

Abstract
Proteogenomics leverages information derived from proteomic data to improve genome annotations. Of particular interest are “novel” peptides that provide direct evidence of protein expression for genomic regions not previously annotated as protein-coding. We present a modular, automated data analysis pipeline aimed at detecting such “novel” peptides in proteomic data sets. This pipeline implements criteria developed by proteomics and genome annotation experts for high-stringency peptide identification and filtering. Our pipeline is based on the OpenMS computational framework; it incorporates multiple database search engines for peptide identification and applies a machine-learning approach (Percolator) to post-process search results. We describe several new and improved software tools that we developed to facilitate proteogenomic analyses that enhance the wealth of tools provided by OpenMS. We demonstrate the application of our pipeline to a human testis tissue data set previously acquired for the Chromosome-Centric Human Proteome Project, which led to the addition of five new gene annotations on the human reference genome.
Keywords: proteogenomics, bioinformatics, workflow, mass spectrometry, genome annotation, testis
Introduction
Proteogenomics is an expanding field of inquiry at the intersection of proteomics and genomics that has been growing in line with the advance of the omics era. Studies in this field analyze proteomic data in search of direct evidence of protein expression to help improve the annotation of protein-coding regions in genomes. In the case of mass spectrometry (MS) data, of particular interest are peptides of “unexpected” origin, i.e., peptides that do not match to a known protein sequence, because these could point to previously unrecognized coding regions. However, for genomes that are already well-characterized (particularly the human genome), novel peptides represent needles in a haystack of peptides matching known proteins. The difficulty of finding them is exacerbated by the importance of avoiding false positive hits that could lead to spurious annotations.1
Several prerequisites are essential for a successful, high-quality proteogenomics endeavor: a suitable proteomics data set, a comprehensive database composed of both known and potential novel protein-coding sequences, and the collaboration of experts for manual genome annotation. Furthermore, a data analysis workflow is needed that reliably and sensitively identifies peptides and filters them according to rigorous criteria. Such a workflow should operate in a reproducible fashion and allow high-throughput processing to enable the analysis of large data sets.
We have recently reported the detection of novel proteins in the human genome based on stringent guidelines for the processing of proteomic MS data for genome annotation efforts.2 Here, we describe an automated processing pipeline based on these principles. Our pipeline is implemented using OpenMS 2.03 as an example of combining task-specific tools into a powerful data analysis workflow. The pipeline introduced here aims to identify novel peptides based on a suitable sequence database. However, due to the great flexibility inherent in the modular workflow approach, this “core” proteogenomics pipeline can be easily adapted to handle different requirements and extended to include additional functionality from the OpenMS toolbox, such as quantification or post-translational modification analysis.
Our approach achieves the goals of modularity, flexibility, and extensibility that are shared by many previously proposed workflows.4−7 Significantly, our pipeline directly implements the quality criteria that were developed in collaboration between proteomics experts and genome annotators and published previously.2 Furthermore, it benefits from the strengths of OpenMS as a mature, reliable platform with an active user and developer base. OpenMS offers high performance suitable for large-scale analyses, supports all major operating systems, and provides integration with various workflow managers. Unlike some alternatives, our pipeline does not generate an amino acid sequence database from genomic data because we prefer a manually curated database that incorporates a variety of sources for proteogenomics. Finally, our pipeline condenses the core of a proteogenomic analysis into a manageable workflow composed of 18 steps, significantly smaller and more straightforward than alternative proteogenomics workflows.
In addition to presenting the proteogenomics pipeline itself, we describe here the contributions that we made in this context to the code base of the OpenMS project and to MascotPercolator.8,9 Finally, we show the application of our pipeline to a relevant published data set, the human testis tissue data set acquired for the Chromosome-Centric Human Proteome Project (C-HPP).10
Materials and Methods
Proteomics Data Set
To demonstrate the capabilities of our proteogenomics pipeline, we applied it to the human testis tissue data set published by Zhang and colleagues.10 We will refer to this as the “C-HPP testis” data set. The data set contains three biological replicates, each fractionated using two different protein separation methods (regular SDS-PAGE and tricine-SDS-PAGE), with six samples in total. Each sample gave rise to 28 (regular) and 22 (tricine) fractions, respectively, which were digested using trypsin and analyzed on an LTQ Orbitrap Velos (Thermo Scientific) mass spectrometer, acquiring fragment ion (MS2) spectra using CID activation. We downloaded the 150 RAW files from these liquid chromatography–tandem mass spectrometry (LC–MS/MS) runs from the PRIDE repository11 (accession PXD002179) and converted them to mzML format, extracting only the MS2 scans, using the “msconvert” program from the ProteoWizard software suite12 (version 3.0.8789). A single RAW file was renamed to fit the naming scheme of the remaining files (“CHPP_Testis_tricine_1010.raw” to “CHPP_TESTIS_Tricine_1010.raw”). Another file, “CHPP_SDS_3003.raw”, turned out to be corrupt, so we downloaded the corresponding MGF file from PRIDE and converted that to mzML. The mzML files were then used as inputs for our OpenMS pipeline.
Sequence Database
The amino acid sequence database against which fragment ion (MS2) spectra are matched is critical for comprehensive identification of peptides. A sequence database for use with our pipeline should consist of four parts: (1) known protein-coding sequences; (2) sequences of common contaminants; (3) prospective protein sequences currently unannotated or presumed noncoding that could potentially contain unrecognized coding regions (e.g., lncRNA transcripts, RNA-Seq models, predicted transcripts, etc.); and (4) decoy sequences. The four parts are concatenated into a single sequence database that is used for database searching of MS2 spectra; parts 1–3 are also used individually to filter the downstream search results. When analyzing data with the aim of finding “novel” peptides, we are interested in high-confidence matches to peptides that are in part 3 of the database but are not also in parts 1 or 2.
For the analysis of the C-HPP testis data, we used the sequence database from Wright et al.2 This database consists of human translated amino acid sequences from the following sources. For part 1, it contains the translated CDS from GENCODE13 v20 and the UniProt14 reference proteome. For part 2, it contains the collection of common contaminant proteins from the Max Planck Institute of Biochemistry, together with sequences of the major histocompatibility complex from the IPD-IMGT/HLA database.15 For part 3, it contains noncoding sequences from GENCODE v20 (pseudogenes, lncRNA, 5′ UTR), gene predictions from AUGUSTUS,16 pseudogene predictions from Pseudogene.org,17 and three-frame translated transcripts from three different large-scale RNA-Seq experiments.13,18,19 For part 4, it contains randomized decoy sequences generated from parts 1–3 using the Mimic software (https://github.com/percolator/mimic). To simplify dealing with the isobaric amino acids leucine and isoleucine, all occurrences of “I” in the sequences were replaced by “L”. In total, the database contains 8 406 627 entries; the FASTA file takes up 1.1 GB of memory (including accessions).
Software
Our data analysis pipeline leverages many existing software tools. We used TOPP tools20 from the OpenMS framework (version 2.0.1; http://openms.org) for most data-processing steps, although in some areas we extended the functionality offered by OpenMS (see below). Importantly, OpenMS provides an easy-to-use graphical interface for designing and running TOPP-based data analysis workflows, called TOPPAS.21 It also offers a mechanism for wrapping non-TOPP command line applications so that they can be included in TOPPAS workflows (“GenericWrapper”).
Central to any proteomics analysis pipeline is the identification of peptides from the MS2 spectra, for which we used the database search engines Mascot22 (version 2.5.1, Matrix Science) and MS-GF+23 (version 10089). The following parameters were used with both engines: 10 ppm precursor mass tolerance, 0.5 Da fragment mass tolerance; trypsin cleavage with full specificity, allowing two missed cleavages; fixed modification: carbamidomethylation of cysteine; variable modifications: oxidation of methionine, deamidation of asparagine and glutamine, N-terminal acetylation, and conversion to pyroglutamic acid of N-terminal glutamine and glutamic acid.
We applied Percolator24 (revision “273ff55” from https://github.com/percolator/percolator) for statistical evaluation and rescoring of the search results (in the Mascot case via an adapted MascotPercolator,8,9 version 2.16; http://www.sanger.ac.uk/science/tools/mascotpercolator). To summarize the results of our pipeline and visualize the data, we used the R software environment for statistical computing (version 3.1.2).25
Results
Software Contributions
We were able to rely on existing applications for the construction of our proteogenomic pipeline and for many of the data processing tasks; however, for several tasks, we developed new tools or refined and extended existing ones. We added wrappers for the search engine MS-GF+23 and for the postprocessing tool Percolator24 to OpenMS. In addition, we adapted several existing TOPP tools and underlying OpenMS library classes, tailoring them for use in an integrated pipeline and for the needs of proteogenomic data analysis. We not only added missing functionality but in several cases also improved the quality (and thus reliability and maintainability) of the source code.
Adapter for the MS-GF+ Search Engine
OpenMS includes TOPP tools that serve as adapters to several widely used database search engines (e.g., Mascot, OMSSA, and X! Tandem). We developed a TOPP tool that wraps the newer search engine MS-GF+ called MSGFPlusAdapter. The adapter takes as input the location of the MS-GF+ Java package, a spectral data file (mzML), a sequence database file (FASTA), and a number of search parameters, including lists of desired fixed and variable modifications. It creates a temporary file containing the user-defined modifications in the format required by MS-GF+. A Java process runs the MS-GF+ program, supplying search parameters and input files. MS-GF+ performs the search and writes results into an mzIdentML file. Because OpenMS internally still uses its own format, idXML, for peptide and protein identification (ID) data, the adapter can optionally convert the mzIdentML file to idXML once the search is complete. This conversion was initially implemented via an intermediary step in which a tabular text file is generated from the mzIdentML file using a function of MS-GF+. Information from this file is then used to fill internal data structures and, from there, written out to idXML. Support for the mzIdentML format in OpenMS has improved in the meantime, so it is now also possible to convert the mzIdentML file to idXML directly. Irrespective of the method used for the conversion, the adapter needs to look up retention time (RT) values for peptide IDs in the spectral data. This information is missing from the mzIdentML file generated by MS-GF+, but it is vital for many downstream applications and analyses in OpenMS. To support this, we developed two related classes for the OpenMS C++ library, SpectrumLookup and SpectrumMetaDataLookup, to handle the recurring task of looking up spectra and their associated information based on different kinds of spectral references (e.g., scan numbers). These classes have now been broadly adopted in OpenMS.
Percolator Wrappers
Percolator is a tool for postprocessing peptide–pectrum matches (PSMs) from sequence database searches of LC–MS/MS data.24 It can improve the number of confident PSMs recovered from the data by applying a semisupervised machine learning approach to distinguish correct from incorrect PSMs. Percolator also calculates statistically meaningful scores for PSMs, i.e., q values (a measure of the false discovery rate, FDR) and posterior error probabilities (PEP).26
Percolator for MS-GF+
To run Percolator on MS-GF+ search results, we created two wrappers using the Generic-Wrapper mechanism provided by OpenMS: one for the “msgf2pin” program packaged with Percolator, which converts mzIdentML files generated by MS-GF+ to Percolator’s input format,27 and one for the Percolator executable itself.
In addition, we implemented a parser for the output files produced by Percolator. Percolator can generate results on the levels of PSMs, peptides, or proteins. We implemented support for PSM-level output in the C++ class PercolatorOutfile. Our changes enable the reading of the corresponding Percolator result files and their conversion to idXML using the TOPP tool IDFileConverter. Internally, the class SpectrumMetaDataLookup is used to annotate peptide IDs with RT values from original spectra.
MascotPercolator
For applying Percolator to Mascot search results, the program MascotPercolator provides a convenient solution that operates directly on the “raw” Mascot search results (.dat files). We adapted MascotPercolator to make it interoperable with OpenMS. To this end, we added the ability to read an idXML file produced by the Mascot search adapter (MascotAdapterOnline) and to extract the ID number of the Mascot search, which MascotPercolator uses to find the corresponding .dat file and perform its analysis. We also modified the format of the input file submitted by MascotPercolator to Percolator itself to ensure that the output would be fully compatible with OpenMS. Specifically, this meant (a) adding annotations of post-translational modifications to the peptide sequences and (b) generating ID strings for the PSMs in a format that would facilitate the lookup of meta information such as retention times, precursor mass-to-charge values, and charge states. With these changes, MascotPercolator could be wrapped using the GenericWrapper approach and thus integrated into our analysis pipeline.
Changes to Existing OpenMS Tools
MascotAdapterOnline
MascotAdapterOnline is the TOPP tool that facilitates running a database search on a remote Mascot server. We contributed a small usability improvement to this adapter by using the name of the input spectra file (mzML) to set the title of the Mascot search, which makes it easier to identify search runs submitted by OpenMS in the Mascot search log. In addition, we made adaptations geared toward interfacing MascotAdapterOnline with MascotPercolator. First, MascotPercolator relies on the Mascot search number to find the .dat file to process, so this number needed to be extracted from the Mascot server’s response to a search query and written to the output file as a metadata entry. Second, because MascotPercolator operates directly on the Mascot .dat file, we do not need to retrieve the search results from MascotAdapterOnline. We thus added a flag to the adapter that allows us to skip the lengthy export process of the search results and to write an essentially empty idXML file containing only the Mascot search number.
ConsensusID
Analyzing a sufficiently large set of MS2 spectra with different peptide and protein identification (ID) engines, including database search engines, generally gives partially different, often complementary results.28 To take advantage of this, OpenMS provides the ConsensusID tool, which combines search results on the PSM level from different ID engines. Given ranked lists of PSMs produced by different engines, several algorithms are available to merge and rescore PSMs derived from the same spectrum: similarity scoring based on sequence or fragmentation pattern similarity,29 ranked voting, or simply using the average or the best score for each peptide. There were, however, problems with the implementations of these algorithms; hence, we rewrote large parts of them, refactored the code into a class hierarchy, and increased the test coverage. These improvements made it easy for us to add new features to ConsensusID. First, a filter allowing the user to specify a minimum fraction of the involved ID engines (e.g., “two out of three”) that must have identified a peptide. Second, a new conservative rescoring algorithm, which assigns to each peptide hit the worst score that it has received from any of the ID engines.
IDFilter
The IDFilter TOPP tool offers a plethora of options for filtering peptide and protein identification data. We added some additional options to complete our proteogenomics pipeline. In preparation for that, we refactored the ID filtering code in the OpenMS library to provide a more-consistent interface, more-descriptive function names, and a cleaner, often more-efficient implementation based on functional programming principles. We realized that most of the filtering options involved checking elements of a list and either removing or keeping those that fulfilled a criterion. Our code is thus built on two aspects. First, simple predicates implemented as functors, e.g., to check if a quality score is above a given threshold. Second, two generic, higher-order functions that filter C++ vectors (e.g., containing identified peptides or proteins) using the erase-remove idiom to either keep or remove elements that match a given predicate. Using these building blocks, we added new filtering options required in our pipeline to the library and exposed them in the IDFilter tool: First, a filter that takes a set of post-translational modifications and removes all peptide hits featuring any of those modifications; this is later used to remove deamidated PSMs (see the Proteogenomics Pipeline section below). Second, a filter that removes all peptide and protein hits matching accessions in a given FASTA file; this functionality is needed for protein-level filtering in our analysis pipeline.
PeptideIndexer
To update the protein references for a set of peptide IDs, the TOPP tool PeptideIndexer can be used. Given an idXML file with peptide IDs and a FASTA file containing amino acid sequences, PeptideIndexer matches the peptide sequences to the database sequences and annotates the peptides with corresponding accessions. Typically, this indexing works in two passes. First, exact string matching using the Aho–Corasick algorithm30 is used to quickly find matches for the majority of peptides. Second, if any peptides remain unmatched, an error-tolerant search using suffix arrays is performed. This step can recover matches to database sequences that contain ambiguity codes for sets of amino acids (“B”: D or N; “Z”: E or Q; and “X”: any), but it may take a long time depending on the number of sequences involved. In our proteogenomics pipeline, we index identified peptides against parts of the full sequence database, in which case we expect that some peptides will not match. We had to make a small change to the PeptideIndexer code to be able to skip the error-tolerant search in these cases, which allowed us to reduce the runtime of the pipeline by several hours per input file.
Additional Tools Not Used in the “Core” Pipeline
MzMLSplitter
There are limits to the size of raw data files suitable for use in our pipeline. On the lower end, they should contain at least several thousand PSMs to allow reliable Percolator training. On the higher end, input files for MascotAdapterOnline should not be much larger than 1 GB. Small raw files can be merged into larger files using the TOPP tool FileMerger; we added a utility called MzMLSplitter for the opposite operation: splitting a large mzML file into multiple equally sized parts. In practice, when dealing with a set of raw data files of widely varying sizes from a fractionation experiment, a useful approach may be to merge all files from one sample using FileMerger and then split the result into manageable 1 GB parts using MzMLSplitter.
IDScoreSwitcher
One important limitation of the idXML format that OpenMS uses to store peptide and protein identification data is that only one primary score statistic can be associated with each peptide or protein hit. Secondary scores can be stored as metadata, but only the primary score is used for ranking and filtering PSMs. Usually only one final, meaningful score for peptides or proteins is required. An exception to this is Percolator, which calculates FDRs (q values) and PEPs, which can be used in conjunction for filtering, e.g., using the common cutoff combination of 1% FDR and 5% PEP. To better support multiple peptide and protein scores in OpenMS, we added a tool called IDScoreSwitcher, which facilitates switching between secondary scores and the primary score. Filtering by two different score types is possible by running IDFilter twice, applying IDScoreSwitcher between the filters.
FidoAdapter
Fido is a protein inference engine that uses a Bayesian probabilistic model to group and score proteins based on PSMs.31 It is freely available under an open-source license. Because OpenMS was lacking a protein inference tool, we decided to add support for Fido via an adapter. To this end, the Fido source code was adapted to work on all platforms that OpenMS supports; the patched version is available on GitHub (https://github.com/hendrikweisser/Fido). The OpenMS adapter we developed, FidoAdapter, receives peptide identification results in idXML format as input. The scores of the peptide hits must be probabilities and the referenced protein hits must be annotated with target and decoy information. This enables the adapter to generate suitable input files for Fido. These files, together with any user-specified parameters, are used to run the Fido executable (typically “FidoChooseParameter”, which includes parameter optimization). The adapter parses the Fido output file containing inferred protein groups and their posterior probabilities, adding the results to the original ID data. The result can be used, for example, as an auxiliary input for the ProteinQuantifier TOPP tool, enabling quantification of protein groups.
Proteogenomics Pipeline
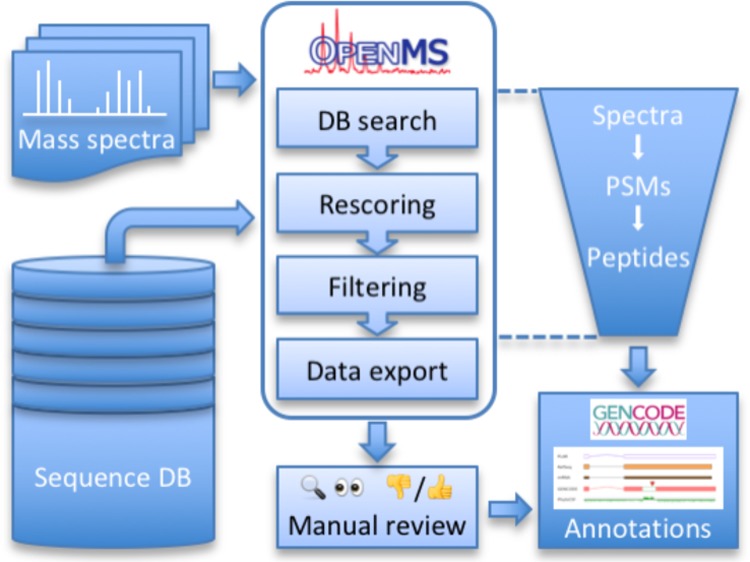
Integrating existing software with tools that we adapted or developed specifically for this purpose, we have designed a pipeline for proteogenomic data analysis. An overview of our approach is shown in Figure 1. The goal of our pipeline is to confidently identify potential “novel” peptides in LC–MS/MS data. Based on conclusions from previous work,2 we define the following list of criteria for peptides and for the PSMs from which they are derived to be designated as “novel”. (1) The peptide must be 7–30 amino acids long. (2) It must be fully tryptic, with no more than two missed cleavages. (3) It must be identified (a) as the joint top hit of two different search engines, (b) with a PEP of 1% or better in both search engine results, and (c) without any deamidation modifications. (4) The peptide must not match to a contaminant and must differ by more than two amino acids from any known protein (to exclude potential matches to known genes with variants and mutations).
Figure 1.

Schematic overview of the OpenMS proteogenomics workflow. Based on a comprehensive sequence database, tandem mass spectra from large proteomic data sets are searched in a competitive target–decoy approach using two search engines, Mascot and MS-GF+. The search results are rescored using Percolator and filtered in multiple stages according to stringent quality criteria. During this process, starting from a large number of spectra and initial PSMs, the set of retained PSMs is refined further and further until in the end, only high-confidence PSMs from novel peptides remain. These are exported and passed on to genome annotators. In a manual review process, novel peptides and other sources of evidence are integrated, in some cases yielding new insights in the form of novel genome annotations.
Figure 2 shows the TOPPAS workflow for our pipeline, which is available at http://openms.org/workflow/proteogenomics. The pipeline works in several stages, which we describe in more detail below: (1) database searching of MS2 spectra; (2) rescoring of search results; (3) filtering on the PSM level; (4) filtering on the protein level; and (5) export of results. Important parameters of the pipeline are listed in Table S1.
Figure 2.

Proteogenomics pipeline, as displayed in the TOPPAS workflow editor. The different stages of the pipeline are indicated using colored boxes. Additional output nodes, which would be used in practice to capture intermediate results at different stages, have been omitted for simplicity. The input file nodes 1–5 contain the following data: 1, MS2 spectra (mzML files); 2, combined target–decoy sequences (FASTA); 3, contaminant sequences (FASTA); 4, known protein sequences (FASTA); and 5, presumed noncoding sequences (FASTA).
Stage 1: Database Searching of MS2 Spectra
At the start of the pipeline, MS2 spectra from experimental data, stored in mzML files, are searched against a combined sequence database in a competitive target–decoy approach. A pair of search engines is used for this purpose, Mascot and MS-GF+, which we chose because of their good performance, especially in combination with Percolator. Including additional search engines in the pipeline would not be difficult; in particular, both an OpenMS adapter and a Percolator converter already exist for X! Tandem.32 Input spectra may have to be centroided, as is required by MS-GF+; for high-resolution spectra in profile mode, centroiding can be performed using the TOPP tool PeakPickerHiRes. The results of this stage are peptide–spectrum matches with associated scores, produced by each search engine.
Stage 2: Rescoring of Search Results
The second stage applies Percolator to the PSMs from stage 1 to enrich for correct matches and obtain statistically meaningful scores for filtering in the next stage. Percolator outputs are converted to the idXML format using IDFileConverter. In the MS-GF+ branch, the corresponding spectral data files are required to associate retention time and precursor mass-to-charge values to PSMs. In the Mascot branch, this is not necessary, as these values are included in the ID column in the Percolator output.
Note that Percolator produces separate outputs for target and decoy PSMs. In the present pipeline, only target hits are utilized. However, to enable the calculation of overall false discovery rates, decoy hits could be retrieved via the “ETool:out_decoy” parameters of the GenericWrapper (Percolator/MascotPercolator) nodes, converted with IDFileConverter and merged with the target hits.
Stage 3: Filtering on the PSM Level
In this stage, the rescored PSMs from Mascot and MS-GF+ are merged and filtered according to our stringent criteria. Using the IDFilter tool, PSMs are filtered by peptide sequence length (7–30 amino acids), PEP score (0.01 or better), and modifications (no deamidation). Deamidated PSMs are removed because they have previously been found to be overrepresented among potentially novel peptides2 and are hence considered unreliable. Finally, the ConsensusID tool is applied to group Mascot and MS-GF+ search hits pertaining to the same spectrum and to filter all cases in which the two search engines did not arrive at the same significant top hit.
Stage 4: Filtering on the Protein Level
During stage 4, PSMs are further filtered based on which proteins (or, more correctly, which entries in the sequence database) match their peptide sequences. The PeptideIndexer tool is repeatedly applied to find matches in each part of the database, followed by IDFilter to remove matching peptides. Initially, all peptides matching contaminant (including HLA) sequences are discarded. Next peptides matching known proteins are removed, allowing up to two amino acid differences in a peptide–protein match to account for possible unknown variants in the proteins of the biological sample. Because this step involves a large part of the sequence database and the required approximate matching is computationally expensive, it proceeds in two phases. First, exact matches are found between the peptide and protein sequences, and corresponding peptides are removed. This is relatively fast and excludes the vast majority of identified peptides from further consideration. Second, an approximate search is performed for the remaining peptides, removing any additional matches. The resultant peptide hits match only to the “presumed noncoding” part of the sequence database. To ascertain to which sequences they match, PeptideIndexer is used again, this time indexing against the “presumed noncoding” part of the database. (If decoy hits were included in the analysis, they could be removed using IDFilter.)
Stage 5: Export of Results
At this point in the pipeline, the PSMs of potentially novel peptides, annotated with accessions from the sequence database, are available in idXML format (one file per input mzML file). The final step of the pipeline applies the TextExporter tool to convert the idXML files into tabular text files (.csv) for further analysis in external tools. It is also possible to convert the idXML files from any stage of the pipeline into the HUPO PSI standard formats mzIdentML33 and mzTab34 using the TOPP tools IDFileConverter and MzTabExporter, respectively.
Testis Data Analysis
We demonstrate the effectiveness of our proteogenomics pipeline on the analysis of the human testis data set generated for the Chromosome-Centric Human Proteome Project by the Liu lab (“C-HPP testis data set”).10 The original analysis of this data by Zhang et al. had focused on the detection of “missing proteins”, i.e., known protein-coding genes for which no direct protein evidence had yet been found. In contrast, our analysis focuses on a disjoint set of protein evidence, “novel” peptides that uniquely map to genomic regions not previously known to be protein-coding genes. To find such peptides, we converted the 150 RAW files containing the LC–MS/MS data to mzML format and used the mzML files as inputs for the TOPPAS workflow shown in Figure 2. The resulting data is available from the PRIDE repository under accession PXD004785.
Runtime Considerations
In practice, we did not run the complete workflow at once but rather performed the Mascot and MS-GF+ searches as separate steps, independent of each other. This was done for efficiency reasons, as the performance characteristics of the two search engines differ. On the C-HPP testis data set, MascotAdapterOnline took between 6 and 32 min (an average of 21 min) per file to run Mascot searches. MSGFPlusAdapter took between 1 and 3 h (an average of 2 h) per file for MS-GF+ searches using four parallel threads. However, the number of Mascot searches that can be run in parallel is limited by the number of Mascot licenses (there is no comparable limit for MS-GF+ searches, provided that adequate computational resources are available). After the database searches, the runtime of the remaining pipeline was between 6 and 18 min per file (an average of 11 min) in our analysis.
Data Processing Summary
A summary of the analysis is presented in Figure 3, which shows the numbers of peptide–spectrum matches generated and retained at each stage of the pipeline. The data set contains 4.2 million MS2 spectra, almost all of which can be assigned peptide sequences by Mascot and MS-GF+. After filtering on the PSM level (1% PEP, peptide length 7–30, no deamidation), we are left with 1.6 million Mascot PSMs (37% of all MS2 spectra) and one million MS-GF+ PSMs (24% of spectra) with a false discovery rate (FDR) of 0.12%. The ConsensusID step further reduces the number of PSMs to 940 000 for which Mascot and MS-GF+ agree in their assignments of the best hit at sufficiently high confidence. 623 000 PSMs and 96 000 PSMs exclusively identified by Mascot and MS-GF+, respectively, are thus removed; at the same time, the fraction of decoy hits decreases, yielding an estimated FDR of 0.006% for the ConsensusID results. Subsequent filtering, removing matches to contaminants and HLA reduces the overall number of PSMs to 880 000. However, almost all of these PSMs (more than 99.9%) are exact matches to known proteins and are thus removed in the next filtering step. Of the remaining 455 PSMs matching noncoding sequences, only 210 pass the final filter, which matches approximately against known protein sequences, allowing up to two amino acid differences per peptide. These 210 PSMs are the result of applying our proteogenomic pipeline to the C-HPP testis data set and contain 47 nonredundant “novel” peptide sequences.
Figure 3.
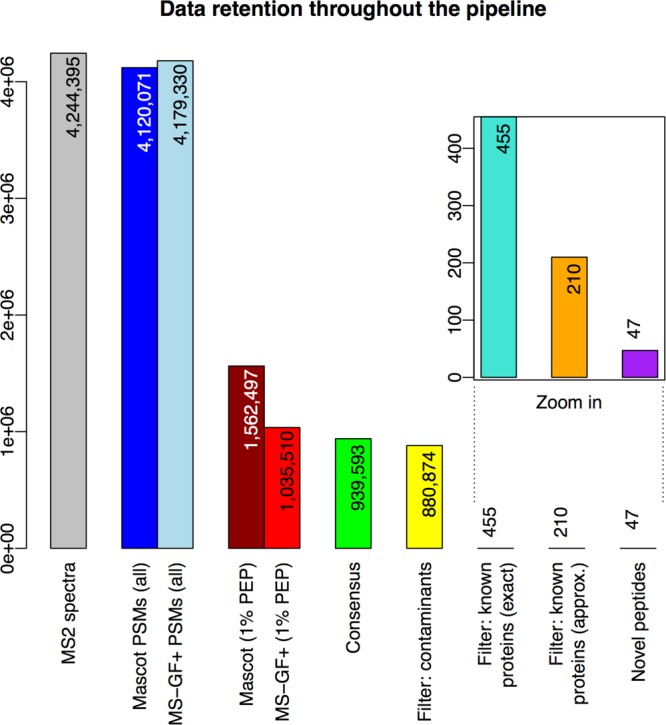
Data retention throughout the pipeline. The bars show the numbers of “data elements” (spectra, PMSs, and peptides) under consideration as these numbers decrease from the start (left) to the end (right) of the proteogenomics pipeline. In detail, the bars represent the following (node numbers refer to the TOPPAS workflow in Figure 2): “MS2 spectra”, input MS2 spectra in the C-HPP testis data set; “Mascot/MS-GF+ PSMs (all)”, spectra that generated PSMs using either search engine; “Mascot/MS-GF+ (1% PEP)”, PSMs after PSM-level filtering (node 15); “Consensus”, PSMs after ConsensusID (node 16); “Filter: contaminants”, PSMs after filtering for contaminants (node 18); “Filter: known proteins (exact)”, PSMs after filtering for exact matches to known proteins (node 20); “Filter: known proteins (approx.)”, PSMs after filtering for approximate matches to known proteins (final set; node 22); and “Novel peptides”, distinct novel peptides identified by the final set of PSMs.
Novel Peptides
We identified 47 potential novel peptides in the C-HPP testis data based on a final set of 210 PSMs generated by our analysis pipeline. We re-evaluated these peptides against an updated, more-comprehensive database of known human proteins composed of RefSeq,35 neXtProt,36 GENCODE v22, and UniProt sequences, using the PeptideIndexer and IDFilter combination from the pipeline and again allowing up to two amino acid mismatches. This removed a further 12 peptides. Spectra for the remaining 35 peptides were manually inspected, and then these peptides were passed on to the manual genome annotators from the GENCODE project. Table S1 lists the 35 peptides together with the outcomes of the manual annotation process. Importantly, eight peptides were used as a source of evidence (together with RNA expression, sequence conservation, gene structure, and other orthogonal evidence) to annotate five new protein-coding genes. These annotations are publicly available in the VEGA database37 and will be incorporated into the next release of the GENCODE gene set. One example is shown in Figure 4. A further 22 peptides were mapped to seven loci that were only recently annotated as new genes2 that would otherwise have counted as novel annotations as well. All of these loci were found to be expressed in testis tissue in the previous study.
Figure 4.

Reannotation of OTTHUMG00000019887 based on proteogenomic analysis. (A) This locus was present in GENCODE v20 as a lincRNA model, and it is currently categorized in this way by RefSeq (orange model) based on mRNA AK056723.1 (brown model) and given the official HGNC gene symbol LINC00961. Furthermore, an equivalent model was generated and classified as a lncRNA by the RNA-Seq-based PLAR pipeline developed by Hezroni et al.38 (purple-outlined model). GENCODE have now converted this model to protein coding (UTRs in red; CDS in green) based on proteogenomic evidence in combination with evolutionary conservation. The conserved region is well resolved by PhyloCSF, with this track being taken from genome.ucsc.edu. Peptide [QEASLFTGPVR] is marked (red triangle). (B) The 75 aa human CDS shows conservation in eutherian mammals, although not outside this group based on available genome alignments. “T. Devil” is Tasmanian devil, and “flying fox” is specifically the black flying fox Pteropus alecto.
Known Proteins
As a proof of concept, we carried out protein inference using Fido (via FidoAdapter) on the set of 880 000 noncontaminant consensus PSMs from the whole data set. Filtering for known proteins resulted in 8679 inferred protein groups. This number is roughly in line with the total of 9597 proteins reported by Zhang et al. in their original analysis, given that our filtering criteria for PSMs and proteins were more stringent than theirs, as required for the reliable identification of novel peptides. Using the same data, we performed spectral counting on the peptide level with OpenMS’ ProteinQuantifier and mapped the peptides to the genome. Further analysis of the known proteins and their expressed peptides is beyond the scope of this study, but we are making our results available in a Track Hub,39 suitable for visualization in genome browsers, at http://ngs.sanger.ac.uk/production/proteogenomics/WTSI_proteomics_CHPP_testis/hub.txt. As an example, our Track Hub can be displayed in the UCSC genome browser, showing tracks for peptides in known proteins, novel peptides, and post-translationally modified peptides, via http://genome.ucsc.edu/cgi-bin/hgTracks?db=hg38&hubUrl=http://ngs.sanger.ac.uk/production/proteogenomics/WTSI_proteomics_CHPP_testis/hub.txt.
Discussion
We present an automated pipeline for proteogenomic data analysis that is implemented within the OpenMS framework for computational mass spectrometry. This pipeline is largely based on existing software (OpenMS, Mascot, MS-GF+, and Percolator) but also benefits from custom extensions and new tools, developed in the context of this project. The aim of our pipeline, given LC–MS/MS data and a suitable sequence database, is to confidently identify peptides that can inform novel genome annotations. We have applied this approach to a relevant data set, the human testis tissue data set generated for the Chromosome-Centric Human Proteome Project. On the basis of 4.2 million MS2 spectra from 150 LC−MS/MS runs, we identified 35 “novel” peptides as candidates for genome annotation. A total of eight of these peptides led to the annotation of five new protein-coding genes for the GENCODE gene set, and a further 12 peptides matched five very recent novel annotations. The fact that our set of results was so highly enriched in peptides that gave rise to new genome annotations demonstrates the efficacy of our approach.
Implementing our pipeline within the OpenMS framework not only allowed us to conveniently reuse a large set of existing computational proteomics algorithms for our purposes but also confers a great amount of flexibility for adapting or extending the pipeline to address different research questions. For example, we have focused on the detection of “novel” peptides in this study, thereby excluding 99.9% of our confident peptide IDs from consideration. For a different research objective, such as comparing tissue proteomes, it would be straightforward to adapt our pipeline to focus on these 99.9% of PSMs instead. As we have demonstrated briefly, protein inference can be carried out using Fido via FidoAdapter. One of several available feature detection algorithms, together with the IDMapper and ProteinQuantifier tools, can add label-free quantification;40 other tools are available for the quantitative analysis of labeled data or for the localization of post-translational modifications. Conversely, because OpenMS is a general-purpose framework for mass-spectrometry-based proteomics and metabolomics, the improvements that we made to its tools will also benefit users in other areas of research.
Adapting our pipeline toward more general-purpose peptide identification would open up avenues for extension that are less-suitable for our current focus on novel peptides. For example, error-tolerant searching could help to identify additional post-translational modifications or sequence variants caused by amino acid substitutions.41 However, such approaches typically restrict the search space to proteins that have already been identified. De novo sequencing could allow us to detect peptides with unexpected sequences;42 however, we would not consider any novel peptides as credible that are based on sequence variants, unless sequencing data shows that those variants are clearly present in the sample. Spectral library searching,43 potentially in combination with spectral clustering,44 has certain advantages over sequence database searching, but it can only assign peptides to spectra if similar spectra have been identified previously. Finally, approaches for identifying cofragmented peptides from “chimeric” spectra may boost identification rates,45 but it is questionable whether PSMs of novel peptides involving chimeric spectra would pass manual validation.
Currently, the mapping of peptides to the genome is part of the manual annotation process, which is based on the inclusion of genomic coordinates in the accessions of the sequence database entries. A recently developed tool that performs the genome mapping could be integrated into our pipeline in the future (Schlaffner et al., manuscript in preparation). Moreover, the availability of paired sequencing data for proteomic samples would make it possible to create custom databases containing the exact sequence variants present in each sample; this would allow the further simplification of the filtering pipeline and increase its sensitivity by obviating the need for an approximate matching step.
To date, we have processed data sets composed of over 55 million MS2 spectra with OpenMS-based proteogenomics workflows, demonstrating the scalability and robustness of our pipeline. Even on the most highly curated genome, this has resulted in over 40 new protein-coding gene annotations for the GENCODE human reference. We anticipate that this pipeline will be particularly useful for the analysis of personalized proteomes and integration with other omics technologies.
Acknowledgments
We thank Christoph Schlaffner for creating the Track Hub of the known and novel peptides identified in this work. We would also like to thank the team of the OpenMS project and all developers who contributed it. We gratefully acknowledge funding from the Wellcome Trust (grant WT098051) and from the National Institutes of Health (grant U41HG007234).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.6b00765.
Author Contributions
H.W. designed the proteogenomics pipeline, developed most of the aforementioned contributions to OpenMS, carried out the data analysis, and wrote the manuscript (with feedback from the other authors). J.C.W. defined the searching and filtering criteria, prepared the sequence database, and implemented adaptations to MascotPercolator. J.M.M. performed the manual genome annotation and created Figure 4. P.G. implemented initial versions of MSGFPlusAdapter and of the Percolator wrappers in OpenMS. J.S.C. initiated and supervised the project and performed the manual review of MS2 spectra. All authors read and approved the final manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Nesvizhskii A. I. Proteogenomics: concepts, applications and computational strategies. Nat. Methods 2014, 11 (11), 1114–1125. 10.1038/nmeth.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright J. C.; Mudge J.; Weisser H.; Barzine M. P.; Gonzalez J. M.; Brazma A.; Choudhary J. S.; Harrow J. Improving GENCODE reference gene annotation using a high-stringency proteogenomics workflow. Nat. Commun. 2016, 7, 11778. 10.1038/ncomms11778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röst H. L.; Sachsenberg T.; Aiche S.; Bielow C.; Weisser H.; Aicheler F.; Andreotti S.; Ehrlich H.-C.; Gutenbrunner P.; Kenar E.; Liang X.; Nahnsen S.; Nilse L.; Pfeuffer J.; Rosenberger G.; Rurik M.; Schmitt U.; Veit J.; Walzer M.; Wojnar D.; Wolski W. E.; Schilling O.; Choudhary J. S.; Malmström L.; Aebersold R.; Reinert K.; Kohlbacher O., OpenMS: A flexible open-source software platform for computational mass spectrometry. Nat. Methods 2016.13741. 10.1038/nmeth.3959 [DOI] [PubMed] [Google Scholar]
- Risk B. A.; Spitzer W. J.; Giddings M. C. Peppy: proteogenomic search software. J. Proteome Res. 2013, 12 (6), 3019–3025. 10.1021/pr400208w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagtap P. D.; Johnson J. E.; Onsongo G.; Sadler F. W.; Murray K.; Wang Y.; Shenykman G. M.; Bandhakavi S.; Smith L. M.; Griffin T. J. Flexible and accessible workflows for improved proteogenomic analysis using the Galaxy framework. J. Proteome Res. 2014, 13 (12), 5898–5908. 10.1021/pr500812t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghali F.; Krishna R.; Perkins S.; Collins A.; Xia D.; Wastling J.; Jones A. R. ProteoAnnotator--open source proteogenomics annotation software supporting PSI standards. Proteomics 2014, 14 (23–24), 2731–2741. 10.1002/pmic.201400265. [DOI] [PubMed] [Google Scholar]
- Nagaraj S. H.; Waddell N.; Madugundu A. K.; Wood S.; Jones A.; Mandyam R. A.; Nones K.; Pearson J. V.; Grimmond S. M. PGTools: A Software Suite for Proteogenomic Data Analysis and Visualization. J. Proteome Res. 2015, 14 (5), 2255–2266. 10.1021/acs.jproteome.5b00029. [DOI] [PubMed] [Google Scholar]
- Brosch M.; Yu L.; Hubbard T.; Choudhary J. Accurate and sensitive peptide identification with Mascot Percolator. J. Proteome Res. 2009, 8 (6), 3176–3181. 10.1021/pr800982s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright J. C.; Collins M. O.; Yu L.; Käll L.; Brosch M.; Choudhary J. S. Enhanced peptide identification by electron transfer dissociation using an improved Mascot Percolator. Mol. Cell. Proteomics 2012, 11 (8), 478–491. 10.1074/mcp.O111.014522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Li Q.; Wu F.; Zhou R.; Qi Y.; Su N.; Chen L.; Xu S.; Jiang T.; Zhang C.; Cheng G.; Chen X.; Kong D.; Wang Y.; Zhang T.; Zi J.; Wei W.; Gao Y.; Zhen B.; Xiong Z.; Wu S.; Yang P.; Wang Q.; Wen B.; He F.; Xu P.; Liu S. Tissue-Based Proteogenomics Reveals that Human Testis Endows Plentiful Missing Proteins. J. Proteome Res. 2015, 14 (9), 3583–3594. 10.1021/acs.jproteome.5b00435. [DOI] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Csordas A.; del Toro N.; Dianes J. A.; Griss J.; Lavidas I.; Mayer G.; Perez-Riverol Y.; Reisinger F.; Ternent T.; Xu Q.-W.; Wang R.; Hermjakob H. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44 (D1), D447–D456. 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers M. C.; Maclean B.; Burke R.; Amodei D.; Ruderman D. L.; Neumann S.; Gatto L.; Fischer B.; Pratt B.; Egertson J.; Hoff K.; Kessner D.; Tasman N.; Shulman N.; Frewen B.; Baker T. A.; Brusniak M.-Y.; Paulse C.; Creasy D.; Flashner L.; Kani K.; Moulding C.; Seymour S. L.; Nuwaysir L. M.; Lefebvre B.; Kuhlmann F.; Roark J.; Rainer P.; Detlev S.; Hemenway T.; Huhmer A.; Langridge J.; Connolly B.; Chadick T.; Holly K.; Eckels J.; Deutsch E. W.; Moritz R. L.; Katz J. E.; Agus D. B.; MacCoss M.; Tabb D. L.; Mallick P. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30 (10), 918–920. 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J.; Frankish A.; Gonzalez J. M.; Tapanari E.; Diekhans M.; Kokocinski F.; Aken B. L.; Barrell D.; Zadissa A.; Searle S.; Barnes I.; Bignell A.; Boychenko V.; Hunt T.; Kay M.; Mukherjee G.; Rajan J.; Despacio-Reyes G.; Saunders G.; Steward C.; Harte R.; Lin M.; Howald C.; Tanzer A.; Derrien T.; Chrast J.; Walters N.; Balasubramanian S.; Pei B.; Tress M.; Rodriguez J. M.; Ezkurdia I.; van Baren J.; Brent M.; Haussler D.; Kellis M.; Valencia A.; Reymond A.; Gerstein M.; Guigó R.; Hubbard T. J. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22 (9), 1760–1774. 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium UniProt: a hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.; Soormally A. R.; Hayhurst J. D.; Marsh S. G. E. The IPD-IMGT/HLA Database - New developments in reporting HLA variation. Hum. Immunol. 2016, 77 (3), 233–237. 10.1016/j.humimm.2016.01.020. [DOI] [PubMed] [Google Scholar]
- Stanke M.; Steinkamp R.; Waack S.; Morgenstern B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, W309–W312. 10.1093/nar/gkh379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karro J. E.; Yan Y.; Zheng D.; Zhang Z.; Carriero N.; Cayting P.; Harrrison P.; Gerstein M. Pseudogene.org: a comprehensive database and comparison platform for pseudogene annotation. Nucleic Acids Res. 2007, 35, D55–D60. 10.1093/nar/gkl851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S.; Davis C. A.; Merkel A.; Dobin A.; Lassmann T.; Mortazavi A.; Tanzer A.; Lagarde J.; Lin W.; Schlesinger F.; Xue C.; Marinov G. K.; Khatun J.; Williams B. A.; Zaleski C.; Rozowsky J.; Röder M.; Kokocinski F.; Abdelhamid R. F.; Alioto T.; Antoshechkin I.; Baer M. T.; Bar N. S.; Batut P.; Bell K.; Bell I.; Chakrabortty S.; Chen X.; Chrast J.; Curado J.; Derrien T.; Drenkow J.; Dumais E.; Dumais J.; Duttagupta R.; Falconnet E.; Fastuca M.; Fejes-Toth K.; Ferreira P.; Foissac S.; Fullwood M. J.; Gao H.; Gonzalez D.; Gordon A.; Gunawardena H.; Howald C.; Jha S.; Johnson R.; Kapranov P.; King B.; Kingswood C.; Luo O. J.; Park E.; Persaud K.; Preall J. B.; Ribeca P.; Risk B.; Robyr D.; Sammeth M.; Schaffer L.; See L.-H.; Shahab A.; Skancke J.; Suzuki A. M.; Takahashi H.; Tilgner H.; Trout D.; Walters N.; Wang H.; Wrobel J.; Yu Y.; Ruan X.; Hayashizaki Y.; Harrow J.; Gerstein M.; Hubbard T.; Reymond A.; Antonarakis S. E.; Hannon G.; Giddings M. C.; Ruan Y.; Wold B.; Carninci P.; Guigó R.; Gingeras T. R. Landscape of transcription in human cells. Nature 2012, 489 (7414), 101–108. 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates A.; Akanni W.; Amode M. R.; Barrell D.; Billis K.; Carvalho-Silva D.; Cummins C.; Clapham P.; Fitzgerald S.; Gil L.; Girón C. G.; Gordon L.; Hourlier T.; Hunt S. E.; Janacek S. H.; Johnson N.; Juettemann T.; Keenan S.; Lavidas I.; Martin F. J.; Maurel T.; McLaren W.; Murphy D. N.; Nag R.; Nuhn M.; Parker A.; Patricio M.; Pignatelli M.; Rahtz M.; Riat H. S.; Sheppard D.; Taylor K.; Thormann A.; Vullo A.; Wilder S. P.; Zadissa A.; Birney E.; Harrow J.; Muffato M.; Perry E.; Ruffier M.; Spudich G.; Trevanion S. J.; Cunningham F.; Aken B. L.; Zerbino D. R.; Flicek P. Ensembl 2016. Nucleic Acids Res. 2016, 44 (D1), D710–D716. 10.1093/nar/gkv1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlbacher O.; Reinert K.; Gröpl C.; Lange E.; Pfeifer N.; Schulz-Trieglaff O.; Sturm M. TOPP--the OpenMS proteomics pipeline. Bioinformatics 2007, 23 (2), e191–e197. 10.1093/bioinformatics/btl299. [DOI] [PubMed] [Google Scholar]
- Junker J.; Bielow C.; Bertsch A.; Sturm M.; Reinert K.; Kohlbacher O. TOPPAS: a graphical workflow editor for the analysis of high-throughput proteomics data. J. Proteome Res. 2012, 11 (7), 3914–3920. 10.1021/pr300187f. [DOI] [PubMed] [Google Scholar]
- Perkins D. N.; Pappin D. J.; Creasy D. M.; Cottrell J. S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20 (18), 3551–3567. . [DOI] [PubMed] [Google Scholar]
- Kim S.; Gupta N.; Pevzner P. A. Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J. Proteome Res. 2008, 7 (8), 3354–3363. 10.1021/pr8001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll L.; Canterbury J. D.; Weston J.; Noble W. S.; MacCoss M. J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 2007, 4 (11), 923–925. 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- R Core Team, R: A language and environment for statistical computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Käll L.; Storey J. D.; MacCoss M. J.; Noble W. S. Posterior error probabilities and false discovery rates: two sides of the same coin. J. Proteome Res. 2008, 7 (1), 40–44. 10.1021/pr700739d. [DOI] [PubMed] [Google Scholar]
- Granholm V.; Kim S.; Navarro J. C. F.; Sjölund E.; Smith R. D.; Käll L. Fast and accurate database searches with MS-GF+Percolator. J. Proteome Res. 2014, 13 (2), 890–897. 10.1021/pr400937n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quandt A.; Espona L.; Balasko A.; Weisser H.; Brusniak M.-Y.; Kunszt P.; Aebersold R.; Malmström L. Using synthetic peptides to benchmark peptide identification software and search parameters for MS/MS data analysis. EuPa Open Proteomics 2014, 5, 21–31. 10.1016/j.euprot.2014.10.001. [DOI] [Google Scholar]
- Nahnsen S.; Bertsch A.; Rahnenführer J.; Nordheim A.; Kohlbacher O. Probabilistic consensus scoring improves tandem mass spectrometry peptide identification. J. Proteome Res. 2011, 10 (8), 3332–3343. 10.1021/pr2002879. [DOI] [PubMed] [Google Scholar]
- Aho A. V.; Corasick M. J. Efficient String Matching: An Aid to Bibliographic Search. Commun. ACM 1975, 18 (6), 333–340. 10.1145/360825.360855. [DOI] [Google Scholar]
- Serang O.; MacCoss M. J.; Noble W. S. Efficient marginalization to compute protein posterior probabilities from shotgun mass spectrometry data. J. Proteome Res. 2010, 9 (10), 5346–5357. 10.1021/pr100594k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig R.; Beavis R. C. TANDEM: matching proteins with tandem mass spectra. Bioinformatics 2004, 20 (9), 1466–1467. 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- Jones A. R.; Eisenacher M.; Mayer G.; Kohlbacher O.; Siepen J.; Hubbard S. J.; Selley J. N.; Searle B. C.; Shofstahl J.; Seymour S. L.; Julian R.; Binz P.-A.; Deutsch E. W.; Hermjakob H.; Reisinger F.; Griss J.; Vizcaíno J. A.; Chambers M.; Pizarro A.; Creasy D. The mzIdentML data standard for mass spectrometry-based proteomics results. Mol. Cell. Proteomics 2012, 11 (7), M111014381. 10.1074/mcp.M111.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griss J.; Jones A. R.; Sachsenberg T.; Walzer M.; Gatto L.; Hartler J.; Thallinger G. G.; Salek R. M.; Steinbeck C.; Neuhauser N.; Cox J.; Neumann S.; Fan J.; Reisinger F.; Xu Q.-W.; Del Toro N.; Pérez-Riverol Y.; Ghali F.; Bandeira N.; Xenarios I.; Kohlbacher O.; Vizcaíno J. A.; Hermjakob H. The mzTab data exchange format: communicating mass-spectrometry-based proteomics and metabolomics experimental results to a wider audience. Mol. Cell. Proteomics 2014, 13 (10), 2765–2775. 10.1074/mcp.O113.036681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt K. D.; Brown G. R.; Hiatt S. M.; Thibaud-Nissen F.; Astashyn A.; Ermolaeva O.; Farrell C. M.; Hart J.; Landrum M. J.; McGarvey K. M.; Murphy M. R.; O’Leary N. A.; Pujar S.; Rajput B.; Rangwala S. H.; Riddick L. D.; Shkeda A.; Sun H.; Tamez P.; Tully R. E.; Wallin C.; Webb D.; Weber J.; Wu W.; DiCuccio M.; Kitts P.; Maglott D. R.; Murphy T. D.; Ostell J. M. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014, 42, D756–D763. 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane L.; Argoud-Puy G.; Britan A.; Cusin I.; Duek P. D.; Evalet O.; Gateau A.; Gaudet P.; Gleizes A.; Masselot A.; Zwahlen C.; Bairoch A. neXtProt: a knowledge platform for human proteins. Nucleic Acids Res. 2012, 40, D76–D83. 10.1093/nar/gkr1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J. L.; Steward C. A.; Frankish A.; Gilbert J. G.; Gonzalez J. M.; Loveland J. E.; Mudge J.; Sheppard D.; Thomas M.; Trevanion S.; Wilming L. G. The Vertebrate Genome Annotation browser 10 years on. Nucleic Acids Res. 2014, 42, D771–D779. 10.1093/nar/gkt1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hezroni H.; Koppstein D.; Schwartz M. G.; Avrutin A.; Bartel D. P.; Ulitsky I. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 2015, 11 (7), 1110–1122. 10.1016/j.celrep.2015.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raney B. J.; Dreszer T. R.; Barber G. P.; Clawson H.; Fujita P. A.; Wang T.; Nguyen N.; Paten B.; Zweig A. S.; Karolchik D.; Kent W. J. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics 2014, 30 (7), 1003–1005. 10.1093/bioinformatics/btt637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisser H.; Nahnsen S.; Grossmann J.; Nilse L.; Quandt A.; Brauer H.; Sturm M.; Kenar E.; Kohlbacher O.; Aebersold R.; Malmström L. An automated pipeline for high-throughput label-free quantitative proteomics. J. Proteome Res. 2013, 12 (4), 1628–1644. 10.1021/pr300992u. [DOI] [PubMed] [Google Scholar]
- Creasy D. M.; Cottrell J. S. Error tolerant searching of uninterpreted tandem mass spectrometry data. Proteomics 2002, 2 (10), 1426–1434. . [DOI] [PubMed] [Google Scholar]
- Frank A.; Pevzner P. PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal. Chem. 2005, 77 (4), 964–973. 10.1021/ac048788h. [DOI] [PubMed] [Google Scholar]
- Lam H.; Deutsch E. W.; Eddes J. S.; Eng J. K.; King N.; Stein S. E.; Aebersold R. Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 2007, 7 (5), 655–667. 10.1002/pmic.200600625. [DOI] [PubMed] [Google Scholar]
- Griss J.; Perez-Riverol Y.; Lewis S.; Tabb D. L.; Dianes J. A.; Del-Toro N.; Rurik M.; Walzer M. W.; Kohlbacher O.; Hermjakob H.; Wang R.; Vizcaíno J. A. Recognizing millions of consistently unidentified spectra across hundreds of shotgun proteomics datasets. Nat. Methods 2016, 13 (8), 651–656. 10.1038/nmeth.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Pirmoradian M.; Chernobrovkin A.; Zubarev R. A. DeMix workflow for efficient identification of cofragmented peptides in high resolution data-dependent tandem mass spectrometry. Mol. Cell. Proteomics 2014, 13 (11), 3211–3223. 10.1074/mcp.O114.038877. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.