presence or absence of symptoms of hypercalcemia may indicate a particular diagnosis, and the urgency with which investigations should be pursued. For example, mild, nonprogressive, asymptomatic hypercalcemia may potentially indicate a diagnosis of familial hypocalciuric hypercalcemia (FHH), whereas severe hypercalcemia associated with fractures and respiratory distress is suggestive of the life‐threatening disorder of neonatal severe primary hyperparathyroidism (NSHPT).7 In addition, a dietary assessment, details of existing medical problems and medications (including over‐the‐counter supplements), and a family history may help to reveal the cause of the hypercalcemia. Physical examination should include an assessment for dysmorphic features, which may reveal a genetic syndrome, and for sequelae of hypercalcemia, such as bony deformities. Finally, the clinical details of the parents should be assessed because the neonate's condition will have been influenced by the in utero environment. In addition, it is important to measure the serum calcium concentrations of the parents, because a hypercalcemic neonate may have inherited FHH from the mother, or be at a high risk of developing transient NSHPT if inheritance of FHH, due to an inactivating calcium‐sensing receptor (CaSR) mutation, is from the father and the mother is normocalcemic.8
Classification of Hypercalcemia and Pathophysiology
There is no formal classification or grading system for defining the severity of hypercalcemia. However, the severity of clinical symptoms are more likely to be associated with greater elevations in plasma calcium concentrations, and hypercalcemia is generally considered to be mild, moderate, and severe for total serum calcium concentrations <12mg/dL (3.00 mmol/L), between 12 and 14 mg/dL (3.00 to 3.50 mmol/L), and >14mg/dL (3.50 mmol/L), respectively.9 A classification of hypercalcemia that is useful in identifying the underlying etiologies can also be based on an understanding of the pathophysiological mechanisms. Thus, hypercalcemia may arise through increased bone resorption (eg, from lytic bone lesions), increased gastrointestinal absorption of calcium (eg, through enhanced 1,25(OH)2D3 production), and decreased renal excretion of calcium (eg, through the action of thiazides) (Fig. 1). Hypercalcemia may result from more than one mechanism; for example, excessive PTH causes increased gut absorption of calcium through enhanced 1,25(OH)2D3 production, and also stimulates calcium resorption in bone and and calcium reabsorption in the renal tubules. The causes of hypercalcemia may also be classified by whether the circulating PTH concentrations are elevated, ie, hypercalcemia that is PTH‐dependent (eg, as occurring in parathyroid tumors), or reduced, ie, hypercalcemia that is PTH‐independent (eg, through excessive production of parathyroid hormone‐related protein [or PTHrP] by a cancer, or an excess production of downstream mediators such as 1,25(OH)2D3) (Table 1, Fig. 2). Primary hyperparathyroidism (PHPT) and malignancy, which account for >90% of hypercalcemia in adults,10 are rare in children and likely account for <5% of hypercalcemia in children, in whom other causes, especially those that are PTH‐independent and due to genetic abnormalities, are more likely (Table 1). A careful history (eg, for vitamin D ingestion, drugs, renal disease) and examination (eg, for dysmorphology, endocrinopathies, granulomatous diseases), together with appropriate investigations will help to establish the diagnosis.
Figure 1.
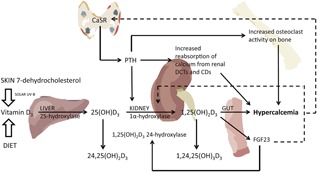
Hormonal regulation of extracellular calcium homeostasis. PTH is the principal calciotropic hormone, and acts to elevate calcium levels by promoting osteoclastic activity on bone, increasing reabsorption of calcium from renal distal tubules and collecting ducts, and stimulating renal enzyme 1α‐hydroxylase conversion of 25(OH)D3 into the active 1,25(OH)2D3. 25(OH)D3 is produced from vitamin D3, formed by the action of solar UVB on the skin or taken in through the diet, by the liver enzyme 25‐hydroxylase. 1,25(OH)2D3 is released into the circulation and stimulates calcium uptake from the gut. Rising serum calcium levels are detected by the CaSR, which facilitates a negative feedback on the parathyroid glands and PTH secretion attenuates to maintain homeostasis. 1,25(OH)2D3 is also regulated by FGF23, the main function of which is to regulate plasma phosphate concentrations. It is secreted by osteocytes in response to an elevated 1,25(OH)2D3 concentration and acts on the kidney proximal tubules to inhibit reabsorption and increase excretion of phosphate. It also inhibits 1α‐hydroxylase, thereby exerting a negative feedback on 1,25(OH)2D3 and promotes the action of 1,25(OH)2D3 24‐hydroxylase, leading to lower levels of 1,25(OH)2D3 and reduced calcium absorption from the gut. 25(OH)D3 = 25‐hydroxyvitamin D3; 1,25(OH)2D3 = 1,25‐dihydroxyvitamin D3; CaSR = calcium‐sensing receptor; FGF23 = fibroblast growth factor‐23; PTH = parathyroid hormone.
Figure 2.
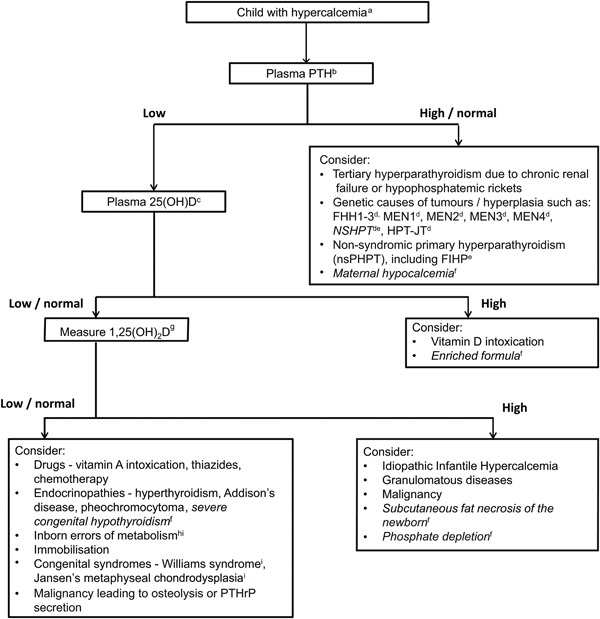
Clinical approach to investigation of causes of hypercalcemia in a child. aConfirm hypercalcemia, defined as plasma (or serum) adjusted calcium > 10.5 mg/dL (2.60 mmol/L) or ionized calcium > 5.25 mg/dL (1.32 mmol/L) (see Table 2). bPTH–parathyroid hormone. c25(OH)D–25‐hydroxyvitamin D. dFHH1‐3– Familial Hypocalciuric Hypercalcemia types 1‐3; MEN1–Multiple Endocrine Neoplasia type 1; MEN2–Multiple Endocrine Neoplasia type 2; MEN3–Multiple Endocrine Neoplasia type 3; MEN4–Multiple Endocrine Neoplasia type 4; NSHPT–Neonatal Severe Primary Hyperparathyroidism; HPT‐JT–Hyperparathyroid‐Jaw Tumour syndrome. eFamilial Isolated Hyperparathyroidism. fConditions affecting neonates (shown in italics). g1,25(OH)2D–1,25‐dihydroxyvitamin D. hInborn errors of metabolism, e.g. Hypophosphatasia, Congenital Lactase Deficiency (CLD) and blue diaper syndrome. iThese syndromes may be associated with dysmorphic features, e.g. Williams syndrome, Jansen's metaphyseal chondrodysplasia, Hypophosphatasia.
PTH‐Independent Hypercalcemia
PTH‐independent hypercalcemia, which is commoner in children than PTH‐dependent hypercalcemia, may be due to many and diverse causes that may be genetic or acquired (Table 1) and include hypervitaminosis D and A, drugs, malignancies, granulomatous disorders, endocrinopathies, renal tubular disorders, chronic inflammatory disorders, infections, immobilization, congenital syndromes, and inborn errors of metabolism. These disorders, some of which may be associated with either high plasma concentrations of 25(OH)D3 or 1,25(OH)2D3 concentrations, are reviewed in the next sections (Fig. 2).
Hypercalcemia associated with high plasma 25(OH)D3 concentrations
Hypercalcemia following vitamin D intoxication may occur due to incorrect prescriptions or accidental overdosing.11, 12, 13, 14 For example, in the summer of 2016, >70 children were reported to have developed hypercalcemia after receiving a vitamin D preparation that contained 75 times higher levels than those recommended.15, 16 Hypercalcemia may also complicate the use of single high‐dose vitamin D therapy (600,000 IU vitamin D3, also known as Stoss [from the German “to shove”] therapy), that is utilized by some centers for the treatment of vitamin D insufficiency or deficiency in children with rickets or cystic fibrosis.17, 18, 19, 20 The precise mechanism by which high doses of 25(OH)D3 can cause hypercalcemia remains unclear. In normal physiology, 25(OH)D3 binds to the vitamin D receptor (VDR) with very low affinity in contrast to its active metabolite, 1,25(OH)2D3. In 25(OH)D3 toxicity, 25(OH)D3 precursors and metabolites are elevated but 1,25(OH)2D3 is usually normal, thereby suggesting that the hypercalcemia is not due to the actions of 1,25(OH)2D3. It has been proposed that the high concentrations of circulating 25(OH)D3 displace 1,25(OH)2D3 from the vitamin D binding protein, thereby increasing the free concentrations of 1,25(OH)2D3, which then stimulate gene transcription via the VDR.21 It is important to note that excessive exposure to sunlight does not pose a risk of vitamin D toxicity because the UVB light stimulates production and destruction of vitamin D3. Thus, 7‐dehydroxyxholesterol is converted to previtamin D3 under UVB, and previtamin D3 is then converted to vitamin D3 at the plasma membrane; however, UVB light also degrades previtamin D3 and vitamin D3, thereby allowing an equilibrium to be reached and thereby preventing excessive vitamin D3 production.22
Hypercalcemia associated with high plasma 1,25(OH)2D3 concentrations
High circulating 1,25(OH)2D3 concentrations may arise because of excessive renal synthesis associated with phosphate depletion, extrarenal activation of the 1α‐hydroxylase enzyme with overproduction of 1,25(OH)2D3, or because of impaired renal catabolism of 1,25(OH)2D3 to its inactive metabolite 1,24,25(OH)3D3 (Fig. 1).
Increased renal synthesis of 1,25(OH)2D3 in association with phosphate depletion
Vitamin D metabolism is affected by phosphate homeostasis and the actions of the phosphate hormone fibroblast growth factor‐23 (FGF23), which are to inhibit and stimulate the activities of the renal 1α‐hydroxylase (CYP27B1) and 1,25‐dihydroxyvitamin D‐24‐hydroxylase (CYP24A1), respectively (Fig. 1). Renal phosphate reabsorption in the proximal tubule involves the sodium‐phosphate cotransporters 2A (NaPi‐IIa) and 2C (NaPi‐IIc), and phosphate reabsorption by NaPi‐IIa is controlled by FGF23 and PTH.23 Loss of phosphate transport activity due to defects of NaPi‐IIa, encoded by the solute carrier 34A1 gene (SLC34A1), results in phosphate depletion with a decrease in circulating FGF23 concentrations, that releases the inhibition of the 1α‐hydroxylase and causes inappropriate excessive production of 1,25(OH)2D3,23 which leads to hypercalcemia, hypercalciuria, and nephrocalcinosis, a combination of features seen in children with idiopathic infantile hypercalcemia (IIH). IIH classically presents in the first year of life with failure to thrive, vomiting, dehydration, and lethargy, and may be fatal. The hypercalcemia usually resolves by 1 year of age, but in some individuals it may persist into adulthood.24 In addition, some patients may later develop hypercalciuria and be at risk of developing renal stone disease and osteoporosis, such that long‐term surveillance is recommended for these patients.25 IIH is an autosomal recessive disorder, and two types of IIH (IIH1 and IIH2) are recognized, and are due to homozygous, or compound heterozygous mutations of the CYP24A1 and SLC34A1 genes.23, 26, 27
Extrarenal synthesis of 1,25(OH)2D3 in malignant and granulomatous diseases
Lymphomas and ovarian dysgerminomas can be extrarenal sites of 1α‐hydroxylase activity, and hypercalcemia due to elevated production of 1,25(OH)2D3, and may occur in 15% and 5% of patients with non‐Hodgkin's and Hodgkin's lymphoma, respectively.28, 29 Similarly, macrophages represent an extrarenal site that can have substantial 1α‐hydroxylase activity. Sequestration of macrophages in granulomatous and inflammatory tissues (eg, sarcoidosis, tuberculosis, human immunodeficiency virus [HIV] immune reconstitution syndrome, leprosy, fungal granuloma including coccidiomycosis, cat scratch fever, Crohn's disease, CMV, histoplasmosis, and subcutaneous fat necrosis of the newborn) can cause dysregulated production of 1,25(OH)2D3, leading to hypercalcemia.30, 31, 32, 33, 34, 35, 36 Subcutaneous fat necrosis of the newborn (SFN) is an unusual form of lobular panniculitis that typically affects newborns born at term or postterm, often with a preceding history of birth trauma or birth asphyxia, and may occur from birth up until the first 6 weeks of life. SFN may also be associated with hypothermia or therapeutic cooling, and is characterized by single or multiple erythematous violaceous plaques and nodules that can evolve into calcifications and tend to occur on the back, face, buttocks, and shoulders.31, 32 It is associated with hypercalcemia that can be life‐threatening and the severity and duration of hypercalcemia are associated with the extent of the skin lesions.31 It has been proposed that an insult on the immature fat cells, such as exposure to cold (eg, therapeutic hypothermia for hypoxia‐ischemia encephalopathy or hypoperfusion),31 may result in the development of necrosis and the development of a granulomatous infiltrate in the necrotic areas. In keeping with this, abundant levels of 1α‐hydroxylase have been found in affected tissues, which may lead to increased production of 1,25(OH)2D3 with associated hypercalcemia as reported in other granulomatous disorders.37
Impaired degradation of 1,25(OH)2D3
Loss‐of‐function mutations of 1,25‐dihydroxyvitamin D3 24‐hydroxylase, encoded by cytochrome P450 family 24 subfamily A member 1 (CYP24A1), resulting in impaired catabolism of 25(OH)D3 and 1,25(OH)2D3 to their inactive metabolites 1,24,25(OH)3D3 and 24,25(OH)2D3, may be associated with elevated circulating levels of the active metabolites and the disorder IIH.25, 38
Hypercalcemia not associated with altered vitamin D concentrations
PTH‐independent hypercalcemia may arise without alterations in circulating 25(OH)D3 or 1,25(OH)2D3 concentrations, and the causes for this form of hypercalcemia include: malignancies that may produce PTHrP; drugs and vitamins; endocrinopathies; renal tubular disorders; congenital and hereditary syndromes and skeletal diseases; inborn errors of metabolism; and specific neonatal disorders.
Drugs and vitamin A toxicity
Drugs such as thiazides and vitamin A (retinol), vitamin A derivatives (eg, its active metabolite, retinoic acid), and inappropriate doses of calcium carbonate and sodium bicarbonate to patients with chronic renal failure resulting in milk alkali syndrome can cause hypercalcemia.39, 40, 41, 42, 43, 44 Thus, thiazides act to increase renal calcium reabsorption, which may cause hypercalcemia or unmask hypercalcemia from other causes that had been compensated for by hypercalciuria.40, 43, 44 Isotretinoin (13‐cis‐retinoic acid), which is used for treatment of severe acne and neuroblastoma, may also cause hypercalcemia by increasing osteoclastic bone resorption.41, 45 However, vitamin A toxicity is a rare cause of hypercalcemia and may occur in children with malabsorptive conditions such as cystic fibrosis39 given supplements containing preformed vitamin A, of which 70% to 90% is absorbed, thereby making children particularly sensitive to overdose. It is important to note that vitamin A toxicity does not occur with high intake of provitamin carotenoids from fruit and vegetables because conversion to the active form of vitamin A is required, and this rarely occurs when large quantities of foods such as fish or animal liver that contain a bioavailable form (retinol) are ingested.
Malignancy and PTHrP
Cancers associated with hypercalcemia in children include hematological malignancies (eg, leukemias, lymphomas, and myeloma), neurological tumors including neuroblastoma, rhabdomyosarcoma, hepatic tumors (eg, hepatoblastoma and hepatocellular carcinoma), and dysgerminomas.46, 47, 48, 49, 50 Hypercalcemia is associated with malignancy in <1% of children,4 and may be caused by: osteolysis due to metastases or leukemias; or osteoclastic bone resorption stimulated by hormones (eg, PTHrP) that are produced by the tumor. PTHrP acts as a paracrine and intracrine hormone to regulate bone development, but some tumors (eg, renal cell carcinomas, squamous cell carcinomas, dysgerminomas, ovarian and breast carcinomas, pheochromocytoma,51, 52 benign congenital mesoblastic nephroma,53 multicystic dysplastic kidney disease,54 and renal dysplasia55, 56) may secrete PTHrP systemically, and the actions of circulating PTHrP on the type 1 PTH/PTHrP receptor cause hypercalcemia.
Endocrinopathies
Endocrine disorders, such as pheochromocytoma, Addison's disease, thyrotoxicosis, and severe congenital hypothyroidism, may be associated with development of hypercalcemia in children. The hypercalcemia associated with pheochromocytoma may be due to secretion of PTHrP.51, 52 In Addison's disease the hypercalcemia may be due to increased intestinal calcium absorption,57 that is possibly aggravated by volume depletion due to lack of mineralocorticoid hormone. Thyroid hormone in children increases bone resorption and skeletal growth; however, in thyrotoxicosis there is also premature fusion of growth plates, resulting in short stature,57 which leads to a negative balance between bone formation and resorption,58 and this may possibly explain the development of hypercalcemia. Severe congenital hypothyroidism in neonates may be associated with mild hypercalcemia in <40% of children,59 although it is rarely symptomatic and the mechanisms remain unknown. Moreover, levothyroxine treatment in children with congenital hypothyroidism59 may also lead to an increase in circulating levels of 1,25(OH)2D3 and hypercalciuria, and hypercalcemia has been reported to only occur in neonates with congenital hypothyroidism who were treated with levothyroxine and vitamin D supplementation, although the hypercalcemia was not correlated with circulating vitamin D concentrations or metabolites of vitamin D.60
Renal tubular disorders
Distal renal tubular acidosis has been reported to be associated with hypercalcemia.61 In addition, Bartter syndrome type 1 (neonatal), which is characterized by metabolic alkalosis, renal hypokalemia, and secondary hyperaldosteronism,62 and is due to mutations in the sodium‐potassium‐chloride co‐transporter‐2 gene (SLC12A1), has also been reported to be associated with hypercalcemia, hypercalciuria, and nephrocalcinosis.(62–64) However, the hypercalcemia was associated with increased circulating PTH concentrations, which may be explained by an inappropriate response to hypocalcemia secondary to hypercalciuria63 and is consistent with marginally increased circulating PTH concentrations that have been reported in other, older children with Bartter syndrome.64
Inborn errors of metabolism
Inborn errors of metabolism that are reported to be associated with hypercalcemia include hypophosphatasia (HPP), congenital lactase deficiency (CLD), disaccharide intolerance, and blue diaper syndrome. HPP is characterized by reduced bone mineralization due to loss‐of‐function mutations in tissue‐nonspecific alkaline phosphatase (TNSALP), encoded by the alkaline phosphatase liver/bone/kidney (ALPL) gene. HPP is classified into six forms, and each is characterized by age of onset and severity of symptoms, ranging from severe perinatal (lethal), where respiratory distress from a hypoplastic chest is the main cause of death, to milder odontohypophosphatasia, where only dental manifestations are seen.65 Patients present perinatally or in childhood and >30% may have hypercalcemia.66 The hypercalcemia is seen in the infantile form of HPP, and may resolve spontaneously within the first year of life,67 or following targeted asfotase alfa enzyme replacement therapy.68 However, hypercalcemia may occur in adults with HPP who have been immobilized.69 In CLD, the severely reduced or absent activity of the gut enzyme lactase, leads to an inability to breakdown dietary lactose, which results in osmotic diarrhea, dehydration, and weight loss shortly after feeding commences, and patients typically have hypercalcemia with nephrocalcinosis. CLD, an autosomal recessive disorder,70, 71, 72, 73 is caused by a mutation of the lactase‐phlorizin hydrolase gene, with a reported incidence of 1:60,000 newborns in Finland,73 although it can occur in other populations.70, 71, 72, 73 Hypercalcemia in CLD usually resolves within weeks after starting a lactose‐free diet, whereas nephrocalcinosis may persist for years.74 The hypercalcemia in CLD may be secondary to, or exacerbated by, dehydration and a metabolic acidosis, or be by a direct action of lactose on the gut.74 Hypercalcemia and nephrocalcinosis may also complicate the presentation of sucrose‐isomaltase deficiency,75 and it is thought that there may be a similar underlying mechanism to CLD. Blue diaper syndrome is due to an abnormality of tryptophan metabolism that results in diarrhea in association with excessive urinary excretion of indole derivatives.76 The characteristic “blue diaper” is caused by the high levels of indican formed when high levels of unabsorbed tryptophan in the intestine are metabolized by bacteria.76 The mechanisms causing hypercalcemia in blue diaper syndrome, which is associated with hypercalciuria and nephrocalcinosis, are not known. Hypercalcemia in the absence of renal dysfunction may also be a rare complication of primary oxalosis77 and has also been reported in IMAGe syndrome (characterized by intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies).78
Immobilization
Immobilization hypercalcemia may occur in 10% to 23% of children with spinal cord injuries, in association with suppressed plasma PTH concentrations, within 4 to 8 weeks of the injury,79 and be preceded by hypercalciuria, which may develop within the first week and continue for up to 18 months.79 Immobilization hypercalcemia also occurs in some children after single‐limb fractures.80 The hypercalcemia is more common in adolescents and males, possibly because of increased bone turnover associated with rapid growth, and a higher bone mass, respectively.79 The mechanism is not understood but is thought to result from increased osteoclastic activity and reduced osteoblastic activity due to a lack of mechanical stimulation.79
Congenital syndromes and diseases
Hypercalcemia is more likely to be genetic in children than in adults, and may occur as part of congenital syndromes that are associated with dysmorphism and/or skeletal abnormalities. These disorders, which include Williams syndrome, Jansen's disease, and Down syndrome, are often detected early in life. However, in some cases dysmorphia may be subtle or missed, and therefore the diagnoses should not necessarily be ruled out on the basis of age alone.
Williams syndrome
Williams syndrome affects approximately 1 in 7,500 to 10,000 individuals81 and is characterized by elfin‐like facies, learning disabilities, supravalvular aortic stenosis, nephrocalcinosis, urinary tract abnormalities, and endocrinopathies, including hypercalcemia, which affects 5% to 50% of patients.81, 82, 83 It usually occurs sporadically, but may be inherited in an autosomal‐dominant manner.84 The mechanisms causing hypercalcemia, which may resolve spontaneously within days to weeks,83 remain unknown, but abnormal 1,25(OH)2D3 metabolism and decreased calcitonin production have been implicated,81, 83, 85 although no abnormality has been consistently demonstrated. Hemizygosity due to a microdeletion of chromosome 7q11.23 involving the ELASTIN and LIM‐KINASE genes, which may explain the respective cardiovascular and neurologic features, have been reported in Williams syndrome. However, the calcitonin receptor gene, located on chromosome 7q21 and close to the region deleted in Williams syndrome, was not involved in the deletion in four patients, indicating that it is unlikely to be implicated in the hypercalcemia of such children. Another, as‐yet‐uncharacterized gene that is within this contiguously deleted region is likely to be involved to explain the abnormalities of calcium metabolism.
Jansen's disease
Jansen's metaphyseal chondrodysplasia is an autosomal‐dominant disease characterized by short‐limbed dwarfism in association with severe hypercalcemia and hypercalciuria, despite normal or undetectable PTH and PTHrP concentrations. These abnormalities are associated with heterozygous mutations of the PTH‐PTHrP receptor, causing constitutive ligand‐independent activation.86
Down syndrome
Down syndrome, which is due to trisomy 21, is one of the most frequent genetic causes of dysmorphism, and occurs in 1 in 690 live births.87 Down syndrome has been reported in association with hypercalcemia and nephrocalcinosis in six patients.88 The mechanisms causing the hypercalcemia are unknown, although the hypercalcemia does appear to respond to dietary calcium restriction suggesting increased intestinal absorption as a possible etiology.88
Acquired causes of neonatal hypercalcemia
Nutritional
Enriched formula and/or phosphate depletion can cause hypercalcemia in preterm newborns89 and, less frequently, in newborns born at term.90 Phosphate depletion may suppress secretion of FGF23, alleviating the inhibitory effect of FGF23 on 1,25(OH)2D3 production (Fig. 1), causing an increase in 1,25(OH)2D3 levels resulting in hypercalcemia. The data on plasma PTH concentrations from such babies is scant and it has been suggested that as the hypercalcemia is associated with hypophosphatemia and increased plasma 1,25(OH)2D3 concentrations, then the plasma PTH is likely to be low.90 The hypercalcemia in these very low birth weight babies fed breast milk, which has a relatively high calcium to phosphate content, can be ameliorated by early administration of phosphate supplements.89
PTH‐Dependent Hypercalcemia
PTH‐dependent hypercalcemia is usually caused by parathyroid tumors, which may give rise to PHPT or tertiary hyperparathyroidism (Table 1, Fig. 2). PHPT usually occurs as an isolated nonsyndromic nonhereditary endocrinopathy and less commonly, as part of inherited complex syndromic disorders such as multiple endocrine neoplasia (MEN) and hyperparathyroid jaw‐tumor syndrome (HPT‐JT).91 Tertiary hyperparathyroidism usually arises in association with chronic renal failure, and may also occur in the treatment of children with hypophosphatemic rickets.92
The main non–parathyroid tumor–related cause of PTH‐dependent hypercalcemia in children is gestational maternal hypocalcemia (Table 1). This acquired cause of hypercalcemia may be apparent from the clinical history and preliminary investigations, whereas the causes associated with parathyroid tumors, which include genetic abnormalities, may be more challenging, and are reviewed further in the next section.
Genetic causes of hypercalcemia
PHPT may occur as a hereditary familial disorder or sporadically, ie, as nonfamilial disease93, 94, 95; however, distinguishing between sporadic and nonfamilial forms may sometimes be difficult. Sporadic PHPT may be the result of a de novo germline mutation in the patient or due to an inherited mutation with an absent family history; eg, if family members have not been investigated or have died before developing symptoms.91 Both de novo and inherited mutations resulting in PHPT will lead to an increased risk of hereditary PHPT in the children of the patient.93, 94, 95, 96 Studies of patients with both syndromic and nonsyndromic forms of PHPT have shown that >10% will harbor a germline mutation in one of 12 genes (Table 3).55, 93, 94, 97, 98 The nonsyndromic forms, which include FHH, NSHPT, and familial isolated primary hyperparathyroidism (FIHP), are likely to be more frequent than the syndromic disorders of MEN and HPT‐JT.
Table 3.
Genetic Disorders Associated With Primary Hyperparathyroidism
| Disorder a | Chromosomal location | Gene |
|---|---|---|
| FHH1 | 3q21.1 | CASR |
| FHH2 | 19p13 | GNA11 |
| FHH3 | 19q13.2–q13.3 | AP2S1 |
| NSHPT | 3q21.1 | CASR |
| nsPHPT | 11p15.3 –p15.1,6p21.2, 9p21, 1p32, 6p24.2 | PTH, b CDKN1A, CDKN2B, CDKN2C, GCM2 c |
| FIHP | 11q13, 1q31.2, 3q21.1, 6p24.2 | MEN1, CDC73, CASR, GCM2 c |
| MEN1 | 11q13 | MEN1 |
| MEN2/MEN3 | 10q11.2 | RET |
| MEN4 | 12p13 | CDKN1B |
| HPT‐JT | 1q31.2 | CDC73 |
FHH1/FHH2/FHH3 = familial hypocalciuric hypercalcemia types 1, 2, and 3; NSHPT = neonatal severe primary hyperparathyroidism; nsPHPT = nonsyndromic primary hyperparathyroidism; FIHP = familial isolated hyperparathyroidism; MEN1/MEN2/MEN3/MEN4 = multiple endocrine neoplasia types 1, 2, 3, and 4; HPT‐JT = hyperparathyroid jaw‐tumor syndrome.
Inheritance of all disorders is autosomal dominant, but NSHPT can be recessive.
A nonsense PTH mutation has been reported in one patient.
Activating mutations of GCM2.
Syndromic PHPT
MEN
MEN is an autosomal dominant disorder in which patients develop two or more endocrine tumors. Four types of MEN (MEN1 to MEN4) are recognized with each associated with a distinct set of endocrine tumors; however, parathyroid tumors occur in all of the MEN syndromes. Thus, in MEN1 patients, parathyroid tumors occur in 95% of patients in association with pancreatic islet cell tumors (∼40% of patients), anterior pituitary tumors (∼30% of patients), and adrenocortical tumors (∼40% of patients).96 In MEN1, parathyroid tumors causing hypercalcemia are the first manifestation of the disease in 90% of patients, and parathyroid tumors may develop as early as 8 years of age, although only 17% of cases below 21 years of age will be symptomatic with urolithiasis, fatigue, and bone pain, with the youngest symptomatic case being aged 8 years with urolithiasis.99 In MEN1, hyperparathyroidism is typically a multigland disease affecting all four parathyroid glands and patients who undergo subtotal parathyroidectomy usually develop recurrent hypercalcemia within a decade.100, 101 In MEN2, parathyroid tumors occur in ∼20% of patients, in association with medullary thyroid carcinoma (MTC) (∼99% of patients) and pheochromocytomas (∼50% of patients).96, 101 In MEN3, MTC and pheochromocytomas are also common, but parathyroid tumors are rarely seen; instead patients have other features such as a Marfanoid habitus, mucosal neuromas, medullated corneal nerve fibers, and intestinal autonomic ganglion dysfunction leading to multiple diverticula and megacolon. Only a few patients with MEN4 have been described; all have parathyroid tumors in association with other tumors affecting the adrenals, pituitary, and gonads.102 MEN1 is caused by a mutation in the tumor suppressor MEN1 gene encoding menin; MEN2 and MEN3 are caused by mutations in the proto‐oncogene RET, encoding a tyrosine kinase receptor; and MEN4 is caused by mutations of CDNK1B, encoding p27.96, 101
HPT‐JT
HPT‐JT syndrome, an autosomal dominant disorder, is characterized by the development of multiple parathyroid tumors, which may be carcinomas, and fibro‐osseous tumors of the maxilla and mandible.94 Some families also have increased risk of developing renal tumors and affected women have greater risk of developing uterine tumors.94 However, in some families the affected individuals may have developed only parathyroid tumors without other tumors or jaw tumors, and this may cause confusion with other hereditary hypercalcemic disorders such as MEN1, FHH, and FIHP.93 HPT‐JT is due to mutations of the cell division cycle 73 (CDC73) gene, which is a tumor suppressor encoding parafibromin that is involved in transcriptional and posttranscriptional pathways.94 Parathyroid tumors with hypercalcemia occur in >70% of individuals with CDC73 mutations, with the onset being typically in late adolescence or early adulthood.103, 104, 105, 106 The youngest patient reported with hypercalcemia is 7 years old,103 and the youngest patient reported with parathyroid carcinoma is 20 years old.107 Germline pathogenic CDC73 mutations have also been reported in patients with apparently sporadic: parathyroid carcinoma; parathyroid adenoma; or ossifying fibromas of the jaw.108 Due to the potential for early onset of tumors, biochemical surveillance for PHPT, starting from age 5 to 10 years, is recommended in individuals known to be at risk.103
Nonsyndromic PHPT
FHH
FHH is characterized by lifelong elevations of serum calcium concentrations, elevated or inappropriately normal plasma PTH concentrations, and low urinary calcium excretion resulting from PTH‐independent reduced calcium excretion in the kidneys.109 The hypercalcemia of FHH is considered to be primarily due to inappropriate conservation of calcium in the kidney rather than being driven by an inappropriate PTH concentration.109 The mean calcium‐to‐creatinine clearance ratio (CCCR) (measured in either molar units or mass units) is typically <0.01 in FHH. However, more than 20% FHH patients have a CCCR >0.01, and such patients are at risk of being misdiagnosed with PHPT.110, 111, 112 In contrast to PHPT, the hypercalcemia in FHH is generally benign and is not corrected by parathyroidectomy; therefore, distinguishing between PHPT and FHH is important to avoid unnecessary surgery in FHH patients.
FHH is an autosomal‐dominant, genetically heterogeneous disorder with three clinically indistinguishable variants (FHH1 to FHH3).111, 113 FHH1 comprises ∼65% of FHH patients and is due to loss‐of‐function mutations of CaSR, a G‐protein‐coupled receptor (GPCR). FHH2 comprises <5% of all FHH patients and is due to loss‐of‐function mutations of GNA11, which encodes G‐protein subunit α‐11 (Gα11).111 FHH3 may occur in ∼20% of FHH patients without CaSR mutations9 and is due to loss‐of‐function mutations of the adaptor‐related protein complex 2, sigma 1 subunit (AP2S1) gene. The AP2S1 gene encodes the adaptor‐protein 2 sigma (AP2σ) subunit that forms a heterotetramer with other subunits, and plays a central role in clathrin‐mediated endocytosis of plasma membrane constituents such as GPCRs.9
As FHH is usually asymptomatic, the age of diagnosis will be variable, with individuals with concurrent medical problems or hypercalcemic symptoms being detected earlier. The youngest age of diagnosis of FHH1 is in a 4.5‐month‐old Greek infant who had a urinary tract infection with incidental hypercalcemia.114 The youngest age for diagnosis of FHH3 is in a Japanese infant who at age 49 days presented with poor weight gain and had an AP2S1 mutation.115 FHH3 children are more likely than FHH1 children to be symptomatic, and to have additional phenotypic features, and thus may present earlier.116 For example, four FHH3 children, aged <1 to 11 years, have been reported to have learning difficulties (LDs); one aged 14 years had LD, pancreatitis, and short stature (SS); and one aged 15 years had LD, an atrial septal defect, and SS.116
NSHPT
NSHPT usually presents at birth with marked hypercalcemia, hypotonia, respiratory distress, and bone demineralization, and is usually fatal by 3 months if untreated. Bone demineralization occurs due to osteoclast overactivity and can result in bone deformities and fractures presenting at birth. Respiratory difficulties may arise from rib cage involvement. Most cases are associated with homozygous or compound heterozygous loss‐of‐function CaSR mutations117 and some may occur in FHH families, either through inheriting two mutated copies of the CASR gene as in consanguineous families, or one copy together with a de novo CASR mutation. Urgent parathyroidectomy is lifesaving and the treatment of choice, but pamidronate or cinacalcet have been used for treatment while awaiting surgery.118 Cinacalcet is a positive allosteric modulator of the CaSR and will ameliorate signaling disturbances associated with most loss‐of‐function CaSR mutations; however, if both CASR alleles have mutations whereby the CaSR is not expressed, eg, with a homozygous deletion in exon 5 (c.1392_1404del13), then cinacalcet will be ineffective.119 Some centers distinguish between NSHPT and neonatal hyperparathyroidism (NHPT) on the basis of homozygous and heterozygous CaSR mutations, respectively. NHPT may be associated with less marked and symptomatically transient hypercalcemia than NSHPT, with some patients developing symptomless FHH, and therefore not requiring parathyroid surgery.120 This highlights the value of CASR mutational analysis in distinguishing NSHPT from NHPT.
FIHP
FIHP is characterized by hereditary PHPT occurring without the association of other tumors and has been described in >100 families.91, 93 The diagnosis of FIHP is based on excluding other hereditary disorders associated with PHPT, such as MEN1, MEN2, MEN4, HPT‐JT, and FHH, and by screening for mutations in known causative genes, such as MEN1, RET, CDKN1B, CDC73, and CASR, respectively.91 In the majority of these families the genetic etiology of FIHP remains unknown.121, 122 However, patients with activating mutations of glial cells missing 2 (GCM2), a parathyroid‐specific transcription factor, have been reported,123 as well as one PHPT patient who had a nonsense mutation of the PTH gene.124 Approximately 10% of patients presenting under the age of 45 years with sporadic PHPT will have a de novo germline MEN1, CDC73, GCM2, or CASR mutation,93, 94, 95, 125, 126 and this has implications for their future management, to ensure appropriate screening for complications associated with the specific syndrome, and for screening first‐degree relatives. The finding of a mutation in MEN1, RET, CDC73, or CDKN1B in an apparent FIHP kindred would lead to a revised diagnosis of one of the associated syndromes with incomplete penetrance93 and, in some families, FIHP may represent an incomplete manifestation of a syndromic form of PHPT caused by an as‐yet‐undiscovered mutation.93, 127, 128 Some patients diagnosed as having FIHP have later been reported to develop features of MEN193, 94, 95, 96, 97, 127 and, in addition, some FIHP kindreds have associated MEN1 mutations and may represent an allelic variant of MEN1.127
FIHP may be distinguished from MEN1, FHH, and HPT‐JT through clinical, histological, and genetic findings. Over 90% of MEN1 and HPT‐JT patients develop hypercalcemia as their first manifestation of the disease, and distinguishing between MEN1 and HPT‐JT patients at this early stage from FIHP patients can be difficult.91, 93, 98 It is important to distinguish FIHP from HPT‐JT because HPT‐JT patients are at higher risk for developing parathyroid carcinomas.129, 130, 131, 132 The presence of ossifying fibromas of the jaw is an important distinguishing feature of HPT‐JT and the identification of renal, pancreatic, thyroid, and testicular abnormalities may also help to identify HPT‐JT patients. The jaw tumors in HPT‐JT differ from the brown tumors observed in some PHPT patients and do not resolve after parathyroidectomy.133 Similarly, the occurrence of pancreatic, pituitary, or adrenal tumors in the patient or family will suggest a diagnosis of MEN1 rather than FIHP. FIHP may be distinguished from FHH on the basis of plasma and urinary calcium findings. In FHH, serum calcium levels are elevated from the neonatal period, whereas in FIHP, hypercalcemia rarely occurs until after the first decade.98, 127 FIHP patients are likely to have associated hypercalciuria, unlike FHH patients who usually have a CCCR <0.01.116 However, there are considerable clinical overlaps between these disorders, and genetic testing is advisable for making the correct diagnosis.
Genetic testing in patients with syndromic and nonsyndromic forms of PHPT
Genetic testing for mutations in PHPT patients is worthwhile because >10% of patients with PHPT will have a mutation in one of 13 genes (Table 3) and therefore benefit from an accurate diagnosis.91 Thus, genetic testing may provide confirmation of the clinical diagnosis (ie, syndromic or nonsyndromic PHPT) and allow appropriate screening for associated tumors to be undertaken, with implementation of appropriate treatment, eg, early parathyroidectomy for HPT‐JT patients who are at increased risk of developing parathyroid carcinomas; appropriate parathyroid surgery in MEN1 patients who generally have multigland disease requiring open neck exploration; and avoidance of unnecessary parathyroidectomies in FHH patients.91 Asymptomatic mutation carriers should receive screening for associated tumors to facilitate appropriate treatment, and the 50% of family members who do not harbor the germline mutation can be reassured and alleviated of the anxiety of developing the condition,93, 94, 95 thereby reducing costs to the individuals, and also to the health services in avoiding unnecessary biochemical and radiological investigations.93, 94, 95, 98
Indications for genetic testing in PHPT patients include: (i) young age of presentation, ie, <45 years of age; (ii) multigland disease; (iii) parathyroid carcinoma or atypical parathyroid adenomas (eg, with fibrous bands or cysts); (iv) being a first‐degree relative of a known mutation carrier; and (v) being an index case with two or more MEN syndrome–associated tumors.93, 94, 95, 98, 126, 134 Patients should be offered genetic counseling prior to testing, and genetic testing should use non‐tumor cells (eg, DNA obtained from leukocytes, salivary cells, skin cells, or hair follicles), because DNA from parathyroid tumors may contain multiple mutations in addition to the germline mutation and therefore are not clinically useful.91 Individuals with PHPT identified to have a germline mutation should enter into an appropriate screening program; eg, for MEN‐associated and HPT‐JT–associated tumors.93, 94 If no genetic abnormalities are found within the 13 genes (Table 3), and clinical manifestations of hereditary or syndromic forms of PHPT are absent, then the likelihood of a MEN syndrome, HPT‐JT or FHH is low (ie, <5%).93, 94, 95, 98 The first‐degree relatives of PHPT patients, including children, with a germline mutation should be offered genetic counseling and appropriate gene testing, and any affected individuals subsequently identified should also enter into an appropriate screening program, even if asymptomatic.93, 94, 95, 134 First‐degree relatives who have not inherited the causative mutation require no further follow‐up and can be reassured.93, 94
Patients who present with PHPT at a later age, ie, >45 years, and who have an underlying syndromic etiology for their PHPT, are more likely to have manifested other associated features that may be revealed during clinical evaluation. A detailed family history for PHPT (ie, FIHP), MEN syndrome, HPT‐JT, or FHH should be undertaken, and gene testing should then be offered to determine the etiology of the PHPT.93, 94, 95, 110, 124, 134 However, up to 5% of patients >45 years of age with sporadic PHPT due to a solitary parathyroid adenoma may have a germline mutation involving CDKN1A, CDKN2B, or CDKN2C,91 and this may have implications for their children. Such first‐degree relatives of a patient with a mutation in CDKN1A, CDKN2B, or CDKN2C who are also found to be mutation carriers should then have periodic screening to detect the onset of hypercalcemia in order to facilitate appropriate earlier treatment aimed at preventing the skeletal and renal complications of PHPT.93, 95, 97, 98
Acquired causes of neonatal hypercalcemia
Neonatal hyperparathyroidism
Neonatal hyperparathyroidism may be an adaptation to maternal hypocalcemia due to hypoparathyroidism, vitamin D deficiency, pseudohypoparathyroidism, or renal tubular acidosis,4 although in the latter, PTH is often high with normal calcium. Maternal hypervitaminosis D does not usually affect the neonate because 1,25(OH)2D3 is inactivated by the placenta, so excessive maternal 1,25(OH)2D3 consumption should not cause hypercalcemia in the newborn. However, there have been case reports of excessive maternal intake of 1,25(OH)2D3 and 25(OH)D3 causing hypercalcemia in the neonate.135, 136
Extracorporeal membrane oxygenation
Extracorporeal membrane oxygenation (ECMO) is a technique used in pediatric resuscitation as a way of providing tissue oxygenation in cases of refractory hypoxemia to allow time for treatment of the underlying cause of the disorder to take effect. It is associated with metabolic disturbances including hypercalcemia,137, 138, 139 although the mechanisms for this are not known, but may be related to increased circulating PTH concentrations.138
Management of Hypercalcemia
The management of hypercalcemia involves establishing the underlying diagnosis in parallel with lowering serum calcium levels. Medications that may cause or exacerbate the hypercalcemia, such as calcium and vitamin D preparations, should be stopped. Symptomatic hypercalcemia may require fluid administration to rectify intravascular volume contraction caused by hypercalcemia‐induced nephrogenic diabetes insipidus.140 Normal (0.9%) saline given intravenously is effective at rehydrating the patient, thereby diluting serum calcium concentrations.5 Moreover, normal saline lowers serum calcium concentrations by promoting urinary calcium excretion from the proximal renal tubule and loop of Henle.140 Loop diuretics such as furosemide can also enhance urinary calcium excretion,140 but should be used cautiously because they may exacerbate the intravascular volume contraction and lead to renal impairment.4 Subcutaneous calcitonin is an effective antiresorptive agent for symptomatic hypercalcemia; however, its effects are short‐lived due to tachyphylaxis.4 The use of i.v. bisphosphonates leads to a more sustained reduction in serum calcium concentrations, and pamidronate (0.5 to 1.0 mg/kg) is used most commonly in children.140 Patients should be adequately hydrated prior to receiving i.v. bisphosphonates in order to minimize the potential risk of nephrotoxicity, which has been widely reported in adults.141 Glucocorticoids such as prednisolone are effective at rectifying hypercalcemia caused by granulomatous disorders, although long‐term use may increase skeletal fragility and impair linear growth.4, 140 Cinacalcet, which is a CaSR‐positive allosteric modulator, has been successfully used to manage life‐threatening hypercalcemia in some NSHPT probands.142 However, the effectiveness of cinacalcet for NSHPT will depend on the underlying CaSR mutation, and patients harboring biallelic truncating CaSR mutations will be unlikely to respond to this calcimimetic agent.143 Parathyroidectomy remains the treatment of choice for children with primary hyperparathyroidism or NSHPT because it represents a curative procedure for these disorders.144 Hypocalcemia due to hungry bone syndrome or postsurgical hypoparathyroidism represents the most common complication of parathyroidectomy, and close monitoring of serum calcium concentrations is required following surgery.140, 144
Conclusions
The majority of causes of hypercalcemia in children are similar to those in adults, but occur at different frequencies across the age spectrum from birth to maturity. Hypercalcemia has a broad differential diagnosis (Table 1), and comprehensive clinical assessment followed by stepwise use of investigations is necessary to elucidate the underlying cause (Fig. 2). Determining the etiology of hypercalcemia is critical for successful treatment and for ensuring the child's growth and development.
Disclosures
All authors state that they have no conflicts of interest.
Acknowledgments
The work in the Academic Endocrine Unit is supported by the United Kingdom Medical Research Council (MRC) programme grants G9825289 and G1000467, and National Institute for Health Research (NIHR) Oxford Biomedical Research Centre Programme. RVT is a Wellcome Trust Investigator and NIHR Senior Investigator. VJS is a Wellcome Trust Clinical Training Fellow.
Authors’ roles: VJS made substantial contributions to the design of the manuscript, participated in drafting the manuscript, revised the manuscript critically for important intellectual content, and approved the final version of the submitted manuscript. MFN made substantial contributions to the design of the manuscript, participated in drafting the manuscript, revised the manuscript critically for important intellectual content, and approved the final version of the submitted manuscript. FMH made substantial contributions to the design of the manuscript, made substantial contributions in revising the manuscript critically for important intellectual content, and approved the final version of the submitted manuscript. RVT made substantial contributions to the conception and design of the manuscript, revised the manuscript critically for important intellectual content, and approved the final version of the submitted manuscript.
References
- 1. McNeilly JD, Boal R, Shaikh MG, Ahmed SF. Frequency and aetiology of hypercalcaemia. Arch Dis Child. 2016; 101(4):344–7. [DOI] [PubMed] [Google Scholar]
- 2. Payne RB, Little AJ, Williams RB, Milner JR. Interpretation of serum calcium in patients with abnormal serum proteins. BMJ. 1973; 4(5893):643–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wandrup J. Critical analytical and clinical aspects of ionized calcium in neonates. Clin Chem. 1989; 35(10):2027–33. [PubMed] [Google Scholar]
- 4. Lietman SA, Germain‐Lee EL, Levine MA. Hypercalcemia in children and adolescents. Curr Opin Pediatr. 2010; 22(4):508–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. El Saleeby CM, Grottkau BE, Friedmann AM, Westra SJ, Sohani AR. Case records of the Massachusetts General Hospital. Case 4‐2011. A 4‐year‐old boy with back pain and hypercalcemia. N Engl J Med. 2011; 364(6):552–62. [DOI] [PubMed] [Google Scholar]
- 6. Babar G, Alemzadeh R. A case of acute psychosis in an adolescent male. Case Rep Endocrinol. 2014; 2014:937631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hannan FM, Thakker RV. Calcium‐sensing receptor (CaSR) mutations and disorders of calcium, electrolyte and water metabolism. Best Pract Res Clin Endocrinol Metab. 2013; 27(3):359–71. [DOI] [PubMed] [Google Scholar]
- 8. Bai M, Pearce SH, Kifor O, et al. In vivo and in vitro characterization of neonatal hyperparathyroidism resulting from a de novo, heterozygous mutation in the Ca2+‐sensing receptor gene: normal maternal calcium homeostasis as a cause of secondary hyperparathyroidism in familial benign hypocalciuric hypercalcemia. J Clin Invest. 1997; 99(1):88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bushinsky DA, Monk RD. Electrolyte quintet: calcium. Lancet. 1998; 352(9124):306–11. [DOI] [PubMed] [Google Scholar]
- 10. Lafferty FW. Differential diagnosis of hypercalcemia. J Bone Miner Res. 1991;6 Suppl 2:S51–9; discussion S61. [DOI] [PubMed] [Google Scholar]
- 11. Gorris MA, Arora H, Lieb DC, Aloi JA. A word of caution when prescribing high dose vitamin D. Am J Med. 2017; 130(4):e129–30. [DOI] [PubMed] [Google Scholar]
- 12. Wani M, Wani I, Banday K, Ashraf M. The other side of vitamin D therapy: a case series of acute kidney injury due to malpractice‐related vitamin D intoxication. Clin Nephrol. 2016; 86(11):236–41. DOI:10.5414/CN108904. [DOI] [PubMed] [Google Scholar]
- 13. Perez‐Barrios C, Hernandez‐Alvarez E, Blanco‐Navarro I, Perez‐Sacristan B, Granado‐Lorencio F. Prevalence of hypercalcemia related to hypervitaminosis D in clinical practice. Clin Nutr. 2016; 35(6):1354–8. [DOI] [PubMed] [Google Scholar]
- 14. Nimesh M, Singh P, Jhamb U, Dubey AP. An unsuspected pharmacological vitamin D toxicity in a child and its brief review of literature. Toxicol Int. 2015; 22(1):167–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rychla L. Danish health authority warns of toxic vitamin D product [Internet]. CPH Post Online; 2016. Jul 26 [cited 2017 Sep 27]. Available from: http://cphpost.dk/news/danish‐health‐authority‐warns‐of‐toxic‐vitamin‐d‐product.html.
- 16.Danish Health Authority. Risk of severe intoxication with vitamin D drops [Internet]. Copenhagen, Denmark; 2016. July 26 [updated 2016 August 01; cited 2017 Sep 27]. Available from: https://www.sst.dk/en/news/2016/risk‐of‐severe‐intoxication‐with‐vitamin‐d‐drops.
- 17. Viswanathan A, Quintos JB. Stosstherapy for treatment of vitamin D deficiency rickets. Hosp Physician. 2006. October; 42(10):39–42. [Google Scholar]
- 18. Shepherd D, Belessis Y, Katz T, Morton J, Field P, Jaffe A. Single high‐dose oral vitamin D3 (stoss) therapy—a solution to vitamin D deficiency in children with cystic fibrosis? J Cyst Fibros. 2013; 12(2):177–82. [DOI] [PubMed] [Google Scholar]
- 19. Emel T, Dogan DA, Erdem G, Faruk O. Therapy strategies in vitamin D deficiency with or without rickets: efficiency of low‐dose stoss therapy. J Pediatr Endocrinol Metab. 2012; 25(1‐2):107–10. [DOI] [PubMed] [Google Scholar]
- 20. Harnot J, Verma S, Singhi S, Sankhyan N, Sachdeva N, Bharti B. Comparison of 300, 000 and 600, 000 IU oral vitamin‐D bolus for vitamin‐D deficiency in young children. Indian J Pediatr. 2017; 84(2):111–6. [DOI] [PubMed] [Google Scholar]
- 21. Jones G. Pharmacokinetics of vitamin D toxicity. Am J Clin Nutr. 2008; 88(2):582s–6s. [DOI] [PubMed] [Google Scholar]
- 22. Holick MF. Vitamin D deficiency. N Engl J Med. 2007; 357(3):266–81. [DOI] [PubMed] [Google Scholar]
- 23. Schlingmann KP, Ruminska J, Kaufmann M, et al. Autosomal‐recessive mutations in SLC34A1 encoding sodium‐phosphate cotransporter 2A cause idiopathic infantile hypercalcemia. J Am Soc Nephrol. 2016; 27(2):604–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang J, Coman D, McTaggart SJ, Burke JR. Long‐term follow‐up of patients with idiopathic infantile hypercalcaemia. Pediatr Nephrol. 2006; 21(11):1676–80. [DOI] [PubMed] [Google Scholar]
- 25. Dinour D, Beckerman P, Ganon L, Tordjman K, Eisenstein Z, Holtzman EJ. Loss‐of‐function mutations of CYP24A1, the vitamin D 24‐hydroxylase gene, cause long‐standing hypercalciuric nephrolithiasis and nephrocalcinosis. J Urol. 2013; 190(2):552–7. [DOI] [PubMed] [Google Scholar]
- 26. Schlingmann KP, Kaufmann M, Weber S, et al. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med. 2011; 365(5):410–21. [DOI] [PubMed] [Google Scholar]
- 27. Pronicka E, Ciara E, Halat P, et al. Biallelic mutations in CYP24A1 or SLC34A1 as a cause of infantile idiopathic hypercalcemia (IIH) with vitamin D hypersensitivity: molecular study of 11 historical IIH cases. J Appl Genet. 2017; 58(3):349–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seymour JF, Gagel RF. Calcitriol: the major humoral mediator of hypercalcemia in Hodgkin's disease and non‐Hodgkin's lymphomas. Blood. 1993; 82(5):1383–94. [PubMed] [Google Scholar]
- 29. Levendoglu‐Tugal O, Kroop S, Rozenblit GN, Weiss R. Primary renal lymphoma and hypercalcemia in a child. Leuk Lymphoma. 2002; 43(5):1141–6. [DOI] [PubMed] [Google Scholar]
- 30. Conron M, Young C, Beynon HL. Calcium metabolism in sarcoidosis and its clinical implications. Rheumatology (Oxford). 2000; 39(7):707–13. [DOI] [PubMed] [Google Scholar]
- 31. Del Pozzo‐Magana BR, Ho N. Subcutaneous fat necrosis of the newborn: a 20‐year retrospective study. Pediatr Dermatol. 2016; 33(6):e353–e5. [DOI] [PubMed] [Google Scholar]
- 32. Sharata H, Postellon DC, Hashimoto K. Subcutaneous fat necrosis, hypercalcemia, and prostaglandin E. Pediatr Dermatol. 1995; 12(1):43–7. [DOI] [PubMed] [Google Scholar]
- 33. Sharma OP. Hypercalcemia in granulomatous disorders: a clinical review. Curr Opin Pulmon Med. 2000; 6(5):442–7. [DOI] [PubMed] [Google Scholar]
- 34. Bosch X. Hypercalcemia due to endogenous overproduction of active vitamin D in identical twins with cat‐scratch disease. JAMA. 1998; 279(7):532–4. [DOI] [PubMed] [Google Scholar]
- 35. Tuohy KA, Steinman TI. Hypercalcemia due to excess 1,25‐dihydroxyvitamin D in Crohn's disease. Am J Kidney Dis. 2005; 45(1):e3–6. [DOI] [PubMed] [Google Scholar]
- 36. Tsao YT, Wu CC, Chu P. Immune reconstitution syndrome‐induced hypercalcemic crisis. Am J Emerg Med. 2011; 29(2):244.e3–6. [DOI] [PubMed] [Google Scholar]
- 37. Farooque A, Moss C, Zehnder D, Hewison M, Shaw NJ. Expression of 25‐hydroxyvitamin D3‐1alpha‐hydroxylase in subcutaneous fat necrosis. Br J Dermatol. 2009; 160(2):423–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dinour D, Davidovits M, Aviner S, et al. Maternal and infantile hypercalcemia caused by vitamin‐D‐hydroxylase mutations and vitamin D intake. Pediatr Nephrol. 2015; 30(1):145–52. [DOI] [PubMed] [Google Scholar]
- 39. Safi KH, Filbrun AG, Nasr SZ. Hypervitaminosis A causing hypercalcemia in cystic fibrosis. Case report and focused review. Ann Am Thorac Soc. 2014; 11(8):1244–7. [DOI] [PubMed] [Google Scholar]
- 40. Goltzman D. Approach to hypercalcemia. In: De Groot LJ, Chrousos G, Dungan K, et al., editors. Endotext. South Dartmouth, MA: MDText.com, Inc.; 2000: pp 1093‐1100.
- 41. Marabelle A, Sapin V, Rousseau R, Periquet B, Demeocq F, Kanold J. Hypercalcemia and 13‐cis‐retinoic acid in post‐consolidation therapy of neuroblastoma. Pediatr Blood Cancer. 2009; 52(2):280–3. [DOI] [PubMed] [Google Scholar]
- 42. Kari JA, Desoky El SM. Milk alkali syndrome in an infant with chronic kidney disease. Pediatric Health, Medicine and Therapeutics. 2012; 2012(3):19–23. DOI:10.2147/PHMT.S30290. [Google Scholar]
- 43. Al Nofal A, Lteif A. Thiazide diuretics in the management of young children with central diabetes insipidus. J Pediatr. 2015; 167(3):658–61. [DOI] [PubMed] [Google Scholar]
- 44. Cervera A, Corral MJ, Gomez Campdera FJ, De Lecea AM, Luque A, Lopez Gomez JM. Idiopathic hypercalciuria in children. Classification, clinical manifestations and outcome. Acta Paediatr Scand. 1987; 76(2):271–8. [DOI] [PubMed] [Google Scholar]
- 45. Valentic JP, Elias AN, Weinstein GD. Hypercalcemia associated with oral isotretinoin in the treatment of severe acne. JAMA. 1983; 250(14):1899–900. [PubMed] [Google Scholar]
- 46. Park HJ, Choi EJ, Kim JK. A successful treatment of hypercalcemia with zoledronic acid in a 15‐year‐old boy with acute lymphoblastic leukemia. Ann Pediatr Endocrinol Metab. 2016; 21(2):99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Watanabe N, Yasuda H, Morishita S, et al. Richter's syndrome with hypercalcemia induced by tumor‐associated production of parathyroid hormone‐related peptide. Case Rep Oncol. 2017; 10(1):123–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grunewald TG, von Luettichau I, Welsch U, et al. First report of ectopic ACTH syndrome and PTHrP‐induced hypercalcemia due to a hepatoblastoma in a child. Eur J Endocrinol. 2010; 162(4):813–8. [DOI] [PubMed] [Google Scholar]
- 49. Nourani M, Manera RB. Pediatric ovarian dysgerminoma presenting with hypercalcemia and chronic constipation: a case report. J Pediatr Hematol Oncol. 2013; 35(7):e272–3. [DOI] [PubMed] [Google Scholar]
- 50. Demirkaya M, Sevinir B, Yalcinkaya U, Yazici Z. Disseminated rhabdomyosarcoma presenting as hypercalcemia. Indian Pediatr. 2012; 49(1):66–7. [PubMed] [Google Scholar]
- 51. Kimura S, Nishimura Y, Yamaguchi K, Nagasaki K, Shimada K, Uchida H. A case of pheochromocytoma producing parathyroid hormone‐related protein and presenting with hypercalcemia. J Clin Endocrinol Metab. 1990; 70(6):1559–63. [DOI] [PubMed] [Google Scholar]
- 52. Takeda K, Hara N, Kawaguchi M, Nishiyama T, Takahashi K. Parathyroid hormone‐related peptide‐producing non‐familial pheochromocytoma in a child. Int J Urol. 2010; 17(7):673–6. [DOI] [PubMed] [Google Scholar]
- 53. Srivastava T, Kats A, Martin TJ, Pompolo S, Alon US. Parathyroid‐hormone‐related protein‐mediated hypercalcemia in benign congenital mesoblastic nephroma. Pediatr Nephrol. 2011; 26(5):799–803. [DOI] [PubMed] [Google Scholar]
- 54. Grob F, Zambrano P, Ibarra X, Reyes ML. Parathyroid hormone‐independent hypercalcemia in an infant with renal dysplasia: possible role of PTHrP. J Pediatr Endocrinol Metab. 2013;26(3‐4):365–7. [DOI] [PubMed] [Google Scholar]
- 55. Al Kalbani N, Frieling M, Teh JC, Harvey E, Geary DF. Idiopathic hypercalcemia in infants with renal dysplasia. Clin Nephrol. 2011; 75(5):466–71. [DOI] [PubMed] [Google Scholar]
- 56. Kodous N, Filler G, Sharma AP, Van Hooren TA. PTHrP‐related hypercalcaemia in infancy and congenital anomalies of the kidney and urinary tract (CAKUT). Can J Kidney Health Dis. 2015; 2:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vasikaran SD, Tallis GA, Braund WJ. Secondary hypoadrenalism presenting with hypercalcaemia. Clin Endocrinol. 1994; 41(2):261–4. [DOI] [PubMed] [Google Scholar]
- 58. Eriksen EF, Mosekilde L, Melsen F. Trabecular bone remodeling and bone balance in hyperthyroidism. Bone. 1985; 6(6):421–8. [DOI] [PubMed] [Google Scholar]
- 59. Laufer J, Noff D, Orda S, Sack J. Effect of short‐term hyperthyroxinemia on vitamin D metabolism in congenital hypothyroidism. Horm Metab Res. 1993; 25(7):386–8. [DOI] [PubMed] [Google Scholar]
- 60. Tau C, Garabedian M, Farriaux JP, Czernichow P, Pomarede R, Balsan S. Hypercalcemia in infants with congenital hypothyroidism and its relation to vitamin D and thyroid hormones. J Pediatr. 1986; 109(5):808–14. [DOI] [PubMed] [Google Scholar]
- 61. Faqeih E, Al‐Akash SI, Sakati N, Teebi PA. Four siblings with distal renal tubular acidosis and nephrocalcinosis, neurobehavioral impairment, short stature, and distinctive facial appearance: a possible new autosomal recessive syndrome. Am J Med Genet A. 2007;143a(17):1951–7. [DOI] [PubMed] [Google Scholar]
- 62. Scheinman SJ, Guay‐Woodford LM, Thakker RV, Warnock DG. Genetic disorders of renal electrolyte transport. N Engl J Med. 1999; 340(15):1177–87. [DOI] [PubMed] [Google Scholar]
- 63. Gross I, Siedner‐Weintraub Y, Simckes A, Gillis D. Antenatal Bartter syndrome presenting as hyperparathyroidism with hypercalcemia and hypercalciuria: a case report and review. J Pediatr Endocrinol Metab. 2015; 28(7–8):943–6. [DOI] [PubMed] [Google Scholar]
- 64. Bettinelli A, Vigano C, Provero MC, et al. Phosphate homeostasis in Bartter syndrome: a case‐control study. Pediatr Nephrol. 2014; 29(11):2133–8. [DOI] [PubMed] [Google Scholar]
- 65. Whyte MP. Hypophosphatasia − aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016; 12(4):233–46. [DOI] [PubMed] [Google Scholar]
- 66. Whyte MP, Madson KL, Phillips D, et al. Asfotase alfa therapy for children with hypophosphatasia. JCI Insight. 2016; 1(9):e85971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mochizuki H, Saito M, Michigami T, et al. Severe hypercalcaemia and respiratory insufficiency associated with infantile hypophosphatasia caused by two novel mutations of the tissue‐nonspecific alkaline phosphatase gene. Eur J Pediatr. 2000; 159(5):375–9. [DOI] [PubMed] [Google Scholar]
- 68. Whyte MP, Greenberg CR, Salman NJ, et al. Enzyme‐replacement therapy in life‐threatening hypophosphatasia. N Engl J Med. 2012; 366(10):904–13. [DOI] [PubMed] [Google Scholar]
- 69. Linglart A, Biosse‐Duplan M. Hypophosphatasia. Curr Osteoporos Rep. 2016; 14(3):95–105. [DOI] [PubMed] [Google Scholar]
- 70. Fazeli W, Kaczmarek S, Kirschstein M, Santer R. A novel mutation within the lactase gene (LCT): the first report of congenital lactase deficiency diagnosed in Central Europe. BMC Gastroenterol. 2015; 15:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Torniainen S, Freddara R, Routi T, et al. Four novel mutations in the lactase gene (LCT) underlying congenital lactase deficiency (CLD). BMC Gastroenterol. 2009; 9:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Uchida N, Sakamoto O, Irie M, et al. Two novel mutations in the lactase gene in a Japanese infant with congenital lactase deficiency. Tohoku J Exp Med. 2012; 227(1):69–72. [DOI] [PubMed] [Google Scholar]
- 73. Jarvela I, Enattah NS, Kokkonen J, Varilo T, Savilahti E, Peltonen L. Assignment of the locus for congenital lactase deficiency to 2q21, in the vicinity of but separate from the lactase‐phlorizin hydrolase gene. Am J Hum Genet. 1998; 63(4):1078–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Saarela T, Simila S, Koivisto M. Hypercalcemia and nephrocalcinosis in patients with congenital lactase deficiency. J Pediatr. 1995; 127(6):920–3. [DOI] [PubMed] [Google Scholar]
- 75. Belmont JW, Reid B, Taylor W, et al. Congenital sucrase‐isomaltase deficiency presenting with failure to thrive, hypercalcemia, and nephrocalcinosis. BMC Pediatr. 2002; 2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Drummond KN, Michael AF, Ulstrom RA, Good RA. The blue diaper syndrome: familial hypercalcemia with nephrocalcinosis and indicanuria; a new familial disease, with definition of the metabolic abnormality. Am J Med. 1964; 37:928–48. [DOI] [PubMed] [Google Scholar]
- 77. Yamaguchi K, Grant J, Noble‐Jamieson G, Jamieson N, Barnes ND, Compston JE. Hypercalcaemia in primary oxalosis: role of increased bone resorption and effects of treatment with pamidronate. Bone. 1995; 16(1):61–7. [PubMed] [Google Scholar]
- 78. Balasubramanian M, Sprigg A, Johnson DS. IMAGe syndrome: case report with a previously unreported feature and review of published literature. Am J Med Genet A. 2010;152a(12):3138–42. [DOI] [PubMed] [Google Scholar]
- 79.Massagli TL, Reyes MRL. Hypercalcemia and spinal cord injury [Internet]. New York: MedScape; 2015. [updated 2015 Apr 16; cited 2017 Sep 28]. Available from: http://emedicine.medscape.com/article/322109‐overview#showall.
- 80. Rosen JF, Wolin DA, Finberg L. Immobilization hypercalcemia after single limb fractures in children and adolescents. Am J Dis Child. 1978; 132(6):560–4. [DOI] [PubMed] [Google Scholar]
- 81. Pober BR. Williams‐Beuren syndrome. N Engl J Med. 2010; 362(3):239–52. [DOI] [PubMed] [Google Scholar]
- 82. Stromme P, Bjornstad PG, Ramstad K. Prevalence estimation of Williams syndrome. J Child Neurol. 2002; 17(4):269–71. [DOI] [PubMed] [Google Scholar]
- 83. Kim YM, Cho JH, Kang E, et al. Endocrine dysfunctions in children with Williams‐Beuren syndrome. Ann Pediatr Endocrinol Metab. 2016; 21(1):15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Morris CA, Thomas IT, Greenberg F. Williams syndrome: autosomal dominant inheritance. Am J Med Genet. 1993; 47(4):478–81. [DOI] [PubMed] [Google Scholar]
- 85. Palacios‐Verdu MG, Segura‐Puimedon M, Borralleras C, et al. Metabolic abnormalities in Williams‐Beuren syndrome. J Med Genet. 2015; 52(4):248–55. [DOI] [PubMed] [Google Scholar]
- 86. Schipani E, Langman C, Hunzelman J, et al. A novel parathyroid hormone (PTH)/PTH‐related peptide receptor mutation in Jansen's metaphyseal chondrodysplasia. J Clin Endocrinol Metab. 1999; 84(9):3052–7. [DOI] [PubMed] [Google Scholar]
- 87. Parker SE, Mai CT, Canfield MA, et al. Updated National Birth Prevalence estimates for selected birth defects in the United States, 2004‐2006. Birth Defects Res A Clin Mol Teratol. 2010; 88(12):1008–16. [DOI] [PubMed] [Google Scholar]
- 88. Tran HA, Song S, Crock PA, Mattes J, Howard K. The A, B, C, D of hypercalcaemia in Down syndrome. BMJ Case Rep. 2009;2009:bcr06.2008.0232. Epub 2009 Mar 5 [cited 2017 Sep 28]. DOI:10.1136/bcr.06.2008.0232. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3030300/. [DOI] [PMC free article] [PubMed]
- 89. Hair AB, Chetta KE, Bruno AM, Hawthorne KM, Abrams SA. Delayed introduction of parenteral phosphorus is associated with hypercalcemia in extremely preterm infants. J Nutr. 2016; 146(6):1212–6. [DOI] [PubMed] [Google Scholar]
- 90. Hazza IA, Ghandour MS, Almardini RI, Haddad RE, Salaita GM. Hypercalcemia, hypercalciuria and nephrocalcinosis in a breast‐fed term newborn: a rare presentation. Saudi J Kidney Dis Transpl. 2014; 25(4):849–53. [DOI] [PubMed] [Google Scholar]
- 91.Thakker RV. Familial and hereditary forms of primary hyperparathyroidism. In: Bilezikian JP, Marcus R, Levine M, Marcocci C, Silverberg SJ, Potts J, editors . The parathyroids: basic and clinical concepts. 3rd ed. Cambridge, MA: Academic Press; 2015. p. 341–63.
- 92. Yavropoulou MP, Kotsa K, Gotzamani Psarrakou A, et al. Cinacalcet in hyperparathyroidism secondary to X‐linked hypophosphatemic rickets: case report and brief literature review. Hormones (Athens). 2010; 9(3):274–8. [DOI] [PubMed] [Google Scholar]
- 93. Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012; 97(9):2990–3011. [DOI] [PubMed] [Google Scholar]
- 94. Newey PJ, Bowl MR, Cranston T, Thakker RV. Cell division cycle protein 73 homolog (CDC73) mutations in the hyperparathyroidism‐jaw tumor syndrome (HPT‐JT) and parathyroid tumors. Hum Mutat. 2010;31(3):295–307. [DOI] [PubMed] [Google Scholar]
- 95. Kloos RT, Eng C, Evans DB, et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid. 2009; 19(6):565–612. [DOI] [PubMed] [Google Scholar]
- 96. Thakker RV. Multiple endocrine neoplasia type 1. In: De Groot L, Jameson JL, editors . Endocrinology. 6th ed. Philadelphia: Elsevier; 2010. p. 2719–41.
- 97. Costa‐Guda J, Soong CP, Parekh VI, Agarwal SK, Arnold A. Germline and somatic mutations in cyclin‐dependent kinase inhibitor genes CDKN1A, CDKN2B, and CDKN2C in sporadic parathyroid adenomas. Horm Cancer. 2013; 4(5):301–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Thakker RV BF, Juppner H. Genetic disorders of calcium homeostasis caused by abnormal regulation of parathyroid hormone secretion or responsiveness. In: De Groot L, Jameson JL, editors . Endocrinology. 6th ed. Philadelphia: Elsevier; 2010. p. 1136–59.
- 99. Goudet P, Dalac A, Le Bras M, et al. MEN1 disease occurring before 21 years old: a 160‐patient cohort study from the Groupe d'etude des Tumeurs Endocrines. J Clin Endocrinol Metab. 2015; 100(4):1568–77. [DOI] [PubMed] [Google Scholar]
- 100. Giri D, McKay V, Weber A, Blair JC. Multiple endocrine neoplasia syndromes 1 and 2: manifestations and management in childhood and adolescence. Arch Dis Child. 2015; 100(10):994–9. [DOI] [PubMed] [Google Scholar]
- 101. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol. 2014;386(1‐2):2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Thakker RV. Genetics of parathyroid tumours. J Intern Med. 2016; 280(6):574–83. [DOI] [PubMed] [Google Scholar]
- 103. Pichardo‐Lowden AR, Manni A, Saunders BD, Baker MJ. Familial hyperparathyroidism due to a germline mutation of the CDC73 gene: implications for management and age‐appropriate testing of relatives at risk. Endocr Pract. 2011; 17(4):602–9. [DOI] [PubMed] [Google Scholar]
- 104. Iacobone M, Masi G, Barzon L, et al. Hyperparathyroidism‐jaw tumor syndrome: a report of three large kindred. Langenbecks Arch Surg. 2009; 394(5):817–25. [DOI] [PubMed] [Google Scholar]
- 105. Cavaco BM, Guerra L, Bradley KJ, et al. Hyperparathyroidism‐jaw tumor syndrome in Roma families from Portugal is due to a founder mutation of the HRPT2 gene. J Clin Endocrinol Metab. 2004; 89(4):1747–52. [DOI] [PubMed] [Google Scholar]
- 106. Cavaco BM, Barros L, Pannett AA, et al. The hyperparathyroidism‐jaw tumour syndrome in a Portuguese kindred. QJM. 2001; 94(4):213–22. [DOI] [PubMed] [Google Scholar]
- 107. Howell VM, Haven CJ, Kahnoski K, et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet. 2003; 40(9):657–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Jackson MA, Rich TA, Hu MI, Perrier ND, Waguespack SG. CDC73‐related disorders. 2008. Dec 31 [updated 2015 Jan 15; cited 2017 Sep 28]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors . GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2017. Available from: https://www.ncbi.nlm.nih.gov/books/NBK3789/.
- 109. Attie MF, Gill JR, Stock JL, et al. Urinary calcium excretion in familial hypocalciuric hypercalcemia. Persistence of relative hypocalciuria after induction of hypoparathyroidism. J Clin Invest. 1983; 72(2):667–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Hannan FM, Nesbit MA, Zhang C, et al. Identification of 70 calcium‐sensing receptor mutations in hyper‐ and hypo‐calcaemic patients: evidence for clustering of extracellular domain mutations at calcium‐binding sites. Hum Mol Genet. 2012; 21(12):2768–78. [DOI] [PubMed] [Google Scholar]
- 111. Nesbit MA, Hannan FM, Howles SA, et al. Mutations affecting G‐protein subunit alpha11 in hypercalcemia and hypocalcemia. N Engl J Med. 2013; 368(26):2476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Christensen SE, Nissen PH, Vestergaard P, Heickendorff L, Brixen K, Mosekilde L. Discriminative power of three indices of renal calcium excretion for the distinction between familial hypocalciuric hypercalcaemia and primary hyperparathyroidism: a follow‐up study on methods. Clin Endocrinol. 2008; 69(5):713–20. [DOI] [PubMed] [Google Scholar]
- 113. Nesbit MA, Hannan FM, Howles SA, et al. Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nat Genet. 2013; 45(1):93–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Papadopoulou A, Gole E, Melachroinou K, Meristoudis C, Siahanidou T, Papadimitriou A. Identification and functional characterization of a calcium‐sensing receptor mutation in an infant with familial hypocalciuric hypercalcemia. J Clin Res Pediatr Endocrinol. 2016; 8(3):341–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Fujisawa Y, Yamaguchi R, Satake E, et al. Identification of AP2S1 mutation and effects of low calcium formula in an infant with hypercalcemia and hypercalciuria. J Clin Endocrinol Metab. 2013; 98(12):E2022–7. [DOI] [PubMed] [Google Scholar]
- 116. Hannan FM, Howles SA, Rogers A, et al. Adaptor protein‐2 sigma subunit mutations causing familial hypocalciuric hypercalcaemia type 3 (FHH3) demonstrate genotype‐phenotype correlations, codon bias and dominant‐negative effects. Hum Mol Genet. 2015; 24(18):5079–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Pollak MR, Brown EM, Chou YH, et al. Mutations in the human Ca(2+)‐sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell. 1993; 75(7):1297–303. [DOI] [PubMed] [Google Scholar]
- 118. Wilhelm‐Bals A, Parvex P, Magdelaine C, Girardin E. Successful use of bisphosphonate and calcimimetic in neonatal severe primary hyperparathyroidism. Pediatrics. 2012; 129(3):e812–6. [DOI] [PubMed] [Google Scholar]
- 119. Garcia Soblechero E, Ferrer Castillo MT, Jimenez Crespo B, Dominguez Quintero ML, Gonzalez Fuentes C. Neonatal hypercalcemia due to a homozygous mutation in the calcium‐sensing receptor: failure of cinacalcet. Neonatology. 2013; 104(2):104–8. [DOI] [PubMed] [Google Scholar]
- 120. Glaudo MLS, Quinkler M, Ulrich B, Elbelt U, Strasburger CJ, Schnabel D, Lankes E, Scheel S, Feldkamp J, Haag C, Schulze E, Frank‐Raue K, Raue F, Mayr B, Schoff C. Heterozygous inactivating CaSR mutations causing neonatal hyperparathyroidism: function, inheritance and phenotype. Eur J Endocrinol. 2016; 175(5):421–31. [DOI] [PubMed] [Google Scholar]
- 121. Simonds WF, Robbins CM, Agarwal SK, Hendy GN, Carpten JD, Marx SJ. Familial isolated hyperparathyroidism is rarely caused by germline mutation in HRPT2, the gene for the hyperparathyroidism‐jaw tumor syndrome. J Clin Endocrinol Metab. 2004; 89(1):96–102. [DOI] [PubMed] [Google Scholar]
- 122. Simonds WF, James‐Newton LA, Agarwal SK, et al. Familial isolated hyperparathyroidism: clinical and genetic characteristics of 36 kindreds. Medicine. 2002; 81(1):1–26. [DOI] [PubMed] [Google Scholar]
- 123. Guan B, Welch JM, Sapp JC, et al. GCM2‐activating mutations in familial isolated hyperparathyroidism. Am J Hum Genet. 2016; 99(5):1034–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Au AY, McDonald K, Gill A, et al. PTH mutation with primary hyperparathyroidism and undetectable intact PTH. N Engl J Med. 2008; 359(11):1184–6. [DOI] [PubMed] [Google Scholar]
- 125. Hannan FM, Nesbit MA, Christie PT, et al. A homozygous inactivating calcium‐sensing receptor mutation, Pro339Thr, is associated with isolated primary hyperparathyroidism: correlation between location of mutations and severity of hypercalcaemia. Clin Endocrinol. 2010; 73(6):715–22. [DOI] [PubMed] [Google Scholar]
- 126. Starker LF, Akerstrom T, Long WD, et al. Frequent germ‐line mutations of the MEN1, CASR, and HRPT2/CDC73 genes in young patients with clinically non‐familial primary hyperparathyroidism. Horm Cancer. 2012; 3(1‐2):44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Hannan FM, Nesbit MA, Christie PT, et al. Familial isolated primary hyperparathyroidism caused by mutations of the MEN1 gene. Nat Clin Pract Endocrinol Metab. 2008; 4(1):53–8. [DOI] [PubMed] [Google Scholar]
- 128. Pannett AA, Kennedy AM, Turner JJ, et al. Multiple endocrine neoplasia type 1 (MEN1) germline mutations in familial isolated primary hyperparathyroidism. Clin Endocrinol. 2003; 58(5):639–46. [DOI] [PubMed] [Google Scholar]
- 129. Wassif WS, Moniz CF, Friedman E, et al. Familial isolated hyperparathyroidism: a distinct genetic entity with an increased risk of parathyroid cancer. J Clin Endocrinol Metab. 1993; 77(6):1485–9. [DOI] [PubMed] [Google Scholar]
- 130. Wassif WS, Farnebo F, Teh BT, et al. Genetic studies of a family with hereditary hyperparathyroidism‐jaw tumour syndrome. Clin Endocrinol. 1999; 50(2):191–6. [DOI] [PubMed] [Google Scholar]
- 131. Weinstein LS, Simonds WF. HRPT2, a marker of parathyroid cancer. N Engl J Med. 2003; 349(18):1691–2. [DOI] [PubMed] [Google Scholar]
- 132. Williamson C, Cavaco BM, Jauch A, et al. Mapping the gene causing hereditary primary hyperparathyroidism in a Portuguese kindred to chromosome 1q22‐q31. J Bone Miner Res. 1999; 14(2):230–9. [DOI] [PubMed] [Google Scholar]
- 133. Costa‐Guda J, Marinoni I, Molatore S, Pellegata NS, Arnold A. Somatic mutation and germline sequence abnormalities in CDKN1B, encoding p27Kip1, in sporadic parathyroid adenomas. J Clin Endocrinol Metab. 2011; 96(4):E701–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Newey PJ, Jeyabalan J, Walls GV, et al. Asymptomatic children with multiple endocrine neoplasia type 1 mutations may harbor nonfunctioning pancreatic neuroendocrine tumors. J Clin Endocrinol Metab. 2009; 94(10):3640–6. [DOI] [PubMed] [Google Scholar]
- 135. Moulis G, Batz A, Durrieu G, Viard C, Decramer S, Montastruc J‐L. Severe neonatal hypercalcemia related to maternal exposure to nutritional supplement containing Spirulina. Eur J Clin Pharmacol. 2012; 68(2):221–2. [DOI] [PubMed] [Google Scholar]
- 136. Huseyin Altunhan AA, Murat Konak, Rahmi Ors, Mervan Bekdas. A severe neonatal hypercalcemia case due to maternal high dose vitamin D usage. Acta Medica Anatolia. 2013; 1(1):33–4. [Google Scholar]
- 137. Rambaud J, Guellec I, Guilbert J, Leger PL, Renolleau S. Calcium homeostasis disorder during and after neonatal extracorporeal membrane oxygenation. Indian J Crit Care Med. 2015; 19(9):513–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Hak EB, Crill CM, Bugnitz MC, Mouser JF, Chesney RW. Increased parathyroid hormone and decreased calcitriol during neonatal extracorporeal membrane oxygenation. Intensive Care Med. 2005; 31(2):264–70. [DOI] [PubMed] [Google Scholar]
- 139. Fridriksson JH, Helmrath MA, Wessel JJ, Warner BW. Hypercalcemia associated with extracorporeal life support in neonates. J Pediatr Surg. 2001; 36(3):493–7. [DOI] [PubMed] [Google Scholar]
- 140. Davies JH, Shaw NJ. Investigation and management of hypercalcaemia in children. Arch Dis Child. 2012; 97(6):533–8. [DOI] [PubMed] [Google Scholar]
- 141. Perazella MA, Markowitz GS. Bisphosphonate nephrotoxicity. Kidney Int. 2008; 74(11):1385–93. [DOI] [PubMed] [Google Scholar]
- 142. Gannon AW, Monk HM, Levine MA. Cinacalcet monotherapy in neonatal severe hyperparathyroidism: a case study and review. J Clin Endocrinol Metab. 2014; 99(1):7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Atay Z, Bereket A, Haliloglu B, et al. Novel homozygous inactivating mutation of the calcium‐sensing receptor gene (CASR) in neonatal severe hyperparathyroidism‐lack of effect of cinacalcet. Bone. 2014; 64:102–7. [DOI] [PubMed] [Google Scholar]
- 144. Alagaratnam S, Kurzawinski TR. Aetiology, diagnosis and surgical treatment of primary hyperparathyroidism in children: new trends. Horm Res Paediatr. 2015; 83(6):365–75. DOI:10.1159/000381622. [DOI] [PubMed] [Google Scholar]