

Abstract
Ruthenium complexes capable of light-triggered cytotoxicity are appealing potential prodrugs for photodynamic therapy (PDT) and photoactivated chemotherapy (PACT). Two groups of Ru(II) polypyridyl complexes with 2-(2-pyridyl)-benzazole ligands were synthesized and investigated for their photochemical properties and anticancer activity to compare strained and unstrained systems that are likely to have different biological mechanisms of action. The structure-activity relationship was focused on the benzazole core bioisosterism and replacement of coligands in Ru(II) complexes. Strained compounds rapidly ejected the 2-(2-pyridyl)-benzazole ligand after light irradiation, and possessed strong toxicity in the HL-60 cell line both under dark and light conditions. In contrast, unstrained Ru(II) complexes were non-toxic in the absence of light, induced cytotoxicity at nanomolar concentrations after light irradiation, and are capable of light-induced DNA damage. The 90−220-fold difference in light and dark IC50 values provides a large potential therapeutic window to allow for selective targeting of cells by exposure to light.
Keywords: synthesis, photochemistry, cytotoxicity, DNA damage, ruthenium
Introduction
Cancer is currently the second leading cause of death in the United States, following heart disease. More than 1.7 million people are estimated to be diagnosed with cancer in 2016.[1] With global cancer morbidity rising, the development of new cancer treatments is crucial. Chemotherapy is used in most treatment regimens for cancer. Since its discovery in the late 1960s, cisplatin and derivatives thereof have achieved great success, and nearly 50% of patients being treated for cancer are given a platinum based drug.[2] Widespread treatment with cisplatin, however, revealed major clinical problems associated with its use. Cisplatin has dose-limiting side effects, such as nephrotoxicity, neurotoxicity, ototoxicity, and myelosuppression.[3] Due to these severe side effects, cisplatin has to be administered at concentrations that might not be lethal to tumor cells, thereby facilitating development of drug resistance. These limitations have driven the investigations of other (non-platinum) transition metal compounds.
In recent years, ruthenium-based complexes have emerged as promising antitumor and antimetastatic agents with potential uses in platinum-resistant tumors.[4] Ruthenium compounds are well suited for medical applications due to a combination of chemical and biological properties: they can form multiple geometries with facile ligand exchange, they can be activated by environmental features or external triggers, and they are capable of mimicking iron binding for transportation.[5] The different oxidation states can be exploited to design prodrugs, where the inactive 3+ ruthenium complexes can be reduced to 2+, creating an active species and a biological effect. The reducing environment of tumors has been associated with the selective activity of ruthenium-based drugs NAMI-A, KP1019, and KP1339, which have been investigated in clinical trials.[6] An alternative prodrug strategy is to use light to transform inert complexes into cytotoxic agents.[7] We have demonstrated that this can be accomplished with strained Ru(II) polypyridyl complexes with distorted octahedral geometry, which photo-decompose via ligand dissociation.[8] The resulting ligand-deficient Ru(II) center can covalently modify DNA or other biomolecules, and induce cytotoxicity.[9]
The application of light-mediated ruthenium complexes can be divided into two categories: photodynamic therapy (PDT) and photoactivated chemotherapy (PACT).[10] PDT relies mainly on the generation of the toxic reactive oxygen species (ROS) such as singlet oxygen (1O2). In contrast, PACT exploits different mechanisms to induce cell death, such as ligand ejection to create metal centers able to form DNA adducts, or photocaging approaches. In this article, we present the investigation of strained and unstrained ruthenium (II) complexes with 2-(2-pyridyl)-benzazole ligands as promising antitumor agents with possible application in both PDT and PACT.
The benzazole moiety was chosen as it combines features including extended conjugation for modulation of the absorption profile, and the potential for intrinsic steric clash within a coordination complex, similar to quinoline-containing ligands.[8b] It also facilitated a systematic investigation as it provided a single point for chemical variation, with a heteroatom (N, O, S) at the 1-position with a nitrogen at the 3-position, or a carbon at the analogous position in indole (Scheme 1). Moreover, benzazole-containing systems exhibit a variety of biological activities and applications. Recently, organometallic systems containing this ligand type have been explored, including half-sandwich ruthenium(II) arene compounds with pyridyl-benzimidazole ligands studied for their DNA binding ability,[11] cyclin-dependent kinase (CDK1) inhibitory effects,[11b] and inhibition of protein tyrosine phosphatase (PTP-1B).[12] The previous investigations of ruthenium complexes with aryl-benzimidazole ligands showed cytotoxic effect at μM concentrations.[11b, 13]
Scheme 1.
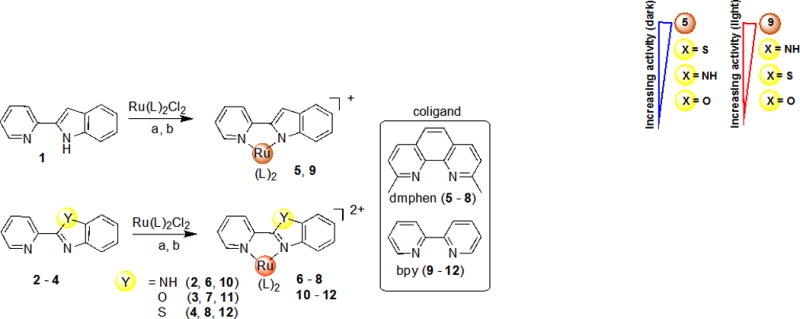
Synthesis of Ru(II) complexes with bioisosteric 2-(2-pyridyl)benzazole ligands. Reagents, conditions and yields: (a) Ru(dmphen)2Cl2 (1.0 eq), ethylene glycol, 100–120 °C for 2 h (5–8), 20–30%; (b) Ru(bpy)2Cl2 (1.0 eq), ethanol-water (1:1), 90 °C for 2 h (9–12), 45–95%.
In this report, we have discovered that coordination of non-cytotoxic 2-(2-pyridyl)-benzazole ligands with a Ru(dmphen)2 (dmphen = 2,9-dimethyl-1,10-phenanthroline) scaffold, forming strained Ru(II) complexes, promoted significant cytotoxic potential of compounds with single μM IC50 values both in dark and light conditions. In contrast, the complexes with 2,2′-bipyridine (bpy) ligands were not active in the absence of light. However, these compounds were effective in killing cells when irradiated, producing nM IC50 values. DNA damage analysis and evaluation of singlet oxygen production confirmed that unstrained compounds generate toxic ROS. However, the disparity in the effective concentration and trends for cytotoxicity (IC50< 1 μM) and singlet oxygen generation (> 10 μM) suggests that these compounds act through some additional, currently unknown, mechanism(s) of action.
Results and Discussion
Synthesis and Characterization
To explore structure-activity relationships (SAR), a small family of heteroleptic Ru(II) complexes (5–12) were synthesized that contained one 2-(2-pyridyl)benzazole type ligand and two strain-inducing dmphen ligands or two bpy ligands as shown in Scheme 1. Four heterocyclic bioisosteres were studied: 2-(2-pyridyl)indole (pi) 1, 2-(2-pyridyl)benzimidazole (pbi) 2, 2-(2-pyridyl)benzoxazole (pbo) 3 and 2-(2-pyridyl)benzothiazole (pbt) 4. These systems were chosen in order to investigate the impact of replacement of one pyridyl-type ligand with a benzazole on the cytotoxicity and photochemical properties of the Ru(II) complexes.
The Ru(II) complexes were synthesized from a racemic mixture of the Δ and Λ enantiomers of Ru(dmphen)2Cl2 or Ru(bpy)2Cl2 and thus form a mixture of enantiomers upon coordination of the pyridyl-benzazole ligands. All complexes were exhaustively purified to ensure no contamination of either free ligands or coordinatively unsaturated Ru(II) centers. As the pyridyl-indole is deprotonated, the complexes carry a +1 charge; all other complexes are +2 charged. The complexes were characterized by 1H NMR spectroscopy, ESI-MS, X-ray and UV (see Figure S6–10, S18–27 in the Supporting Information). The strained complexes 5–8 were synthesized and characterized for the first time; the unstrained complexes 9–12 have been described previously.[14] In contrast to the described 1H NMR spectra (300 MHz, CD3CN) for 11 and 12,[14b] we observed that some resonances for H4 and H5 of bpy coligands were resolved as doublet of doublets of doublets (ddd).[15]
X-ray Crystallography
The structures of complexes 6–8 were determined by X-ray crystallography and are shown in Figure 1. Selected bond lengths and angles are listed in Table 1.
Figure 1.
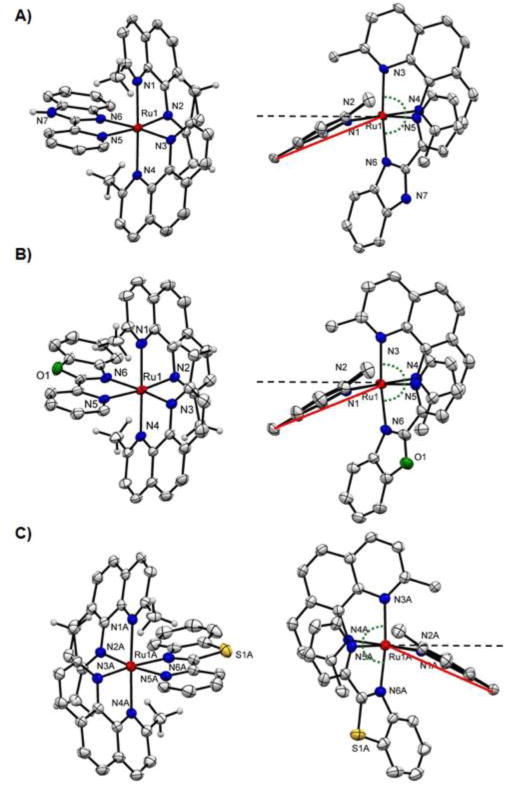
Ellipsoid plot of ruthenium complexes A) (Δ)-6, B) (Δ)-7, C) (Λ)-8 at 50% probability with H atoms omitted for clarity. Right column: side views highlighting the distortion of the dmphen ligand. The black dashed lined indicates the normal plane and the angle between red and black dash line represents the ligand bend. Note: for 8 only one cation of the asymmetric unit is shown.
Table 1.
Selected bond lengths (Å), bond angles (°) and torsion angles (°) of 6–8.
| 6 | 7 | 8 | |
|---|---|---|---|
| Bond Lengths (Å) | |||
| Ru-N1 | 2.114(3) | 2.114(3) | 2.114(2) |
| Ru-N2 | 2.097(3) | 2.103(3) | 2.102(2) |
| Ru-N3 | 2.100(3) | 2.090(3) | 2.097(2) |
| Ru-N4 | 2.120(3) | 2.105(3) | 2.108(2) |
| Ru-N5 | 2.109(3) | 2.106(3) | 2.097(2) |
| Ru-N6 | 2.099(3) | 2.112(3) | 2.112(2) |
| Bond Angles (°) | |||
| N1-Ru-N2 | 79.45(11) | 79.73(10) | 79.06(9) |
| N1-Ru-N3 | 100.6(1) | 100.96(11) | 101.40(9) |
| N1-Ru-N4 | 178.35(11) | 178.86(11) | 177.52(9) |
| N1-Ru-N5 | 95.9(1) | 95.99(10) | 96.07(9) |
| N1-Ru-N6 | 81.86(10) | 80.24(11) | 82.05(9) |
| N2-Ru-N3 | 94.57(10) | 94.56(10) | 93.35(9) |
| N2-Ru-N4 | 102.1(1) | 100.78(10) | 103.19(9) |
| N2-Ru-N5 | 170.3(1) | 171.59(10) | 171.30(9) |
| N2-Ru-N6 | 92.78(10) | 94.05(10) | 94.03(9) |
| N3-Ru-N4 | 79.88(11) | 80.04(11) | 79.62(9) |
| N3-Ru-N5 | 94.66(10) | 93.36(10) | 94.69(9) |
| N3-Ru-N6 | 172.56(10) | 171.39(10) | 172.34(9) |
| N4-Ru-N5 | 82.49(10) | 83.38(10) | 81.57(9) |
| N4-Ru-N6 | 97.46(10) | 98.69(10) | 96.67(9) |
| N5-Ru-N6 | 78.06(10) | 78.02(10) | 78.07(10) |
| Torsion Angles (°) | |||
| N1-C6-C7-N2 | 2.1(4) | 1.2(5) | −2.2(4) |
| Ru-N1-C2-C3 | −167.4(3) | −167.1(3) | 163.2(2) |
| Ru-N2-C11-C10 | 164.0(2) | 165.9(3) | −162.8(2) |
| N3-C20-C21-N4 | 1.8(5) | 2.0(4) | −4.0(4) |
| Ru-N3-C16-C17 | 169.4(3) | 168.7(3) | −171.0(2) |
| Ru-N4-C25-C24 | −165.6(2) | −167.8(2) | 162.3(2) |
| N5-C33-C34-N6 | 1.8(4) | 3.0(5) | −2.3(4) |
| Ru-N5-C29-C30 | 176.0(3) | 175.3(3) | −177.4(2) |
| L1 bend a | 22.6 | 22.6 | 22.7 |
| L2 bend b | 21.4 | 19.5 | 21.6 |
L1 bend = average angle (N3-Ru-C13/C14) – 90°; L1 – dmphen where L is N1 and N2
L2 bend = average angle (N2-Ru-C27/C28) – 90°; L2 – dmphen where L is N3 and N4
As expected, complexes 6–8 exhibited distorted octahedral geometries. Incorporation of two dmphen ligands resulted in the Ru−N bond lengthening to 2.108 Å (average value for 6), 2.103 Å (average value for 7), and 2.105 Å (average value for 8), in comparison with 2.040–2.059 Å for the corresponding complexes with bpy coligands.[14b, 16] The bond length to the pyridine ring (Ru-N5) is shorter in the pbo and pbt ligands than the bond to the benzazole ring, while the Ru-N6 bond to the benzimidazole is shorter than to the pyridine ring in 6 (Table 1). In contrast to complexes containing the 2,2′-biquinoline ligand,[8b] the two ring systems in the benzazole-containing ligands are essentially co-planar, and do not contribute significantly to the distortion in the complexes.
The bond angles between dmphen ligands are nonequivalent, with the largest distortion from the ideal 90° and 180° for complex 8. These deviations are larger than for unstrained compound 11 (Figure S5).[14b] Both the dmphen ligands (L1 and L2, Figure 1, Table 1) for each compound 6–8 are considerably bent from the normal plane, with deviations of 19.5–22.7°. While the bend angle for L1 is the same for all complexes, the bends of L2 are not equivalent for 6–8, creating variations in strain in the molecules that could cause the difference in photoejection kinetics (Table 2).
Table 2.
Photophysical and photochemical properties for 5–12 for various reaction conditions.
| Compound | λmax (nm) | t1/2 (min) | ||
|---|---|---|---|---|
|
| ||||
| water | Opti-MEM | water | Opti-MEM | |
| 5 | 475 | 475 | 47.3±2.80 | n.r.* |
| 6 | 455 | 455 | 1.17±0.08 | 10.38±0.33 |
| 7 | 450 | 450 | 1.03±0.09 | 1.42±0.04 |
| 8 | 440 | 440 | 0.34±0.04 | 0.66±0.002 |
| 9 | 490 | 490 | 66.02±13.11 | 66.11±5.95 |
| 10 | 455 | 435 | n.r. | n.r. |
| 11 | 450 | 450 | n.r. | n.r. |
| 12 | 445 | 450 | n.r. | n.r. |
n.r. = no reaction
Photochemistry
The photochemical reaction of strained Ru(II) complexes 5–9 were monitored by absorption spectroscopy, and exhibited selective photoejection of one ligand when irradiated with >450 nm light, as shown in Figure 2A and Figures S6–10. The presence of an isobestic point indicated the direct conversion to a single product (Figure 2A). The half-life (t1/2) of ligand ejection in water for 6–8 is 40–140× faster than for 5.
Figure 2.
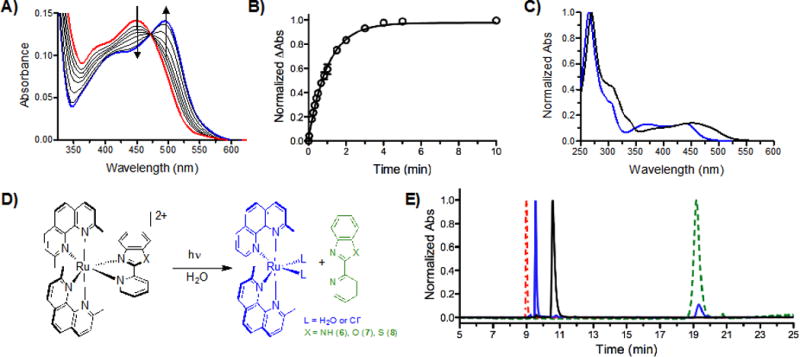
A) Photoejection of 7 (30 μM) in water for 0–240 min irradiation followed by UV/Vis absorption; red = initial, blue = final. B) The photoejection kinetics for 7; reaction was complete in less than 5 min. Determination of photoejection products by HPLC: C) Absorption profile of 7 (black, retention time = 10.56 min) and the photochemical product (blue, retention time = 9.55 min); note that the presence of CH3CN changes the absorption profile; see Figure S28). D) Photoejection reaction scheme for 6–8, showing the photochemical products. E) HPLC chromatogram of 7 before (black) and after irradiation for 1 min with the Indigo LED (blue), in comparison with started ligands: dmphen (red) and pbo (green). The same light dose was used in the cell studies.
Complex 8 exhibited the fastest ejection, and also the largest bend of dmphen ligand (L2, Table 1), indicating a correlation between the strain in the complex and the photochemical properties. The half-life was also found to be sensitive to the environment, as compound 6 demonstrated a 9-fold slower ligand ejection in Opti-MEM, the media used in tissue culture experiments, than in water (Table 2).
The selective ejection of the 2-(2-pyridyl)benzazole ligands after irradiation of 6–8 in water was confirmed by HPLC by comparison with starting complex and ligands (Figure 2E; the same light dose was used as in the cell experiments). Most unstrained Ru(II) complexes with bpy coligands (10–12) did not eject after 4 h irradiation, but complex 9 gave a t1/2 of 66 min (Table 2).
Cytotoxicity, SAR and DNA Damage
An SAR study was performed for 2-(2-pyridyl)benzazole ligands based on benzazole core bioisosterism and the corresponding Ru(II) complexes with dmphen or bpy coligands. None of the free ligands exhibited activity against a leukemic cell line (HL60 human promyelocytic leukemia) up to 100 μM concentrations (Figure 3, Table 3). Compounds 5–8 were 20–300-fold more potent against HL60 cell line than parent ligands, with IC50 values ranging from 0.34–4.55 μM. Unexpectedly, the photoreactive compounds 6–8 exhibited the same range of activity under dark and light conditions. However, the strained Ru(II) complexes exhibited a steeper dose response when light activated, and caused essentially complete cell death in lower concentrations (Figure 3A).
Figure 3.
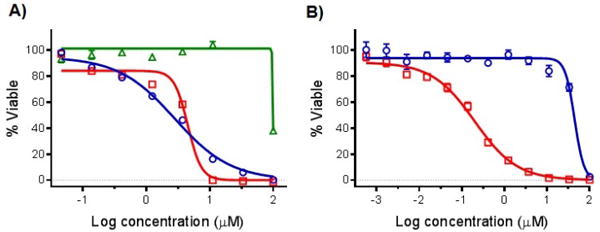
Cytotoxicity dose responses of ruthenium complexes and parent ligand on HL60 cells: A) 2 and 6; B) 10. Dark conditions (circles, blue line); irradiated samples, 1 min >450 nm light using the Indigo LED (29.1 J/cm2; squares, red line); ligand 2 (triangles, green line). (n = 3).
Table 3.
Cytotoxicity IC50 Values for 2-(2-Pyridyl)Benzazole Ligands and Ru Complexes in the HL60 Cancer Cell Line
| Comp. | Coligand | Y | Dark IC50 [μM] | Lighta IC50 [μM] | PI |
|---|---|---|---|---|---|
| 1 | – | CH | >100 | n.d.b | n.d. |
| 2 | – | NH | >100 | n.d. | n.d. |
| 3 | – | O | >100 | n.d. | n.d. |
| 4 | – | S | >100 | n.d. | n.d. |
| 5 | dmphen | CH | 0.34 (±0.008) | 0.034 (±0.001) | 10 |
| 6 | dmphen | NH | 2.79 (±0.31) | 4.49 (±0.07) | 0.6c |
| 7 | dmphen | O | 4.54 (±1.37) | 3.43 (±0.75) | 1.3 |
| 8 | dmphen | S | 1.55 (±0.41) | 4.55 (±1.73) | 0.3c |
| 9 | bpy | CH | 1.24 (±0.008) | 0.18 (±0.001) | 7.3 |
| 10 | bpy | NH | 44.8 (±0.4) | 0.20 (±0.017) | 224 |
| 11 | bpy | O | 63.5 (±8.3) | 5.18 (±0.047) | 12.2 |
| 12 | bpy | S | 83.3 (±1.2) | 0.94 (±0.09) | 88 |
| Cisplatin | – | – | 3.1 (±0.2) | 3.1 (±0.2) | 1 |
Using the Loctite Indigo LED array (light dose of 29.1 J/cm2)
n.d. – not determined.
The PI values reflect differences in the slopes of the dose response curves, and likely indicate that different cytotoxicity mechanisms are responsible for cell death in the dark vs. light conditions.
For the photoejecting systems, the largest Phototoxicity Index (PI) value was found for complex 5, which produced a 10-fold enhanced activity upon irradiation, with a 34 nM IC50 value (Table 3). The highest PI values were found for 10–12, which contain the Ru(bpy)2 scaffold. After irradiation, compounds 9, 10, and 12 produced submicromolar IC50 values, with 7–220-fold differences in the light and dark, and demonstrated 3–17-fold greater potencies than cisplatin. The 88-fold (12, Figure S13) and 224-fold (10, Figure 3B) difference in light and dark IC50 values provides a large potential therapeutic window to allow for selective targeting of cells by exposure to light.
The SAR study revealed the following: (1) coordination of 2-(2-pyridyl)benzazole ligands with the Ru(II) scaffolds is crucial for potency; (2) the cytotoxic effect is sensitive to the coligands (dmphen vs bpy) – replacement of dmphen with bpy decreased the potency in the dark, but promoted the nM activity after light irradiation (compounds 9, 10, 12) and provided a large potential therapeutic window (88 for 12 and >220 for 10); (3) the nature of a benzazole core had an influence on the antitumor activity, with complexes 5 and 9 exhibiting 5–35-fold greater potency under dark conditions in comparison to other compounds from strained and unstrained groups, respectively (Table 3, Figure 4). It should be noted that complexes 5 and 9 carry a +1 charge, while all other compounds were +2.
Figure 4.
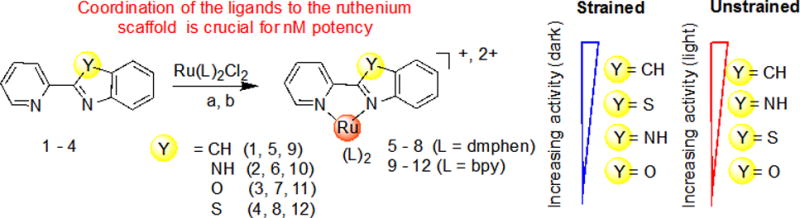
SAR Analysis for Antitumor Ru(II) Complexes with 2-(2-Pyridyl)Benzazole Ligands
The effect of the compounds 8 and 12 on cell cycle was analyzed at the IC50 of the compounds over several time points (Figures S15,16). At the 24 h time point, the sub-G1 phase had increased to 45% of the population for 8 and 27% for 12 when the compounds had been irradiated. No significant increase in the population of apoptotic cells and the G1, S or G2/M populations occurred for 8 and 12 in the dark. Thus, neither compound induced cell cycle arrest under dark conditions or upon irradiation.
In an attempt to determine a potential mechanism of action, DNA damage was assessed by agarose gel electrophoresis. Supercoiled pUC19 plasmid was incubated with each complex in dose response and kept in the dark or exposed to 470 nm light for one hour (Figure 5). The irradiated samples revealed significant differences in damage profiles. The strained photoactive Ru(II) complexes 6 and 8 exhibited a combination of DNA photocleavage and DNA photobinding (Figure 5A, B). Covalent adducts were visualized by the reduced mobility on the agarose gel with increasing concentration of Ru(II) complex, as well as loss of EtBr signal. Unstrained complex 12 induced single-strand breaks in the DNA when irradiated with light, likely due to the photogeneration of 1O2. This was visualized by the conversion from supercoiled DNA to relaxed circle (Figure 5D). Unexpectedly, unstrained 10 produced fewer single-strand breaks than 12 based on the small ratio between relaxed circle and supercoiled DNA (Figure 5C). Precipitation of the DNA with the complexes 5, 8, and 9 was observed at concentrations above 125 μM.
Figure 5.
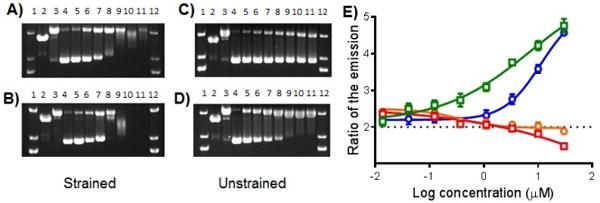
Agarose gel electrophoresis of 40 μg/mL pUC19 plasmid (10 mM phosphate buffer, pH 7.5) with light-activated Ru(II) compounds. Dose response profiles: (A) 6; (B) 8; (C) 10; (D) 12; Lanes 1 and 12, DNA molecular weight standard; lane 2, linear pUC19; lane 3, relaxed circle (Cu(phen)2 reaction with pUC19); lanes 4−11, 0, 7.8, 15.6, 31.25, 62.5, 125, 250, and 500 μM compound. E) Singlet oxygen generation dose responses of ruthenium complexes: 6 (circles, orange line); 8 (squares, red line); 10 (circles, blue line); and 12 (squares, green line). (n = 2). The data is shown as a ratio of the emission of the sensor after irradiation with the compound vs. without irradiation. The slight downward curve for 8 suggests this compound quenches singlet oxygen at high doses.
Despite the difference in their ability to inflict DNA damage, both the unstrained compounds 10 and 12 induced submicromolar cytotoxicity after light irradiation and exhibited large PI values. In an attempt to confirm or disprove the involvement of light-activated generation of 1O2 in the biological mechanism of action, dose responses of compounds were performed with Singlet Oxygen Sensor Green reagent with light irradiation (Figure 5E; Figure S15). As anticipated, photogeneration of 1O2 was observed for both unstrained complexes 10 and 12, in contrast to corresponding strained compounds 6 and 8. Consistent with the DNA damage gels, compound 12 exhibited greater potency for 1O2 generation; however, there is a large discrepancy between the concentrations needed to produce 1O2 or induce strand breaks in the DNA compared to cytotoxicity IC50 values. This suggests that 1O2 alone cannot be responsible for the potent effects in cells.[17] Moreover, the pyridyl-indole based complexes (5 and 9) possessed the highest cytotoxicity and did not generate 1O2 upon irradiation (Figure S15).
CONCLUSIONS
Eight heteroleptic Ru(II) complexes were synthesized in order to explore structure−activity relationships. The complexes contained one 2-(2-pyridyl)benzazole type ligand combined with either two dmphen ligands to make intrinsically strained complexes, or two 2,2′-bipyridine ligands to form unstrained complexes. While the free benzazole type ligands 1–4 were not toxic in the investigated concentration range, the Ru(II) complexes exhibited marked cytotoxicity. The most potent compounds, 5 and 9, contained the 2-(2-pyridyl)indole ligand, and were highly effective in killing leukemic cells when irradiated, with IC50 values less than 0.04 and 0.2 μM. However, the observed high toxicity in the dark could be a limitation for their potential application as PDT agents.
In contrast, large therapeutic windows were found for complexes 12 and 10 (with 88- and 224-fold differences in light and dark IC50 values), which demonstrated 3–15-fold greater potency than cisplatin. The unstrained compounds are capable of generating singlet oxygen, but the significant disparity in the effective concentration for cytotoxicity, 1O2 production, and DNA cleavage suggests that some other, currently unknown, mechanisms of action could be involved for anticancer activity. This may involve different species of ROS.
Considering the promising dark-cytotoxicity of strained complexes and light-induced antitumor potential of unstrained compounds, we are currently modifying these complexes, aiming to generate more potent anticancer agents with possible application in both standard chemotherapy and photodynamic therapy.
EXPERIMENTAL SECTION
Materials and Methods
The starting 2-(2-pyridyl)benzazole ligands were obtained from commercial sources (2,3) or were synthesized according to the methods described previously (1,4).[18] Complexes 10–12 were synthesized using previously established procedures.[14]
All 1H NMR spectra were obtained on a Varian Mercury spectrometer (400 MHz) with chemical shifts reported relative to the residual solvent peak of acetonitrile at δ 1.94. Electrospray ionization mass spectra were obtained on a Varian 1200L mass spectrometer. Absorption spectra were obtained on an Agilent Cary 60 spectrophotometer or a BMG Labtech FLUOstar Omega microplate reader. Photoejection, DNA damage, and singlet oxygen generation experiments were performed using a 470 nm LED array from Elixa, and a Loctite Indigo LED array (for cell cytotoxicity studies and HPLC photoejection analysis). All synthesized compounds were isolated in >95% purity, as determined by analytical HPLC. For HPLC analysis, the ruthenium complexes were injected on an Agilent 1100 series HPLC equipped with a model G1311 quaternary pump, G1315B UV diode array detector, and ChemStation software version B.01.03. Chromatographic conditions were optimized on a Column Technologies Inc. C18, 120 Å (250 mm × 4.6 mm inner diameter, 5 μM) fitted with a Phenomenex C18 (4 mm × 3 mm) guard column. Injection volumes of 15 μL of 100 μM solutions of the complex were used. The detection wavelength was 280 nm. Mobile phases were: mobile phase A, 0.1% formic acid in dH2O; mobile phase B, 0.1% formic acid in HPLC grade acetonitrile. The mobile phase flow rate was 1.0 mL/min. The following mobile phase gradient was used: 98−95% A (containing 2− 5% B) from 0 to 5 min; 95−70% A (5−30% B) from 5 to 15 min; 70−40% A (30−60% B) from 15 to 20 min; 40−5% A (60−95% B) from 20 to 30 min; 5−98% A (95−2% B) from 30 to 35 min; reequilibration at 98% A (2% B) from 35 to 40 min.
General procedure for synthesis of Ru(dmphen)2L complexes with 2-(2-pyridyl)benzazole ligands
Ru(dmphen)2Cl2 (1 eq) and 2-(2-pyridyl)benzazole (1.1 eq) were added to 4 mL of ethylene glycol in a 15 mL pressure tube. The mixture was heated at 100–120 °C for 2 h while protected from light. The dark brown (5) or orange solution (6–8) was allowed to cool to room temperature and poured into 50 mL of dH2O. Addition of a saturated aq. KPF6 solution (ca. 1 mL) produced a brown or red-orange precipitate that was collected by vacuum filtration. The purification of the solid was carried out by flash chromatography (silica gel, loaded in 0.1% KNO3, 5%H2O in MeCN). A gradient was run, and the pure complex eluted at 0.2% KNO3, 5–10% H2O in MeCN. The product fractions were concentrated under reduced pressure, and a saturated aq solution of KPF6 was added, followed by extraction of the complex into CH2Cl2. The solvent was removed under reduced pressure to give a solid.
5 Rf=0.63 (0.1% KNO3, 5%H2O in MeCN); 1H NMR (CD3CN): δ 8.59 (d, J = 8.2 Hz, 1H), 8.52 (d, J = 8.3 Hz, 1H), 8.21 (d, J = 8.3 Hz, 1H), 816 (d, J = 8.7 Hz, 1H), 8.02–8.06 (m, 3H), 7.81 (d, J = 8.7 Hz, 1H), 7.74 (d, J = 8.3 Hz, 1H), 7.70 (d, J = 8.0 Hz, 1H), 7.64 (d, J = 8.3 Hz, 1H), 7.44 (t, J = 7.8 Hz, 1H), 7.32 (d, J = 8.3 Hz, 1H), 7.28 (d, J = 8.3 Hz, 1H), 7.20 (d, J = 8.0 Hz, 1H), 6.93 (s, 1H), 6.57 (d, J = 5.6 Hz, 1H), 6.43–6.47 (m, 2H), 6.13 (d, J = 7.5 Hz, 1H), 4.47 (d, J = 8.5 Hz, 1H), 2.02 (s, 3H), 1.98 (s, 3H), 1.85 (s, 3H), 1.82 (s, 3H); purity by HPLC = 97 %; ESI MS calcd for C41H33N6Ru [M]+ 711.18, found 711.3 [M]+; UV/Vis (CH3CN): λmax (ε) 490 nm (8400 mol−1dm3cm−1).
6 Rf=0.38 (0.1% KNO3, 5%H2O in MeCN); 1H NMR (CD3CN): δ 8.69 (d, J = 8.3 Hz, 1H), 8.62 (d, J = 8.3 Hz, 1H), 8.30 (d, J = 8.3 Hz, 1H), 8.23 (d, J = 8.8 Hz, 1H), 8.09–8.15 (m, 3H), 8.00 (d, J = 7.9 Hz, 1H), 7.82–7.88 (m, 3H), 7.72 (d, J = 8.3 Hz, 1H), 7.36–7.43 (m, 3H), 7.15 (t, J = 7.4 Hz, 1H), 6.93–7.00 (m, 3H), 6.72 (ddd, J = 8.8, 7.3, 0.9 Hz, 1H), 4.91 (d, J = 8.5 Hz, 1H), 1.98 (s, 3H), 1.95 (s, 3H), 1.91 (s, 3H), 1.88 (s, 3H); purity by HPLC = 97 %; ESI MS calcd for C40H33N7Ru [M]2+ 356.59; found 356.7 [M]2+; UV/Vis (CH3CN): λmax (ε) 455 nm (11800 mol−1dm3cm−1).
7 Rf=0.52 (0.1% KNO3, 5%H2O in MeCN); 1H NMR (CD3CN): δ 8.74 (d, J = 8.3 Hz, 1H), 8.68 (d, J = 8.3 Hz, 1H), 8.35 (d, J = 8.3 Hz, 1H), 8.27 (d, J = 8.7 Hz, 1H), 8.23 (d, J = 8.3 Hz, 1H), 8.13–8.20 (m, 3H), 7.94–7.98 (m, 2H), 7.86 (d, J = 8.3 Hz, 1H), 7.78 (d, J = 8.4 Hz, 1H), 7.66 (d, J = 8.5 Hz, 1H), 7.40–7.46 (m, 3H), 7.16 (ddd, J = 8.0, 5.8, 1.5 Hz 1H), 7.00–7.04 (m, 2H), 5.12 (d, J = 8.3 Hz, 1H), 2.11 (s, 3H), 2.00 (s, 3H), 1.98 (s, 3H), 1.90 (s, 3H); purity by HPLC = 98 %; ESI MS calcd for C40H32N6ORu [M2+•PF6−]+ 859.13, [M]2+ 357.09; found 859.3 [M2+•PF6−]+, 356.9 [M]2+; UV/Vis (CH3CN): λmax (ε) 445 nm (7700 mol−1dm3cm−1).
8 Rf=0.51 (0.1% KNO3, 5%H2O in MeCN);1H NMR (CD3CN): δ 8.72 (d, J = 8.3 Hz, 1H), 8.67 (d, J = 8.3 Hz, 1H), 8.41 (d, J = 8.3 Hz, 1H), 8.28 (d, J = 8.8 Hz, 1H), 8.23 (d, J = 8.4 Hz, 1H), 8.16–8.20 (m, 2H), 8.08 (d, J = 8.8 Hz, 1H), 7.91–7.95 (m, 2H), 7.89 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 8.7 Hz, 1H), 7.73 (d, J = 8.4 Hz, 1H), 7.49 (d, J = 8.3 Hz, 1H), 7.40 (d, J = 8.4 Hz, 1H), 7.34 (ddd, J = 8.8, 7.4, 1.0 Hz, 1H), 7.09–7.14 (m, 2H), 6.92 (ddd, J = 8.8, 7.1, 1.2 Hz, 1H), 5.37 (d, J = 8.6 Hz, 1H), 2.10 (s, 3H), 2.01 (s, 3H), 1.85 (s, 3H), 1.84 (s, 3H); purity by HPLC = 99 %; ESI MS calcd for C40H32N6RuS [M2+•PF6−]+ 875.11, [M]2+ 365.08; found 875.3 [M2+•PF6−]+, 365.1 [M]2+; UV/Vis (CH3CN): λmax (ε) 445 nm (8900 mol−1dm3cm−1).
General Preparation of Ru(bpy)2L Complexes
Ru(bpy)2Cl2•2H2O (120 mg, 0.23 mmol) and 2-(2-pyridyl)benzazole (0.27 mmol) were added to 6 mL of 50:50 EtOH:H2O in a 15 mL pressure tube. The mixture was heated at 90 °C for 2 h, after which the orange solution was allowed to cool to room temperature. Addition of a saturated aq KPF6 solution resulted in precipitation of the complex, which was extracted into methylene chloride. Purification of the orange solid was carried out by flash chromatography (silica gel, loaded in 0.1% KNO3, 5% H2O in MeCN). The pure complex eluted at 0.2% KNO3, 10% H2O in MeCN, and the product fractions were concentrated under reduced pressure. A saturated aq solution of KPF6 was added, and the complex was extracted into CH2Cl2, followed by removal of the solvent under reduced pressure to give an orange solid.
9 Rf=0.60 (0.1% KNO3, 5%H2O in MeCN);1H NMR (CD3CN): δ 8.40–8.44 (m, 3H), 8.30 (d, J = 8.2 Hz, 1H), 7.92–8.01 (m, 5H), 7.88 (td, J = 8.0, 1.5 Hz, 1H), 7.75–7.90 (m, 2H), 7.69 (ddd, J = 8.2, 7.5, 1.6 Hz, 1H), 7.55 (ddd, J = 6.0, 1.6, 0.8 Hz, 1H), 7.46 (dt, J = 8.0, 1.0 Hz, 1H), 7.31–7.35 (m, 3H), 7.19–7.27 (m, 3H), 6.89 (ddd, J = 8.0, 5.8, 1.6 Hz, 1H), 6.68 (ddd, J = 8.0, 6.8, 0.8 Hz, 1H), 6.47 (ddd, J = 8.8, 6.8, 1.2 Hz, 1H), 5.38 (d, J = 7.5 Hz, 1H); purity by HPLC = 98 %; ESI MS calcd for C33H25N6Ru [M] + 607.12; found 607.1 [M]+; UV/Vis (CH3CN): λmax (ε) 480 nm (8600 mol−1dm3cm−1).
10 Rf=0.38 (0.1% KNO3, 5%H2O in MeCN);1H NMR (CD3CN): δ 8.51–8.54 (m, 3H), 8.47 (d, J = 8.1 Hz, 1H), 8.44 (d, J = 8.1 Hz, 1H), 7.96–8.15 (m, 6H), 7.80–7.87 (m, 3H), 7.71–7.75 (m, 2H), 7.47 (ddd, J = 8.0, 5.6, 1.3 Hz, 1H), 7.37–7.44 (m, 4H), 7.34 (ddd, J = 8.0, 5.6, 1.2 Hz, 1H), 7.05 (ddd, J = 8.8, 7.4, 1.1 Hz, 1H), 5.82 (d, J = 8.3 Hz, 1H); purity by HPLC = 99 %; ESI MS calcd for C32H25N7Ru [M]2+ 304.56; found 304.6 [M]2+; UV/Vis (CH3CN): λmax (ε) 455 nm (13100 mol−1dm3cm−1).
11 Rf=0.45 (0.1% KNO3, 5%H2O in MeCN);1H NMR (CD3CN): δ 8.51–8.54 (m, 3H), 8.48 (d, J = 7.9 Hz, 1H), 8.45 (d, J = 8.2 Hz, 1H), 8.01–8.19 (m, 6H), 7.94 (d, J = 5.4 Hz, 1H), 7.88 (d, J = 8.5 Hz, 1H), 7.77–7.82 (m, 3H), 7.61 (ddd, J = 8.8, 7.5, 1.1 Hz, 1H), 7.55 (ddd, J = 8.0, 5.6, 1.3 Hz, 1H), 7.42–7.49 (m, 3H), 7.39 (ddd, J = 8.0, 5.6, 1.2 Hz, 1H), 7.28 (ddd, J = 8.8, 7.6, 0.9 Hz, 1H), 5.96 (d, J = 8.2 Hz, 1H); purity by HPLC = 95 %; ESI MS calcd for C32H25N6ORu [M]2+ 305.06; found 305.1 [M]2+; UV/Vis (CH3CN): λmax (ε) 450 nm (12600 mol−1dm3cm−1).
12 Rf=0.45 (0.1% KNO3, 5%H2O in MeCN);1H NMR (CD3CN): δ 8.51–8.54 (m, 4H), 8.44 (d, J = 8.1 Hz, 1H), 8.20 (d, J = 8.1 Hz, 1H), 8.04–8.16 (m, 4H), 8.01 (td, J = 8.0, 1.4 Hz, 1H), 7.92 (d, J = 5.8 Hz, 1H), 7.84 (d, J = 5.2 Hz, 1H), 7.74 (d, J = 5.7 Hz, 1H), 7.69 (d, J = 5.5 Hz, 1H), 7.67 (d, J = 5.3 Hz, 1H), 7.57 (ddd, J = 8.4, 7.2, 1.0 Hz, 1H), 7.45–7.49 (m, 2H), 7.37–7.42 (m, 2H), 7.35 (ddd, J = 8.0, 5.6, 1.2 Hz, 1H), 7.27 (ddd, J = 8.8, 7.3, 1.1 Hz, 1H), 6.31 (d, J = 8.5 Hz, 1H); purity by HPLC = 98 %; ESI MS calcd for C32H24N6RuS [M2+•PF6−]+ 771.05, [M]2+ 313.04; found 771.2 [M2+•PF6−]+, 313.1 [M]2+; UV/Vis (CH3CN): λmax (ε) 445 nm (13100 mol−1dm3cm−1).
Counter ion exchange
Compounds 5–12 were converted to Cl− salts by dissolving 5–20 mg of product in 1–2 mL methanol. The dissolved product was loaded onto an Amberlite IRA-410 chloride ion exchange column, eluted with methanol, and the solvent was removed in vacuo.
Cytotoxicity Assay
HL60 cells were plated at 30,000 cell per well in Opti-MEM media with 1% FBS and pen-strep in 96 well plates. Compounds were serially diluted in opti-MEM with 1% FBS and pen-strep in a 96 well plate and then added to the cells. They were then irradiated with 29.1 J/cm2 light (>450 nm using the Indigo LED) for 1 minute or kept in the dark. The cells were incubated with the compounds for 72 h followed by the addition of resazurin. The plates were incubated for 3 h and then read on a SpectraFluor Plus plate reader with an excitation filter of 535 nm and emission of 595 nm.
DNA Gel Electrophoresis
Compounds were mixed with 40 μg/mL pUC19 plasmid DNA in 10 mM potassium phosphate buffer, pH 7.4. To determine the effect of light, samples were irradiated with a 470 nm LED for a total light dose of 46.8 J/cm2. Samples were then incubated for 12 h at room temperature in the dark. Single- and double-strand DNA break controls were prepared, and the DNA samples were resolved on agarose gels, as described previously.[8a] In brief, samples were resolved on a 1% agarose gels prepared in tris-acetate buffer with 0.3 μg of plasmid/lane. The gels were stained with 0.5 μg/mL ethidium bromide in tris-acetate buffer at room temperature for 40 min, destained with tris-acetate buffer, and imaged on a ChemiDoc MP System (Bio-Rad).
Singlet Oxygen Assay
Compounds were serially diluted in 10 mM potassium phosphate buffer, pH 7.4, with ~5μM Singlet Oxygen Sensor Green reagent in 96 well plates. The plates were read on a SpectraFluor Plus plate reader with an excitation filter of 485 nm and emission of 535 nm in a dark and after 1h irradiation with a 470 nm LED for total light dose of 46.8 J/cm2.
Cell Cycle Analysis
HL60 cells were plated in opti-MEM with 1% FBS at a density of 500,000 cells/ml in 6-well plates. The compounds were added and incubated with the cells from 0 to 12 h. For each time point the cells were transferred to FACS tubes, pelleted, washed with PBS, followed by the addition of cold 70% ethanol and incubated on ice for one hour to fix the cells. Cells were then centrifuged at 2000 rpm for 5 minutes, and resuspended in 1 ml PBS for each tube. The tubes were centrifuged at 2000 rpm for 5 minutes, the supernatant was aspirated and the cells were resuspended in 0.5 mL PI staining buffer (20 mg/mL PI in PBS, 0.2 mg/mL RNAse, 0.1% TritonX-100) and incubated at room temperature for 30 minutes. Samples were run through the flow cytometer and data was analyzed with ModFit and FlowJo.
Crystallography
Single crystals of compounds 6–8 were grown from methylene chloride or acetone by vapor diffusion of diethyl ether. They were mounted in inert oil and transferred to the cold gas stream of the diffractometer. X-ray diffraction data were collected at 90.0(2) K on either a Nonius kappaCCD diffractometer using MoKα X-rays or on a Bruker-Nonius X8 Proteum diffractometer with graded-multilayer focused CuKα X-rays. Raw data were integrated, scaled, merged and corrected for Lorentz-polarization effects using either the HKL-SMN package[19] or the APEX2 package.[20] Corrections for absorption were applied using SADABS[21] and XABS2.[22] The structures were solved by SHELXT,[23] and refined against F2 by weighted full-matrix least-squares using SHELXL-2014.[24] For compound 8 the SQUEEZE routine[25] was used to treat disordered solvent. Hydrogen atoms were placed at calculated positions and refined using a riding model. Non-hydrogen atoms were refined with anisotropic displacement parameters. Structures were checked using check CIF tools in Platon[26] and by an R-tensor.[27] Crystal data and relevant details of the structure determinations are summarized below and selected geometrical parameters are given in Table 1.
Crystal data (6)
C41H35Cl2F12N7P2Ru, Mr = 1087.67, Monoclinic, P21/c, a = 12.3286(2) Å, b = 18.7316(3)Å, c = 18.1692(3) Å, β = 94.943(1)°, V = 4180.29(12)Å3, Z = 4, ρ = 1.728 mg m−3, μ = 5.802 mm−1, F(000) = 2184, crystal size = 0.300×0.120×0.060 mm, θ(max) = 68.373°, 56392 reflections collected, 7596 unique reflections (Rint = 0.0433), GOF = 1.065, R1 = 0.0408 and wR2 = 0.0933 [I > 2σ(I)], R1 = 0.0436 and wR2 = 0.0949 (all indices), largest difference peak/hole = 1.531/−1.465 eÅ−3.
Crystal data (7)
C48.29H50.15F12N6O3.50P2Ru, Mr = 1161.55, Monoclinic, C2/c, a = 23.0734(5) Å, b = 19.9646(5) Å, c = 22.6098(5) Å, β = 108.547(1)°, V = 9874.3(4) Å3, Z = 8, ρ = 1.563 mg m−3, μ = 4.028 mm−1, F(000) = 4735, crystal size = 0.230×0.180×0.030 mm, θ(max) = 68.355°, 60921 reflections collected, 8945 unique reflections (Rint = 0.0643), GOF = 1.036, R1 = 0.0409 and wR2 = 0.0985 [I > 2σ(I)], R1 = 0.0561 and wR2 = 0.1067 (all indices), largest difference peak/hole = 0.631/−0.544 eÅ−3.
Crystal data (8)
C89H82F24N12O3P4Ru2S2, Mr = 2213.80, Triclinic, P-1, a = 14.2840(2) Å, b = 17.5165(2)Å, c = 20.0585(3)Å, α = 91.5547(8)°, β = 90.7709(8)°, γ = 110.9785(7)°, V = 4682.91(11)Å3, Z = 2, ρ = 1.570 mg m−3, μ = 0.539 mm−1, F(000) = 2240, crystal size = 0.320×0.280×0.270 mm, θ(max) = 27.509°, 136315 reflections collected, 21482 unique reflections (Rint = 0.0402), GOF = 1.054, R1 = 0.0445 and wR2 = 0.1153 [I > 2σ(I)], R1 = 0.0688 and wR2 = 0.1297 (all indices), largest difference peak/hole = 1.247/−0.749 eÅ−3.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (5R01GM107586). The X8 Proteum was funded by the NSF (MRI CHE-0319176). Mass spectroscopy analysis was performed at the University of Kentucky Environmental Research Training Laboratory (ERTL).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Siegel RL, Miller KD, Jemal A. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Galanski M, Jakupec MA, Keppler BK. Curr Med Chem. 2005;12:2075–2094. doi: 10.2174/0929867054637626. [DOI] [PubMed] [Google Scholar]
- 3.a) Hannon MJ. Pure and Applied Chemistry. 2007;79:2243–2261. [Google Scholar]; b) Griffin AM, Butow PN, Coates AS, Childs AM, Ellis PM, Dunn SM, Tattersall MH. Ann Oncol. 1996;7:189–195. doi: 10.1093/oxfordjournals.annonc.a010548. [DOI] [PubMed] [Google Scholar]
- 4.a) Antonarakis ES, Emadi A. Cancer Chemother Pharmacol. 2010;66:1–9. doi: 10.1007/s00280-010-1293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Barry NP, Sadler PJ. Chem Commun (Camb) 2013;49:5106–5131. doi: 10.1039/c3cc41143e. [DOI] [PubMed] [Google Scholar]
- 5.Bergamo A, Gaiddon C, Schellens JH, Beijnen JH, Sava G. J Inorg Biochem. 2012;106:90–99. doi: 10.1016/j.jinorgbio.2011.09.030. [DOI] [PubMed] [Google Scholar]
- 6.Ang WH, Casini A, Sava G, Dyson PJ. Journal of Organometallic Chemistry. 2011;696:989–998. [Google Scholar]
- 7.Glazer EC. Israel Journal of Chemistry. 2013;53:391–400. [Google Scholar]
- 8.a) Howerton BS, Heidary DK, Glazer EC. J Am Chem Soc. 2012;134:8324–8327. doi: 10.1021/ja3009677. [DOI] [PubMed] [Google Scholar]; b) Wachter E, Heidary DK, Howerton BS, Parkin S, Glazer EC. Chem Commun. 2012;48:9649–9651. doi: 10.1039/c2cc33359g. [DOI] [PubMed] [Google Scholar]; c) Hidayatullah AN, Wachter E, Heidary DK, Parkin S, Glazer EC. Inorg Chem. 2014;53:10030–10032. doi: 10.1021/ic5017164. [DOI] [PubMed] [Google Scholar]
- 9.Heidary DK, Glazer EC. Chembiochem. 2014;15:507–511. doi: 10.1002/cbic.201300681. [DOI] [PubMed] [Google Scholar]
- 10.a) Farrer NJ, Salassa L, Sadler PJ. Dalton Trans. 2009:10690–10701. doi: 10.1039/b917753a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mari C, Pierroz V, Ferrari S, Gasser G. Chemical Science. 2016;6:2660–2668. doi: 10.1039/c4sc03759f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) N M-A, Busto M, Leal JM, Rodríguez AM, Domínguez F, Acuña MI, Espino G, García B. Organometallics. 2015;34:319–327. [Google Scholar]; b) Martinez-Alonso M, Busto N, Jalon FA, Manzano BR, Leal JM, Rodriguez AM, Garcia B, Espino G. Inorg Chem. 2014;53:11274–11288. doi: 10.1021/ic501865h. [DOI] [PubMed] [Google Scholar]
- 12.Ong JX, Yap CW, Ang WH. Inorg Chem. 2012;51:12483–12492. doi: 10.1021/ic301884j. [DOI] [PubMed] [Google Scholar]
- 13.Yellol GS, Donaire A, Yellol JG, Vasylyeva V, Janiak C, Ruiz J. Chem Commun (Camb) 2013;49:11533–11535. doi: 10.1039/c3cc46239k. [DOI] [PubMed] [Google Scholar]
- 14.a) Wu F, Chamchoumis CM, Thummel RP. Inorg Chem. 2000;39:584–590. doi: 10.1021/ic990445s. [DOI] [PubMed] [Google Scholar]; b) Richardson CMFC, Keene FR, Steel PJ. Australian Journal of Chemistry. 2008;61:183–188. [Google Scholar]; c) Yi H, Crayston JA, Irvine JTS. Dalton Trans. 2003:685–691. [Google Scholar]
- 15.Shen WZ, Trotscher-Kaus G, Lippert B, Trans Dalton. 2009:8203–8214. doi: 10.1039/b904173g. [DOI] [PubMed] [Google Scholar]
- 16.Liu YJ, Chao H, Yu HJ, Yuan YX, Ji LN. ActaCrystallogr Sect E: Struct REp Online. 2006;62:m585. [Google Scholar]
- 17.Unfortunately, attempts to quantify the singlet oxygen using 1,3-diphenylisobenzofuran (DPBF) failed due to poor stability of the reagent under irradiation. The procedure used was reported by; Zhang W, Li B, Ma H, et al. ACS Appl Mater Interfaces. 2016;8(33):21465–71. doi: 10.1021/acsami.6b05817. [DOI] [PubMed] [Google Scholar]
- 18.a) Cravotto G, Demartin F, Palmisano G, Penoni A, Radice T, Tollari S. J Organomet Chem. 2005;690:2017–2026. [Google Scholar]; b) Sedachat N, Naimi-Jamal MR, Mokhtari J. Current Chemistry Letters. 2014;3:57–62. [Google Scholar]
- 19.O Z, Minor W. In: Processing of x-ray diffraction data collected in oscillation mode. Carter JRMSCW, editor. Vol. 276. Academic Press; 1997. [DOI] [PubMed] [Google Scholar]
- 20.APEX2. Programs for data collection and data reduction. Bruker-Nonius; Madison WI. USA: 2012. [Google Scholar]
- 21.Krause L, Herbst-Irmer R, Sheldrick GM, Stalke D. J Appl Cryst. 2015;48:3–10. doi: 10.1107/S1600576714022985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parkin S, Moezzi B, Hope H. J Appl Cryst. 1995;28:53–56. [Google Scholar]
- 23.Sheldrick GM. Acta Cryst. 2015;A71:3–8. [Google Scholar]
- 24.Sheldrick GM. Acta Cryst. 2015;C71 [Google Scholar]
- 25.van der Sluis P, Spek AL. Acta Cryst. 1990;A46:194–201. [Google Scholar]
- 26.Spek AL. Acta Cyst. 2009;D65:148–155. [Google Scholar]
- 27.Parkin S. Acta Cyst. 2000;A56:157–162. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.