

Abstract
Purpose
The goal of this study was to determine whether single-nucleotide polymorphisms (SNPs) in genes involved in gemcitabine metabolism, DNA damage repair, multidrug resistance (MDR) and alkylator detoxification influence the clinical outcome of patients with refractory/relapsed lymphoid malignancies receiving high-dose gemcitabine/busulfan/melphalan (Gem/Bu/Mel) with autologous stem-cell support.
Experimental Design
We evaluated 21 germline SNPs of the gemcitabine metabolism genes CDA, dCK, and hCNT3, DNA damage repair genes RECQL, XRCC1, RAD54L, ATM, ATR, MLH1, MSH2, MSH3, TREX1, EXO1, and TP73, and multidrug resistance genes MRP2 and MRP5, as well as glutathione-S-transferase GSTP1 in 153 patients with relapsed or refractory lymphoma or myeloma receiving Gem/Bu/Mel. We studied the association of genotypes with overall survival (OS), progression-free survival (PFS) and nonhematological grade 3–4 toxicity.
Results
CDA C111T and TREX1 Ex14-460C>T genotypes had a significant effect on OS (P=0.007 and P=0.005, respectively), and CDA C111T, ATR C340T and EXO1 P757L genotypes were significant predictors for severe toxicity (P=0.037, P=0.024 and P=0.025, respectively) in multivariable models that adjusted for clinical variables. The multi-SNP risk score analysis identified the combined genotypes of TREX1 Ex14-460 TT and hCNT3 Ex5 +25A>G AA as a significant predictor for OS and the combination of MRP2 Ex10 +40 GG/GA and MLH1 IVS12-169 TT as significant predictor for PFS.
Conclusions
Polymorphic variants of certain genes involved in gemcitabine metabolism and DNA damage repair pathways may be potential biomarkers for clinical outcome in patients with refractory/relapsed lymphoid tumors receiving Gem/Bu/Mel.
Keywords: Single nucleotide polymorphisms, gemcitabine metabolism, DNA damage repair, drug resistance gene, glutathione-S-transferases
INTRODUCTION
Gemcitabine is a pyrimidine nucleoside analogue with broad antitumor activity and wide clinical use. This prodrug requires cellular uptake and intracellular phosphorylation before incorporation into DNA, which is believed to be its mechanism of cytotoxicity.1, 2 The profile of gemcitabine, with dose-dependent cytotoxicity and few nonhematological side effects, has prompted its study at high doses with autologous stem-cell support.3 Since gemcitabine inhibits DNA damage repair,4 combinations of this agent with alkylators should have synergistic or additive antitumor activity and a favorable therapeutic index.
We have developed a new high-dose combination of infusional gemcitabine with busulfan and melphalan (Gem/Bu/Mel) for lymphoid tumors, with promising results in relapsed Hodgkin’s and diffuse large B-cell lymphoma (DLBCL).5,6 The extramedullary toxicity profile of Gem/Bu/Mel includes mucositis, skin rash and transaminase elevation. Since these nonhematological side effects can be severe, it would be helpful to predict their occurrence for a given patient. Unfortunately, no patient or clinical features have been associated with toxicity.5
The cellular pharmacodynamic effect of gemcitabine depends on multiple enzymes, such as those involved in its intracellular metabolism, DNA damage repair and multidrug resistance mechanisms, whose activity may depend on their genetic polymorphic variants. We have previously identified single nucleotide polymorphisms (SNPs) of key enzymes in these pathways with a major impact on clinical outcome or toxicity on patients with pancreatic cancer undergoing gemcitabine-based chemoradiation7,8,9,10,11,12,13,14 In contrast, the pharmacogenomics of high-dose gemcitabine have not been adequately studied. Since the effect of gemcitabine on normal and tumor cells is greater at higher doses, it is conceivable that the impact of polymorphic genetic variation of relevant enzymes may be greater in the transplant setting.
The electrophilic alkylators busulfan and melphalan are detoxified inside the cell by reduced glutathione (GSH). GSH conjugation of alkylating agents is mediated by glutathione S-transferase (GST), whose activity also depends on polymorphic variations.15,16,17 GST pi 1 (GSTP1) is the most abundant GST class found in many normal cell and malignant tissues.18,19 The GSTP1 Ile105Val polymorphism has been associated with improved outcomes in patients with myeloma receiving high-dose melphalan.20
We hypothesized that polymorphic variations of genes involved in gemcitabine metabolism, DNA damage repair, multidrug resistance and glutathione detoxification correlate with the toxicity and outcome of patients with relapsed/refractory lymphoid tumors receiving Gem/Bu/Mel.
Materials and Methods
Patient recruitment and data collection
This prospective study involved patients with relapsed/refractory lymphoid malignancies, including Hodgkin’s lymphoma or DLBCL and myeloma, with refractory or poor-risk features that made them eligible for clinical trials of Gem/Bu/Mel with autologous stem-cell transplantation at our institution.5,6 This laboratory study was approved by the Institutional Review Board and all patients provided informed consent prior to enrollment. All patients received the same treatment doses and schema of Gem/Bu/Mel, as previously described.5 Overall survival (OS) and progression-free survival (PFS) were calculated from the date of diagnosis to date of death and progression/death, respectively. Living patients and patients without progression at the last follow up time were censored. Nonhematological toxicities, including mucositis, skin rash and transaminase elevation, were graded according to the Common Terminology Criteria for Adverse Events (CTCAE 3.0).21
DNA extraction and genotyping
We selected 21 SNPs of the deoxycytidine deaminase (CDA), deoxycytidine kinase (dCK), human concentrative nucleotide transporter (hCNT3), RECQL, X-ray repair complementing (XRCC)1, RAD54L, ATM, ATM and Rad3-related (ATR), mutL homolog (MLH)1, mutS homolog (MSH)2, MSH3, three prime repair exonuclease (TREX)1, exonuclease I (EXO1), tumor protein (TP)73, multidrug resistance-associated protein (MRP)2, MRP5, and GSTP1 genes according to the following criteria: 1) Minor allele frequency of the SNP >15% among Caucasians, 2) coding SNPs including nonsynonymous or synonymous SNPs, and 3) association with cancer risk or clinical outcome in previous studies. The genes, chromosome locations, nucleotide substitutions, function (such as encoding amino acid changes), reference SNP identification numbers, and minor allele frequencies of the 21 SNPs evaluated in this study are summarized in Table 1.
Table 1.
Single nucleotide polymorphisms (SNPs) evaluated
| Gene | Chromosome | SNP | RS No. | Minor Allele Frequency Observeda | Minor Allele Frequency Reportedb |
|---|---|---|---|---|---|
| CDA | 1p36.12b | Ex4 +111C>T, T145T | 1048977 | 0.31 | 0.28 |
| Ex2 − 76A>C, K27Q | 2072671 | 0.30 | 0.44 | ||
| dCK | 4q13.3b | IVS6 − 1205C>T | 4694362 | 0.41 | 0.45 |
| IVS2 +9846A>G | 12648166 | 0.43 | 0.43 | ||
| hCNT3 | 9q21.32c | Ex14 − 69C>T, L461L | 7853758 | 0.18 | 0.15 |
| Ex5 +25A>G, T89T | 7867504 | 0.44 | 0.39 | ||
| RECQL | 12p12 | Ex15 +159A>C | 13035 | 0.40 | 0. 28 |
| XRCC1 | 19q13.2 | Ex6 − 22C>T, R194W | 1799782 | 0.09 | 0.15 |
| RAD54L | 1p32 | Ex18 +157C>T, A730A | 1048771 | 0.10 | 0.15 |
| ATM | 11q22 - q23 | IVS22 − 77 C>T | 664677 | 0.43 | 0.29 |
| Ex38+61A>G, D1853N | 1801516 | 0.07 | 0.16 | ||
| ATR | 3q22 - q24 | Ex4 +340C>T, T211M | 2227928 | 0.47 | 0.38 |
| MLH1 | 3q21.3 | IVS12 − 169C>T | 2286940 | 0.37 | 0.41 |
| MSH2 | 2p22 - p21 | IVS12 − 6T>C | 2303428 | 0.10 | 0.15 |
| MSH3 | 5q11 - q12 | Ex4 − 100G>A, P231P | 1805355 | 0.14 | 0.15 |
| TREX1 | 3p21 | Ex14 − 460C>T | 11797 | 0.38 | 0.43 |
| EXO1 | 1q42 - q43 | Ex15 +59C>T, P757L | 9350 | 0.20 | 0.18 |
| TP73 | 1p36.3 | Ex2 +4G>A | 2273953 | 0.23 | 0. 26 |
| MRP2 | 10q24.2c | Ex10 +40G>A, V417I | 2273697 | 0.19 | 0.25 |
| MRP5 | 3q27.1b | Ex10 − 2A>G, Q382Q | 7636910 | 0.36 | 0.35 |
| GSTP1 | 11q13 | Ex5 − 24A>G, I105V | 1695 | 0.35 | 0.46 |
Abbreviation: SNP, single-nucleotide polymorphism; RS No., reference SNP identification number.
The data observed in current study.
The reported minor allele frequency was from SNP500 cancer database.
DNA was extracted from peripheral-blood lymphocytes in a single 10-cc blood sample of patients using Qiagen DNA isolation kits (Valencia, CA). Genotyping was performed using the Taqman 5′ nuclease assay. Primers and TaqMan MGB probes were provided by TaqMan SNP Genotyping Assay Services (Applied Biosystems, Foster City, Calif). The probes were labeled with the fluorescent dye VIC or FAM for each allele at the 5′ end. Polymerase chain reaction (PCR) was performed in a 5-μL total volume consisting of TaqMan Universal PCR Master Mix, 20 ng of genomic DNA (diluted with dH2O), and TaqMan SNP Genotyping Assay Mix. Allele discrimination was accomplished by running endpoint detection using the ABI Prism 7900HT Sequence Detection System and SDS 2.3 software (Applied Biosystems). Twenty percent of the samples were analyzed in duplicate, with 100% concordance in genotype calling.
Statistical analysis
The distribution of genotypes was tested for Hardy-Weinberg equilibrium with the goodness-of-fit χ2 test. The association of clinical factors and genotypes with OS and PFS was evaluated using log-rank test and Kaplan-Meier methods. Hazard rations (HRs) and 95% confidence intervals (CIs) were estimated using univariable or multivariate Cox proportional hazard models. The association of genotypes and severe toxicity was estimated with odds ratios (OR) using univariable or multivariate logistic regression. Multivariate analyses of OS adjusted for age, number of prior chemotherapy lines, progression, and severe toxicity in this study. The effect of genotype on severe toxicity was adjusted for age, number of prior chemotherapy lines, and progression.
We estimated the false-positive report probability (FPRP) for the observed statistically significant associations using the Wacholder method.22 FPRP is the probability of no true association between a genetic variant and a phenotype given a statistically significant finding. It depends on the observed P value, on the prior probability that the association between the genetic variant and the phenotype is real, as well as on the statistical power of the test. In the current study, we set the HR and OR values of 2.0–4.0 as a likely threshold value. The prior probability used was 0.25 for all SNPs. The FPRP value for noteworthiness was set at 0.2.
To better assess combined genotype effects, we also conducted the risk score analysis as previously described.23,24 Briefly, we selected SNPs with P ≤0.15 in likelihood ratio test derived from Cox or logistic regression models. We generated a risk score for these SNPs with deleterious genotype(s) as “1” and the reference genotype(s) as “0”. We explored all possible combinations of multiple SNPs to find the best-fitting models with consideration of the model likelihood and C statistic (Cox regression) or Area Under the Curve (AUC) (logistic regression).25 In parallel, each selected SNP went through stepwise selection (α = 0.05) in 1,000 bootstraps. The SNPs surviving >50% bootstraps were most likely to be selected to the best-fitting model. A final multi-SNP risk score was developed based on the best-fitting SNPs through summation of risk scores. We compared the performance of the clinical factor based model, the SNP only based model and the clinical factor and SNP combined model using C-statistics or AUC. All statistical testing used SPSS Statistics, v17.0 (SPSS Inc., Chicago, IL) and R v.3.1.0 software packages. Statistical significance was defined as P<0.05.
RESULTS
Patient Characteristics and genotype frequencies
This prospective study included 153 patients with relapsed/refractory Hodgkin’s (N=54), DLBCL (N=64) and myeloma (N=35), enrolled in clinical trials of Gem/Bu/Mel from 08/2006 to 04/2011 and followed through 04/2014. Their median age was 47 years (range, 18–66 years). Their median number of pretransplant lines of treatment was 2 (range, 1–10). Their racial distribution was white (N=137), black (N=11), and Asian (N=5). Median follow-up time was 22 months (range, 1–42). The number of prior chemotherapy lines was significantly associated with PFS (P=0.006) and OS (P=0.002).
The 21 genotypes of interest were successfully amplified in 100% of the samples. No homozygous TT genotype was detected for RAD54L C157T SNP. The observed allele frequencies in this study population were comparable to previously reported allele frequencies in the general population (Table 1). Genotype frequencies of the 21 SNPs were found to be in Hardy-Weinberg equilibrium (χ2=0.006–3.304; P=0.069–0.936) except ATM A61G (χ2=15.118; P=0.0001) and MSH3 P231P (χ2=7.903; P=0.005).
Associations of genotypes with overall and progression-free survival
In the univariate analyses, 3 of the 21 SNPs evaluated (hCNT3 A25G, TREX1 Ex14-460C>T and MRP2 G40A) showed significant associations (P<0.05) with OS, and 7 SNPs (CDA C111T, dCK A9846G, hCNT3 C-69T, ATM C77T, ATM A61G, ATR C340T and GSTP1 Ex5-24A>G) showed nonsignificant associations with OS (Table 2). The association of CDA C111T and TREX1 Ex14-460C>T genotypes with OS remained statistically significant after adjusting for age, number of prior chemo lines, progression and severe toxicity (P=0.007 and P=0.005, respectively). The FPRP was 0.086 for CDA and 0.026 for TREX1, respectively, given a prior probability of 25%. Both are below the threshold of 0.20 indicating noteworthiness.
Table 2.
Characteristics of the study
| Variable | No. of Patients | No. of Deaths (%) |
P (log-rank) |
No. of Progression (%) |
P (log-rank) |
|---|---|---|---|---|---|
| Age, years | 0.013 | 0.846 | |||
| ≦50 | 87 | 19 (21.8) | 44 (50.6) | ||
| 51–60 | 37 | 5 (13.5) | 22 (59.5) | ||
| 61–70 | 29 | 12 (41.4) | 15 (51.7) | ||
| Diagnosis | 0.102 | 0.744 | |||
| Hodgkin’s lymphoma | 54 | 7 (13.0) | 27 (50.0) | ||
| Diffuse large B-cell lymphoma | 64 | 18 (28.1) | 32 (50.0) | ||
| Myeloma | 35 | 11 (31.4) | 22 (62.9) | ||
| Progression | <0.001 | – | |||
| Yes (Progression) | 81 | 36 (44.4) | 81 (100) | ||
| No (No Progression) | 72 | 0 (0) | 72 (100) | ||
| Number of prior chemo lines | 0.002 | 0.006 | |||
| 1 | 5 | 0 (0) | 2 (40.0) | ||
| 2 | 73 | 12 (16.4) | 31 (42.5) | ||
| 3 | 41 | 10 (24.4) | 24 (58.5) | ||
| 4 | 17 | 5 (29.4) | 10 (58.8) | ||
| 5 | 8 | 4 (50.0) | 7 (87.5) | ||
| 6 | 4 | 2 (50.0) | 3 (75.0) | ||
| 7 | 4 | 2 (50.0) | 3 (75.0) | ||
| 10 | 1 | 1 (100) | 1 (100) | ||
| Grade 3–4 toxicity | 0.591 | 0.633 | |||
| Yes | 47 | 12 (25.5) | 58 (54.7) | ||
| No | 106 | 24 (22.6) | 23 (48.9) |
To find the best multi-SNP predictor for OS and PFS, we conducted a risk score analysis. We first compared clinical factor-based, SNP-based and clinical factor and SNP combined model to find the best-fitting model (Table S1). SNP alone model had the highest predicting power compared to the other models. (Table S2). We found that the best multi-SNP risk score for OS is the TREX1 Ex14-460 TT genotype which had C-statistics as high as 0.91 in SNP alone model and 0.84 in combined model (Table S2). Patients carrying at-risk genotypes had HR=2.77 (95% CI, 1.26–6.11) (P=0.011) (Figure 1A). Furthermore, the best SNP combination of MRP2 Ex10 +40 GG/GA and MLH1 IVS12-169 TT remained a significant predictor of PFS after adjusting age, number of chemotherapy lines, and severe toxicity, with HR=2.06 (95% CI, 1.35–3.16) for patients with one or more variant alleles compared with those with no variant allele (P<0.001) (Figure 1B).
Figure 1.
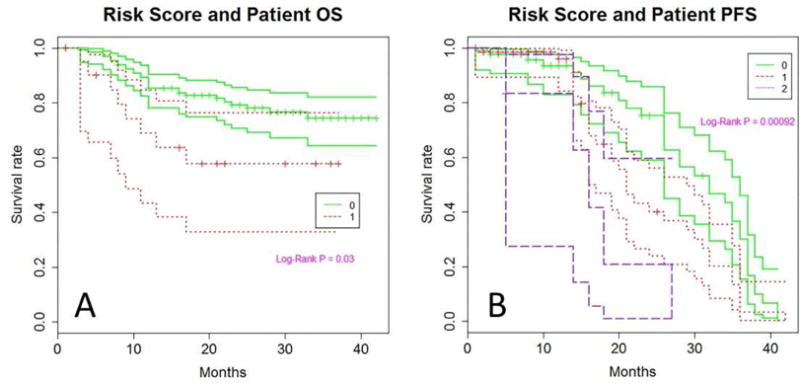
Kaplan Meier plot of overall survival (panel A) and progression free survival (panel B) by risk score in the entire study population. The number 0 or 1 in panel A indicates the absence or presence of the at-risk TT genotype of TREX1 Ex14-460 (rs11797), respectively. The number 0–2 in panel B indicates the number of at-risk genotypes of MRP2 Ex10 +40 GG/GA and MLH1 IVS12-169 TT genotypes. The three lines for each genotype represent the survival curve and their 95% confidence intervals.
Subgroup analysis by disease
Risk score analyses in patients with HL and DLBCL showed a significant association of those 2-SNP genotypes with PFS within those patient subgroups [HR=2.08 (95% CI, 1.26–3.46), P=0.005], as well as a nonsignificant association with OS [HR=1.63 (95% CI, 0.88–3.03), P=0.1]. A borderline significant association of the risk score with PFS was also observed in patients with myeloma [HR=4.4 (95% CI, 0.92–21.15), P =0.064].
Association of genotypes with toxicity
The ATR C340T and EXO1 P757L genotypes showed significant associations with grade 3–4 nonhematologic toxicity, whereas CDA C111T, MRP2 G40A, and GSTP1 Ex5-24A>G had non-significant associations with severe toxicity (Table 4). The CDA C111T, ATR C340T and EXO1 P757L genotypes remained as significant predictors after adjusting for age, number of prior chemo lines, and progression (P=0.037, P=0.024 and P=0.025, respectively). The FPRP was 0.207 for CDA, 0.133 for ATR, and 0.137 for EXO1.
Table 4.
Extramedullary toxicity and genotype
| Genotype | Grade 3–4 Toxicity, n (%)
|
P (χ2) | Univariate
|
Multivariate
|
||||
|---|---|---|---|---|---|---|---|---|
| No | Yes | OR (95% CI) | P | OR (95% CI)* | P | |||
| Gemcitabine metabolic gene | ||||||||
| CDA C111T (T145T) | 0.139 | |||||||
| CC | 45 (61.6) | 28 (38.4) | 1.0 | 1.0 | ||||
| CT | 49 (75.4) | 16 (24.5) | 0.53 (0.25–1.10) | 0.086 | 0.49 (0.23–1.06) | 0.070 | ||
| TT | 12 (80.0) | 3 (20.0) | 0.40 (0.10–1.55) | 0.186 | 0.34 (0.80–1.40) | 0.134 | ||
| CC vs CT/TT | 0.50 (0.25–1.01) | 0.052 | 0.46 (0.22–0.95) | 0.037 (0.043) | ||||
| CDA A-76C (K27Q) | 0.559 | |||||||
| AA | 51 (65.4) | 27 (34.6) | 1.0 | 1.0 | ||||
| AC | 43 (72.9) | 16 (27.1) | 0.70 (0.34–1.47) | 0.350 | 0.60 (0.28–1.31) | 0.207 | ||
| CC | 12 (75.0) | 4 (25.0) | 0.63 (0.19–2.14) | 0.459 | 0.56 (0.16–2.01) | 0.380 | ||
| AA vs AC/CC | 0.69 (0.34–1.37) | 0.288 | 0.60 (0.29–1.24) | 0.169 | ||||
| hCWT3 A25G (T89T) | 0.528 | |||||||
| TT | 38 (73.1) | 14 (26.9) | 1.0 | 1.0 | ||||
| CT | 47 (70.1) | 20 (29.9) | 1.16 (0.52–2.59) | 0.726 | 1.03 (0.45–2.40) | 0.897 | ||
| CC | 21 (61.8) | 13 (38.2) | 1.68 (0.67–4.23) | 0.271 | 2.04 (0.68–5.47) | 0.155 | ||
| TT/CT vs CC | 1.55 (0.70–2.44) | 0.283 | 2.01 (0.84–4.82) | 0.119 | ||||
|
| ||||||||
| DNA repair gene | ||||||||
| RECQL A159C | 0.398 | |||||||
| AA | 35 (66.0) | 18 (34.0) | 1.0 | 1.0 | ||||
| AC | 57 (74.0) | 20 (26.0) | 0.33 (0.32–1.46) | 0.326 | 0.66 (0.30–1.45) | 0.296 | ||
| CC | 14 (60.9) | 9 (39.1) | 1.25 (0.45–3.44) | 0.666 | 1.24 (0.41–3.69) | 0.706 | ||
| AA/CC vs AC | 0.64 (0.32–1.27) | 0.202 | 0.61 (0.30–1.26) | 0.186 | ||||
| XRCC1194 | 0.306 | |||||||
| CC | 90 (69.8) | 39 (30.2) | 1.0 | 1.0 | ||||
| CT | 15 (71.4) | 6 (28.6) | 0.92 (0.33–2.56) | 0.878 | 0.97 (0.34–2.83) | 0.960 | ||
| TT | 1 (33.3) | 2 (66.7) | 4.62 (0.41–52.4) | 0.217 | 6.24 (0.42–92.6) | 0.183 | ||
| CC/CT vs TT | 4.67 (0.41–52.8) | 0.213 | 6.27 (0.42–92.5) | 0.182 | ||||
| ATR C340T | 0.087 | |||||||
| CC | 27 (60.0) | 18 (40.0) | 1.0 | |||||
| CT | 50 (68.5) | 23 (31.5) | 0.69 (0.32–1.50) | 0.348 | 0.60 (0.27–1.35) | 0.219 | ||
| TT | 29 (82.9) | 6(17.1) | 0.31 (0.11–0.90) | 0.031 | 0.28 (0.10–0.84) | 0.024 | ||
| CC/CT vs TT | 0.39 (0.15–1.01) | 0.053 | 0.39 (0.15–1.03) | 0.057 | ||||
| EXO1 P757L | 0.032 | |||||||
| CC | 63 (62.4) | 38 (37.6) | 1.0 | 1.0 | ||||
| CT | 35 (81.4) | 8(18.6) | 0.38 (0.16–0.90) | 0.028 | 0.38 (0.16–0.92) | 0.032 | ||
| TT | 8 (88.9) | 1 (11.1) | 0.21 (0.03–1.72) | 0.145 | 0.24 (0.03–2.10) | 0.195 | ||
| CC vs CT/TT | 0.35 (0.15–0.79) | 0.012 | 0.38 (0.16–0.88) | 0.0258) | ||||
| TP73 Ex2+4G>A | 0.342 | |||||||
| GG | 65 (69.9) | 28 (30.1) | 1.0 | 1.0 | ||||
| GA | 33 (64.7) | 18 (35.3) | 1.27 (0.61–2.62) | 0.524 | 1.32 (0.63–2.77) | 0.467 | ||
| AA | 8 (88.9) | 1 (21.1) | 0.29 (0.04–2.43) | 0.254 | 0.27 (0.03–2.16) | 0.206 | ||
| GG/GA vs AA | 0.27 (0.03–2.19) | 0.219 | 0.22 (0.03–1.94) | 0.174 | ||||
|
| ||||||||
| Drug resistance gene | ||||||||
| MRP2 G40A | 0.083 | |||||||
| GG | 73 (73.7) | 26 (26.3) | 1.0 | 1.0 | ||||
| AG | 30 (61.2) | 19 (38.8) | 1.78 (0.86–3.68) | 0.121 | 1.72 (0.81–3.63) | 0.158 | ||
| AA | 3 (60.0) | 2 (40.0) | 1.87 (0.30–11.8) | 0.505 | 2.06 (0.28–15.3) | 0.479 | ||
| GG vs AG/AA | 1.79 (0.88–3.62) | 0.108 | 1.74 (0.84–3.63) | 0.139 | ||||
| GSTP1 Ex5-24A>G | 0.321 | |||||||
| AA | 46 (74.2) | 16 (25.8) | 1.0 | 1.0 | ||||
| AG | 47 (63.5) | 27 (36.5) | 1.65 (0.79–3.46) | 0.184 | 1.73 (0.80–3.73) | 0.165 | ||
| GG | 13 (76.5) | 4 (23.5) | 0.89 (0.25–3.11) | 0.848 | 0.67 (0.17–2.56) | 0.554 | ||
| AA/GG vs AG | 1.70 (0.85–3.39) | 0.136 | 1.87 (0.90–3.88) | 0.092 | ||||
Multi-SNP risk score analysis showed the clinical factor and SNP combined model moderately increased the power compared to clinical factor or SNP alone model (Table S2). The combined genotypes of EXO1 rs9350 (Ex15 +59C>T, P757L) and CDA rs1048977 (Ex4 +111C>T, T145T) had a HR of 2.47 (95% CI: 1.40–4.35, P = 0.0018) (Table 5).
Table 5.
Risk estimates and 95% confidence intervals for the best multi-SNP score models
| Outcome | Regression | Model type | SNPs for Score | HR/OR (95% CI) | P |
|---|---|---|---|---|---|
| OS | Cox | 1-SNP | TREX1 rs11797 | 2.77 (1.26–6.11)a | 0.011 |
| PFS | Cox | 2-SNP | MRP2 rs2273697, MLH1 rs2286940 | 2.06 (1.35–3.16)b | 0.0009 |
| Toxicity | Logistic | 2-SNP | EXO1 rs9350, CDA rs1048977 | 2.47 (1.4–4.35)c | 0.0018 |
OS (overall survival) adjusted for progression free survival.
PFS (progression free survival) adjusted for age and number of chemotherapy lines.
Toxicity adjusted for PFS and number of chemotherapy lines.
DISCUSSION
In this study, we evaluated the effect of polymorphic variants of genes involved in gemcitabine metabolism, DNA damage repair, drug resistance and glutathione detoxification on patient outcomes. The study was conducted in patients enrolled in clinical trials of our new HDC regimen Gem/Bu/Mel. Out of the 21 SNPs evaluated, CDA C111T and TREX1 Ex14-460C>T genotypes were independently associated with OS. The CDA C111T, ATR C340T and EXO1 P757L genotypes were independent predictors for severe toxicity. Furthermore, risk score analysis showed that the TREX1 Ex14-460 TT genotype and the combined genotypes of MRP2 Ex10 +40 GG/GA and MLH1 IVS12-169 TT were significant predictors for OS and PFS, respectively. These findings suggest that genetic variations in drug metabolism and DNA damage repair have value as prognostic biomarkers.
The selection of SNPs in this study was based on our previous observations in patients with pancreatic cancer receiving gemcitabine-based chemoradiation.1–8 CDA, an enzyme involved in the salvage pathway of pyrimidine, is the major gemcitabine inactivation enzyme. Three potentially functional SNPs of the CDA gene, i.e. C111T (T145T), A-76C (K27Q), and G208A (A70T) have been clinically investigated in patients receiving gemcitabine.11,13, 26,27 The CDA C111T was significantly associated with severe toxicity in pancreatic cancer patients,5 as well as fewer tumor responses and worse outcomes in advanced non-small cell lung cancer.28, 29 Although CDA exon 4 C111T is a synonymous SNP that does not result in amino acid change, bioinformatics analysis predicted possible changes in splicing regulation and transcriptional regulation.30 It is possible that the variant allele confers a reduced enzyme activity, making the variant allele carriers more susceptible to drug toxicity. Further functional studies are required to elucidate the mechanisms underlying the observed associations.
ATR, TREX1 and EXO1 are all DNA damage response genes that were significantly associated with either outcome or toxicity in the current study. Gemcitabine incorporation causes DNA replication arrest, and ATR/Chk1 signaling pathway plays a crucial role in the cellular response to the stalled DNA replication fork.31 Cells lacking ATR or Chek1 genes have been shown to be more sensitive to gemcitabine. The ATR gene encodes a protein kinase that is critically important in maintaining the integrity of the replication apparatus following damage that arrests the progression of the complex.32 ATR C340T (rs2227928) is a nonsynonymous SNP, and the replacement of threonine to methionine could have an impact on transcriptional regulation and post-translation consequence as predicted by bioinformatic models.33 A lower level of expression or activity of ATR could explain the increased toxicity in patients with the variant allele observed in the current study. TREX1 is a major 3 prime exonuclease in mammalian cells. Loss of TREX1 leads to reduce the phosphorylation of the Chk1 gene in cells exposed to hydroxyurea,34 which suggests a compromised ATR signaling pathway function. The TREX1 SNP (rs17971) investigated in the current study is an expression quantitative trait locus (eQTL).34 As in or previous study, we saw a significant association of TREX1 Ex14-460C>T genotype with outcome. Thus, TREX1 is a critical determinant of efficacy of gemcitabine-induced DNA damage. EXO1 is a 5′′3′ exonuclease involved in the DNA mismatch repair and other DNA metabolic pathways affecting genomic stability, including homologous recombination and DNA damage repair.35,36,37 EXO1 stability is dependent on ATR signaling.38 The current study found a significant association of EXO1 P757L genotype with drug toxicity. The EXO1 P757L is a nonsynonymous SNP that result in replacement of amino acids, possibly affecting the protein functions.
In addition to the individual SNP effects, we have observed significant associations of the combined at-risk alleles of the TREX1, hCNT3 (involved in gemcitabine intracellular uptake), MRP2 (involved in exporting bilirubin and glucuronides of certain anticancer drugs) and MLH1 (DNA mismatch repair enzyme) genes with outcomes and toxicity. Although many of the at-risk alleles showed non-significant mild effect individually, the combined genotype had a strong effect on the clinical outcome, even within the disease subgroups. These observations support the concept that genes act in concert, and that the combined action of many genes exerts a greater influence on phenotype than individual SNPs. For future clinical applications, a battery of several genes/SNPs involved in the same pathway may have a better predicting power than relying on single gene/SNP.
Limitations to the present study include its moderate sample size and the heterogeneity of diagnoses. While the effect of the relevant SNPs was similar across patient diagnoses, our findings should be confirmed in disease-specific studies. Although our sample size is moderate and some observations might have occurred by chance, the consistency with previously reported associations, the functional basis of the observed associations, and the good performance of the risk scores argue for their potential importance.
In conclusion, we observed an important effect of polymorphic variants of genes involved in gemcitabine metabolism, DNA repair and multidrug resistance in a population of patients with lymphoid tumors receiving homogeneous HDC with Gem/Bu/Mel. The ultimate goal of this research is to identify genetic profiles that can be used in the clinic as predictors for therapy response or prognosis. If these findings are replicated in additional patient populations, such information may be helpful in stratifying patients for a more individualized high-dose cancer therapy.
Supplementary Material
Table 3.
Association of genotypes with overall survival
| Genotype | No. of Cases | No. of Deaths |
P (LR) |
Univariate
|
Multivariate
|
||
|---|---|---|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI)* | P | ||||
| Gemcitabine metabolic gene | |||||||
| CDA C111T (T145T) | 0.210 | ||||||
| CC | 73 | 15 | 1.0 | 1.0 | |||
| CT | 65 | 19 | 1.61 (0.82–3.17) | 0.168 | 2.35 (1.11–4.97) | 0.025 | |
| TT | 15 | 2 | 0.61 (0.14–2.69) | 0.517 | 0.51 (0.11–2.31) | 0.381 | |
| CC/TT vs CT | 1.73 (0.90–3.33) | 0.101 | 2.67 (1.31–5.43) | 0.007 | |||
| dCK A9846G | 0.313 | ||||||
| GG | 51 | 13 | 1.0 | 1.0 | |||
| AG | 72 | 13 | 0.72 (0.33–1.55) | 0.396 | 1.21 (0.53–2.77) | 0.653 | |
| AA | 30 | 10 | 1.34 (0.59–3.06) | 0.487 | 1.38 (0.58–3.25) | 0.464 | |
| GG/AA vs AG | 0.64 (0.32–1.26) | 0.195 | 1.05 (0.51–2.18) | 0.889 | |||
| hCNT3 C-69T (L461L) | 0.332 | ||||||
| CC | 105 | 28 | 1.0 | 1.0 | |||
| CT | 42 | 7 | 0.58 (0.25–1.32) | 0.190 | 0.63 (0.27–1.48) | 0.288 | |
| TT | 6 | 1 | 0.47 (0.06–3.48) | 0.461 | 0.27 (0.04–2.03) | 0.202 | |
| CC vs CT/TT | 0.56 (0.23–1.23) | 0.147 | 0.54 (0.24–1.19) | 0.124 | |||
| hCNT3 A25G (T89T) | 0.081 | ||||||
| TT | 52 | 16 | 1.0 | 1.0 | |||
| CT | 67 | 10 | 0.42 (0.19–0.93) | 0.033 | 0.53 (0.22–1.27) | 0.153 | |
| CC | 34 | 10 | 0.84 (0.38–1.85) | 0.663 | 0.77 (0.34–1.78) | 0.546 | |
| TT/CC vs CT | 0.46 (0.22–0.94) | 0.034 | 0.60 (0.28–1.30) | 0.196 | |||
|
| |||||||
| DNA repair gene | |||||||
| ATM C77T | 0.224 | ||||||
| TT | 47 | 10 | 1.0 | 1.0 | |||
| TC | 81 | 17 | 1.00 (0.46–2.18) | 0.996 | 1.10 (0.49–2.44) | 0.819 | |
| CC | 25 | 9 | 1.91 (0.78–4.71) | 0.158 | 1.85 (0.71–4.81) | 0.210 | |
| TT/TC vs CC | 1.92 (0.90–4.08) | 0.091 | 1.74 (0.78–3.88) | 0.178 | |||
| ATMA61G | 0.376 | ||||||
| GG | 135 | 30 | 1.0 | 1.0 | |||
| GA | 14 | 5 | 1.93 (0.74–4.98) | 0.177 | 1.72 (0.62–4.73) | 0.297 | |
| AA | 4 | 1 | 1.35 (0.18–9.89) | 0.771 | 0.97 (0.12–7.67) | 0.976 | |
| GG/AA vs GA | 1.91 (0.74–4.93) | 0.181 | 1.72 (0.62–4.73) | 0.297 | |||
| ATR C340T | 0.326 | ||||||
| CC | 45 | 14 | 1.0 | 1.0 | |||
| CT | 73 | 16 | 0.67 (0.33–1.38) | 0.281 | 0.96 (0.46–2.03) | 0.922 | |
| TT | 35 | 6 | 0.52 (0.20–1.35) | 0.177 | 0.80 (0.30–2.14) | 0.661 | |
| CC vs CT/TT | 0.62 (0.32–1.22) | 0.166 | 0.91 (0.46–1.81) | 0.790 | |||
| TREX1 Ex14-460C>T | 0.078 | ||||||
| CC | 59 | 11 | 1.0 | 1.0 | |||
| CT | 73 | 17 | 1.29 (0.61–2.76) | 0.506 | 0.87 (0.38–1.96) | 0.732 | |
| TT | 21 | 8 | 2.69 (1.08–6.69) | 0.033 | 3.02 (1.17–7.81) | 0.023 | |
| CC/CT vs TT | 2.32 (1.06–5.09) | 0.036 | 3.28 (1.44–7.51) | 0.005 | |||
|
| |||||||
| Drug resistance gene | |||||||
| MRP2 G40A | 0.057 | ||||||
| GG | 99 | 23 | 1.0 | 1.0 | |||
| AG | 49 | 10 | 0.93 (0.44–1.95) | 0.839 | 0.92 (0.42–2.03) | 0.834 | |
| AA | 5 | 3 | 3.71 (1.11–12.4) | 0.034 | 1.80 (0.52–6.22) | 0.353 | |
| GG/AG vs AA | 3.80 (1.16–12.5) | 0.028 | 1.85 (0.55–6.22) | 0.320 | |||
| GSTP1 Ex5–24A>G | 0.216 | ||||||
| AA | 62 | 18 | 1.0 | 1.0 | |||
| AG | 74 | 13 | 0.54 (0.26–1.10) | 0.090 | 0.65 (0.31–1.36) | 0.253 | |
| GG | 17 | 5 | 0.88 (0.33–2.37) | 0.799 | 0.73 (0.25–2.14) | 0.571 | |
| AA/GG vs AG | 0.56 (0.28–1.10) | 0.091 | 0.67 (0.34–1.31) | 0.243 | |||
Abbreviations: P(LR), log-rank P; HR, hazard ratio; CI, confidence interval.
HR was from multivariate Cox regression model with adjustment for age, number of prior chemo lines, progression and severe toxicity.
HIGHLIGHTS.
This prospective study shows that polymorphic variants of enzymes involved in gemcitabine metabolism, DNA damage repair and multidrug resistance pathways are associated with outcome and severe toxicity in patients with refractory/relapsed lymphoma or myeloma receiving high-dose chemotherapy with gemcitabine/busulfan/melphalan.
These findings suggest the value of pharmacogenomics in stratifying patients for individualized high-dose cancer therapy.
Acknowledgments
KS is the recipient of a research fellowship from the Uehara Memorial Foundation, Tokyo, Japan.
FINANCIAL SUPPORT: Supported by a MD Anderson Cancer Center Institutional Research Grant (YN)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
POTENTIAL CONFLICTS OF INTEREST: None.
References
- 1.Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol. 2006;17(Suppl 5):v7–12. doi: 10.1093/annonc/mdj941. [DOI] [PubMed] [Google Scholar]
- 2.Ueno H, Kiyosawa K, Kaniwa N. Pharmacogenomics of gemcitabine: can genetic studies lead to tailor-made therapy? Br J Cancer. 2007;97:145–151. doi: 10.1038/sj.bjc.6603860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang E, Gulbis A, Hart JW, Nieto Y. The Emerging Role of Gemcitabine in Conditioning Regimens for Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant. 2014;20:1382–1389. doi: 10.1016/j.bbmt.2014.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Plunkett W, Huang P, Searcy CE, Gandhi V. Gemcitabine: preclinical pharmacology and mechanisms of action. Semin Oncol. 1996;23:3–15. [PubMed] [Google Scholar]
- 5.Nieto Y, Thall P, Valdez B, et al. High-dose infusional gemcitabine combined with busulfan and melphalan with autologous stem-cell transplantation in patients with refractory lymphoid malignancies. Biol Blood Marrow Transplant. 2012;18:1677–1686. doi: 10.1016/j.bbmt.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nieto Y, Popat U, Anderlini P, et al. Autologous stem cell transplantation for refractory or poor-risk relapsed Hodgkin’s lymphoma: effect of the specific high-dose chemotherapy regimen on outcome. Biol Blood Marrow Transplant. 2013;19:410–417. doi: 10.1016/j.bbmt.2012.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong X, Jiao L, Li Y, et al. Significant associations of mismatch repair gene polymorphisms with clinical outcome of pancreatic cancer. J Clin Oncol. 2009;27:1592–1599. doi: 10.1200/JCO.2008.20.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong X, Li Y, Hess KR, Abbruzzese JL, Li D. DNA mismatch repair gene polymorphisms affect survival in pancreatic cancer. The Oncologist. 2011;16:61–70. doi: 10.1634/theoncologist.2010-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li D, Frazier M, Evans DB, et al. Single nucleotide polymorphisms of RecQ1, RAD54L, and ATM genes are associated with reduced survival of pancreatic cancer. J Clin Oncol. 2006;24:1720–8. doi: 10.1200/JCO.2005.04.4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li D, Liu H, Jiao L, et al. Significant effect of homologous recombination DNA repair gene polymorphisms on pancreatic cancer survival. Cancer Res. 2006;66:3323–30. doi: 10.1158/0008-5472.CAN-05-3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okazaki T, Javle M, Tanaka M, Abbruzzese JL, Li D. Single nucleotide polymorphisms of gemcitabine metabolic genes and pancreatic cancer survival and drug toxicity. Clin Can Res. 2010;16:320–9. doi: 10.1158/1078-0432.CCR-09-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okazaki T, Jiao L, Chang P, Evans DB, Abbruzzese JL, Li D. Single-nucleotide polymorphisms of DNA damage response genes are associated with overall survival in patients with pancreatic cancer. Clin Can Res. 2008;14:2042–8. doi: 10.1158/1078-0432.CCR-07-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka M, Javle M, Dong X, Eng C, Abbruzzese JL, Li D. Gemcitabine metabolic and transporter gene polymorphisms are associated with drug toxicity and efficacy in patients with locally advanced pancreatic cancer. Cancer. 2010;116:5325–35. doi: 10.1002/cncr.25282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka M, Okazaki T, Suzuki H, Abbruzzese JL, Li D. Association of multi-drug resistance gene polymorphisms with pancreatic cancer outcome. Cancer. 2011;117:744–51. doi: 10.1002/cncr.25510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Awasthi S, Bajpai KK, Piper JT, et al. Interactions of melphalan with glutathione and the role of glutathione S-transferase. Drug Metab Dispos. 1996;24:371–4. [PubMed] [Google Scholar]
- 16.Czerwinski M, Gibbs JP, Slattery JT. Busulfan conjugation by glutathione S-transferases alpha, mu, and pi. Drug Metab Dispos. 1996;24:1015–9. [PubMed] [Google Scholar]
- 17.Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol. 1995;30:445–600. doi: 10.3109/10409239509083491. [DOI] [PubMed] [Google Scholar]
- 18.Di Simone D, Galimberti S, Mattii L, Petrini M. c-Jun and GST-pi expression in human plasma cells. Haematologica. 1997;82:69–70. [PubMed] [Google Scholar]
- 19.Lourenco GJ, Neri IA, Sforni VC, Kameo R, Lorand-Metze I, Lima CS. Polymorphisms of glutathione S-transferase Mu 1, glutathione S-transferase theta 1 and glutathione S-transferase Pi 1 genes in Hodgkin’s lymphoma susceptibility and progression. Leuk Lymphoma. 2009;50:1005–9. doi: 10.1080/10428190902878455. [DOI] [PubMed] [Google Scholar]
- 20.Dasgupta RK, Adamson PJ, Davies FE, et al. Polymorphic variation in GSTP1 modulates outcome following therapy for multiple myeloma. Blood. 2003;102:2345–50. doi: 10.1182/blood-2003-02-0444. [DOI] [PubMed] [Google Scholar]
- 21.NCI Common Toxicity Critera v3.0. Available at: http://ctep.cancer.gov/protocoldevelopment/electronic…/ctcaev3.pdf.
- 22.Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, Rothman N. Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J Natl Cancer Inst. 2004;96:434–42. doi: 10.1093/jnci/djh075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Habermann TM, Wang SS, Maurer MJ, et al. Host immune gene polymorphisms in combination with clinical and demographic factors predict late survival in diffuse large B-cell lymphoma patients in the pre-rituximab era. Blood. 2008;112:2694–702. doi: 10.1182/blood-2007-09-111658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cerhan JR, Wang S, Maurer MJ, et al. Prognostic significance of host immune gene polymorphisms in follicular lymphoma survival. Blood. 2007;109:5439–46. doi: 10.1182/blood-2006-11-058040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Begg CB, Cramer LD, Venkatraman ES, Rosai J. Comparing tumour staging and grading systems: a case study and a review of the issues, using thymoma as a model. Statistics in medicine. 2000;19:1997–2014. doi: 10.1002/1097-0258(20000815)19:15<1997::aid-sim511>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 26.Li L, Schaid DJ, Fridley BL, et al. Gemcitabine metabolic pathway genetic polymorphisms and response in patients with non-small cell lung cancer. Pharmacogenet Genomics. 2012;22:105–16. doi: 10.1097/FPC.0b013e32834dd7e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ludovini V, Floriani I, Pistola L, et al. Association of cytidine deaminase and xeroderma pigmentosum group D polymorphisms with response, toxicity, and survival in cisplatin/gemcitabine-treated advanced non-small cell lung cancer patients. J Thorac Oncol. 2011;6:2018–26. doi: 10.1097/JTO.0b013e3182307e1f. [DOI] [PubMed] [Google Scholar]
- 28.Soo RA, Wang LZ, Ng SS, et al. Distribution of gemcitabine pathway genotypes in ethnic Asians and their association with outcome in non-small cell lung cancer patients. Lung Cancer. 2009;63:121–7. doi: 10.1016/j.lungcan.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 29.Tibaldi C, Giovannetti E, Tiseo M, et al. Correlation of cytidine deaminase polymorphisms and activity with clinical outcome in gemcitabine-/platinum-treated advanced non-small-cell lung cancer patients. Ann Oncol. 2012;23:670–7. doi: 10.1093/annonc/mdr280. [DOI] [PubMed] [Google Scholar]
- 30.Lee PH, Shatkay H. An integrative scoring system for ranking SNPs by their potential deleterious effects. Bioinformatics. 2009;25:1048–55. doi: 10.1093/bioinformatics/btp103. [DOI] [PubMed] [Google Scholar]
- 31.Karnitz LM, Flatten KS, Wagner JM, et al. Gemcitabine-induced activation of checkpoint signaling pathways that affect tumor cell survival. Mol Pharmacol. 2005;68:1636–44. doi: 10.1124/mol.105.012716. [DOI] [PubMed] [Google Scholar]
- 32.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–9. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 33.Montgomery SB, Sammeth M, Gutierrez-Arcelus M, et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–7. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell. 2007;131:873–86. doi: 10.1016/j.cell.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 35.Liberti SE, Rasmussen LJ. Is hEXO1 a cancer predisposing gene? Mol Cancer Res. 2004;2:427–32. [PubMed] [Google Scholar]
- 36.Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–4. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmutte C, Sadoff MM, Shim KS, Acharya S, Fishel R. The interaction of DNA mismatch repair proteins with human exonuclease I. J Biol Chem. 2001;276:33011–8. doi: 10.1074/jbc.M102670200. [DOI] [PubMed] [Google Scholar]
- 38.El-Shemerly M, Hess D, Pyakurel AK, Moselhy S, Ferrari S. ATR-dependent pathways control hEXO1 stability in response to stalled forks. Nucleic Acids Res. 2008;36:511–9. doi: 10.1093/nar/gkm1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.