

ABSTRACT
In the United States, prostate cancer is the second leading cause of cancer-related deaths among men with an approximately 220,000 patients diagnosed with the disease in 2015. Prostate cancer is a hormone-driven tumor, and a common therapy is androgen-deprivation therapy (ADT) that involves anti-androgen treatments and/or castration therapy. Understanding the molecular basis for androgen-independent tumors is crucial toward developing new therapies for these patients. Understanding how androgen receptor itself functions is an important step in elucidating this process. Androgen receptor (AR), NR3C4, is a nuclear hormone receptor and functions as a DNA-binding transcription factor that regulates the expression of protein-coding genes. Translocation of AR to improper gene promoter elements or DNA-binding sites can result in an alteration in gene expression and thus normal prostate function. Therefore, it is crucial to understand which AR-promoter interactions are drivers of disease, as compared to promiscuous or benign AR-binding interactions. While a large portion of our genome is considered a gene desert, it is now appreciated that these regions of the genome contain non-coding RNA genes such as microRNAs (miRNAs). These non-coding RNAs have enormous regulatory potential, as they post-transcriptionally regulate gene expression by binding to messenger RNAs (mRNAs) to promote degradation or intervention of translational processes. In this review, we focus specifically on the notion that mis-regulation of non-coding RNAs such as miRNAs by improper AR–DNA binding are an important component that promotes prostate cancer. We also highlight the role of miR-206 and the interaction of miR-206 and AR within this process, given this is a miRNA known to be regulated by hormones in both breast and prostate cancer.
KEYWORDS: androgen receptor, biomarker, microRNA, miR-206, prostate cancer, therapy, treatment resistance, tumor suppressor
Introduction
Prostate cancer
Prostate cancer is a leading cause of cancer-related deaths in men over the age of 50.1 While much of prostate cancer is curable, the challenge is treating those with castrate-resistance prostate cancer (CRPC). Currently, there is an enormous research effort underway to understand the mechanism by which prostate cancer initiates, how prostate tumors grow independent of androgen, and to develop cures for castration-resistant disease. Currently, prostate-specific antigen (PSA) is the major biomarker used to determine a patient's risk for developing prostate cancer.2 PSA is a serine protease produced by the prostate and is released into circulation as a consequence of glandular disruption. Those with elevated PSA levels typically undergo a biopsy to determine the histopathological grading of the disease, defined as a Gleason score. Patients are then treated by various approaches such as radical prostatectomy, external beam radiation therapy, or androgen deprivation therapy depending on the nature or aggressiveness of the disease.
Unfortunately, PSA as a stand-alone biomarker is not reliable and generates high false positive rates that are the result of benign inflammatory disorders. PSA also is not able to discriminate between indolent and aggressive disease, important given in many cases patients positive for PSA screening harbor low grade or benign prostate tumors that may not require surgery.3 This has called into question whether clinicians are over treating patients based on PSA testing. In fact, the spike in prostate-cancer related incidence in the late 1990s was due to the introduction of the PSA test rather than a rise in prostate cancer incidence, which only slightly increased in the subsequent 5 years.1 Conversely, identifying patients with high-risk disease through screening efforts increases the curative potential of local treatment in men 75 years of age or younger.4 Therefore, while PSA is still a major and crucial development in the prostate cancer field, there is a need for more effective biomarkers for prostate cancer, especially for those with castrate-resistant and/or advanced prostate cancer. Investigators have focused on protein biomarkers such as MUC1, which has a strong correlation with Gleason scoring and androgen-independent prostate cancer (AIPC) onset.5 However, there are limitations to non-enzymatic proteins as circulating biomarkers. In this review, we highlight the role of non-coding RNAs as potential biomarkers in androgen-dependent prostate cancer (ADPC) and AIDPC. To highlight this point, a recent study by Shulda et al.6 elucidated the role of a long non-coding RNA, PCAT14, as an effective biomarker for low-grade prostate cancer while serving as a prognostic indicator for disease aggressiveness and recurrence.
Biomarker development
The most effective way to develop effective biomarkers for those with advanced prostate cancer is to in fact elucidate the basic mechanisms that underlie the initiation and progression of the disease. Prostate cancer develops from glandular cells of the prostate and is characterized by uncontrolled cell growth and proliferation. Despite the controversy surrounding PSA as a reliable biomarker in the prostate cancer field, PSA testing is still considered an important clinical diagnostic marker to screen patients for risk of cancer development and/or progression. Furthermore, there is considerable evidence that inflammation itself plays an important role in the initiation and development of prostate cancer.7 TMPRSS2, an AR-regulated gene in prostate cancer, is frequently mutated and contributes to oncogenic processes. Moreover, inflammatory cytokines, such as TNFα, can induce TMPRSS2-ERG gene fusions in androgen-responsive cell lines. Additionally, utilizing an in vivo air-pouch model LPS promoted similar gene fusions. While it is unclear how TMPRSS2-ERG gene fusions contribute to prostate cancer development, the overall model in the field is that pro-inflammatory cues promote ROS-induced DNA damage and chromatin breaks, and androgen signaling recruits AR (Androgen Receptor) to these DNA break points inducing AR-related non-homologous end-joining (NHEJ) and ETS-related gene fusions. This model has been supported by a number of investigators and potentially explains the cellular mechanisms that push normal prostate cells from a pro-inflammatory state to a prostatic intraepithelial neoplasia, a precursor to invasive prostate cancer.
There is also a clear role for pluripotent stem cell factors in driving prostate cancer development. Recently, the role of Trp53 has been implicated in anti-androgen resistance and prostate cancer metastasis using a PBCre4:Ptenf/f:Rbf/+ mouse model.8,9 In these models, the notion is that genes promoting stemness such as Sox2 are aberrantly expressed and in a mutant TP53 background can induce prostatic neoplasia. This also indicates that, along with other tumor types, an underlying genomic instability mediated by loss of P53 function is required for tumor formation. Therefore, numerous mechanisms are at play when discussing how prostate cancer develops.
Function of AR
In prostate cancer, a known genetic driver of the disease is the AR. AR belongs to a super family of nuclear hormone receptors that function by binding to DNA consensus sequences in gene promoter regions.10,11 This activity is ligand-dependent meaning androgen–AR binding induces a conformational change in the protein structure that allows AR to engage chromatin elements and promote transcription (Fig. 1). AR contains seven domains some of which are structural/regulatory domains and includes an activation function domain is important for transcriptional activity of AR (noted AF1, AF2, and AF5), the hinge region that contains the nuclear localization signal, and the C-terminal domain. AR also harbors three functional domain structures: an NTD (N-terminal domain), a central DBD (DNA-binding domain), and a C-terminal LBD (ligand-binding domain).12 While the DBD interacts with target genes through DNA binding at gene promoters, the LBD is where androgen and androgen agonists physically engage AR, and where dimerization of two AR proteins occurs to promote gene transcription. The NTD is a less well studied region of AR; however, it contains the most potent regulatory capacity of AR given the NTD is responsible for proper transactivation of AR.13,14 Furthermore, certain AR splice variants may lack portions of the NTD, which becomes important when attempting to target this region of the protein to block or abrogate AR function in patients with prostate cancer. Some investigators are testing a small-molecule inhibitor derived from marine sponge extracts called EPI-506, which binds to the AR NTD, and is currently the first compound of its kind in clinical development.15,16 Further investigation is warranted to determine if EPI-506 can target all AR isoforms, or only specific isoforms.17 This is relevant given AR-V7 is an AR isoform lacking the LBD and is detectable in circulating tumor cell populations from AIPC samples refractory to enzalutamide and abiraterone. Therefore, identifying novel drugs that can downregulate these ligand-dependent AR isoforms will be of great clinical utility.
Figure 1.
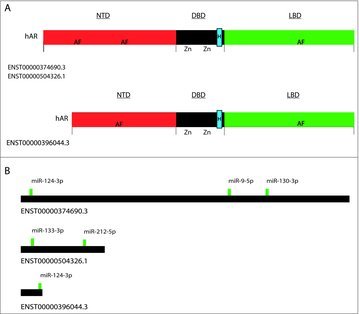
Depiction of androgen receptor isoforms. Panel (A) depicts protein isoforms and highlights the three crucial domains of hAR for proper protein function. The shorter isoform lacks a portion of the N-terminal domain (NTD) and could play an important role in androgen-resistance.96 The other domains DBD and LBD are depicted in both isoforms. The nuclear localization signal (NLS) is contained within the DBD and denoted as a blue square. The NLS is also located in the hinge portion of the DBD and is denoted with a letter H. (B) Depiction of the 3′-UTRs of the three hAR RNA isoforms, as well as the most well conserved miRNA binding sites. These sites are considered putative sites via TargetScan.
AR functions normally by interacting with specific gene promoter regions at precise times during cellular development and cell cycle, and occurs in response to a coordinated set of cell signaling and regulatory cues. When these spatio-temporal regulatory pathways are disrupted, improper gene regulation occurs resulting in mis-expression of AR-dependent genes, ultimately resulting in diseases such as cancer (Fig. 2). Many well-known AR-downstream genes include IL8 and EGR.18 However, there is increasing evidence that AR itself can bind and regulate miRNA genes. This raises the interesting prospect that miRNAs can become therapies if proper miRNA–AR target interaction are identified.
Figure 2.
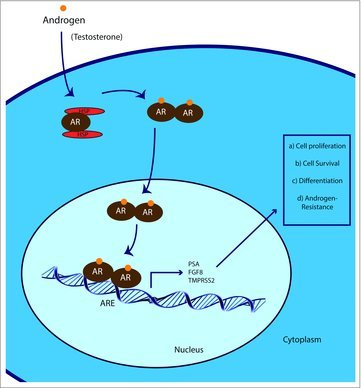
Depiction of AR interaction and function. Upon testosterone and androgen derivatives binding to AR, a conformational change in the protein structure occurs that involves release of chaperone proteins and homodimerization to the AR receptor, translocation into the nucleus, and binding to specific AR response elements (AREs) within the genome. Upon AR binding, recruitment of co-activator proteins and RNA PolII results in the initiation of transcription. Bona fide AR responsive genes include PSA and TMPRSS2. AR responsive genes promote cell proliferation and cell survival, but in other cases regulate cell differentiation and androgen-resistance.
Another consequence of inappropriate AR–DNA binding is during the transition from ADPC to AIPC. Numerous mechanisms are involved in this transition, but in AIPC AR is not properly localized to regions of the genome where normal AR activity is expected. Chip-seq studies determined that differential AR genome binding events occur in AIPC versus ADPC.10 Using a LNCaP cell model, where normal androgen-sensitive LNCaP cells and androgen-independent LNCaP-AI cells were subjected to Chip-Seq experiments, it was determined that in LNCaP-AI cells the frequency of AR binding to gene transcriptional start sites (TSS) is significantly reduced. Furthermore, only 28% of the AR-binding sites were shared between these two cell lines, indicating that in LNCaP-AI cells AR binds and regulates a distinct genetic network independent than that of hormone-sensitive LNCaP cells. This indicates that AIPC is not necessarily driven by lack of AR transcription, but instead a re-wiring of the AR program that allows for tumor growth and survival independent of androgen. Interestingly, some of the differentially regulated genes were involved in cell death pathways, indicating that AR itself promotes pro-apoptotic signaling in AIPC, and therefore a feedback mechanism may be in place to dampen AR-activity to support tumorigenesis in these situations.
Androgen resistance in prostate disease
Understanding the mechanisms that promote AIPC or other androgen insensitivity syndromes is of major clinical interest. In general, the observation is that in AIPC, PSA levels are continuously high, and counterintuitively AR expression and activity also appear elevated. Closer examination of this phenomena indicates that there are ligand-dependent mechanisms at play where AR is overexpressed and/or interacts with enhanced testosterone/dihydroxytestoterone (T/DHT) levels produced by the prostate, or adrenal glands if the patient underwent radical prostatectomy.19 There are also ligand-independent mechanisms of AR activation that include AR splice variants, AR mutations, cross-talk between cellular signaling pathways, and elucidated the expression or activity of AR co-regulator proteins such as PSAP and CTBP1.20,21,22
Many therapies involve targeting the LBD of AR with anti-androgens to block or inhibit AR function. In AIPC, this approach is not feasible given cells do not respond to androgen signaling. Therefore, understanding the structure of AR will allow for elucidation of AR function and open the door for additional therapeutic strategies to target AR itself. In fact, targeting the DBD of AR would be of interest given in AIPC AR dysregulation promotes improper gene regulation (Fig. 1). Crystallography studies indicate the DBD of AR contains two zinc DNA binding regions flanked by three α helices, one of which contains a P-box element, the physical region where AR binds to the DNA major groove.23 Therefore, proper protein folding of this region is crucial not only for DNA binding, but for proper dimerization of AR. In fact, mutation analysis of AR indicates that certain DBD mutations such as Ser597 enhance the binding efficacy of AR to AREs independent of AR agonists. This suggests certain mutations promote promiscuous AR binding and support the notion that AIPC is driven by dysregulated AR genome binding rather than a lack of overall AR signaling. In fact, patients with partial androgen insensitivity syndrome (PAIS) harbor mutations of residue Ser597.24,25 Therefore, small molecule-based therapies targeting the AR-DBD either to completely abrogate AR-ARE binding or to reduce AR-ARE binding potential to induce more selective AR–DNA binding in PAIS or AIPC would be of clinical relevance, and a novel therapeutic strategy.
In prostate cancer, much of the cell signaling and pro-proliferation pathways linked to tumorigenesis occur via AR signaling. Therefore, the treatment of prostate cancer largely involves testosterone deprivation therapy.26 In many cases, this effect is temporary and eventually tumors proliferate independent of androgen. One of the mechanisms described earlier was that there is significant cross-talk between AR/DHT signaling and growth factor receptor signaling such as the EGF/EGFR pathway. Similar phenomena occur in breast cancer where ERα signaling becomes dampened by EGF signaling and the subsequent development of tamoxifen-resistant breast cancer, or triple-negative-breast cancer.27 Mechanistic studies involving the treatment of LNCaP cells with androgen indicates transcriptional activity of AR-dependent genes such as FKBP5 and AR itself increases 30 minutes after treatment.28 Additionally, a dramatic decrease in phospho-MAPK and concomitant increase in pAKT activity occurs as well. This indicates that AR signaling not only promotes androgen-related pathways, but in fact dampens non-steroidogenic pathways such as EGF/MAPK signaling.29
MicroRNAs
MicroRNAs (miRNAs) are short (18–25 nucleotides (nt) long) single-stranded non-coding RNAs30 that post-transcriptionally regulate gene expression by binding to messenger RNAs (mRNAs) to promote degradation of mRNA or inhibition of translational processes.31,32 These endogenous small RNAs are transcribed by RNA polymerase II, 5′ capped and 3′ polyadenylated, and are processed by the proteins DROSHA and DICER into shorter 19–22-nt mature miRNAs. miRNAs guide the RNA-induced silencing complex (RISC) to the 3′-untranslated region (3′-UTR) of target mRNAs via sequence complementarity, resulting in degradation/destabilization or translational repression.33,34 By some estimates, miRNAs regulate more than 60% of human protein-coding genes, and therefore aberrant expression of a single miRNA can have a profound influence on fundamental cell processes such as development, proliferation, survival, and death.30,35
The miRNAs deemed to be overexpressed in cancer that concomitantly repress tumor suppressor genes are described as oncogenic miRNAs, while those miRNAs under expressed in cancer and target known oncogenes are defined as tumor suppressive miRNAs.36,37 Successful initiation of tumorigenesis requires cells to develop sustained cellular proliferation, evasion of apoptosis, initiate replicative immortality, and evade growth suppressors.38 miRNAs target genes crucial in each of these processes or pathways, making miRNAs an essential regulator of the tumorigenic process. Furthermore, miRNAs regulate gene products while reciprocally being regulated by signaling pathways that dampen or promote miRNA function.39,40,41 These miRNA feedback loops result in the fine-tuning of gene expression important for normal cellular function, and the disruption of these loops contributes to disease pathogenesis.
miRNAs in prostate cancer
Comprehensive analysis of the prostate cancer genome indicates that while allelic loss of tumor suppressor genes such as PTEN occur, there is little evidence that point mutations exist in well-studied oncogenes and tumor suppressors.42,43 Therefore, the gain of gene function at the transcriptome level is primarily linked to prostate cancer development. Non coding RNA control gene expression at the post-transcriptional level, and tune gene dosage in a temporal manner. Therefore, certain noncoding RNA such as miRNAs have garnered attention over the past decade as potential therapeutics and to elucidate their involvement in disease pathogenesis. Numerous studies have highlighted the role of miRNAs in prostate cancer.19,44
Numerous miRNAs are known to play an important role in prostate cancer, see Walter et al. for review.45 Recently, a novel miRNA, miR-1207-3p, was found to regulate the AR pathway in prostate cancer by targeting fibronectin type III domain containing 1 (FNDC1) and fibronectin 1 (FN1) genes.46 A comprehensive study was performed where RNA samples were profiled from 40 prostate cancer samples, as well as normal epithelium and tumor-associated stroma. A total of 18 miRNAs were identified as being expressed at significantly lower levels in tumor samples compared with normal prostate epithelium. In this study, they identified several miRNAs dysregulated in prostate cancer compared with normal epithelium, including miR-30c, miR-181a, miR-181c, and miR-146b-5p. One of the most highly expressed miRNAs was miR-10b, and the most significantly upregulated miRNA was miR-30c. Both miRNAs are known oncogenic miRNAs in breast and lung cancer, indicating that these miRNAs may serve as useful biomarkers for prostate cancer. In fact, Kachakova et al. identified that miRNA expression from circulating serum sources such as miR-30c and let-7 have prognostic value in prostate cancer patients.47
Interestingly, when comparing prostate cancer samples to the stromal fraction, the expression of miR-206 and miR-125b was significantly upregulated in the tumor-associated stromal fraction.45 While many miRNAs have known oncogenic roles in a variety of tumors including prostate cancer, miR-206 and miR-125b are known to function as tumor suppressors in solid tumors.48,49 Therefore, it is interesting that these miRNAs were elevated in the stromal fraction of the tumor, and highlights the important role of stromal infiltration in tumors. Understanding this dynamic between the stroma and epithelium in tumors is important given a common approach in the field is to develop antisense technologies to target miRNAs overexpressed in disease, such as miR-146b-5p. As an example, in the context of prostate cancer, developing antisense sense approaches to a particular miRNA target may harbor more therapeutic efficacy in those that have advanced disease.
Despite these profiling studies,50 common miRNAs have been heavily investigated for their role in prostate cancer development. miR-21 is a well-studied oncogene, located on chromosome 17q23.2, which is a region known for genomic amplification in cancer.51 miR-21 targets PDCD4 and MARCKS,52,53,54 tumor suppressor genes known to control cell motility and invasiveness. Furthermore, miR-21 harbors a bona fide ARE in the promoter region indicating that androgen singling controls the expression of miR-21. Additionally, miR-221 is a miRNA associated with pathological stage, Gleason score, and capsular invasion in prostate cancer samples.54 Further mechanistic studies indicated that miR-221 targets DVL2 and drives metastatic AIPC, further supporting the pro-tumorigenic function of miR-221 in prostate cancer.52
Finally, through expression profiling studies, miR-23-3p and miR-17/92 are found to be elevated in prostate cancer versus normal samples. These miRNAs have numerous cellular targets; however, in prostate cancer, these miRNAs have been linked to regulating metabolic pathways related to prostate cancer development. miR-23b-3p targets PX/PRODH, which controls proline and glutamine metabolism in PC3 prostate cancer cell lines.54 The miR-17/92 family targets PPARA, which controls lipogenesis in LNCaP cell lines.55 Therefore, regulating the metabolic pathways that promotes tumorigenic formation by regulating the activity or expression of miRNAs is a novel way to treat prostate cancer.
miRNAs regulating AR
As mentioned, AR is an important regulator of prostate gland development as well as a culprit in prostate cancer development. Identifying miRNAs that regulate AR will help in developing future therapeutics. The 3′-UTR of AR is approximately 4 kb in length and is AU rich, similar to most steroid hormone receptors. This property increases the frequency at which miRNAs interact with the AR mRNA to regulate its expression.19,56 Supporting this notion, Östling et al. identified 71 unique miRNAs that control or alter the expression of AR in PC cell lines. Among these miRNAs include the miR-34/miR-449 family of miRNAs, which are known tumor suppressors in a wide array of cancer types.56 miR-34a is known to be inversely correlated with AR expression in prostate cancer and inversely linked to PSA levels and prostate cancer disease pathogenesis.57,58 Additionally, miR-634, miR-9, and miR-205 were also shown to interact with the AR 3′-UTR via luciferase assays, and forced expression of these miRNAs can suppress AR expression and function. Not surprisingly, most of these miRNAs operate as tumor suppressors in a broad range of cancer types. The fact that these miRNAs target AR supports the notion that the AR is a pro-oncogenic factor in prostate cancer, and that loss of regulatory miRNAs allows for aberrantly high expression of AR. It should be noted that while miRNAs regulate target gene expression through binding the 3′-UTR of the respective mRNA, it is known that miRNAs can bind the coding region or the 5′-UTR of target genes.59,60 We did not focus our efforts on these regions as it requires a more complex bioinformatic analysis to identify these binding sites.
AR regulation of miRNAs
Given the primary function of AR is as a steroid-regulated transcription factor, a question being investigated is whether certain miRNAs are transcriptionally regulated by AR. This was reviewed by Mo et al., where miRNAs such as miR-19a, miR-27a, and miR-133b contained AREs upstream of the TSS. Most of these sites were confirmed through luciferase assays and qPCR studies.61 Additionally, oncogenic miRNAs such as miR-32, miR-148a, miR-99a, and miR-21 have bona fide AREs upstream of these respective non-coding RNA genes. miR-32 harbors an ARE 14 kb upstream of the miR-32 TSS and may be a predictor of poor outcome in patients post prostatectomy. miR-32 also regulates AR-interacting genes such as BTG2 in cellular models of AIPC,62,63 indicating that miR-32 is intricately tied to AR signaling in prostate cancer.64 miR-99a harbors five unique AREs 50 kb upstream of the TSS, and it was shown that androgen-dependent AR binding occurs at two of the sites in AR-positive prostate cancer cell lines.65,66 Mechanistically, AR binding to the miR-99a/let-7c/miR-125b-2 cluster induces a repressive response whereby AR downregulates miR-99a levels. Furthermore, reducing miR-99a levels through antisense treatment promotes a growth advantage in AR-positive prostate cancer cells. It was also shown that AR-induced target genes such as IGF1R are also targets of miR-99a, indicating miR-99a and AR are parts of an interactive androgen signaling network that controls AR signaling in AR-positive prostate cancer cells. Finally, miR-21 is genomically located in a region frequently associated with amplification in hormone refractory prostate cancer.67 The expression of miR-21 is stimulated in response to androgen, and harbors a conserved ARE in the gene promoter.68 However, miR-21 is highly expressed in both hormone-dependent and hormone-independent prostate cancers.69 Mechanistically, this can be explained in a prostate cancer xenograft mouse model, where the forced expression of miR-21 supports tumor growth post-castration. Therefore, miR-21 in prostate cancer operates in a similar manner as miR-206 in breast cancer,70 and is further evidence that aberrant expression of miRNAs can support the development of hormone-independent tumors.
The role of miR-206 in prostate cancer
Initially, miR-206 was identified as a myogenic miRNA since miR-206 is highly expressed in skeletal muscle tissue and actively supports myogenesis when expressed at the appropriate times.71,72 This normal developmental function of miR-206 is supported mechanistically, where forced expression miR-206 controls C2C12 myoblast proliferation and differentiation.73 miR-206 is located on chromosomal region 6p12.274 and has been linked with several genetic disorders, including cancer. miR-206 belongs to a family of miRNAs including miR-1, miR-133, miR-145, and miR-206, each containing the same seed sequence, but having a slightly varied 3′ end sequence that may result in differential mRNA target recognition and regulatory activity. The miR-1/133a cluster was reported as a candidate tumor suppressor gene in prostate cancer,75 due to the low expression of this miRNA cluster in prostate cancer cell lines and tumor samples, but also given forced expression of miR-1/133 supports tumor regression in xenograft mouse models. However, the genomic location of the miR-1 cluster is separate and distinct from that of miR-206. This means that miR-206 is under a different regulatory potential and operates under a discrete mechanism than that of the other miRNA-1 family members.
To highlight this point, Walter et al. determined that miR-206 was upregulated in high-grade prostate tumors (i.e., Gleason score 8–9) as compared with normal epithelium, and not miR-133b.45 This indicates that miR-206 may function as an oncogene in prostate cancer, though it is unclear as to whether this miRNA is a driver of the disease. As mentioned earlier, miR-206 is expressed in both the prostate tumor samples and the surrounding stroma, interesting given miR-1/206 is known to be expressed in skeletal muscle tissue,71 as well as the surrounding myoepithelium of glandular tissues.76 This is a crucial point to mention since more aggressive tumors tend to harbor greater stromal infiltration, and suggests miR-206 could be a superior marker for more invasive tumors. In fact, Walter et al. only found miR-206 significantly differentially expressed between normal and tumor samples in prostate cancer samples of high Gleason scores.45
Other groups have also identified miR-206 to be highly associated with invasive disease. Specifically, miR-206 expression is higher in ERα-negative breast tumors as compared with ERα-positive breast cancer,77,78 and the forced expression of miR-206 in MCF-7 cells directly targets and represses ERα and promotes an estrogen-independent state.27 However, miR-206 in prostate cancer operates mostly as a tumor suppressor since miR-206 targets known proto-oncogenes such as Cyclin D1 (CCND1) and RARB.73,79 Furthermore, transfection of prostate cancer cells with miR-206 mimic reduced CCND1 protein expression as well as colony formation. Additionally, miR-206 targets three genes in the pentose phosphate pathway (glucose-6-phosphate dehydrogenase, G6PD; phosphogluconate dehydrogenase, PGD; and transketolase, TKT), a metabolic pathway hijacked by tumors that supports proliferation and cell survival.80 Furthermore, the transcription factor NRF2 enhances tumor metabolism by altering redox homeostasis and inducing nucleotide synthesis to support the accelerated proliferation required of tumors. NRF2 does this in part by enhancing the activity of the pentose phosphate pathway by repressing the expression of miR-206 that subsequently attenuates the repressive regulatory action on G6PD, PGD, and TKT.
Therefore, the mechanism of miR-206 function is cell context dependent. In some instances, miR-206 functions as a tumor suppressor and in others as an oncogene. For instance, miR-206 targets NOTCH2, activates apoptosis, and inhibits migration in cervical cancer.81 In support of the latter, miR-206 is associated with inflammatory processes as well as inflammatory disorders, interesting given inflammation is a precursor for prostatic tumorigenesis. In a study by Wu et al., miR-206 was associated with ulcerative colitis (UC) and identified as a regulator of adenosine A3 receptor (A3AR).82 While the role of A3AR in prostate cancer is unclear, in UC, A3AR levels are decreased compared with normal tissue specimens. Conversely, miR-206 levels are elevated in UC specimens, and via luciferase and western blot assays forced expression of miR-206 was shown to directly downregulate A3AR expression. Moreover, miR-206 expression in HT-29 cells promotes IL8 expression and NF-κβ translocation into the nucleus upon pro-inflammatory cues such as TNFα treatment. In mouse models of UC, DSS treatment induces miR-206 expression. Therefore, in this model, it is quite clear that miR-206 is part of a feed-forward mechanism that supports inflammatory cues required in promoting disease. Therefore, one might speculate that in certain cellular contexts miR-206 can function as a pro-oncogenic miRNA by converting normal epithelial cells to a pre-neoplastic legion. For instance, we identified several miR-206 binding sites in the PTEN 3′-UTR, the primary tumor suppressor of clinical relevance in prostate cancer (unpublished observations). To stress this point, we and others have shown that miR-206 action as a putative oncogene operates at the earliest stages of tumor development, and moreover, the regulatory actions of miR-206 in the stromal compartment of the tumor may alter the microenvironment and aid in establishing a proper niche for tumor development.
Evidence for an AR miR-206 interaction
Given the role of miRNAs in endocrine related-tumors, a question that remains is whether in prostate cancer, an endocrine-regulated miRNA such as miR-206 is regulated by the predominate steroid hormone receptor AR. In general, there are three ways for a miRNA gene to be regulated by a steroid hormone receptor such as AR: (1) direct binding of AR to the promoter regions of miRNA genes, (2) indirect regulation by AR-responsive transcription factors that in turn bind to the miRNA gene promoter, and (3) AR-regulated RNA-binding proteins that alter the stability of the miRNA.30 Therefore, identifying putative AREs within miR-206 is crucial in understanding whether AR regulates miR-206.
AR itself recognizes a degenerate set of response elements with inverted repeats of hexameric half sites with a 3 bp spacer, or sequential repeats of these response elements.83 The initial crystallography studies indicated that AR binds to AREs in a head-to-head fashion meaning that the consensus AREs are normally found in a symmetric arrangement. Since the original identification of an ARE consensus sequence to be that of 5′-GGA/TACANNNTGTTCT-3′ in 1992 by Roche et al., other groups have been successfully cloning and functionally characterizing additional sequences.11 One of the most specific binding sites includes a defined sequence of 5′-GGCTCTNNNAGTTCT-3′.84,85 While the presence of an ARE does not determine that a gene is regulated by AR, if presented in the proper genomic context it does however give evidence as being a putative AR-regulated gene.
To directly address the question regarding AR regulation of miR-206, we performed in silico analysis to identify putative AR-binding sites within the miR-206 promoter (Fig. 3B). We scanned the promoter region of the miR-206 gene by analyzing a dataset on the UCSC genome Browser,86,87 and found two candidate AREs 1.5 kb and 2 kb upstream of the miR-206 transcriptional start site (TSS). The location of these two sites are interesting given that one of the miR-206 family members, miR-133b is also downstream of these site, and indicates more than one of the miR-206 family members may be regulated by AR. Furthermore, these sites are within close proximity to the initiation of transcription as determined by a separate UCSC genome browser track. Therefore, there is sufficient evidence to propose miR-206 as an AR-regulated miRNA. In fact, upon further investigation we identified over 20 ARE half-sites along the 5 kb of promoter sequence upstream of miR-206, and an additional 10 ARE half-sites 2kb downstream of miR-206 gene. The study by Bolton et al. indicated that approximately 42% of AREs are in the promoter region of genes, with the remaining 42% of sites being intragenic, supporting the idea that the AREs we identified could be bona fide miR-206-related AREs.18 Furthermore, it is now well established that chromosomal looping occurs quite readily in the transcriptome, and is induced by steroid hormone receptor binding and recruitment of co-activators and other enhancer elements to induce hormone-dependent gene transcription.88,89 Therefore, it is easy to speculate that AR and the subsequent recruitment of co-activator proteins can induce a looping event that brings into proximity these ARE half-sites to generate a full AR-mediated initiation of miR-206 transcription.
Figure 3.
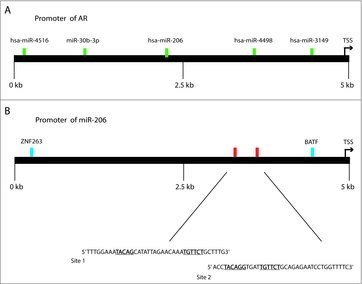
Locations within the AR promoter that harbor putative miR-206-binding sites (A), and locations within the miR-206 gene that contain putative AR-binding sites (B). miRNA-binding sites are highlighted in green. AR consensus sequences are highlighted in red and are expanded to show actual sequence. Gene promoters are denoted as black lines. TF-binding sites are denoted in blue. AR-binding locations are derived from the hg19 genome track.
Since it is common for miRNAs to operate in feedback loops with steroid hormone receptor signaling, we performed in silico analysis to understand if a potential negative feedback loop was present between AR and miR-206 in prostate cancer cell lines. Specifically, we screened for miR-206 binding sites along the entire 3′-UTR of the AR gene (Fig. 3A). We identified a miR-206 binding region approximately 2 kb within the sequence. The site was not well conserved across species; however, the site was classified as an 8mer in TargetScan 6.0, which means the seed sequence of the miRNA is fully bound to the 3′-UTR sequence. We also identified numerous miR-206 binding sites within the coding region of the AR gene, and in the 5′-UTR of AR. This signifies to us the potential for and auto-regulatory functions between these two entities that should be further explored. The further elucidation of this mechanism is beyond the scope of this review and will include knockout studies by removing the AREs upstream of the miR-206 TSS. However, highlighting this mechanism is crucial to re-invigorate investigators to explore the functions of miR-206 in a variety of tumor models, beyond that of breast and other endocrine-related tumors.
The future of miR-206 as a biomarker and therapeutic
miRNA therapeutics have garnered much attention over the last 5 years, with the advent of new delivery technologies, RNA as a therapy is being widely accepted and practiced in the clinic in the form of Phase I and Phase II clinical trials. miR-206 specifically as a therapeutic may be challenging given the cell context-dependent nature of its functions in certain disorders. However, as noted before, miR-206 in general harbors tumor suppressive functions, while the expression of miR-206 associates with more aggressive disease. Therefore, the development of miR-206 as a therapeutic may be limited, and the use of miR-206 as a biomarker may be more relevant.
miR-206 has already been shown to serve as a functional biomarker for a variety of diseases and disorders. Since miR-206 has an important role in muscle development, it is not surprising that miR-206 is a biomarker for Rhabdomyosarcoma (RMS), a soft tissue sarcoma.90 In clinical trials, miR-206 was identified as a diagnostic marker where levels of miR-206 were elevated in RMS patients as compared with healthy controls. However, in noncancerous diseases, miR-206 also serves as a potential biomarker for disease progression. miR-206 levels are elevated in patients with amyotrophic lateral sclerosis (ALS)91 as well as those with Alzheimer disease.92 Additionally, miR-206 has been implicated in scleroderma, liver steatosis, COPD, and cardiomyopathy. Therefore, miR-206 is a theranostic marker for many diseases.
In this review, we highlighted the potential miR-206 could play in advanced prostate cancer, a devastating disease that has few curative treatment options. Therefore, there is a need for developing effective biomarkers to detect the progression of benign prostate cancer to AIPC. To this end, there are efforts underway to screen for miRNA levels in prostatic secretions as well as from urine samples, with the hope that circulating miRNA levels in these biofluids can become a biomarker for prostate-related disorders.47 In fact, investigators have successfully performed in situ hybridization techniques on tissue microarrays as well as RNA expression analysis on expressed prostatic secretions in urine to determine prostate cancer patient prognosis and outcome.93,94 Additionally, there has been an impetus for developing more effective biopsy tools to measure and detect prostate cancer. The development of whole-transcriptome assays on needle core biopsy specimens has remarkably advanced the biomarker field.95 It can be imagined that small RNA sequencing can be developed on similar specimens given miRNAs have proven a reliable biomarker for disease. Overall, it is quite feasible that within the next 5–10 years, noncoding RNA expression from small needle biopsies, or additional sources such as circulating biofluids will aid in the development of prostate cancer diagnosis and treatment.
Disclosure of potential conflicts of interest
B.D.A. holds patent interests with, and consults with AUM LifeTech. The other authors have no conflicts of interest to disclose.
Acknowledgments
We thank Jun Liu and Jessica Tytell for the critical reading of this manuscript.
Funding
This work was supported by grants to B.D.A. by the Research Foundation, and through start-up funds by the RNA Institute and the State of New York.
References
- [1].Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. Cancer J Clin 2015; 65:5-29; PMID:25559415; https://doi.org/ 10.3322/caac.21254 [DOI] [PubMed] [Google Scholar]
- [2].Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev 2010; 24:1967-2000; PMID:20844012; https://doi.org/ 10.1101/gad.1965810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wolf AM, Wender RC, Etzioni RB, et al. American Cancer Society guideline for the early detection of prostate cancer: update 2010. CA Cancer J Clin 2010; 60:70; PMID:20200110; https://doi.org/ 10.3322/caac.20066 [DOI] [PubMed] [Google Scholar]
- [4].Cooperberg MR, Carroll PR. Trends in management for patients with localized prostate cancer, 1990-2013. JAMA 2015; 314(1):80-82; PMID:26151271; https://doi.org/ 10.1001/jama.2015.6036 [DOI] [PubMed] [Google Scholar]
- [5].Eminaga O, Wei W, Hawley SJ, Auman H, Newcomb LF, Simko J, Hurtado-Coll A, Troyer DA, Carroll P, Gleave ME et al.. MUC1 expression by immunohistochemistry is associated with adverse pathologic features in prostate cancer: a multi-institutional study. PLoS ONE 2006; 11(11):e0165236; PMID:27846218; https://doi.org/ 10.1371/journal.pone.0165236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shukla S, Zhang X, Niknafs YS, Xiao L, Mehra R, Cieślik M, Ross A, Schaeffer E, Malik B et al.. identification and validation of PCAT14 ad a Prognostic Biomarker in Prostate Cancer. Neoplasia 2016; 18(8):489-499; PMID:27566105; https://doi.org/ 10.1016/j.neo.2016.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mani RS, Amin MA, Li X, Kalyana-Sundaram S, Veeneman BA, Wang L, Ghosh A, Aslam A, Ramanand SG, Rabquer BJ et al.. Inflammation induced oxidative stress mediates gene fusion formation in prostate cancer. Cell Rep 2016; 17(10):2620-2631; PMID:27926866; https://doi.org/ 10.1016/j.celrep.2016.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbe DP, Gomez EC, Wang J et al.. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastatsis, and antiandrogen resistance. Science 2017; 355(6320):78-83; PMID:28059767; https://doi.org/ 10.1126/science.aah4199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, Wongvipat J, Ku SY, Gao D, Cao Z et al.. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53-and RB1-defecient prostate cancer. Science 2017; 355(6320):84-88; PMID:28059768; https://doi.org/ 10.1126/science.aah4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cheng Y, Yu P, Duan X, Liu C, Xu S, Chen Y, Yunnian T, Qiang Y, Shen J, Tao Z. Genome-wide analysis of androgen receptor binding sites in prostate cancer cells. Exp Thera Med 2015; 9:2319-2324; PMID:26136980; https://doi.org/ 10.3892/etm.2015.2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Roche PJ, Hoarse SA, Parker MG. A consensus DNA-binding site for the androgen receptor. Mol Endocrinol 1992; 6(12):2229-2235; PMID:1491700; https://doi.org/ 10.1210/mend.6.12.1491700 [DOI] [PubMed] [Google Scholar]
- [12].Lavery DN, Mcewani IJ. . Structure and function of steroid receptor AF1 transactivation domains: induction of active conformations. Biochem J 2005; 391:449-464; PMID:16238547; https://doi.org/ 10.1042/BJ20050872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jenster G, van der Korput HA, van Vroonhoven C, van der Kwast TH, Trapman J, Brinkmann AO. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol Endocrinol 1991; 5:1396-1404; PMID:1775129; https://doi.org/ 10.1210/mend-5-10-1396 [DOI] [PubMed] [Google Scholar]
- [14].Simental JA, Sar M, Lane MV, French FS, Wilson EM. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J Biol Chem 1991; 266:510-518; PMID:1985913 [PubMed] [Google Scholar]
- [15].Antonarakis ES, Chandhasin C, Osbourne E, Luo J, Sadar MD, Perabo F. Targeting the N-terminal domain of the androgen receptor: A new approach for the treatment of advanced prostate cancer. Oncologist 2016; 21:1-9; PMID:26712959; https://doi.org/ 10.1634/theoncologist.2016-0161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, Watt K, Tam T, Yang YC, Bañuelos CA et al.. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 2010; 17:535-546; PMID:20541699; https://doi.org/ 10.1016/j.ccr.2010.04.027 [DOI] [PubMed] [Google Scholar]
- [17].Emmanuel S, Antonarakis MD, Changxue L, Wang H, Luber B, Nakazawa M, Eisenberger M, Luo J. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014; 371:1028-1038; PMID:25184630; https://doi.org/ 10.1056/NEJMoa1315815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bolton EC, So AY, Chaivorapol C, Hagg CM, Li H, Yamamoto KR. Cell-and gene-specific regulation of primry target genes by the androgen receptor. Gene Dev 2007; 21(16):2005-2017; PMID:17699749; https://doi.org/ 10.1101/gad.1564207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shih JW, Wang LY, Hung CL, Kung HJ, Hsieh CL. Non-coding RNAs in castration-resistant prostate cancer: regulation of androgen receptor signaling and cancer metabolism. Int J Mol Sci 2015; 16(12):28943-28978; PMID:26690121; https://doi.org/ 10.3390/ijms161226138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Takayama K, Horie-Inoue K, Katayama S, Suzuki T, Tsutsumi S, Ikeda K, Urano T, Fujimura T, Takagi K, Takahashi S et al.. Androgen-responsive long noncoding RNA CTBP1-as promotes prostate cancer. EMBO J 2013; 32:1665-1680; PMID:23644382; https://doi.org/ 10.1038/emboj.2013.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Boll K, Reiche K, Kasack K, Morbt N, Kretzschmar AK, Tomm JM, Verhaegh G, Schalken J, von Bergen M, Horn F et al.. miR-130a, miR-203 and miR-205 jointly repress key oncogenic pathways and are downregulated in prostate carcinoma. Oncogene 2013; 32:277-285; PMID:22391564; https://doi.org/ 10.1038/onc.2012.55 [DOI] [PubMed] [Google Scholar]
- [22].Verrijdt G, Schoenmakers E, Haelens A, Peeters B, Verhoeven G, Rombauts W, Claessens F. Change of specificity mutations in androgen-selective enhancers. Evidence for a role of differential DNA binding by the androgen receptor. J Biol Chem 2000; 275:12298-12305; PMID:10766869; https://doi.org/ 10.1074/jbc.275.16.12298 [DOI] [PubMed] [Google Scholar]
- [23].Verrijdt G, Tanner T, Moehren U, Callewaert L, Haelens A, Claessens F. The androgen receptor DNA-binding domain determines androgen selectivity of transcriptional response. Biochem Soc Trans 2006; 34:1089-1094; PMID:17073757; https://doi.org/ 10.1042/BST0341089 [DOI] [PubMed] [Google Scholar]
- [24].Giwercman YL, Ivarsson S, Richthoff J, Lundin KB, and Gewircman A. A novel mutation in the D-box of the androgen receptor gene (S597R) in two unrelated individuals Is associated with both normal phenotype and severe PAIS. Horm Res 2004; 61(2):58-62; PMID:14646391; https://doi.org/ 10.1159/000075240 [DOI] [PubMed] [Google Scholar]
- [25].Nordenskjöld A, Friedman E, Tapper-Persson M, Söderhäll C, Leviav A, Svensson J, Anvret M. Screening for mutations in candidate genes for hypospadias. Urol Res 1999; 27(1):49-55; PMID:10092153; https://doi.org/ 10.1007/s002400050088 [DOI] [PubMed] [Google Scholar]
- [26].Wang X, Yin L, Rao P, Stein R, Harsch KM, Lee Z, Heston WD. Targeted treatment of prostate cancer. J Cell Biochem 2007; 102:571-579; PMID:17685433; https://doi.org/ 10.1002/jcb.21491 [DOI] [PubMed] [Google Scholar]
- [27].Adams BD, Cowee DM, White BA. The role of miR-206 in the epidermal growth factor (EGF) induced repression of estrogen receptor-α (ERα) signaling and a luminal phenotype in MCF-7 breast cancer cells. Mol Endocrinol 2009; 23(8):1215-1230; PMID:19423651; https://doi.org/ 10.1210/me.2009-0062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lin HK, Hu YC, Yang L, Altuwaijri S, Chen YT, Kang HY, Chang C. Suppression versus induction of androgen receptor functions by the phosphatidylinositol 3-kinase/akt pathway in prostate cancer LNCaP cells with different passage numbers. J Biol Chem 2003; 278:50902-50907; PMID:14555644; https://doi.org/ 10.1074/jbc.M300676200 [DOI] [PubMed] [Google Scholar]
- [29].Zhu ML, Kyprianou N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr Relat Cancer 2008; 15:841-849; PMID:18667687; https://doi.org/ 10.1677/ERC-08-0084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yang Z, Wang L. Regulation of microRNA expression and function by nuclear receptor signaling. Cell Biosci 2011; 1:1-9; PMID:21711586; https://doi.org/ 10.1186/2045-3701-1-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Deng JH, Deng Q, Kuo CH, Delaney SW, Ying SY. MiRNA targets of prostate cancer. Methods in Molecular Biology 2012; 936:357-369; https://doi.org/ 10.1007/978-1-62703-083-0_27 [DOI] [PubMed] [Google Scholar]
- [32].Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005; 120:15-20; PMID:15652477; https://doi.org/ 10.1016/j.cell.2004.12.035 [DOI] [PubMed] [Google Scholar]
- [33].Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 2009; 11(3):228-234; PMID:19255566; https://doi.org/ 10.1038/ncb0309-228 [DOI] [PubMed] [Google Scholar]
- [34].Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem 2010; 79:351-379; PMID:20533884; https://doi.org/ 10.1146/annurev-biochem-060308-103103 [DOI] [PubMed] [Google Scholar]
- [35].Adams BD, Parsons C, Zhang WC, Slack FJ. Targeting noncoding RNAs in disease. J Clin Invest 2017; 127(3):761-771; PMID:28248199; https://doi.org/ 10.1172/JCI84424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Esquela-Kerscher A, Slack FJ. Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer 2006; 6:259-269; PMID:16557279; https://doi.org/ 10.1038/nrc1840 [DOI] [PubMed] [Google Scholar]
- [37].Nohata N, Hanazawa T, Enokida H, Seki N. microRNA-1/133a and microRNA-206/133b clusters: dysregulation and functional roles in human cancers. Oncotarget 2012; 3(1):9-21; PMID:22308266; https://doi.org/ 10.18632/oncotarget.424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100(1):57-70; PMID:10647931; https://doi.org/ 10.1016/S0092-8674(00)81683-9 [DOI] [PubMed] [Google Scholar]
- [39].Herranz H, Cohen SM. MicroRNAs and gene regulatory networks: managing the impact of noise in biological systems. Genes Dev 2010; 24(13):1339-1344; PMID:20595229; https://doi.org/ 10.1101/gad.1937010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Martinez NJ, Walhout AJ. The interplay between transcription factors and microRNAs in genome-scale regulatory networks. Bioessays 2009; 31(4):435-445; PMID:19274664; https://doi.org/ 10.1002/bies.200800212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tsang J, Zhu J, van Oudenaarden A. MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol Cell 2007; 26(5):753-767; PMID:17560377; https://doi.org/ 10.1016/j.molcel.2007.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA et al.. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013; 499:214-218; PMID:23770567; https://doi.org/ 10.1038/nature12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Roychowdhury S, Chinnalyan AM. Translating cancer genomes and transcriptomes for precision oncology. CA Cancer J Clin 2016; 66(1):75-88; PMID:26528881; https://doi.org/ 10.3322/caac.21329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet 2009; 10:704-714; PMID:19763153; https://doi.org/ 10.1038/nrg2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Walter BA, Valera VA, Pinto PA, Merino MJ. Comprehensive microRNA profiling of prostate cancer. J Cancer 2013; 4(5):350-357; PMID:23781281; https://doi.org/ 10.7150/jca.6394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Das DK, Naidoo M, Ilboudo A, Park JY, Ali T, Krampis K, Robinson BD, Osborne JR, Ogunwobi OO. miR-1207-3p regulates the androgen receptor in prostate cancer via FNDC1/fibronectin. Exp Cell Res 348(2):190-200; PMID:27693493; https://doi.org/ 10.1016/j.yexcr.2016.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kachakova D, Mitkova A, Popov E, Popov I, Vlahova A, Dikov T, Christova S, Mitev V, Slavov C, Kaneva R. Combinations of serum prostate-specific antigen and plasma expression levels of let-7c, miR-30c, miR-141, and miR-375 as potential better diagnostic biomarkers for prostate cancer. DNA Cell Biol 2015; 34:189-200; PMID:25521481; https://doi.org/ 10.1089/dna.2014.2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science 2001; 294:853-858; PMID:11679670; https://doi.org/ 10.1126/science.1064921 [DOI] [PubMed] [Google Scholar]
- [49].Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol 2008; 9:219-230; PMID:18270516; https://doi.org/ 10.1038/nrm2347 [DOI] [PubMed] [Google Scholar]
- [50].Ambs S, Prueitt RL, Yi M, Hudson RS, Howe TM, Petrocca F, Wallace TA, Liu CG, Volinia S, Calin GA et al.. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res 2008; 68:6162-6170; PMID:18676839; https://doi.org/ 10.1158/0008-5472.CAN-08-0144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov 2010; 9:775-789; PMID:20885409; https://doi.org/ 10.1038/nrd3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Buscaglia LE, Li Y. Apoptosis and the target genes of microRNA-21. Chin J Cancer 2011; 30:371-380; PMID:21627859; https://doi.org/ 10.5732/cjc.30.0371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Shen J, Hruby GW, McKiernan JM, Gurvich I, Lipsky M, Benson MC, Santella RM. Dysregulation of circulating microRNAs and prediction of aggressive prostate cancer. Prostate 2012; 72:1469-1477; PMID:22298119; https://doi.org/ 10.1002/pros.22499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Liu W, Le A, Hancock C, Lane AN, Dang CV, Fan TW, Phang JM. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-Myc. Proc Natl Acad Sci USA 2012; 109:8983-8988; PMID:22615405; https://doi.org/ 10.1073/pnas.1203244109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wang WL, Welsh J, Tenniswood M. 1,25-dihydroxyvitaminD3modulateslipidmetabolisminprostate cancer cells through miRNA mediated regulation of PPARA. J Steroid Biochem Mol Biol 2013; 136:247-251; PMID:23059473; https://doi.org/ 10.1016/j.jsbmb.2012.09.033 [DOI] [PubMed] [Google Scholar]
- [56].Ostling P, Leivonen SK, Aakula A, Kohonen P, Makela R, Hagman Z, Edsjo A, Kangaspeska S, Edgren H, Nicorici D et al.. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res 2011; 71:1956-1967; PMID:21343391; https://doi.org/ 10.1158/0008-5472.CAN-10-2421 [DOI] [PubMed] [Google Scholar]
- [57].Kong D, Heath E, Chen W, Cher M, Powell I, Heilbrun L, Li Y, Ali S, Sethi S, Hassan O et al.. Epigenetic silencing of miR-34a in human prostate cancer cells and tumor tissue specimens can be reversed by BR-dim treatment. Am J Transl Res 2012; 4:14-23; PMID:22347519 [PMC free article] [PubMed] [Google Scholar]
- [58].Hagman Z, Haflidadottir BS, Ceder JA, Larne O, Bjartell A, Lilja H, Edsjo A, Ceder Y. miR-205 negatively regulates the androgen receptor and is associated with adverse outcome of prostate cancer patients. Br J Cancer 2013; 108:1668-1676; PMID:23571738; https://doi.org/ 10.1038/bjc.2013.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Helwak A, Kudla G, Dudnakova T, Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013; 153:654-665; PMID:23622248; https://doi.org/ 10.1016/j.cell.2013.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Argarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015; 4:e05005; PMID:26267216; https://doi.org/ 10.7554/eLife.05005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mo W, Zhang J, Li X, Meng D, Gao Y, Yang S, Wan X, Zhou C, Guo F, Huang Y et al.. Identification of novel AR-targeted MicroRNAs mediating androgen signaling through critical pathways to regulate cell viability in prostate Cancer. PLos One 8(2):e56592; PMID:23451058; https://doi.org/ 10.1371/journal.pone.0056592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ficazzola MA, Fraiman M, Gitlin J, Woo K, Melamed J, Rubin MA, Walden PD. Antiproliferative B cell translocation gene 2 protein is down-regulated post-transcriptionally as an early event in prostate carcinogenesis. Carcinogenesis 2001; 22:1271-1279; PMID:11470758; https://doi.org/ 10.1093/carcin/22.8.1271 [DOI] [PubMed] [Google Scholar]
- [63].Coppola V, Musumeci M, Patrizii M, Cannistraci A, Addario A, Maugeri-Sacca M, Biffoni M, Francescangeli F, Cordenonsi M, Piccolo S et al.. BTG2 loss and miR-21 upregulation contribute to prostate cell transformation by inducing luminal markers expression and epithelial-mesenchymal transition. Oncogene 2013; 32:1843-1853; PMID:22614007; https://doi.org/ 10.1038/onc.2012.194 [DOI] [PubMed] [Google Scholar]
- [64].Jalava SE, Urbanucci A, Latonen L, Waltering KK, Sahu B, Janne OA, Seppala J, Lahdesmaki H, Tammela TL, Visakorpi T. Androgen-regulated miR-32 targets BTG2 and is overexpressed in castration-resistant prostate cancer. Oncogene 2012; 31:4460-4471; PMID:22266859; https://doi.org/ 10.1038/onc.2011.624 [DOI] [PubMed] [Google Scholar]
- [65].Sun D, Lee YS, Malhotra A, Kim HK, Matecic M, Evans C, Jensen RV, Moskaluk CA, Dutta A. miR-99 family of microRNAs suppresses the expression of prostate-specific antigen and prostate cancer cell proliferation. Cancer Res 2011; 71:1313-1324; PMID:21212412; https://doi.org/ 10.1158/0008-5472.CAN-10-1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sun D, Layer R, Mueller AC, Cichewicz MA, Negishi M, Paschal BM, Dutta A. Regulation of several androgen-induced genes through the repression of the miR-99a/let-7c/miR-125B-2 miRNA cluster in prostate cancer cells. Oncogene 2014; 33:1448-1457; PMID:23503464; https://doi.org/ 10.1038/onc.2013.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Andersen CL, Monni O, Wagner U, Kononen J, Barlund M, Bucher C, Haas P, Nocito A, Bissig H, Sauter G et al.. High-throughput copy number analysis of 17q23 in 3520 tissue specimens by fluorescence in situ hybridization to tissue microarrays. Am J Pathol 2002; 161:73-79; PMID:12107091; https://doi.org/ 10.1016/S0002-9440(10)64158-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ribas J, Ni X, Haffner M, Wentzel EA, Salmasi AH, Chowdhury WH, Kudrolli TA, Yegnasubramanian S, Luo J, Rodriguez R et al.. miR-21: An androgen receptor-regulated microRNA that promotes hormone-dependent and hormone-independent prostate cancer growth. Cancer Res 2009; 69:7165-7169; PMID:19738047; https://doi.org/ 10.1158/0008-5472.CAN-09-1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Li T, Li RS, Li YH, Zhong S, Chen YY, Zhang CM, Hu MM, Shen ZJ. miR-21asanindependent biochemical recurrence predictor and potential therapeutic target for prostate cancer. J Urol 2012; 187:1466-1472; PMID:22341810; https://doi.org/ 10.1016/j.juro.2011.11.082 [DOI] [PubMed] [Google Scholar]
- [70].Adams BD, Furneaux H, White BA. The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-α (ERα) and represses ERα messenger RNA and protein expression in breast cancer cell lines. Mol Endocrinol 2007; 2(5):1132-1147; PMID:17312270; https://doi.org/ 10.1210/me.2007-0022 [DOI] [PubMed] [Google Scholar]
- [71].Kim HK, Lee YS, Sivaprasad U, Malhotra A, Dutta A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J Cell Biol 2006; 174(5):677-687; PMID:16923828; https://doi.org/ 10.1083/jcb.200603008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Stahlhut C, Suárez Y, Lu J, Mishima Y, Giraldez AJ. miR-1 and miR-206 regulate angiogenesis by modulating VegfA expression in zebrafish. Development 2012; 139:4356-4365; PMID:23132244; https://doi.org/ 10.1242/dev.083774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Goljanek-Whysall K, Pais H, Rathjen T, Sweetman D, Dalmay T, Münsterberg A. Regulation of multiple target genes by miR-1 and miR-206 is pivotal for C2C12 myoblast differentiation. J Cell Sci 2012; 125(Pt 15):3590-3600; PMID:22595520; https://doi.org/ 10.1242/jcs.101758 [DOI] [PubMed] [Google Scholar]
- [74].Lin CY, Lee HC, Fu CY, Ding YY, Chen JS, Lee MH, Huang WJ, Tsai HJ. miR-1 and miR-206 target different genes to have opposing roles during angiogenesis in zebrafish embryos. Nat Commun 2013; 4:28229; PMID:24264597; https://doi.org/ 10.1038/ncomms3829 [DOI] [PubMed] [Google Scholar]
- [75].Hudson R, Yi M, Esposito D, Watkins S, Hurwitz A, Yfantis H, Lee DH, Borin J, Naslund M, Alexander R et al.. MicroRNA-1 is a candidate tumor suppressor and prognostic marker in human prostate cancer. Nucleic Acids Res 2011; 40(8):3689-3703; PMID:22210864; https://doi.org/ 10.1093/nar/gkr1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ritland JC, Zhang F, Pederson T. MicroRNA-206 colocalizes with ribosome-rich regions in both the nucleolus and cytoplasm of rat myogenic cells. PNAS 2006; 103(50):18957-18962; PMID:17135348; https://doi.org/ 10.1073/pnas.0609466103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Kondo N, Toyama T, Sugiura H, Fujii Y, Yamashita H. miR-206 expression is down-regulated in estrogen receptor α–positive human breast cancer. Cancer Res 2008; 68(13):5004-5008; PMID:18593897; https://doi.org/ 10.1158/0008-5472.CAN-08-0180 [DOI] [PubMed] [Google Scholar]
- [78].Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M et al.. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA 2006; 103:2257-2261; PMID:16461460; https://doi.org/ 10.1073/pnas.0510565103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Elliman SJ, Howley BV, Mehta DS, Fearnhead HO, Kemp HM, Barkley LR. Selective repression of the oncogene cyclin D1 by the tumor suppressor miR-206 in cancers. Oncogenesis 2014; 3:e113; PMID:25111862; https://doi.org/ 10.1038/oncsis.2014.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Singh A, Happel C, Manna SK, Acquaah-Mensah G, Carrerero J, Kumar S, Nasipuri P, Krausz KW, Wakabayashi N, Dewi R et al.. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J Clin Investig 2013; 123:2921-2934; PMID:23921124; https://doi.org/ 10.1172/JCI66353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Song G, Zhang Y, Wang L. MicroRNA-206 targets notch3, activates apoptosis, and inhibits tumor cell migration and focus formation. JBC 2009; 284(46):31921-31927; PMID:19723635; https://doi.org/ 10.1074/jbc.M109.046862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Wu W, He Y, Feng X, Ye S, Wang H, Tan W, Yu C, Hu J, Zheng R, Zhou Y. MicroRNA-206 is involved in the pathogenesis of ulcerative colitis via regulation of adenosine A3 receptor. Oncotarget 2017; 8(1):705-721; PMID:27893428; https://doi.org/ 10.18632/oncotarget.13525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Shaffer PL, Jivan A, Dollins DE, Claessens F, Gewirth DT. Structural basis of androgen receptor binding to selective androgen response elements. PNAS 2004; 101(14):4758-4763; PMID:15037741; https://doi.org/ 10.1073/pnas.0401123101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Denaver S, Helsen C, Thorrez L, Haelens A, Claessens F. The rules of DNA recognition by the androgen receptor. Mol Endocrinol 2010; 24(5):898-913; PMID:20304998; https://doi.org/ 10.1210/me.2009-0310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Haelens A, Verrijdt G, Callewaert L, Christiaens V, Schauwaers K, Peeters B, Rombauts W, Claessens F. DNA recognition by the androgen receptor: evidence for an alternative DNA-dependent dimerization, and an active role of sequences flanking the response element on transactivation. Biochem J 2003; 369:141-151; PMID:12350223; https://doi.org/ 10.1042/bj20020912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res 2002; 12(6):996-1006; PMID:12045153; https://doi.org/ 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kent WJ. BLAT - the BLAST-like alignment tool. Genome Res 2002; 12(4):656-664; PMID:11932250; https://doi.org/ 10.1101/gr.229202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Pan YF, Wansa KD, Liu MH, Zhao B, Hong SZ, Tan PY, Lim KS, Bourque G, Liu ET, Cheung E. Regulation of estrogen receptor-mediated long range transcription via evolutionarily conserved distal response elements. J Biol Chem 2008; 283(47):32977-32988; PMID:18728018; https://doi.org/ 10.1074/jbc.M802024200 [DOI] [PubMed] [Google Scholar]
- [89].Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH et al.. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009; 462(7269):58-64; PMID:19890323; https://doi.org/ 10.1038/nature08497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Miyachi M, Tsuchiya K, Yoshida H, Yagyu S, Kikuchi K, Misawa A, Iehara T, Hosoi H. Circulating muscle-specific microRNA, miR-206, as a potential diagnostic marker for rhabdomyosarcoma. Biochem Biophys Res Commun 2010; 400:89-93; PMID:20696132; https://doi.org/ 10.1016/j.bbrc.2010.08.015 [DOI] [PubMed] [Google Scholar]
- [91].Bruneteau G, Simonet T, Bauche S, Mandjee N, Malfatti E, Girard E, Tanguy ML, Behin A, Khiami F, Sariali E et al.. Muscle histone deacetylase 4 upregulation in amyotrophic lateral sclerosis: potential role in reinnervation ability and disease progression. Brain 2013; 136:2359-2368; PMID:23824486; https://doi.org/ 10.1093/brain/awt164 [DOI] [PubMed] [Google Scholar]
- [92].Novák J, Kružliak P, Bienertová-Vašků J, Slabý O, Novák M. microRNA-206: a promising theranostic marker. Theranostics 2014; 4(2):119-133; PMID:24465270; https://doi.org/ 10.7150/thno.7552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Blume-Jensen P, Berman DM, Rimm DL, Shipitsin M, Putzi M, Nifong TP, Small C, Choudhury S, Capela T, Coupal L et al.. Development and Clinical Validation of an in situ biopsy-based multimarker assay for risk stratification in prostate cancer. Clin Cancer Res 2015; 21(11):2591-2600; PMID:25733599; https://doi.org/ 10.1158/1078-0432.CCR-14-2603 [DOI] [PubMed] [Google Scholar]
- [94].Lewis H, Lance R, Troyer D, Beydoun H, Hadley M, Orians J, Benzine T, Madric K, Semmes OJ, Drake R et al.. miR-888 is an expressed prostatic secretions-derived microRNA that promotes prostate cell growth and migration. Cell Cycle 2014; 13(2):227-239; PMID:24200968; https://doi.org/ 10.4161/cc.26984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Knudsen BS, Kim HL, Erho N, Shin H, Alshalalfa M, Lam LL, Tenggara I, Chadwich K, Kwast TV, Fleshner N et al.. Application of a clinical whole-transcriptome assay for staging and prognosis of prostate cancer diagnosed in needle core biopsy specimens. J Mol Diagn 2016; 18(3):395-406; PMID:26945428; https://doi.org/ 10.1016/j.jmoldx.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Jenster G, van der Korput HA, Trapman J, Brinkmann AO. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J Biol Chem 1995; 270:7341-7346; PMID:7706276; https://doi.org/ 10.1074/jbc.270.13.7341 [DOI] [PubMed] [Google Scholar]