

ABSTRACT
Dengue virus (DV) is the etiologic agent of dengue fever, the most significant mosquito-borne viral disease in humans. Most DV vaccine approaches are focused on generating antibody mediated responses; one such DV vaccine is approved for use in humans but its efficacy is limited. While it is clear that T cell responses play important role in DV infection and subsequent disease manifestations, fewer studies are aimed at developing vaccines that induce robust T cells responses. Potent T cell based vaccines require 2 critical components: the identification of specific T cell stimulating MHC associated peptides, and an optimized vaccine delivery vehicle capable of simultaneously delivering the antigens and any required adjuvants. We have previously identified and characterized DV specific HLA-A2 and -A24 binding DV serotypes conserved epitopes, and the feasibility of an epitope based vaccine for DV infection. In this study, we build on those previous studies and describe an investigational DV vaccine using T cell epitopes incorporated into a calcium phosphate nanoparticle (CaPNP) delivery system. This study presents a comprehensive analysis of functional immunogenicity of DV CaPNP/multipeptide formulations in vitro and in vivo and demonstrates the CaPNP/multipeptide vaccine is capable of inducing T cell responses against all 4 serotypes of DV. This synthetic vaccine is also cost effective, straightforward to manufacture, and stable at room temperature in a lyophilized form. This formulation may serve as an effective candidate DV vaccine that protects against all 4 serotypes as either a prophylactic or therapeutic vaccine.
KEYWORDS: calcium phosphate nanoparticles, CTLs, dengue virus, peptide vaccine
Introduction
Dengue virus (DV) is a member of the Flaviviridae family and is characterized by a single stranded RNA genome enclosed within a spherical enveloped virion. Four distinct serotypes of DV circulate globally with most endemic countries reporting circulation of all 4 serotypes.1 The incidence of DV infections has spread dramatically around the world in recent decades; although recent estimates indicate that roughly 390 million people are infected with DV each year,2 over 3 billion people are actually at risk of being infected.3 As the climate continues to warm and the mosquito vector of DV continues to move northward4,5 both the number of infections and the number of people at risk of infection will continue to rise. Therefore, developing efficacious anti-viral treatments and/or vaccines to prevent infection from these viruses is of the utmost importance.
One of the most challenging aspects of DV immunotherapy is that although recovery from infection by one DV serotype may provide lifelong immunity against that particular strain, cross-protective immunity to other serotypes is only partial and temporary. Subsequent infections by other serotypes increase the risk of developing severe dengue fever, or dengue hemorrhagic fever, mediated by antibody dependent enhancement (ADE) of infection.6,7 This is compounded by the fact that there are no specific anti-viral treatments of DV infection. Clinical management of infection is based only on supportive therapy. Recent improvements in case management have reduced the fatality rates in hospitalized dengue illness to less than 1%.8 Other primary forms of combating the virus have targeted the viral vector,9 though the data supporting positive impact on incidence of DV infection is limited.10 In terms of prophylactic measures, there is one live attenuated tetravalent dengue vaccine (CYD-TDV, or Dengvaxia) first approved for use in Mexico, Philippines and Brazil in 2015 and many other countries thereafter. CYD-TVD induces neutralizing antibodies against all 4 DV serotypes where induction of high-titer neutralizing antibodies can provide temporary cross-protection to these serotypes, lasting about 2 years.11,12 However, CYD-TVD shows some shortcomings. First, the efficacy of CYD-TVD for confirmed dengue cases was surprisingly lower in seronegative individuals than in seropositive individuals,13 perhaps reflecting a “boosting” phenomenon that temporality provides additional cross-protection. Furthermore, the rate of hospitalization of sero-negative individuals was considerably higher, especially among children younger than 9 years old.14 This observation was attributed to CYD-TDV inducing non-protective dengue antibodies that enhance infection.15 As such, there still remains a significant need to develop efficacious prophylactics and immunotherapies for DV infections.
There are several variables important to developing a successful DV vaccine. Inducing both humoral and cellular immunity will be essential in forming a safe and effective cross-protective DV vaccine. Therefore it is necessary to use tools that identify conserved B-cell and T-cell epitopes within viral proteins that stimulate protective immune responses16 but not the immune amplification17,18 observed during antibody mediated enhancement of DV infection. T cell based vaccines are an attractive alternative strategy as they can be used as ‘stand-alone’ vaccines or be paired with current and future anti-viral treatments and/or the CYD-TDV vaccine. CD8+ cytotoxic T lymphocytes (CTLs) are a major contributor of protection against DV infection.6,7,19 DV specific CD8+ T cells were detected in patients after natural infection20-24 and after attempts at vaccination25 with some level of cross-reactivity between the strains. Studies in children have indicated that CD8+ T cell mediated secretion of IFN-γ and TNF-α was more robust in asymptomatic or subclinical infections compared with symptomatic or severe disease.16 In line with those observations, a murine model of DV infection demonstrated that CD8+ T cells play a major role in viral clearance as depletion of these cells significantly increased viral titers in the infected animals.26 Development of an efficacious DV vaccine would require CD4+ and/or CD8+ T-cell responses, not only to successfully protect against infection by each serotype but also against ADE. As discussed above, a major limitation in DV vaccines which solely depend on induction of neutralizing antibodies is the potential for non-neutralizing antibody mediated enhancement of DV infection. Zellweger et al. (2014) demonstrated that DV specific CD8+ T cells were able to prevent ADE in mice infected with DV,27 thus, it is plausible to hypothesize a similar effect in humans infected with DV. Ideally after vaccination, the T cell memory response should eliminate the infected cells, halting the spread of infection that would be enhanced by ADE. In vitro studies from our laboratory have demonstrated that CD8+ T cells that recognize epitopes shared across all 4 DV serotypes could be generated.28 We have also shown that these T cells are able to be activated via selected HLA-A2+ and HLA-A24+ in both healthy, seronegative, individuals and in seropositive individuals who have been previously infected with DV.29 These studies suggested that a vaccine that activates a CTL response against DV and another that can be paired with an efficient humoral immune-stimulating vaccine could be used to induce sustained immunity against DV.
The rationale for prophylactic vaccination against DV begins with the observations that natural infection protects against exogenous re-infection with the same serotype.Although some studies report that the NS3 protein is an immunodominant target of CD8+ T-cell responses to natural infection, little is known about T-cell epitopes from this nonstructural protein.30,31 Several groups have attempted to identify T cell epitopes by either screening overlapping peptides from structural and nonstructural DV proteins, including preM, E, and NS3, or by predicting major histocompatibility complex (MHC) peptide binding motifs of DV proteins,32,33 NS1, NS2A.25,34 While these studies have revealed a few potential candidate epitopes, as we have also accomplished,28 a comprehensive analysis of naturally presented epitopes on the infected cells has not been undertaken or reported to the best of our knowledge.
Implementation of the approach to produce broad, cross-protective immunity involves the identification of conserved CD8+ T cell epitopes that can be induced in most members of the population and that can maintain epitope-specific CD8+ T cells capable of controlling the infection. Activation of T cells depends on complex interactions between the innate and adaptive immune systems; enhancing innate immune responses is thought to drive more robust adaptive immunity, and a primary method of enhancing the innate immune activation during vaccination is through the use of adjuvants. Currently, only a few adjuvants have been approved for use in humans in the US including alum and limited use of a few lipid based emulsions.35 To the best of our knowledge, there is no adjuvant that is licensed for use in peptide based antigens to date. In addition, a variety of vaccine delivery systems are available but many have issues with stability which directly impacts effectiveness of a vaccine.36 An ideal vaccine delivery system activates a strong, broad, and persistent cellular and humoral immunity, while also stimulating an improved immunological memory. An ideal vaccine must also improve immune responses in people with reduced or suppressed immunity, and be capable of broadening the immune response to allow recognition of pathogenic strain variants, while remaining cost effective. Use of adjuvants and/or antigen delivery systems are considered to provide dose-sparing to allow immunization of more people using smaller amount of antigen, reduce the need for booster dosing, improve vaccine efficacy, and consequently to reduce the cost of vaccination. Many vaccine delivery systems, including liposomal formulations,37,38 virus-like particle (VLP) vaccines, DNA vaccines, viral vector-based vaccines,39 and synthetic gold nanoparticles (NP),40 are currently being investigated in an effort to achieve long-term protection against a broad range of viral subtypes. More recently, liposomes,41,42 VLP43 and gold NP44 have been conjugated with synthetic peptides and tested in transgenic mice models, which show effective CTL immune response. Several delivery systems, such as immunostimulatory complex (ISCOM-based adjuvants),45 virosomes (viral vector)46 and liposomes,47 have been investigated further as potential vaccine formulations with peptides in stage I clinical trials, which all produce a potent T cell immunity. Clincial studies with HLA-restricted peptides, selected by virtue of being naturally presented by cancer, when administered in combination with Granulocyte-macrophage colony-stimulating factor (GM-CSF), elicit specific vaccine-induced immune responses in renal cell cancer that are associated with clinical benefit.48 Those studies warrant further clinical development of the vaccine delivery systems for synthetic peptide antigens.
Calcium phosphate nanoparticles (CaPNPs) have shown promise for use both as an adjuvant,49,50 and as a drug delivery vehicle.51-55 In preclinical safety and toxicity studies,49,53 CaPNPs were shown to be safe for administration via intramuscular, subcutaneous, oral, or inhalation routes. Although the clinical safety data have not been published to date yet, the company who developed the CaPNP technology has reported in 2 press releases that CaPNP indicated no toxicity, inflammation, or allergic reaction in Phase I safety/toxicity studies conducted in the US (unpublished). Therefore, there was great interest in investigating the potential of CaPNPs for development of vaccines against DV infection. Building on our prior work of antigenic peptides discovered from DV,28,29 we aimed to investigate the beneficial properties of CaPNPs in terms of stability, capacity to incorporate peptide antigens, simplicity of formulation protocols, and T cell responses. The HLA-A2+ DV peptides selected for this work were previously identified through a comprehensive analysis of naturally presented epitopes on infected cells using an immunoproteomic approach.28,29 These novel HLA-A2+ DV-specific peptides are derived from conserved regions of DV proteins. We previously demonstrated that DV peptides derived from conserved regions are capable of inducing cross-reactive T cell responses,28 a benefit when designing a vaccine to protect against multiple strains. We formulated the vaccine by incorporating the characterized antigenic peptides to pre-formed CaPNPs through physical adsorption. We assessed the ability of the CaPNP/peptide formulation to induce CD8+ T cell responses, in both in vitro and in vivo experiments, for the potential development of a T cell vaccine against DV infection.
Results
Selection of calcium phosphate nanoparticle (CaPNP) for vaccine formulation
In previous investigations, influenza A (H1N1) vaccines formulated with a fixed antigen dose and various concentrations of CaPNPs (0.15% – 0.8%) indicated the maximal virus-specific antibody response for the vaccine containing 0.3% CaPNPs (there was no significant difference between the 0.3% and higher concentration of CaPNPs). Importantly, the antibody response produced by the 0.3% CaPNP was approximately 4-fold higher than that of vaccine formulations containing no adjuvant.56 The vaccine formulated with 0.3% CaPNPs has also provided greater than 80% protection against infection with a fatal dose of A/CA/04/2009 (H1N1pdm) virus in challenge studies.56 Using this data as a guide, we investigated the optimal CaPNP concentration suitable for peptide antigen delivery and functional T cell activation. We used a well characterized peptide, SIINFEKL (SIIN) from ovalbumin,57,58 as the model. SIIN formulated with 0.3% or 0.8% CaPNPs were pulsed onto Lkb fibroblasts and the ability of the cells to internalize, process, and present the SIIN peptide was assessed by flow cytometry and T cell hybridoma assays as described elsewhere.48,49 The following formulations were tested at 3 different concentrations of SIIN (0.1, 1, and 2 µg/ml): 1) 0.8% CaPNPs alone, 2) 0.8% CaPNPs/SIIN, 3) 0.3% CaPNPs alone, 4) 0.3% CaPNPs/SIIN, and as controls, 5) free SIIN peptide, and 6) peptide unpulsed samples. Angel antibody, an antibody that recognize the SIIN peptide MHC-I complex,59 staining of Lkb cells pulsed with the various formulations demonstrated that CaPNP/peptide conjugates were taken up and processed, and SIIN peptide was presented in conjunction with the Kb MHC-I molecules (Supplementary Fig 1A and 1C). There were no significant differences observed in levels of SIIN/Kb complexes on the cell surface between formulations nor were there differences observed using various concentrations of SIIN peptide (Fig. S1A).
Figure 1.
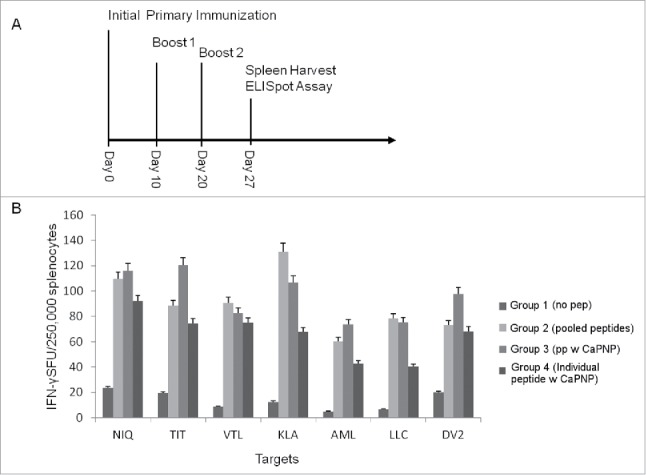
(A) Immunization scheme for vaccine injections. Timeline for mice immunization and sample analysis for CTL responses generated in vivo using HLA-A2+ transgenic mice. All the in vivo experiments were performed with the protocol depicted. (B) DV specific CaPNP/multipeptide formulation stimulate CD8+ T cell activation in vivo. Mice were grouped (n = 3) as: 1) unimmunized (PBS control); 2) pooled peptide emulsified in ISA 51; 3) CaPNP/multipeptide formulation with GlcNAc; or 4) pooled formulations of CaPNP/individual peptides with GlcNAc. Spleens were harvested and splenocytes were isolated then co-cultured with HepG2 targets pulsed with either individual peptides (peptides depicted as the first 3 residues: NIQ, TIT, VTL, KLA, AML, LLC) or infected with DV2 for use in the ELISpot assay. Data represented as mean ± S.D (n = 3) of SFU per 2.5 × 105 splenocytes.
Similarly, the processing and presentation of the CaPNP/SIIN formulations resulted in SIIN-specific T cell activation (Fig. S1B) as measured by the standard B3Z T cell hybridoma assay.59 All CaPNP/SIIN formulations also induced higher levels of T cell activation than the free peptide control. Therefore, we used 0.3% CaPNP concentration in all experiments moving forward.
DV specific CaPNP/multipeptide vaccine formulation
Our previous studies demonstrated that DV specific free peptides can activate CD8+T cells in vitro.28,29 Here, we evaluated if the same peptides formulated with nanoparticles could enhance DV specific T cell response. A total of 6 HLA-A2+, A2+/A24+ dual binding DV peptides (Table 1) were selected and formulated in 0.3% CaPNP particle suspension either individually or combined as a pool of multiple peptides. N-acetylglucosamine (GlcNAc) was also included in the formulations for 2 reasons: first, as a surface modifying agent to increase surface-binding of peptides to CaPNPs,51,60 and second, to act as an adjuvant for targeting the mannose receptors on antigen presenting cells to facilitate antigen uptake and processing.61 We first tested the peptide formulations in vivo using HLA-A2 transgenic mice (Fig. 1A). Mice were divided into test groups and immunized 3 times over the period of 3 weeks: 1) unimmunized (PBS injection as control), 2) pooled free peptides emulsified with Montanide ISA 51 (ISA 51, an adjuvant carrier, has been proved to enhance antigen specific antibody titers and CTL response in therapeutic and prophylactic clinical trials62), 3) pooled formulations of individual peptides formulated in CaPNPs (CaPNP/individual peptide formulation), and 4) pool of free peptides formulated together in CaPNP (CaPNP/multipeptide formulation). On day 27 post-last immunizations, spleens were harvested and the splenocytes were isolated to assess antigen specific T cell responses. Splenocytes were cultured with HepG2 target cells that were either pulsed with individual peptides (Table 1) or infected with DV serotype 2 (DV2) in IFN-γ ELISpot assays (Fig. 1B). The data indicate that the CaPNP formulations induced robust CD8+ T cell activation, both as individual peptide formulations pooled together or as a pool of individual peptides formulated together (referred as CaPNP/multipeptide here after). Importantly, CaPNP/multipeptide formulation induced the highest levels of IFN-γ secretion against DV2 infected HepG2 targets and against some individual peptide pulsed HepG2 target groups (depicted as first 3 residues – NIQ, TIT, and AML) when compared with pooled formulations of CaPNP/individual peptides and the free peptides emulsified with ISA 51 adjuvant (positive control). Therefore, the CaPNP/multipeptide formulation was selected for further optimization and use in subsequent in vitro and in vivo functional characterization studies.
Table 1.
Dengue virus specific MHC class I associated peptides identified previously by immunoproteomics.28,29
To further optimize the CaPNP/multipeptide formulation, we evaluated different concentrations of peptides necessary to induce optimal T cell responses in an HLA-A2+ transgenic mouse model. The mice were immunized in 5 groups: 1) unimmunized (PBS control), 2) 10 µg pooled free peptides (10 µg each peptide/150 µL per mouse) emulsified with ISA51, 3) 50 µg pooled free peptides (50 µg each peptide/150 µL per mouse) emulsified with ISA51, 4) 10 µg CaPNP/multipeptide formulation (10 µg each peptide/150 µL CaPNP per mouse), and 5) 50 µg CaPNP/multipeptide formulation (50 µg each peptide/150 µl CaPNP per mouse). As described previously, the mice were immunized 3 times with the designated vaccines and then the spleens were harvested. The splenocytes were cultured with HepG2 target cells pulsed with pooled peptides (Table 1) or infected with DV2 in an IFN-γ ELISPOT assay. As shown in Fig. 2, there were significant differences in T cell responses against DV2 infected HepG2 targets between the treatment groups as compared with the control group. Specifically, the 50 µg free peptides/ISA 51 group and the 10 µg CaPNP/multipeptide formulation (groups 3 and 4, respectively) elicited higher T cell responses when compared with the unimmunized group (group 1; P<0.001, P<0.0001 respectively; n = 3, one way ANOVA). Furthermore, the 10 µg CaPNP/multipeptide formulation (group 4) demonstrated a higher CTL response than 10 µg pooled free peptides emulsified with ISA 51 group (group 2), which underscores the efficacy of the CaPNP delivery system (P<0.01, n = 3). Overall, the lower concentration of CaPNP/multipeptide formulation (10 µg each peptide/mice) (group 4) generated the highest T cell responses against DV2 infected targets when compared with the higher 50 µg CaPNP/multipeptide formulation group (group 5; P < 0.05, n = 3). In contrast, the free peptides formulated with ISA 51 generated the highest response at higher peptide concentration (50 µg) compared with the lower concentration (10 µg). However, since the CaPNP formulated group with lower concentration showed higher response, we selected the 10 µg CaPNP/multipeptide formulation for further characterization.
Figure 2.
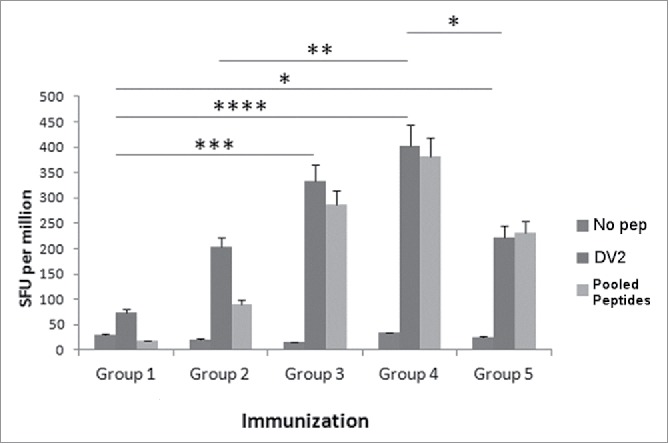
Various concentrations of DV CaPNP/multipeptide formulations stimulate CD8+ T cell activation in vivo. HLA-A2+ transgenic mice were immunized as in Fig. 1A with the following groups: Group 1) unimmunized (PBS control); Group 2) 10µg peptide/150µl PBS emulsified in ISA 51 per mouse; Group 3) 50µg peptide/150µl PBS emulsified in ISA 51 per mouse; Group 4) 10µg peptide/150 µl CaPNP with 1XGlcNAc per mouse; Group 5) 50µg peptide/ 150 µl CaPNP with 1XGlcNAc per mouse. Splenocytes were harvested and co-cultured with HepG2 targets that were either pulsed with no peptides (negative control), pooled peptides (PP including NIQ, TIT, VTL, KLA, AML, LLC) or infected with DV2 for use in the ELISpot assay. Data represented as mean ± S.D (n = 3) of SFU per 1 million splenocytes. * Represents P values: * P<0.05, ** P<0.01, ***P<0.001, **** P<0.0001.
Next, we compared the T cell responses generated by CaPNP/multipeptide formulation in the presence of various concentrations of GlcNAc in HLA-A2+ transgenic mice to determine the effect of adjuvant in the formulation. The mice were immunized in 5 groups: 1) unimmunized (PBS control), 2) pooled free peptides (10 µg each peptide/150 µl per mouse) emulsified with ISA 51, 3) CaPNP/multipeptide formulation (10 µg each peptide/150 µl CaPNP per mouse), 4) CaPNP/multipeptide formulation with 1 × GlcNAc (10 µg each peptide/150 µl CaPNP per mouse); and 5) CaPNP/multipeptide formulation with 3 × GlcNAc (10 µg each peptide/150 µl CaPNP per mouse). Using the IFN-γ ELISPOT assay, as described previously, we evaluated CTL activation in all treatment groups directed against DV2 infected HepG2 targets. Results are shown in Fig. 3 (P<0.05, n = 3). Although there was a relatively high background signal in control groups against DV2 infected HepG2 targets, the T cell response generated by CaPNP/multipeptide formulations against the infected targets were significantly higher. The CaPNP/multipeptide formulations containing GlcNAc (1 × or 3 ×) showed significantly higher responses than the un-immunized control (groups 4 or 5 vs. group 1; P<0.01, P<0.0001, respectively). Our data indicated that the addition of GlcNAc to the CaPNP/multipeptide formulations stimulates statistically significant higher T cell responses in comparison to formulations without GlcNAc (group 3 vs. groups 4 or 5: P<0.05, P<0.001, respectively). However, there was no significant difference between the 1X or 3X GlcNAc formulation groups. In summary, data in Fig. 2 and 3 demonstrated that CaPNP/multipeptide formulations used to immunize mice generated not only peptide specific T cell responses (observed with CTLs cultured with pooled peptide pulsed HepG2 targets), but also generated robust DV specific T cell responses (observed with CTLs cultured with DV2 infected HepG2 targets).
Figure 3.
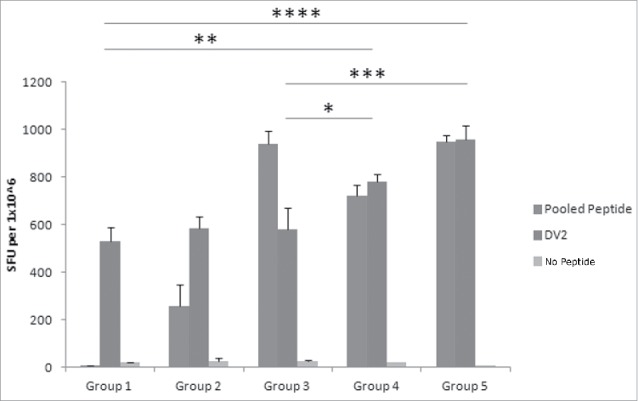
Different GlcNAc concentrations in DV CaPNP/multipeptide formulations stimulate CD8+ T cell activation in vivo. Following the protocol in Fig 1A, CTL responses were generated with the following groups in HLA-A2+ transgenic mice: Group1) unimmunized (PBS control); Group 2) 10µg peptide/150µL PBS per mouse emulsified in ISA 51; Group 3) 10µg peptide/150µl CaPNP per mouse; Group 4) 10µg peptide/150µl CaPNP with 1XGlcNAc per mouse; Group 5) 10µg peptide/150µl CaPNP with 3XGlcNAc per mouse. Splenocytes were harvested and co-cultured with HepG2 targets that were pulsed with either no peptides (negative control), or pooled free peptides (NIQ, TIT, VTL, KLA, AML, LLC) or infected with DV2 for use in the ELISpot assay. Data represented as mean ± S.D (n = 3) of SFU per 1 million splenocytes. * Represents P values: * P<0.05, ** P<0.01, ***P<0.001, **** P<0.0001.
In vitro functional analysis of DV CaPNP/multipeptide vaccine formulation
Our previous studies provided evidence that DV specific MHC class I associated peptides are relevant candidates for a universal (multi-serotype) DV vaccine since they are able to activate CD8+ T cell responses against all 4 serotypes in both healthy, seronegative and seropositive individuals who had previously been infected with a DV serotype.28,29 To assess whether the CaPNP/multipeptide formulations elicit DV specific human T cell responses, we characterized the T cell response with in vitro studies. In the first study, we stimulated peripheral blood mononuclear cells (PBMCs) from HLA-A2+ healthy donors with the DV CaPNP/multipeptide formulations, with or without GlcNAc, to generate DV specific T cells. Immature dendritic cells (DCs) were generated from the adherent monocyte population and were assessed for expression of DC phenotype surface markers (Fig. S2). Then the DCs were co-cultured with PBMCs in the presence of 1) pooled free peptides, or 2) CaPNP/multipeptide formulation with GlcNAc, or 3) CaPNP/multipeptide formulation without GlcNAc. T cells were cultured in vitro and stimulated 3 times before testing DV specific T cell responses. In these T cell activation assays, we used HepG2 targets that were either pulsed with individual peptides (Table 1) or infected with DV2. The data shown in Fig. 4 demonstrate the T cell responses generated against the DV2 infected cells as measured by: IFN-γ secretion in an ELISpot assay (Fig. 4A), by the secretion of granzyme B as measured by MagPIX (Fig. 4B), and upregulation of CD107a determined by flow cytometry (Fig. 4C). All CaPNP/multipeptide formulations, with or without GlcNAc, elicited strong T cell responses against DV2 infected targets. A slightly increased response in the T cell group stimulated without GlcNAc was observed but that was not statistically significant (Fig. 4A). However, we did observe a higher in vivo response with the CaPNP/multipeptide formulation with GlcNAc (Fig. 3). Furthermore, the granzyme B (Fig. 4B) and CD107a marker expression (Fig. 4C) data showed a slight increase in the group which was treated with CaPNP/multipeptide formulation containing GlcNAc, which was consistent with the previous in vivo findings (Fig. 3).
Figure 4.
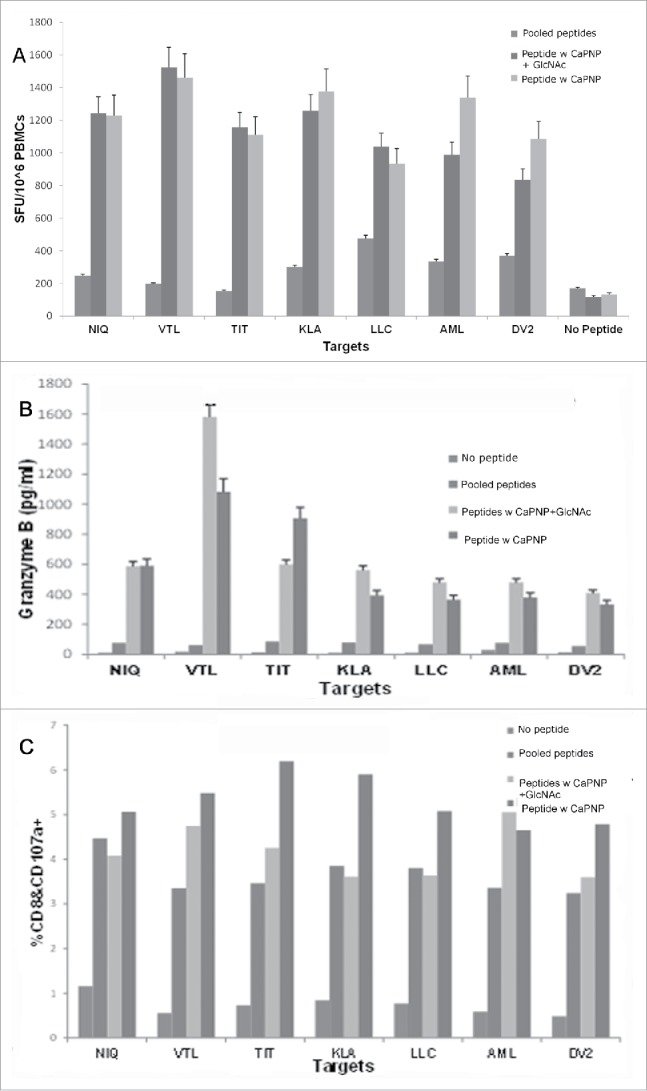
DV CaPNP/multipeptide formulations stimulate CD8+ T cell activation in vitro: (A) IFN-γ ELISpot assay: Using peripheral blood from healthy donors' PBMCs were stimulated in vitro with: 1) pooled peptides, or 2) CaPNP/multipeptide formulation with GlcNAc, or 3) CaPNP/multipeptide formulation without GlcNAc. The activated PBMCs containing the epitope specific CTLs were harvested, washed, and co-cultured overnight with HepG2 targets that were either pulsed with individual peptides (2µg each peptide/mL) or infected with DV2 in an IFN-γ ELISpot assay. Data represented as mean ± S.D (n = 3) of SFU per 1 million PBMCs. (B) Cytolytic response: Identical cultures were set up as in (A) in 96 well round bottom plates. The next day supernatant was harvested and used to detect cytolytic molecules (Granzyme B) using Luminex magnetic bead technology (MagPix). Data represented as mean ± S.D (n = 3) pg/mL. (C) Activation marker analysis: The cells were stained with CD8 and CD107a antibody at the end of the assay culture period to analyze the CTLs for the expression of degranulation marker, CD107a, by flow cytometry. Data represented as mean ± SD (n = 3) percent double positive (both CD8+ and CD107a+ staining) cells.
Stability and efficacy of DV CaPNP/multipeptide vaccine formulation
One of the ultimate goals of this work is to develop a T cell activating DV vaccine that is stable and does not require cold storage for use in DV endemic areas. To test if the CaPNP/multipeptide/GlcNAc formulation can be lyophilized and stored as dry powder without requiring cold storage and without losing functional and structural integrity, we lyophilized the formulation and monitored stability at 4°C or room temperature (RT) for varying lengths of time. We periodically tested the lyophilized formulation upon re-dispersion in suspension with respect to particle size and functional activity by in vitro and in vivo studies. For particle size analysis, the CaPNP/multipeptide/GlcNAc was stored in the following conditions: 1) maintained as a particulate suspension stored at 4°C (standard procedure), 2) lyophilized and then stored as powder at 4°C, 3) lyophilized and stored as powder at RT. The data from particle size analysis at various time points (0, 30, 60, and 90 d post-formulation) is shown in Fig. 5A. The data clearly indicates that formulations were physically stable at all 3 conditions for at least 90 d (P > 0.05, n = 3). There was no significant difference in particle size between 4°C or RT storage for the lyophilized formulations (P > 0.05, n = 3). Although there was an increasing trend in particle size for lyophilized formulations stored at RT (93.4 ± 26.0 nm at day 30, 125.65 ± 45.04 nm at day 60, 103.1 ± 51.9 nm at day 90, mean ± SD, n = 3), the data did not show any statistical difference (P > 0.05, n = 3).
Figure 5.
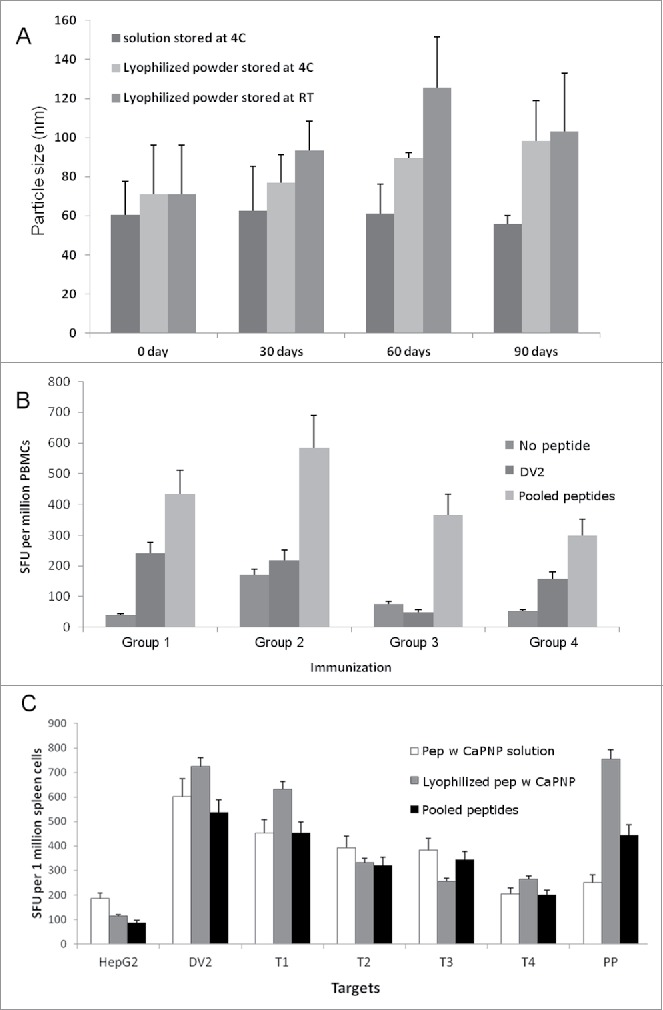
Stability and efficacy of CaPNP/multipeptide formulation. (A) Formulation integrity as measured by particle size over a time of 3 months (days 0, 30, 60 and 90) under different storage conditions (4°C or RT). Data represented as mean ± SD (n = 3) of particle size. (B) Using PBMCs, in vitro CTL responses to formulations in varied storage conditions were generated. Four groups were tested: 1) pooled peptide emulsified in ISA 51; 2) CaPNP/multipeptide formulation with GlcNAc stored at 4°C; 3) lyophilized CaPNP/multipeptide formulation with GlcNAc stored at 4°C; or 4) lyophilized CaPNP/multipeptide formulation with GlcNAc stored at RT. CTL responses were assessed by co-culturing stimulated PBMCs with HepG2 targets that were pulsed with no peptide, or pooled peptides, or infected with DV2. IFNγ expression was measured by ELISpot assay. Data represented as mean ± S.D (n = 3) of SFU per 1 million PBMCs. (C) Using HLA-A2+ transgenic mice, in vivo CTL responses to varied storage conditions were generated. Three groups were tested: 1) pooled peptide emulsified in ISA 51, 2) CaPNP/multipeptide formulation with GlcNAc stored at 4°C, or 3) lyophilized CaPNP/multipeptide formulation with GlcNAc stored at 4°C. Mice were immunized as described in Fig. 1A. CTL responses were assessed by co-culturing splenocytes with HepG2 targets that were either no peptide (HepG2), or pulsed with pooled peptides, or infected with DV2, or infected with Thai isolates of 4 different DV serotypes (Thai isolates of DV serotype-1 [T1], -serotype 2 [T2], -serotype 3 [T3], -serotype 4[T4]). Data represented as mean ± S.D (n = 3) of SFU per 1 million splenocytes.
The functional stability, of lyophilized CaPNP/multipeptide/GlcNAc formulations was assessed, in vitro and in vivo, after 30 d of storage. In vitro studies were performed using HLA-A2+ healthy donor PBMCs and in vivo studies were conducted in HLA-A2+ transgenic mice as described above. Peptide specific T cells were generated in vitro by stimulating peripheral blood T cells with the following test formulations: 1) pooled free peptides, 2) CaPNP/multipeptide/GlcNAc formulation maintained as a particulate suspension and then stored at 4°C (standard procedure), 3) CaPNP/multipeptide/GlcNAc formulation lyophilized and then stored at 4°C, and 4) CaPNP/multipeptide/GlcNAc formulation lyophilized and then stored at RT. HepG2 target cells were pulsed with no peptide, pooled peptides (Table 1), or infected with DV2, and these targets were cocultured to the stimulated T cells. In vitro T cell responses were analyzed using an IFN-γ ELISpot assay. As shown in Fig. 5B, all CaPNP/multipeptide/GlcNAc formulations (group 2, 3 and4) induced a higher levels of IFN-γ against pooled peptide pulsed HepG2 target cells, which indicated the functional stability of all CaPNP/multipeptide/GlcNAc formulations during storage. Similar to data obtained from previous in vitro studies (Fig. 4A), when presented with DV2 infected HepG2 target cells, all CaPNP/multipeptide/GlcNAc formulations at all storage conditions (group 2, 3 and4) demonstrated either equal (group 2 vs. 1) or slightly lower (group 3, 4 vs. 1) T cell responses as compared with the pooled peptide group (group 1), which was not statistically different (P > 0.05, n = 3). Overall, the CaPNP/multipeptide formulations that were lyophilized and then stored at either 4°C or RT elicited peptide and DV specific T cell responses in vitro.
For in vivo evaluation of lyophilized CaPNP/multipeptide/GlcNAc, 3 groups of HLA-A2+ transgenic mice were immunized with the following test formulations: 1) pooled peptides emulsified with ISA 51, 2) CaPNP/multipeptide/GlcNAc formulation stored at 4°C as a particulate solution, and 3) lyophilized CaPNP/multipeptide/GlcNAc formulation stored at 4°C. The T cell responses were induced using HepG2 target cells that were either pulsed with pooled peptide (Table 1), or infected with DV2, or infected with various Thai isolates of DV serotypes (Thai isolate of DV-serotype 1 [T1], -serotype 2 [T2], -serotype 3 [T3], -serotype 4[T4]). Results presented in Fig. 5C demonstrate that CaPNP/multipeptide/GlcNAc formulations (group 2 and 3) yielded in vivo T cell responses against peptide pulsed or DV infected HepG2 target cells, which is consistent with the in vitro results presented in Fig. 5B. Furthermore, the lyophilized CaPNP/multipeptide/GlcNAc formulation (group 3) induced the highest T cell activation against pooled peptide and DV2 infected HepG2 targets compared with all treatment groups. Among the HepG2 targets infected individually with the 4 Thai isolates of DV serotypes (T1-T4), IFN-γ expression was upregulated in both pooled peptides/ISA 51 and CaPNP/multipeptide/GlcNAc formulation groups as measured by ELISpot assay. This indicates the activation of CTL against all the DV serotypes.
Toxicology and safety study of DV CaPNP/multipeptide/GlcNAc vaccine formulation
We performed in vivo toxicology and safety studies in HLA-A2+ transgenic mice. The following 3 groups (n = 10/group) were included in the study: 1) CaPNP alone (vehicle control), 2) CaPNP/multipeptide /GlcNAc formulation (10 µg each peptide/150 µl CaPNP per mouse; treatment), 3) CaPNP/multipeptide/ GlcNAc formulation (50 µg each peptide/150 µl of 0.6% [2x] CaPNP per mouse; treatment). Three doses of the respective formulations were administered by combination of intradermal (i.d.) and subcutaneous (s.c.) routes on days 1, 8, and 15. Mice were observed for clinical and pathological outcomes at initiation (day 0), throughout the treatment (days 1–15) and termination (day 22). Food consumption, body weights and other treatment related clinical effects were closely monitored. On the termination day, blood samples were collected from 5 mice in each group and tested for hematology (supplement table 1) and clinical chemistry (supplement table 2). All animals survived until terminal necropsy. We only observed limited clinical signs such as welts and /or scabs at the base of the tail at the following frequencies: welts were observed in 6 out of 10 animals in group 1 (control), 3 out of 10 in group 2 (10 µg/150 µl, treatment); and scabs in 2 out 10 in group 2 (10 µg/150 µl, treatment), 1 out of 10 in group 3 (50 µg/150 µl, treatment). These findings were located at the injection site and were considered due to the volume of injected fluid.
Fig. S3 shows the data derived from the observations of body weight, change in body weights, and spleen weights for the 3 groups. Results indicated no statistically significant decreases in mean body weights between the treatment groups and the vehicle control group (P > 0.05, n = 10). Weight gain in the treatment groups was comparable to the control group during the 21 day dosing/observation period, however it was not statistically significant (P > 0.05, n = 10). We also examined the external surface of the body including: all orifices, the cranial, thoracic, peritoneal cavities, and their contents after being killed. Observations of gross necropsy consisted of subcutaneous dorsal focus/foci in one mouse from group 2 (10 µg treatment) and 2 mice from group 3 (50 µg treatment). Additionally, a lesion was noted in one of the mice in group 1 which was considered incidental (control).. Within clinical chemistry parameters (Table S2), we observed some changes which consisted of increased potassium and decreased triglycerides for group 3 (50 µg treatment) in comparison to the group 1 (control group); however, neither of these changes were considered treatment-related. Although not statistically significant, alanine aminotransferase (ALT – IU/L serum) levels were increased in group 3 and aspartate aminotransferase (AST – IU/L serum) levels were increased in group 2 (10 µg treatment) and group 3 (50 µg treatment) when compared with the vehicle control group. The toxicological significance of these findings remains unknown. Overall, there was no mortality, no treatment related clinical signs of toxicity, or any other adverse effects in treatment groups when compared with the vehicle control group.
Discussion
Conventional vaccines normally target DV structural proteins to provide protection against all 4 dengue serotypes.63 All of the candidate vaccines currently under development or in clinical trials are designed to stimulate robust humoral immune responses. Yet several concerns related to dengue pathogenesis caused antibody-dependent enhancement or insufficient responses in younger age groups have raised challenges for the development of a robust, broad, and multi-functional DV vaccine. Viral clearance is largely mediated by robust CD8+ T cell responses. Therefore, effective vaccines that induce a broad, multi-functional T cell response with substantial cross-reactivity between all virus serotypes should have major impacts on reducing infection rates and infection related complications. An additional common challenge to vaccine design is delivering small peptides or macromolecules to the target site to establish an appropriate immune response.64 To address these challenges, we developed a synthetic calcium phosphate nanoparticle based multi-peptide T cell vaccine for DV infection which can be used as a standalone vaccine or in combination treatments of disease prevention or therapy.
Since natural infection protects against exogenous re-infection with the same serotype, a prophylactic vaccine must include both T and B cell responses as generated by the natural infection. Implementation of the approach to produce a broad and cross-protective T cell immunity involves the identification of conserved CD8+ T cell epitopes that can be induced in most members of the population and development of a vaccine that can maintain the MHC class I associated epitope/peptide-specific CD8+ T cells in a highly active state to control the infection. Until recently, vaccine research and development almost exclusively focused on antigens selected to trigger a specific immune response in the body to protect against a particular disease. It is now widely accepted that adjuvants,65,66 and antigen delivery systems37 can contribute, substantially, to immune responses induced by a vaccine.
It is becoming increasingly clear that nanoparticles may be an ideal antigen delivery system to target cells or organs for therapeutic purposes.67 A number of previous studies show that nanoparticles can be conjugated with drugs or biomolecules such as peptides or antibodies.49,50,53 The average particle size of the DV CaPNP/multipeptide vaccine formulations used in this study was below 100nm, which is likely small enough to enter the lymphatic network and target antigen presenting cells to generate potent immune responses. More importantly, this vaccine formulation is biodegradable, due to the characteristics of the calcium phosphate nanoparticle, and has a good safety profile.56 Intradermal (i.d.) and subcutaneous (s.c.) injection of CaPNP/multipeptide formulation in HLA-A2+ transgenic mice at lower (10 µg each peptide/150 µL CaPNP per mouse) and higher dose levels (50 µg each peptide/150 µL CaPNP per mouse) did not result in any adverse effects, treatment-related clinical signs of toxicity, or effects on body weight (Fig. S3, Table S1 and 2). However, we did observe the insignificant increasing level of ALT in higher dose and AST in lower dose group, both of which are indicators of liver function. Albeit the toxicological significance of this is unknown, nanoparticles are known to accumulate in liver.68 Based on our animal toxicology study results, it will be prudent to monitor liver function markers during human clinical studies. In addition to its safety, our CaPNP/multipeptide formulation proved to be stable and active during long-term storage at either 4°C or RT as assessed by both in vitro and in vivo studies (Fig. 5). Although it is not uncommon for nanoparticles to show poor long-term stability due to their physical and chemical instability,67 the CaPNP particles used in this study have been shown to be stable for many years at room temperature, either as particulate suspension, as a lyophilized powder, or as spray-dried powder.53 In our study, over the course of a 3 month observation period, the physical integrity of lyophilized CaPNP/multipeptide formulation (4°C or RT) remained stable with respect to particle size (Fig. 5, P > 0.05). Furthermore, the lyophilized vaccine formulations were functionally active and immunogenic over this time period as tested by both in vitro and in vivo studies. These results (Fig. 5) provide evidence that lyophilization is an option for long-term cold chain-independent storage for CaPNP/multipeptide vaccine. Thus, we suggest that this vaccine formulation is suitable for transportation and use in regions where dengue is endemic.
In order for a vaccine to be efficacious, the formulation must include a combination of antigens/epitopes that can induce robust and broad immune responses. In designing the vaccine formulation described in this report, we were interested in specific epitopes that are capable of activating CD8+ T cell responses. More importantly, we were interested in epitopes that are physiologically relevant- in other words, naturally processed and presented by infected cells during an infection. To identify these physiologically relevant epitopes, we used an immunoproteomics approach that identifies MHC class I restricted epitopes presented by DV infected cells.28,29 Although this approach has significant upside, peptides for each HLA supertype need to be identified and developed to be included in the vaccine. The most feasible approach to deal with this limitation is to identify peptides that associate with major HLA supertypes covering the majority of the population, specifically in endemic areas. The epitopes described in our study would cover both HLA-A2+ (> 50% of the world population) and HLA-A24+ (> 50% of the dengue endemic population) supertypes providing potential broad population coverage for vaccine development. Estimates indicate that 5–6 HLA supertypes cover >90% of the world population59 and therefore the opportunity to develop this peptide based universal vaccine is highly feasible. Importantly, these novel MHC class I restricted epitopes were conserved within 4 DV serotypes, covering a wide range of viral proteins including capsid, NS2A, NS4B and NS5. Among these characterized peptides, some of them had HLA-A2 and A2/A24 dual binding motifs and were able to induce CTL activation in vitro.28,29 In this study, we extensively characterized our multipeptide vaccine formulation containing 6 HLA-A2 and A2/A24 peptides (Table 1) using both in vitro and in vivo methods.
CaPNPs can be co-formulated with one or multiple antigens either adsorbed on the surface or co-precipitated with CaP during particle synthesis.49,52 We formulated 6 HLA-A2 and A2/A24 peptides with pre-formed CaPNPs (average particle size of < 100 nm) either individually or as combined in a multipeptide format. Individually formulated 6 CaPNP/peptide formulations were then pooled together and compared with CaPNP/multipeptide formulation. The mean particle size of all formulation remained in the 80–100 nm size range (Fig. 5A), which is smaller than previously reported CaPNP-vaccine formulations and could produce a better performance in terms of immune cells uptake and immune response.49,52,53 As anticipated, both formulations were capable of inducing CTL activation in vivo (Fig. 1). Interestingly, the CaPNP/multipeptide formulations induced higher level of IFN-γ secretion from CD8+ T cells. In our previous studies,28,29 we established that these DV peptides were from conserved regions of DV serotypes and induce potent T cell responses against all 4 serotypes of DV infected cells in vitro, which would be critical to the development of a universally immunogenic vaccine. In the current study, we extend our findings with in vivo studies and demonstrated that the overall immunogenic performance of DV-peptide vaccine can be improved by formulating a pool of DV-peptides with CaPNP when compared with peptides mixed with the standard adjuvant-ISA 51 (Fig. 1, 2, 3). We also showed that the DV CaPNP/multipeptide formulation is antigen sparing, meaning lower doses of CaPNP/multipeptide formulations (10 µg each peptide) can induce higher T cell responses than that of CaPNP/multipeptide formulations containing peptides at higher dose levels (50 µg each peptide) (Fig. 2). It has been known that subdominant T cell epitopes at the low concentration on inoculation activates high avidity T cell responses that are capable of recognizing low endogenous levels of peptide presented on the target cells (i.e. infected cells).69 This suggests that CaPNP is acting both as an antigen delivery system and partially as an adjuvant to facilitate the antigen uptake by APCs, triggering APC activation and inducing high avidity T cell activation.49,64
A major advantage in using an adjuvant clinically is to reduce the required dose of antigens needed to elicit a robust innate and adaptive immune response to the vaccine.64 CaPNP should be considered a preferred adjuvanated delivery system due to its smaller in size (nanometer range),70,71 its high loading capacity for actives,72 and because it is a natural component of body, it is biodegradable/biocompatible,73 and has the ability to induce an effective vaccine immune response.49,71 Our CaPNP/multipeptide vaccine design includes naturally presented DV-specific peptides with confirmed biologic functions formulated as nanoparticles and is a novel vaccine strategy. While it is biologically relevant, our vaccine formulation is also superior to alternatives in terms of ease of manufacturing. Compared to the more widely studied gold nanoparticles, which are constructed by reducing a gold salt74 in the presence of thiol functionalized synthetic neoglyco-conjugates, the manufacturing of CaPNPs involves a simple chemistry using a combination of calcium and phosphate salts and does not require the addition of thiol or any linkers. Manufacturing of CaPNP/multipeptide formulation is also straightforward and involves simple physical mixing methods.51-54 Our results demonstrated that the CaPNP/multipeptide formulations are stable and can either be stored as suspension at 4°C or lyophilized as powder to store at room temperature (Fig. 5) to be reconstituted before use. We showed that all formulations of CaPNP/multipeptide remain biologically active for at least 3 months. Furthermore, we included a bacterial carbohydrate, GlcNAc, in effort to enhance innate immunity generated by the CaPNP formulations and to increase peptide attachment to particles. Previously, GlcNAc has been used in pharmaceutical formulations as delivery tool.51,60,75,76 GlcNAc incorporated in CaPNP/multipeptide formulation is expected to act as a danger signal for the immune system, which often plays a role in initiation of the immune recognition but not necessarily the immune response.77 Activation of innate immune response is considered very important so that it can prime the immune system that involves APCs activation.75 In our study, we observed higher in vivo CTL activation in the presence of GlcNAc which may be due to the danger signal being more prevalent in vivo with antigen spreading and activation of polyclonal T cell response.41,78
Lastly, data from our current study also demonstrated that the CaPNP/multipeptide formulation used to immunize mice could generate not only a peptide specific response (through observation of activation upon recognition of peptide pulsed targets),but also a DV virus specific response (through observation of activation upon recognition of targets infected with either DV2 or Thai isolates of DV serotypes [T1, T2, T3, and T4]) as opposed to the free, pooled peptides emulsified in ISA 51 immunize mice. These were indicated, respectively, by the activation upon recognition of peptide pulsed targets and by the activation upon recognition of targets infected either with DV2 or Thai isolates of DV serotypes T1-T4. Our data also clearly indicate that naturally MHC presented subdominant peptides, when delivered in a targeted antigen delivery system, could generate high avidity CTLs that recognize the endogenously processed and presented antigens on the infected cells at very low concentrations, as was also shown elsewhere.69 These are more functionally and clinically relevant than the dominant T cell epitopes at higher concentrations, which activates low avidity T cells that fail to recognize endogenously presented epitopes on targets.69 Our in vivo findings support this observation where we showed that CaPNP/multipeptide formulation at lower peptide concentrations induced higher CTL responses in mice against DV2 infected targets, indicating activation of high avidity CTLs (Fig. 2).
In summary, we described the development of a synthetic universal DV vaccine based on shared multiple T cell epitopes incorporated in CaPNPs. Although the DV CaPNP/multipeptide formulation focused on 2 of the major HLA supertypes (HLA-A2 and –A24), if successful, this approach could quickly be extended to other major HLA supertypes specific peptides that can be identified and incorporated in subsequent vaccine formulations to increase population coverage. Most importantly, the success of this vaccine will lead to a novel universal vaccine technology platform that could transform the clinical success of prophylactic vaccine strategies for heterologous viruses and that could be applied to therapeutic vaccines for chronic viral infections. From the manufacturing and cost effectiveness perspective, a synthetic universal vaccine strategy would have a significant positive impact.
Materials and methods
Virus
DV serotype 2 (DV2) (strain 16681) was generously provided by Dr. Alex Birk (Cornell University, New York), and Thai isolates of all 4 DV serotypes were a gift from Dr. Guey Chuen (Emory University, Atlanta), were propagated in Vero cells and collected at 4 d post infection. Titer was determined using a plaque assay in Vero cells.. All cell infections were performed at an MOI of 5 for one hour at 37°C and 5% CO2 in complete media containing 1% FBS. Then virus was removed and the cells were washed extensively and cultured with fresh complete media (Dulbecco's Modified Eagle Medium including 10% FBS) for an additional 72 hours before downstream assays.
Cell lines
The HLA-A2+ liver hepatoblastoma cell line HepG2 was obtained from American Type Culture Collection (ATCC). HepG2 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) medium supplemented with 10% fetal bovine serum, L-glutamine (300 mg/mL), nonessential amino acids (1x concentration), 0.5 mM sodium pyruvate, and antibiotic/antimycotic (1x concentration, CellGro, Corning) (complete medium). All cell lines were maintained at 37°C in a humidified incubator with 5% CO2.
Generation of primary human dendritic cells
Dendritic cells (DCs) were generated from leukopheresis obtained from HLA-A2+ healthy donors (Biological Specialty Corp, Colmar, PA). In brief, PBMCs were isolated using lymphocyte separation medium (Mediatech, Flemington, NJ) using differential centrifugation according to standard methods. Adherent cells from overnight cultures of the PBMCs were treated with 100 ng/mL interleukin 4 (IL-4) and 25 ng/mL granulocyte macrophage colony-stimulating factor (GM-CSF) (Peprotech, Rocky Hill, NJ) for 6 d before their infection with DV2.79 Each experiment was performed at least 3 times with different donors. Fresh buffy coats from healthy HLA-A2+ positive donors were obtained and processed for each experiment following standard protocols.
Mice
Six to 8-week old female and male HLA-A2+ transgenic mice were purchased from Taconic (Strain HLA-A2.1, CB6F1-Tg(HLA-A*0201/H2-Kb)A*0201) and housed at Lampire Biologicals (Pipersville, PA). All animal experiments were conducted in adherence to the Guide for Care and use of Laboratory Animals of the NIH. Experimental protocols were approved by the Institutional Animal Care and Use Committee of Lampire Biologicals.
Calcium phosphate nanoparticle (CaPNP) formulation and vaccine generation
The CaPNPs were synthetically manufactured by CaPtivate Pharmaceuticals. Briefly, under aseptic conditions, inorganic salt solutions of calcium and phosphates were mixed at pre-determined ratios under constant mixing according to SOPs developed by CaPtivate. The process yields a stable nano-suspension of calcium phosphate with average particle sizes of 80 nm or smaller as determined by Photon Correlation Spectroscopy (PCS) using a Coulter N4 Plus Submicron Particle Sizer. The storage stability of the particles was monitored at room temperature for up to 6 months. There was no significant change in particle size observed during that period.
Peptides were synthesized by China Peptide Ltd. (Shanghai, China) and dissolved in DMSO with concentration of 10 mg/ml. Peptides were added to particle suspension with N-acetylglucosamine (GlcNAc) (0.093 mol/L, 1 × or 0.279 mol/L, 3 ×) (EMDMillipore, Darmstadt, Germany) and ultra-pure water, then mixed on a rocker for 4 hours at room temperature. The CaPNP/multipeptide formulation was assessed for particle size and stored at 4°C.
In vitro analysis of DV CaPNP/multipeptide formulation
Generation of epitope specific CTLs in vitro
Free peptides and CaPNP/multipeptide formulations were used to generate peptide specific CD8+ T cells (CTLs) as described previously.28,29 Heparinized blood from healthy HLA-A2+ donors was purchased from Biological Specialty Corp. Briefly, PBMCs were purified using lymphocyte separation medium (Corning, Corning, NY) using differential centrifugation following standard methods. PBMCs were cultured in complete RPMI 1640 in 6-well tissue culture plates (BD) overnight. Non-adherent cells were removed and saved. Plastic adherent cells were pulsed with various antigen conditions as specified in individual experiment in complete medium. After 2 hours of incubation, non-adherent cells were added to the plates along with cytokine-rich (5 µg/mL KLH; Sigma-Aldrich, 5ng/mL IL-7, 25ng/mL GM-CSF, and 50ng/mL IL-4) complete RPMI-1640 media (total volume 5 mL). Plates were incubated at 37°C with 5% CO2. T cells were re-stimulated 12 d after initial stimulation with autologous PBMCs depleted of CD4+ and CD8+ T cells by magnetic negative depletion (Dynal beads, Invitrogen, Grand Island, NY) and pulsed with the same starting condition groups and human β2-microglobulin (1.5 µg/mL). Restimulated cells were cultured in complete RPMI-1640 medium supplemented with IL-15 (5ng/mL), GM-CSF (12.5ng/mL) and IL-4 (50ng/mL) for 5 d. Media was exchanged by cautiously removing 2mL media from each well and resupplying complete media supplemented with IL-15 and IL-2 (both 100 µg/mL). The cultures were maintained at 37°C with 5% CO2 for 2 d until the next restimulation or functional assay. Restimulations with accompanying media exchanges were performed a total of 3 times each before CTL functional assays. T cell stimulations and restimulations were completed at various concentrations (50, 25, 10, and 10 µg respectively). Unless otherwise noted, all cytokines and growth factors were purchased from eBiosciences (San Diego, CA).
ELISPOT assay
96 well PVDF-membrane plates (Millipore) were coated with IFN-γ capture antibody overnight at 4°C. On the day of the assay, the plates were blocked for 2 hours in RPMI-1640 complete medium and washed before use in the ELISpot assay. The activated PBMCs generated after stimulations were cultured overnight with appropriate antigen presenting target cells (HepG2 cells) that were specified in individual experiment. Throughout the experiment, PBMCs were left unstimulated and were used in the ELISpot assay (as negative control to assess the basal IFN-γ levels). These unstimulated cells were co-cultured along with the aforementioned HepG2 target cells in the ELISpot. After the overnight co-culture of un-stimulated and activated PBMCs and targets with an effector to target ratio of 10:1, the assay was developed according to the manufactures instructions (BD Biosciences, San Jose, CA). Developed spots were quantified using the ELISpot Reader System (AID, San Diego, CA). Data was normalized to be representative of spot forming units (SFU) per varying numbers of effector cells (example, SFU per 2.5 × 105 or 1 million PBMCs), and the data are also represented as the average of 3 replicates ± the standard deviation.
MagPIX cytokine detection
Activated PBMCs cytokine secretion was measured using a Milliplex magnetic bead assay customized to detect Granzyme-B for T cell characterization (Millipore) as described before.29 Briefly, plates were set up with activated PBMCs concurrently with ELISpot assays and the supernatants were harvested from stimulated PBMCs after an overnight co-culture with HepG2 target cells either pulsed with individual peptides or infected with DV2. In all experiments, the unstimulated cells were the negative controls (basal PBMC cytokine expression levels). Supernatants were cleared of cellular debris by centrifugation and 25 µl of samples, standards, and controls were added to a 96 well plate with assay buffer (1:1 dilution). Magnetic beads coated with antibodies against the specified analyte was added to each well and the plate was incubated overnight on a plate shaker at 4°C. The next morning, the plate was washed twice with wash buffer and biotinylated detection antibodies were added to each well for additional one hour incubation. Streptavidin-PE was added for an additional 30 minutes, the plate was washed twice with wash buffer, loaded with drive fluid, and read on the MagPIX system. Data was analyzed using the Milliplex Analyst software package (Luminex, Austin, TX).
Flow cytometry analysis
Epitope specific CD8+ T cells generated in vitro or in vivo were assessed for cytotoxic capabilities via CD8+ and degranulation marker, CD107a+ staining as described before.29 Briefly, the activated cells were washed and stained with anti-CD8 and anti-CD107a (BD Biosciences, Franklin Lakes, NJ) antibodies. Cells were washed extensively and resuspended in PBS/0.1%BSA for analysis. In general, the assay for CD107a marker expression is performed with activated T cells either during the course of activation or at the end of the activation period.80 In this study, we stained with CD107a antibody at the end of the activation of T cells, which may have not measured the full extent of degranulation during the culture.81 The protocol followed in this study provides, at best, the final expression and not the transient expression during the culture period, which may underestimate the total expression levels. All flow cytometry was performed using Guava 8HT EasyCYTE system (Millipore, Billerica, MA) and data analyzed using the InSight software (Millipore).
In vivo analysis of DV multi-peptide CaPNP
HLA-A2+ transgenic mice were used for in vivo studies. The mice were injected with specified vaccine formulations at 3 sites: 2 intradermal (i.d.) injections and one subcutaneous (s.c.) injection. Injections were repeated twice at 10-day intervals (days 10 and 20). One week after the third injection (day 27), mice were killed and spleens were harvested for functional analysis. Briefly, splenocytes were harvested, homogenized, and then RBCs lysed. Next, cells were pulsed, consistent with each assay, and used in ELISpot assay at an E:T ratio of 10:1 with uninfected or DV infected HepG2 target cells and peptide-loaded HepG2 targets.
Statistical methods
Descriptive statistics (mean and standard deviation) were calculated and data was analyzed for statistical significance. For all analyses, if the data set was normally distributed and of equal variance, statistical comparisons were conducted using a one-way analysis of variance (ANOVA) with post hoc comparisons made (if necessary) using Dunnett's test (GraphPad Prism, GraphPad, CA). A minimum significance level of p < 0.05 was used for the statistical comparisons in this study.
Supplementary Material
Abbreviations
- HLA
human leukocyte antigen
- MHC
major histocompatibility complex
- CTL
CD8+, cytotoxic T lymphocyte
- DV
dengue virus
- ADE
antibody dependent enhancement
- CaPNP
calcium phosphate nanoparticle
- VLP
virus like particle
- PBL
peripheral blood
- PBMC
peripheral blood mononuclear cell
- GlcNAc
N-acetylglucosamine
- ISA 51
Montanide ISA 51
- MUG
fluorescent-β-Galatosidase
- i.d.
intradermal
- s.c.
subcutaneous
- DC
dendritic cell
Disclosure of potential conflicts of interest
The authors report no conflict of interest.
Acknowledgments
The authors would like to thank Dr. Alex Birk for providing the dengue virus 2 (DV2) strain 16681 and Dr. Guey Chuen for providing Thai isolates of all 4 DV serotypes.
Funding
This work was supported by the National Institutes of Health Small Business Innovation Research (2 R44 A1062177–03)
References
- [1].Restrepo AC, Baker P, Clements AC. National spatial and temporal patterns of notified dengue cases, Colombia 2007–2010. Trop Med Int Health. 2014;19:863-71. doi: 10.1111/tmi.12325. PMID:24862214 [DOI] [PubMed] [Google Scholar]
- [2].Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, et al.. The global distribution and burden of dengue. Nature. 2013;496:504-7. doi: 10.1038/nature12060. PMID:23563266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Brady OJ, Gething PW, Bhatt S, Messina JP, Brownstein JS, Hoen AG, Moyes CL, Farlow AW, Scott TW, Hay SI, et al.. Refining the global spatial limits of dengue virus transmission by evidence-based consensus. PLoS Negl Trop Dis. 2012;6:e1760. doi: 10.1371/journal.pntd.0001760. PMID:22880140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ebi KL, Nealon J. Dengue in a changing climate. Environ Res. 2016;151:115-23. doi: 10.1016/j.envres.2016.07.026. PMID:27475051 [DOI] [PubMed] [Google Scholar]
- [5].Colon-Gonzalez FJ, Fezzi C, Lake IR, Hunter PR. The effects of weather and climate change on dengue. PLoS Negl Trop Dis. 2013;7:e2503. doi: 10.1371/journal.pntd.0002503. PMID:24244765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Malavige GN, Ogg GS. T cell responses in dengue viral infections. J Clin Virol. 2013;58:605-11. doi: 10.1016/j.jcv.2013.10.023. PMID:24220605 [DOI] [PubMed] [Google Scholar]
- [7].Weiskopf D, Sette A. T-cell immunity to infection with dengue virus in humans. Front Immunol. 2014;5:93. doi: 10.3389/fimmu.2014.00093. PMID:24639680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Simmons CP, McPherson K, Van Vinh Chau N, Hoai Tam DT, Young P, Mackenzie J, Wills B. Recent advances in dengue pathogenesis and clinical management. Vaccine. 2015;33:7061-8. doi: 10.1016/j.vaccine.2015.09.103. PMID:26458808 [DOI] [PubMed] [Google Scholar]
- [9].WHO Dengue: guidelines for diagnosis, treatment, prevention and control – New edition. 2009. Available from: http://www.who.int/tdr/publications/documents/dengue-diagnosis.pdf?ua=1. [PubMed] [Google Scholar]
- [10].Achee NL, Gould F, Perkins TA, Reiner RC Jr, Morrison AC, Ritchie SA, Gubler DJ, Teyssou R, Scott TW. A critical assessment of vector control for dengue prevention. PLoS Negl Trop Dis. 2015;9:e0003655. doi: 10.1371/journal.pntd.0003655. PMID:25951103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rodrigo WW, Block OK, Lane C, Sukupolvi-Petty S, Goncalvez AP, Johnson S, Diamond MS, Lai CJ, Rose RC, Jin X, et al.. Dengue virus neutralization is modulated by IgG antibody subclass and Fcgamma receptor subtype. Virology. 2009;394:175-82. doi: 10.1016/j.virol.2009.09.024. PMID:19833371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wu RS, Chan KR, Tan HC, Chow A, Allen JC Jr, Ooi EE. Neutralization of dengue virus in the presence of Fc receptor-mediated phagocytosis distinguishes serotype-specific from cross-neutralizing antibodies. Antiviral Res. 2012;96:340-3. doi: 10.1016/j.antiviral.2012.09.018. PMID:23041143 [DOI] [PubMed] [Google Scholar]
- [13].Aguiar M, Stollenwerk N, Halstead SB. The risks behind Dengvaxia recommendation. Lancet Infect Dis. 2016;16:882-3. doi: 10.1016/S1473-3099(16)30168-2. PMID:27477967 [DOI] [PubMed] [Google Scholar]
- [14].Halstead SB, Russell PK. Protective and immunological behavior of chimeric yellow fever dengue vaccine. Vaccine. 2016;34:1643-7. doi: 10.1016/j.vaccine.2016.02.004. PMID:26873054 [DOI] [PubMed] [Google Scholar]
- [15].Hadinegoro SR, Arredondo-García JL, Capeding MR, Deseda C, Chotpitayasunondh T, Dietze R, Muhammad Ismail HI, Reynales H, Limkittikul K, Rivera-Medina DM, et al.. Efficacy and long-term safety of a dengue vaccine in regions of endemic disease. N Engl J Med. 2015;373:1195-206. doi: 10.1056/NEJMoa1506223. PMID:26214039 [DOI] [PubMed] [Google Scholar]
- [16].Arnon R, Horwitz RJ. Synthetic peptides as vaccines. Curr Opin Immunol. 1992;4:449-53. doi: 10.1016/S0952-7915(06)80037-3. PMID:1382452 [DOI] [PubMed] [Google Scholar]
- [17].Halstead SB. In vivo enhancement of dengue virus infection in rhesus monkeys by passively transferred antibody. J Infect Dis. 1979;140:527-33. doi: 10.1093/infdis/140.4.527. PMID:117061 [DOI] [PubMed] [Google Scholar]
- [18].Halstead SB, O'Rourke EJ. Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J Exp Med. 1977;146:201-17. doi: 10.1084/jem.146.1.201. PMID:406347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mathew A, Rothman AL. Understanding the contribution of cellular immunity to dengue disease pathogenesis. Immunol Rev. 2008;225:300-13. doi: 10.1111/j.1600-065X.2008.00678.x. PMID:18837790 [DOI] [PubMed] [Google Scholar]
- [20].Bukowski JF, Kurane I, Lai CJ, Bray M, Falgout B, Ennis FA. Dengue virus-specific cross-reactive CD8+ human cytotoxic T lymphocytes. J Virol. 1989;63:5086-91. PMID:2511337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Simmons CP, Dong T, Chau NV, Dung NT, Chau TN, Thao le TT, Dung NT, Hien TT, Rowland-Jones S, Farrar J. Early T-cell responses to dengue virus epitopes in Vietnamese adults with secondary dengue virus infections. J Virol. 2005;79:5665-75. doi: 10.1128/JVI.79.9.5665-5675.2005. PMID:15827181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nascimento EJ, Mailliard RB, Khan AM, Sidney J, Sette A, Guzman N, Paulaitis M, de Melo AB, Cordeiro MT, Gil LV, et al.. Identification of conserved and HLA promiscuous DENV3 T-cell epitopes. PLoS Negl Trop Dis. 2013;7:e2497. doi: 10.1371/journal.pntd.0002497. PMID:24130917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].de Melo AB, Nascimento EJ, Braga-Neto U, Dhalia R, Silva AM, Oelke M, Schneck JP, Sidney J, Sette A, Montenegro SM, et al.. T-cell memory responses elicited by yellow fever vaccine are targeted to overlapping epitopes containing multiple HLA-I and -II binding motifs. PLoS Negl Trop Dis. 2013;7:e1938. doi: 10.1371/journal.pntd.0001938. PMID:23383350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Piazza P, Campbell D, Marques E, Hildebrand WH, Buchli R, Mailliard R, Rinaldo CR. Dengue virus-infected human dendritic cells reveal hierarchies of naturally expressed novel NS3 CD8 T cell epitopes. Clin Exp Immunol. 2014;177:696-702. doi: 10.1111/cei.12373. PMID:24816171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mathew A, Kurane I, Rothman AL, Zeng LL, Brinton MA, Ennis FA. Dominant recognition by human CD8+ cytotoxic T lymphocytes of dengue virus nonstructural proteins NS3 and NS1.2a. J Clin Invest. 1996;98:1684-91. doi: 10.1172/JCI118964. PMID:8833919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yauch LE, Zellweger RM, Kotturi MF, Qutubuddin A, Sidney J, Peters B, Prestwood TR, Sette A, Shresta S. A protective role for dengue virus-specific CD8+ T cells. J Immunol. 2009;182:4865-73. doi: 10.4049/jimmunol.0801974. PMID:19342665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zellweger RM, Eddy WE, Tang WW, Miller R, Shresta S. CD8+ T cells prevent antigen-induced antibody-dependent enhancement of dengue disease in mice. J Immunol. 2014;193:4117-24. doi: 10.4049/jimmunol.1401597. PMID:25217165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Testa JS, Shetty V, Sinnathamby G, Nickens Z, Hafner J, Kamal S, Zhang X, Jett M, Philip R. Conserved MHC class I-presented dengue virus epitopes identified by immunoproteomics analysis are targets for cross-serotype reactive T-cell response. J Infect Dis. 2012;205:647-55. doi: 10.1093/infdis/jir814. PMID:22246683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Comber JD, Karabudak A, Huang X, Piazza PA, Marques ET, Philip R. Dengue virus specific dual HLA binding T cell epitopes induce CD8+ T cell responses in seropositive individuals. Hum Vaccin Immunother. 2014;10:3531-43. doi: 10.4161/21645515.2014.980210. PMID:25668665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Duangchinda T, Dejnirattisai W, Vasanawathana S, Limpitikul W, Tangthawornchaikul N, Malasit P, Mongkolsapaya J, Screaton G. Immunodominant T-cell responses to dengue virus NS3 are associated with DHF. Proc Natl Acad Sci U S A. 2010;107:16922-7. doi: 10.1073/pnas.1010867107. PMID:20837518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mathew A, Townsley E, Ennis FA. Elucidating the role of T cells in protection against and pathogenesis of dengue virus infections. Future Microbiol. 2014;9:411-25. doi: 10.2217/fmb.13.171. PMID:24762312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Leclerc C, Dériaud E, Megret F, Briand JP, Van Regenmortel MH, Deubel V. Identification of helper T cell epitopes of dengue virus E-protein. Mol Immunol. 1993;30:613-25. doi: 10.1016/0161-5890(93)90072-J. PMID:7683752 [DOI] [PubMed] [Google Scholar]
- [33].Gagnon SJ, Zeng W, Kurane I, Ennis FA. Identification of two epitopes on the dengue 4 virus capsid protein recognized by a serotype-specific and a panel of serotype-cross-reactive human CD4+ cytotoxic T-lymphocyte clones. J Virol. 1996;70:141-7. PMID:8523518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Rothman AL, Kurane I, Lai CJ, Bray M, Falgout B, Men R, Ennis FA. Dengue virus protein recognition by virus-specific murine CD8+ cytotoxic T lymphocytes. J Virol. 1993;67:801-6. PMID:7678307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lee S, Nguyen MT. Recent advances of vaccine adjuvants for infectious diseases. Immune Netw. 2015;15:51-7. doi: 10.4110/in.2015.15.2.51. PMID:25922593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gregory AE, Titball R, Williamson D. Vaccine delivery using nanoparticles. Front Cell Infect Microbiol. 2013;3:13. doi: 10.3389/fcimb.2013.00013. PMID:23532930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Babai I, Barenholz Y, Zakay-Rones Z, Greenbaum E, Samira S, Hayon I, Rochman M, Kedar E. A novel liposomal influenza vaccine (INFLUSOME-VAC) containing hemagglutinin-neuraminidase and IL-2 or GM-CSF induces protective anti-neuraminidase antibodies cross-reacting with a wide spectrum of influenza A viral strains. Vaccine. 2001;20:505-15. doi: 10.1016/S0264-410X(01)00326-7. PMID:11672916 [DOI] [PubMed] [Google Scholar]
- [38].Agger EM, Rosenkrands I, Hansen J, Brahimi K, Vandahl BS, Aagaard C, Werninghaus K, Kirschning C, Lang R, Christensen D, et al.. Cationic liposomes formulated with synthetic mycobacterial cordfactor (CAF01): a versatile adjuvant for vaccines with different immunological requirements. PLoS One. 2008;3:e3116. doi: 10.1371/journal.pone.0003116. PMID:18776936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pandey A, Singh N, Sambhara S, Mittal SK. Egg-independent vaccine strategies for highly pathogenic H5N1 influenza viruses. Hum Vaccin. 2010;6:178-88. doi: 10.4161/hv.6.2.9899. PMID:19875936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O'Neil CP, Lee LK, Swartz MA, Hubbell JA. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol. 2007;25:1159-64. doi: 10.1038/nbt1332. PMID:17873867 [DOI] [PubMed] [Google Scholar]
- [41].Ohno S, Kohyama S, Taneichi M, Moriya O, Hayashi H, Oda H, Mori M, Kobayashi A, Akatsuka T, Uchida T, et al.. Synthetic peptides coupled to the surface of liposomes effectively induce SARS coronavirus-specific cytotoxic T lymphocytes and viral clearance in HLA-A*0201 transgenic mice. Vaccine. 2009;27:3912-20. doi: 10.1016/j.vaccine.2009.04.001. PMID:19490987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Karkada M, Weir GM, Quinton T, Sammatur L, MacDonald LD, Grant A, Liwski R, Juskevicius R, Sinnathamby G, Philip R, et al.. A novel breast/ovarian cancer peptide vaccine platform that promotes specific type-1 but not Treg/Tr1-type responses. J Immunother. 2010;33:250-61. doi: 10.1097/CJI.0b013e3181c1f1e9. PMID:20445345 [DOI] [PubMed] [Google Scholar]
- [43].Pejawar-Gaddy S, Rajawat Y, Hilioti Z, Xue J, Gaddy DF, Finn OJ, Viscidi RP, Bossis I. Generation of a tumor vaccine candidate based on conjugation of a MUC1 peptide to polyionic papillomavirus virus-like particles. Cancer Immunol Immunother. 2010;59:1685-96. doi: 10.1007/s00262-010-0895-0. PMID:20652244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Almeida JP, Lin AY, Figueroa ER, Foster AE, Drezek RA. In vivo gold nanoparticle delivery of peptide vaccine induces anti-tumor immune response in prophylactic and therapeutic tumor models. Small. 2015;11:1453-9. doi: 10.1002/smll.201402179. PMID:25354691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Davis ID, Chen W, Jackson H, Parente P, Shackleton M, Hopkins W, Chen Q, Dimopoulos N, Luke T, Murphy R, et al.. Recombinant NY-ESO-1 protein with ISCOMATRIX adjuvant induces broad integrated antibody and CD4(+) and CD8(+) T cell responses in humans. Proc Natl Acad Sci U S A. 2004;101:10697-702. doi: 10.1073/pnas.0403572101. PMID:15252201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Genton B, Pluschke G, Degen L, Kammer AR, Westerfeld N, Okitsu SL, Schroller S, Vounatsou P, Mueller MM, Tanner M, et al.. A randomized placebo-controlled phase Ia malaria vaccine trial of two virosome-formulated synthetic peptides in healthy adult volunteers. PLoS One. 2007;2:e1018. doi: 10.1371/journal.pone.0001018. PMID:17925866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Berinstein NL, Karkada M, Morse MA, Nemunaitis JJ, Chatta G, Kaufman H, Odunsi K, Nigam R, Sammatur L, MacDonald LD, et al.. First-in-man application of a novel therapeutic cancer vaccine formulation with the capacity to induce multi-functional T cell responses in ovarian, breast and prostate cancer patients. J Transl Med. 2012;10:156. doi: 10.1186/1479-5876-10-156. PMID:22862954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich PY, Mendrzyk R, et al.. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. 2012;18:1254-61. doi: 10.1038/nm.2883. PMID:22842478 [DOI] [PubMed] [Google Scholar]
- [49].He Q, Mitchell AR, Johnson SL, Wagner-Bartak C, Morcol T, Bell SJ. Calcium phosphate nanoparticle adjuvant. Clin Diagn Lab Immunol. 2000;7:899-903. PMID:11063495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].He Q, Mitchell A, Morcol T, Bell SJ. Calcium phosphate nanoparticles induce mucosal immunity and protection against herpes simplex virus type 2. Clin Diagn Lab Immunol. 2002;9:1021-4. PMID:12204953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bell SJD, Morcol T, He Q. Therapeutic calcium phosphate particles and methods of manufacture and use. (Google Patents, 2013). [Google Scholar]
- [52].Morcol T, Nagappan P, Nerenbaum L, Mitchell A, Bell SJ. Calcium phosphate-PEG-insulin-casein (CAPIC) particles as oral delivery systems for insulin. Int J Pharm. 2004;277:91-7. doi: 10.1016/j.ijpharm.2003.07.015. PMID:15158972 [DOI] [PubMed] [Google Scholar]
- [53].Morcol TNP, Nerenbaum L, Mitchel AR, Bell SJD. Particulate drug delivery systems for protein drugs In R K. editors. Handbook of particulate drug delivery. Vol. 2 American Scientific Publishers: Stevenson Ranch; 2008. p. 223-41. [Google Scholar]
- [54].Garcia-Contreras L, Morcol T, Bell SJ, Hickey AJ. Evaluation of novel particles as pulmonary delivery systems for insulin in rats. AAPS PharmSci. 2003;5:E9. doi: 10.1208/ps050209. PMID:12866936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Morcol T, Weidner JM, Mehta A, Bell SJD, Block T. Calcium phosphate particles as pulmonary delivery system for interferon-α in Mice. AAPS PharmSciTech. (In press, 2017). doi: 10.1208/s12249-017-0847-5. PMID:28752471 [DOI] [PubMed] [Google Scholar]
- [56].Morcol T, Hurst BL, Tarbet EB. Calcium Phosphate Nanoparticle (CaPNP) for dose-sparing of inactivated whole virus pandemic influenza A (H1N1) 2009 vaccine in mice. Vaccine. 2017;35:4569-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Testa JS, Apcher GS, Comber JD, Eisenlohr LC. Exosome-driven antigen transfer for MHC class II presentation facilitated by the receptor binding activity of influenza hemagglutinin. J Immunol. 2010;185:6608-16. doi: 10.4049/jimmunol.1001768. PMID:21048109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wherry EJ, Puorro KA, Porgador A, Eisenlohr LC. The induction of virus-specific CTL as a function of increasing epitope expression: responses rise steadily until excessively high levels of epitope are attained. J Immunol. 1999;163:3735-45. PMID:10490969 [PubMed] [Google Scholar]
- [59].Karttunen J, Sanderson S, Shastri N. Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc Natl Acad Sci U S A. 1992;89:6020-4. doi: 10.1073/pnas.89.13.6020. PMID:1378619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Bell SJD, He Q MT. Therapeutic calcium phosphate particles and methods of manufacture and use. Vol. 6, 355,271 (ed. patent, U.) (U.S.A., 2002). [Google Scholar]
- [61].Zamze S, Martinez-Pomares L, Jones H, Taylor PR, Stillion RJ, Gordon S, Wong SY. Recognition of bacterial capsular polysaccharides and lipopolysaccharides by the macrophage mannose receptor. J Biol Chem. 2002;277:41613-23. doi: 10.1074/jbc.M207057200. PMID:12196537 [DOI] [PubMed] [Google Scholar]
- [62].van Doorn E, Liu H, Huckriede A, Hak E. Safety and tolerability evaluation of the use of Montanide ISA51 as vaccine adjuvant: A systematic review. Hum Vaccin Immunother. 2016;12:159-69. doi: 10.1080/21645515.2015.1071455. PMID:26378866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Liu Y, Liu J, Cheng G. Vaccines and immunization strategies for dengue prevention. Emerg Microbes Infect. 2016;5:e77. doi: 10.1038/emi.2016.74. PMID:27436365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Coffman RL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010;33:492-503. doi: 10.1016/j.immuni.2010.10.002. PMID:21029960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Leroux-Roels I, Bernhard R, Gérard P, Dramé M, Hanon E, Leroux-Roels G. Broad Clade 2 cross-reactive immunity induced by an adjuvanted clade 1 rH5N1 pandemic influenza vaccine. PLoS One. 2008;3:e1665. doi: 10.1371/journal.pone.0001665. PMID:18301743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Baras B, Stittelaar KJ, Simon JH, Thoolen RJ, Mossman SP, Pistoor FH, van Amerongen G, Wettendorff MA, Hanon E, Osterhaus AD. Cross-protection against lethal H5N1 challenge in ferrets with an adjuvanted pandemic influenza vaccine. PLoS One. 2008;3:e1401. doi: 10.1371/journal.pone.0001401. PMID:18167560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Abdelwahed W, Degobert G, Stainmesse S, Fessi H. Freeze-drying of nanoparticles: formulation, process and storage considerations. Adv Drug Deliv Rev. 2006;58:1688-713. doi: 10.1016/j.addr.2006.09.017. PMID:17118485 [DOI] [PubMed] [Google Scholar]
- [68].Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33:941-51. doi: 10.1038/nbt.3330. PMID:26348965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].McKee MD, Roszkowski JJ, Nishimura MI. T cell avidity and tumor recognition: implications and therapeutic strategies. J Transl Med. 2005;3:35. doi: 10.1186/1479-5876-3-35. PMID:16174302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Knuschke T, Bayer W, Rotan O, Sokolova V, Wadwa M, Kirschning CJ, Hansen W, Dittmer U, Epple M, Buer J, et al.. Prophylactic and therapeutic vaccination with a nanoparticle-based peptide vaccine induces efficient protective immunity during acute and chronic retroviral infection. Nanomedicine. 2014;10:1787-98. doi: 10.1016/j.nano.2014.06.014. PMID:25014891 [DOI] [PubMed] [Google Scholar]
- [71].Knuschke T, Sokolova V, Rotan O, Wadwa M, Tenbusch M, Hansen W, Staeheli P, Epple M, Buer J, Westendorf AM. Immunization with biodegradable nanoparticles efficiently induces cellular immunity and protects against influenza virus infection. J Immunol. 2013;190:6221-9. doi: 10.4049/jimmunol.1202654. PMID:23667109 [DOI] [PubMed] [Google Scholar]
- [72].Riehemann K, Schneider SW, Luger TA, Godin B, Ferrari M, Fuchs H. Nanomedicine–challenge and perspectives. Angew Chem Int Ed Engl. 2009;48:872-97. doi: 10.1002/anie.200802585. PMID:19142939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Sokolova V, Knuschke T, Kovtun A, Buer J, Epple M, Westendorf AM. The use of calcium phosphate nanoparticles encapsulating Toll-like receptor ligands and the antigen hemagglutinin to induce dendritic cell maturation and T cell activation. Biomaterials. 2010;31:5627-33. doi: 10.1016/j.biomaterials.2010.03.067. PMID:20417963 [DOI] [PubMed] [Google Scholar]
- [74].Brust M, Walker M, Bethell D, Schiffrin DJ, Whyman R. Synthesis of thiol derivatised gold nanoparticles in a two-phase liquid/liquid system. Chem Soc Chem Commun. 1994;6:801-2. doi: 10.1039/C39940000801 [DOI] [Google Scholar]
- [75].Maitre N, Brown JM, Demcheva M, Kelley JR, Lockett MA, Vournakis J, Cole DJ. Primary T-cell and activated macrophage response associated with tumor protection using peptide/poly-N-acetyl glucosamine vaccination. Clin Cancer Res. 1999;5:1173-82. PMID:10353754 [PubMed] [Google Scholar]
- [76].Pedrali A, Bleve M, Capra P, Jonsson T, Massolini G, Perugini P, Marrubini G. Determination of N-acetylglucosamine in cosmetic formulations and skin test samples by hydrophilic interaction liquid chromatography and UV detection. J Pharm Biomed Anal. 2015;107:125-30. doi: 10.1016/j.jpba.2014.12.014. PMID:25589383 [DOI] [PubMed] [Google Scholar]
- [77].Maverakis E, Kim K, Shimoda M, Gershwin ME, Patel F, Wilken R, Raychaudhuri S, Ruhaak LR, Lebrilla CB. Glycans in the immune system and The altered glycan theory of autoimmunity: a critical review. J Autoimmun. 2015;57:1-13. doi: 10.1016/j.jaut.2014.12.002. PMID:25578468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Demaria S, Bhardwaj N, McBride WH, Formenti SC. Combining radiotherapy and immunotherapy: a revived partnership. Int J Radiat Oncol Biol Phys. 2005;63:655-66. doi: 10.1016/j.ijrobp.2005.06.032. PMID:16199306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Shetty V, Sinnathamby G, Nickens Z, Shah P, Hafner J, Mariello L, Kamal S, Vlahović G, Lyerly HK, Morse MA, et al.. MHC class I-presented lung cancer-associated tumor antigens identified by immunoproteomics analysis are targets for cancer-specific T cell response. J Proteomics. 2011;74:728-43. doi: 10.1016/j.jprot.2011.02.020. PMID:21362506 [DOI] [PubMed] [Google Scholar]
- [80].Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004;294:15-22. doi: 10.1016/j.jim.2004.08.008. PMID:15604012 [DOI] [PubMed] [Google Scholar]
- [81].Fukuda M. Lysosomal membrane glycoproteins. Structure, biosynthesis, and intracellular trafficking. J Biol Chem. 1991;266:21327-30. PMID:1939168 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.