

ABSTRACT
Influenza A and B viruses are the causative agents of annual influenza epidemics that can be severe, and influenza A viruses intermittently cause pandemics. Sequence information from influenza virus genomes is instrumental in determining mechanisms underpinning antigenic evolution and antiviral resistance. However, due to sequence diversity and the dynamics of influenza virus evolution, rapid and high-throughput sequencing of influenza viruses remains a challenge. We developed a single-reaction influenza A/B virus (FluA/B) multiplex reverse transcription-PCR (RT-PCR) method that amplifies the most critical genomic segments (hemagglutinin [HA], neuraminidase [NA], and matrix [M]) of seasonal influenza A and B viruses for next-generation sequencing, regardless of viral type, subtype, or lineage. Herein, we demonstrate that the strategy is highly sensitive and robust. The strategy was validated on thousands of seasonal influenza A and B virus-positive specimens using multiple next-generation sequencing platforms.
KEYWORDS: NGS, RT-PCR, influenza, surveillance
INTRODUCTION
Annual influenza epidemics are caused by 4 cocirculating influenza virus subtypes and lineages: A(H1N1)pdm09, which displaced the previously circulating A(H1N1) viruses after the 2009 pandemic, and A(H3N2), B/Yamagata, and B/Victoria, resulting in substantial morbidity, mortality, and economic loss (1–3). For disease surveillance and vaccine strain selection, it is necessary to monitor thousands of circulating strains to identify antigenic drift and antiviral resistance, which are encoded by viral hemagglutinin (HA), neuraminidase (NA), and matrix (M) gene segments (4). Currently, most surveillance sequencing is carried out using subtype- and gene-specific primers to amplify the relevant gene segments before using them as the templates for Sanger or next-generation sequencing (NGS) (5–10). However, one or more drawbacks are inherent to those approaches: (i) conducting multiple reverse transcription-PCRs (RT-PCRs) for one virus/gene is expensive and labor-intensive; (ii) prior knowledge of the viral genotype is required to allow the correct selection of conserved predesigned primer sets; (iii) even when the conserved primer set is selected, sequence mismatch is inevitable in some influenza seasons due to the highly mutable nature of influenza viruses; and (iv) the robustness and sensitivity of the assays are not extensively optimized and validated with primary human specimens.
Sequence-independent random priming methods have been developed to amplify influenza viral genomes for NGS (11, 12). However, host DNA/RNA amplification by nonspecific priming and uneven amplification of viral genomes limited their use for the surveillance of large numbers of human clinical samples. Two techniques that specifically amplify the complete genomes of either influenza A virus or B virus in individual reactions (13, 14) have been widely used for the sequencing of thousands of influenza virus genomes from specimens of human, avian, swine, and other species. Nevertheless, those two methods are influenza virus type specific, and two separate primer sets with different optimal RT-PCR conditions are required. In addition, for antigenic drift and antiviral resistance surveillance, selective sequencing of HA, NA, and M segments provides the essential information needed at higher throughput and lower cost than whole-genome sequencing.
Therefore, to expedite the surveillance of viruses responsible for annual influenza epidemics, we aimed to develop a high-throughput sequencing strategy that overcomes the aforementioned challenges and is universally applicable to diverse circulating epidemic influenza viruses. Herein, we developed and validated a highly sensitive and robust single-reaction influenza A/B virus surveillance multiplex RT-PCR strategy (FluA/B RT-PCR) that enables simultaneous amplification of the HA, NA, and M gene segments of any A(H1N1), A(H1N1)pdm09, A(H3N2), B/Yamagata, or B/Victoria influenza virus directly from clinical specimens or virus isolates for sequencing. The process is ideally suited for NGS; the analysis pipeline we developed streamlines NGS analysis for laboratories that have minimal bioinformatics support and enables the detection of mixed infections with different influenza viruses.
RESULTS
Strategy for universal amplification of seasonal influenza viruses.
We analyzed all full-length HA, NA, and M sequences of human A(H1N1) {including viruses prior to and after 2009 [A(H1N1)pdm09]}, A(H3N2), and influenza B viruses deposited in GenBank by 2013 (15) when this study was initiated, and we found the termini of the HA, NA, and M segments to be extremely conserved (Fig. 1). Harnessing this conservation, oligonucleotide primers were designed for full-length amplification of the HA, NA, and M gene segments (Table 1). As the conserved regions are relatively short, nucleotide tails were added to the 5′ termini of the primers to increase the annealing temperatures for improved PCR efficiency (Table 1). The A-HA-UniF/R, A-NA-UniF/R, and A-M-UniF/R primer pairs are designed to amplify the full-length HA, NA, and M segments of all seasonal A(H1N1)pdm09 or A(H3N2) viruses. The B-HANA-UniF/R primer pair is designed to amplify the full-length HA and NA segments of both the B/Yamagata and B/Victoria lineages of viruses. The M segment of influenza B viruses was not targeted for amplification, since it is not an antiviral target.
FIG 1.
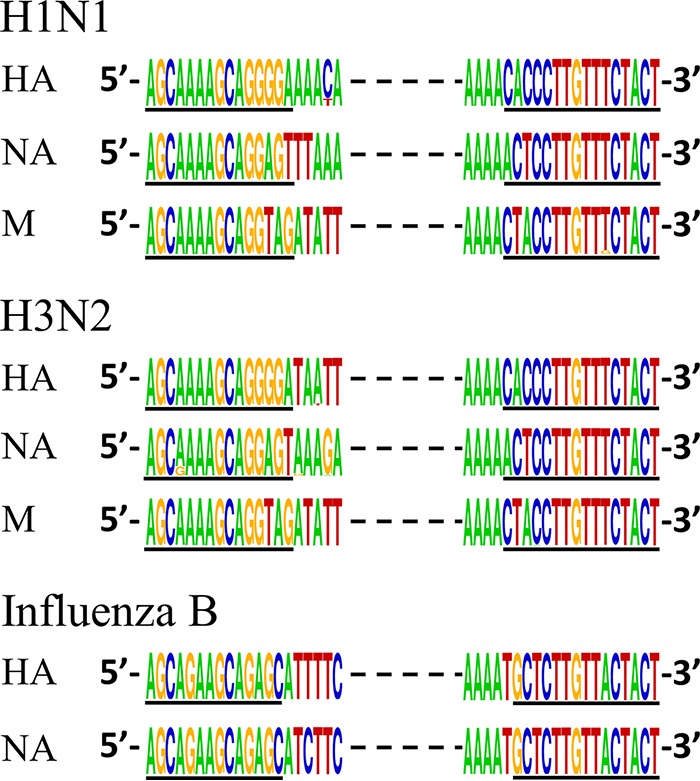
Conserved terminal sequences of seasonal influenza viruses. All publicly available full-length sequences (in cDNA sense) of human influenza A(H3N2), A(H1N1), and influenza B viruses were aligned for each gene segment of interest [9,472 HA, 6,948 NA, and 3,635 M for A(H1N1); 6,745 HA, 6,363 NA, and 3,766 M for A(H3N2); and 3,055 HA and 2,034 NA for influenza B virus. Full-length sequences are defined as sequences that have complete coding regions). The heights of nucleotides at the 20 terminal positions at each end of each segment are representative of their relative frequencies at a specific position using the WebLogo application (35). Underlined nucleotides correspond to the primer binding regions.
TABLE 1.
Universal FluA/B primer cocktail
| Primer | Sequence (5′ to 3′)a | Amt (μl) from each 10 μM oligonucleotideb |
|---|---|---|
| A-HA-UniF | GGGGGGAGCAAAAGCAGGGGA | 40 |
| A-HA-UniR | CCGGGTTATTAGTAGAAACAAGGGTG | 40 |
| A-NA-UniF | GGGGGGAGCAAAAGCAGGAGT | 60 |
| A-NA-UniR | CCGGGTTATTAGTAGAAACAAGGAGT | 60 |
| A-M-UniFc | GGGGGGAGCAAAAGCAGGTAG | 20 |
| A-M-UniRc | CCGGGTTATTAGTAGAAACAAGGTAG | 20 |
| B-HANA-Uni3F | GGGGGAGCAGAAGCAGAGC | 80 |
| B-HANA-Uni3R | CCGGGATATTAGTAGTAACAAGAGC | 80 |
Underlined nucleotides correspond to the conserved genomic termini.
For highest RT-PCR efficiency, the working stock should be made fresh each time.
The primers for the M segment could be left out of the cocktail if the M segment is not needed for the identification of antiviral resistant mutations.
Optimization of the FluA/B surveillance multiplex RT-PCR.
To optimize the sensitivity and robustness of the FluA/B RT-PCR, we tested and analyzed multiple variables, including different 5′ tail sequences, primer ratios, concentrations, and/or thermocycling parameters. The final optimized primer sequences and RT-PCR reaction parameters are listed in Table 1 and described in Materials and Methods.
Aiming to develop techniques that amplify the HA, NA, and M segments evenly and with high sensitivity for all contemporary epidemic influenza viruses, we used several human influenza virus strains for the entire optimization process, including an A(H1N1)pdm09 virus (A/New York/1682/2009), an A(H3N2) virus (A/New York/238/2005), and an influenza B virus (B/Brisbane/60-10/2010). Using the optimized conditions, an RNA template concentration equivalent to 1 50% tissue culture infective dose (TCID50)/RT-PCR was sufficient to generate the desired amplicons for A(H1N1)pdm09 and A(H3N2) viruses (HA, NA, and M) and for influenza B virus (HA and NA) (Fig. 2). All the desired segments were amplified relatively evenly, independent of the concentration of viral RNA template, which is critical for uniform nucleotide coverage in NGS. The NS segment from influenza A virus was also amplified due to its conserved terminal sequences with the HA/NA segments (e.g., the A-HA-UniR primer matches the 3′ terminus of the NS segment precisely).
FIG 2.
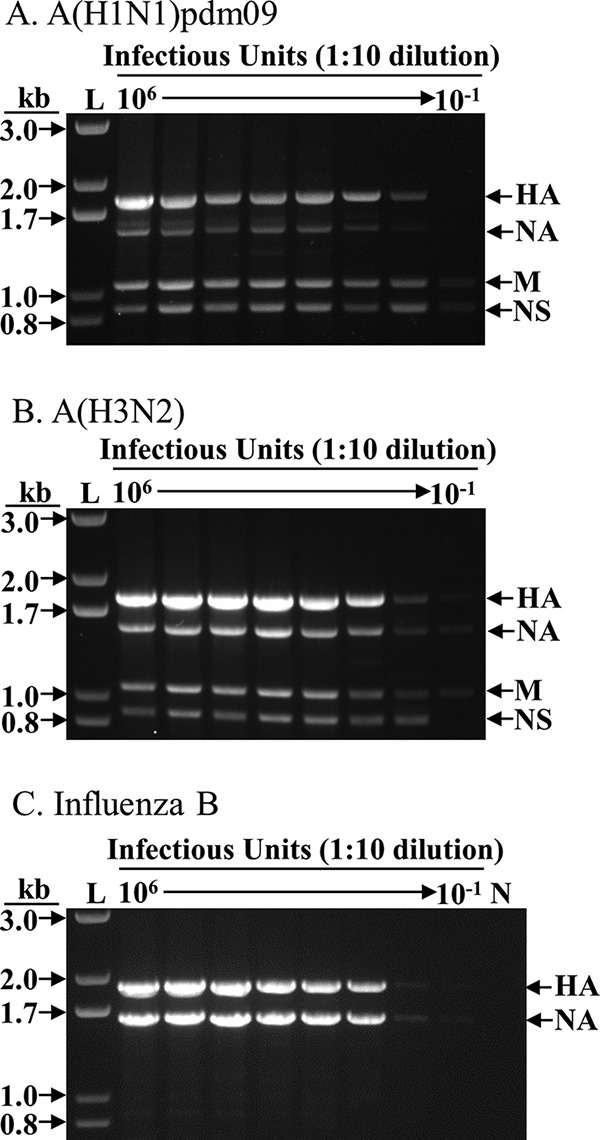
Sensitivity of FluA/B surveillance multiplex RT-PCR. RNA was extracted from 107 TCID50 of each A(H1N1)pdm09, A(H3N2), and influenza B virus, eluted in 30 μl of nuclease-free water, and diluted in a 1:10 series; 3 μl was used as the template for each RT-PCR. The equivalent amount of virus (TCID50) used in each RT-PCR is shown at the top of each lane. For each 25-μl reaction mixture, 3 μl was used for electrophoresis in agarose gel. L, 1-kb Plus ladder (Thermo Fisher Scientific, Inc.); N, negative control (no template). (A) A/New York/NY1682/2009 (H1N1pdm09). (B) A/New York/238/2005 (H3N2). (C) B/Brisbane/60-10/2010 (influenza B virus). The particular A(H1N1)pdm09 virus used here was rescued in an earlier study (13) and contains a “G” at the fourth position of the NA segment (refer to Fig. 1), which may have resulted in the less-robust amplification of the NA segment, a phenomenon we have not noticed in the many wild-type A(H1N1)pdm09 viruses we processed (Table 2).
Of note, the widely used prototype viruses A/Puerto Rico/8/1934 (PR8) and B/Lee/1940 (B/Lee) viruses are not ideal candidates to optimize the FluA/B RT-PCR assay. The NA of PR8 contains a “G” at position 13 of the 5′ end (cRNA sense), whereas almost all other NA of human A(H1N1) [including A(H1N1)pdm09] viruses contain an “A” at that position (Fig. 1). Similarly, the HA of B/Lee contains the unique GTT instead of the conserved AGA at positions 10 to 12 of the 5′ end (Fig. 1). As both PR8 and B/Lee are very old laboratory-adapted isolates, these mismatches are not of concern when using the FluA/B RT-PCR method for the surveillance of contemporary human influenza viruses.
Performance of FluA/B RT-PCR for diverse seasonal influenza viruses.
We then validated independently the performance of the FluA/B RT-PCR on primary and/or culture isolated specimens at 4 institutions: the J. Craig Venter Institute, WHO Collaborating Centre (WHOCC)-Melbourne, CDC Influenza Division, and New York University (Table 2). When results from different sites were combined, 1,761 out of 1,761 cultured specimens (100%) and 286 out of 344 primary specimens (83%) were successfully amplified (and/or sequenced) (Table 2). Our data indicate that the FluA/B RT-PCR is sensitive and robustly amplifies A(H1N1)pdm09, A(H3N2), and influenza B viruses, with threshold cycle (CT) values as high as 30 to 38, when dilutions of A/New York/1682/2009, A/New York/238/2005, and B/Brisbane/60-10/2010 viruses were tested (data not shown). A limited number of primary specimens were also performed with real-time RT-PCR, but more data are needed to determine the cutoff CT value that can be reliably amplified with the FluA/B RT-PCR method.
TABLE 2.
Summary of influenza A and B samples amplified and sequenced in this study
| Sequencing platform | Sample format | Virus type/subtype/lineage | No. of successful samples/total no. of samples (%)a |
|---|---|---|---|
| Ion Torrent PGM | Cultured specimens | A(H1N1)pdm09 | 152/152 (100) |
| A(H3N2) | 1,502/1,502 (100) | ||
| B/Yamagata | 61/61 (100) | ||
| B/Victoria | 46/46 (100) | ||
| Ion Torrent PGM; Illumina MiSeq | Primary specimens | A(H1N1)pdm09b | 68/83 (82) |
| A(H3N2) | 158/189 (84) | ||
| B/Yamagata | 10/11 (91) | ||
| B/Victoria | 14/17 (82) | ||
| B/lineage-not-performed | 36/44 (82) |
Successful samples are either evidenced by RT-PCR amplification of all desired amplicons (for FluA, HA, NA, and M; for FluB, HA and NA) or by sequencing of complete target genes (for FluA, HA, NA, and M; for FluB, HA and NA).
Two of the samples were coinfected with A(H1N1)pdm09 and A(H3N2), or with A(H1N1)pdm09 and influenza B virus.
FluA/B RT-PCR amplicons were then sequenced with either the Ion Torrent PGM or Illumina MiSeq platforms (Table 2). Importantly, when the sequencing reads were mapped to the whole genome, all the desired segments (HA, NA, and M for influenza A virus; HA and NA for influenza B virus) were represented at high coverage, whereas the other segments had minimal, if any, sequence coverage (Fig. 3 and 4). Hence, the sequencing capacity of the platform is focused on the targets that are most critical for antigenic and antiviral resistance analyses. Using this strategy, we have also successfully identified specimens from individuals coinfected with influenza A and B viruses or coinfected with A(H1N1)pdm09 and A(H3N2) viruses (Table 2).
FIG 3.
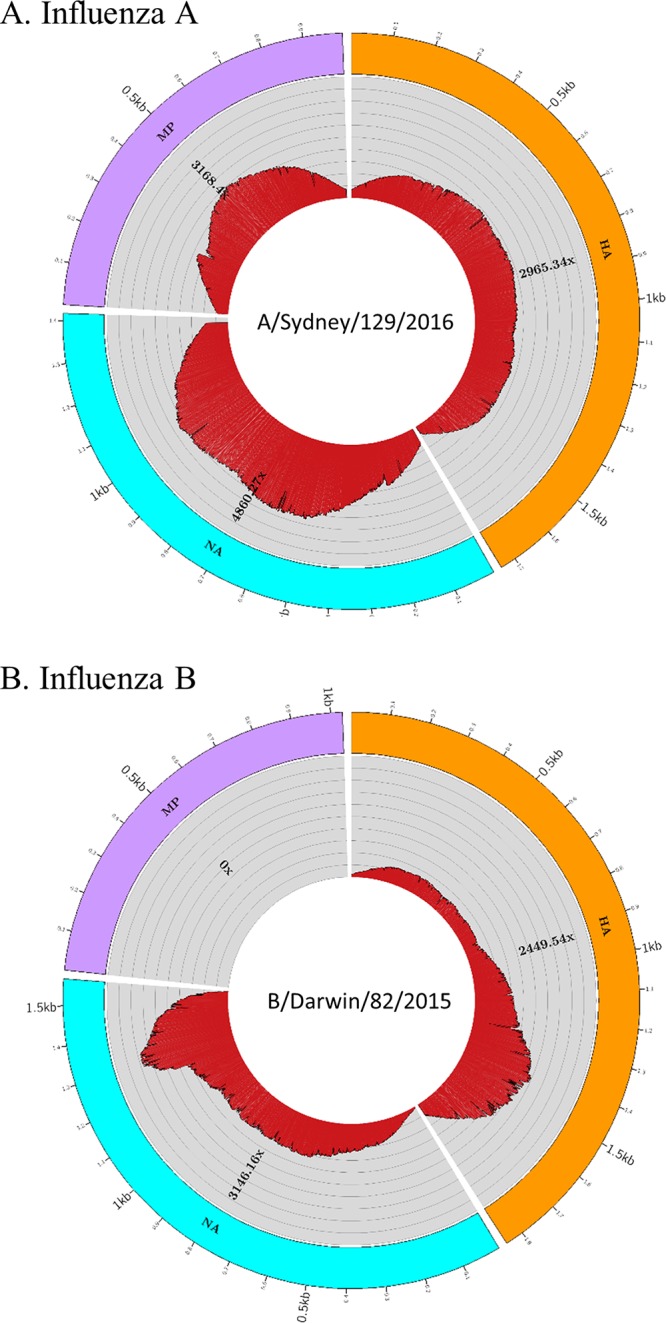
NGS analysis results generated from FluLINE. Influenza virus samples processed at WHOCC-Melbourne were sequenced on the Ion Torrent PGM platform, and data were analyzed using the newly developed FluLINE pipeline described in this study. NGS sequencing coverage and depth for representative influenza A virus (A) and influenza B virus (B) are shown. The outermost circle represents the gene segments: HA (orange), NA (blue), and MP (purple). The depths of coverage are represented by the concentric rings, emanating from the center, and each concentric ring represents 1,000× coverage.
FIG 4.

NGS analysis results generated from IRMA. Influenza virus samples processed at the CDC were sequenced on the Illumina MiSeq platform, and data were analyzed using the IRMA pipeline. Representative graphs of an influenza A virus are shown for the percentage of read counts and the sequencing coverage and depth. (A) Percentages of total read counts. Assembled, influenza virus reads in final assemblies; quality control (QC) filtered, did not pass length/median quality thresholds; other, nonflu, and contaminant/poor flu signal. (B) Percentages of assembled, merged-pair read counts of influenza virus HA, NA, and M segments. (C) Sequencing coverage and depth based on the reads mapped to the HA, NA, and M segments.
Optimization of the influenza virus NGS analysis pipeline.
The NGS data of the FluA/B RT-PCR amplicons can be analyzed with various analysis pipelines. To optimize viral genome assembly and variant calling from NGS data, the Iterative Refinement Meta-Assembler (IRMA) pipeline was developed recently at the CDC (16) and was used to analyze all samples processed at CDC.
Meanwhile, an alternative NGS analysis pipeline, FluLINE (Fig. 5), was developed to analyze all samples processed at the WHOCC-Melbourne (Table 2). FluLINE takes FASTQ files from Ion Torrent or Illumina sequencing to generate influenza virus consensus sequences iteratively and then maps the reads to the final consensus sequences for single nucleotide variant (SNV) calling and coverage visualization (Fig. 3). To assess the accuracy of the NGS and FluLINE, 10 viruses of each subtype were sequenced by both Sanger and Ion Torrent PGM; in all cases, the Sanger sequencing data matched identically with the NGS pipeline-generated consensus sequences from the Ion Torrent PGM data (data not shown).
FIG 5.

Main steps in the NGS analysis pipeline. (i) Filter the sequencing reads by cutadapt and FastQC, (ii) find the nearest sequence in NCBI database for each read, (iii) cluster and identify the viral species, (iv) generate consensus genomic sequence iteratively, (v) map the reads to the final consensus genome, and (vi) identify SNVs and visualize the coverage of the genome. min, minimum.
DISCUSSION
Capturing antigenic evolution and antiviral resistance is essential for molecular surveillance of seasonal human influenza epidemics. Sequencing of the HA, NA, and M gene segments of influenza A virus, as well as the HA and NA gene segments of influenza B virus, would complement and reduce reliance on the traditional hemagglutinin inhibition and phenotypic antiviral resistance assays for a large number of specimens. These assays would then only need to be used to phenotypically characterize viruses representative of each genetic group. However, the sequence diversity within and across the 4 epidemic influenza virus subtypes and lineages presents a major hurdle to designing an efficient universal strategy for conversion and amplification of the viral RNA to double-stranded DNA (dsDNA), a step that dramatically increases sequence coverage and reduces costs for all current sequencing platforms.
Compared to the universal genome amplification methods we previously developed, which are specific for influenza A viruses (13) or influenza B viruses (14), the single-reaction FluA/B surveillance multiplex RT-PCR method overcomes the evolutionary sequence barrier between type A and type B influenza viruses. This method amplifies the genomes of any seasonal epidemic influenza virus(es) present in clinical samples without prior knowledge of the type, subtype, or lineage of the virus. The FluA/B RT-PCR is highly sensitive (1 to 10 TCID50/RT-PCR) and robust, and we have applied it to successfully amplify (and sequence) the desired viral segments of hundreds of contemporary influenza A and B viruses (Table 2). This strategy is a powerful genetic surveillance assay that can be used on specimens from patients with influenza-like illness, and it has the potential to be used for diagnostic NGS, with an overall turnaround time of 2 to 4 days. The distinctive amplicon products visualized after agarose gel electrophoresis or other size fractionation processes will readily identify the type of influenza virus, and sequencing of the amplicons will generate the necessary information required for in-depth phylogenetic analyses. By focusing on the gene segments crucial for antigenic and antiviral surveillance (influenza A virus, HA, NA, and M; influenza B virus, HA and NA), sequencing depth is increased by 3- to 4-fold on these genes, which could be especially helpful in analyzing minor variants. Alternatively, more samples could be multiplexed per sequencing run for higher throughput and reduced cost. Since most of the contemporary epidemic human influenza viruses are resistant to adamantanes (17–19), if desired, the M primers can be removed from the FluA/B primer cocktail to further simplify the reaction and increase the sequencing coverage depth and/or the number of viruses that can be sequenced in a specific sequencing run.
The FluA/B RT-PCR strategy was developed primarily for NGS of contemporary epidemic influenza A and B viruses and could be deployed for a range of activities, including surveillance or diagnostic sequencing. The primers were designed based on the results of an analysis of all human influenza viruses before 2013, and the assay was validated with more than 1,000 human influenza viruses primarily collected after 2013. In addition, the FluA/B RT-PCR primers bind the recently emerged A(H3N2) variant viruses (H3N2v) (20, 21), since they also have the same conserved terminal sequences (data not shown). Given the conservation of the primer binding regions of epidemic seasonal influenza viruses over the past 100 years, we deem it highly unlikely that these regions will change in the foreseeable future. If a new subtype or animal-origin HA/NA is introduced into the human population, current influenza virus surveillance will immediately detect it, and sequencing of these regions will readily indicate if the FluA/B RT-PCR primers should be updated.
We previously also attempted to develop a single-reaction complete genomic amplification approach that works universally for all eight segments of both influenza A and B viruses. We combined the primers and integrated the methods described in an influenza A virus genomic amplification study (13) and an influenza B virus genomic amplification study (14). However, that approach suffered from significant loss of sensitivity: at least 1,000 to 10,000 TCID50/RT-PCR was needed for reliable genomic amplification. In contrast, the FluA/B surveillance multiplex RT-PCR strategy described herein amplifies viral RNA template as low as 1 to 10 TCID50/RT-PCR, which is roughly equivalent to 100 to 1,000 TCID50/ml of virus. We consider 10 TCID50/RT-PCR as the sensitivity level that a practical RT-PCR amplification method should have, as the viral load in many human clinical specimens is in the order of 1,000 (but varies from less than 102 to more than 106) TCID50/ml (22, 23). In addition, some clinical specimens may contain RT-PCR inhibitory factors, degraded viral genomes, and defective interfering particles (24), which further decrease the success rate of the assays. Recently, using degenerate primers, a universal method targeting the whole genome of both influenza A and B viruses was reported (25). However, with a lab-cultured H5N1 virus, at least 1.8 × 105 TCID50/ml of virus (10−4 dilution of the 1.8 × 109 TCID50/ml of virus stock) was needed to sequence all eight segments of the viral genome (25), further illustrating the difficulty in developing a universal whole-genome amplification method that works for both influenza A and B viruses.
Downstream analysis of NGS data is challenging for many laboratories because of the lack of bioinformatics expertise. Therefore, in addition to the recently reported IRMA pipeline developed at CDC (16), FluLINE was specifically developed for influenza A and B virus NGS analysis at the WHOCC-Melbourne. FluLINE can be used for data output from any NGS platform, as it uses raw fastq as the input data format. This set of scripts was written using open source programs; hence, this semiautomated pipeline is freely available to anyone with minimal bioinformatics training and a Linux operating system installed on a contemporary computer.
Overall, the FluA/B RT-PCR approach coupled with NGS and the analysis pipelines were successfully used to sequence the complete HA and NA (and M) gene segments of greater than 1,000 influenza A and B viruses in a fast, user-friendly, and cost-effective way. Finally, the method is easily adopted by laboratories using a variety of NGS platforms.
MATERIALS AND METHODS
Viruses.
Altogether, 2,105 influenza A and B viruses collected between 2005 and 2015 were used in this study, including 344 primary clinical specimens, 1,751 cell culture-isolated viruses, and 10 embryonated chicken egg-isolated viruses. The viruses are mainly collected from the United States, Australia, and other regions within the WHO Global Influenza Surveillance and Response System (WHO GISRS). RNA was extracted from primary or cultured specimens using the RNeasy minikit (Qiagen), QIAamp viral RNA minikit (Qiagen), or QIAxtractor automated system (Qiagen), according to the established protocols at each institution. The presence of influenza viruses was confirmed with real-time reverse transcription-PCR (rRT-PCR) using the CDC Influenza virus real-time RT-PCR assay (catalog no. NR-15592; BEI Resources) or using the FilmArray multiplex PCR system (bioMérieux, Inc.), according to the manufacturers' recommendations.
FluA/B RT-PCR.
Standard desalted oligonucleotide primers were ordered from Integrated DNA Technologies, Inc., and 10 μM working stocks were made with nuclease-free water, aliquoted, and stored at −20°C. The FluA/B surveillance multiplex RT-PCR (FluA/B RT-PCR) primer cocktail should be made fresh for each use, as specified in Table 1, avoiding repeated freeze-and-thaw cycles, which could decrease the RT-PCR efficiency.
The FluA/B RT-PCR reaction was set up as follows: 6 μl of nuclease-free water, 12.5 μl of 2× RT-PCR buffer, 3 μl of FluA/B RT-PCR primer cocktail, 3 μl of RNA, and 0.5 μl of RT/HiFi enzyme were mixed in a 25-μl reaction mixture using the SuperScript III one-step RT-PCR system with Platinum Taq high-fidelity DNA polymerase (Thermo Fisher Scientific, Inc.). The temperature cycle parameters were 45°C for 45 min, 55°C for 15 min, 94°C for 2 min, and then 5 cycles of 94°C for 20 s, 44°C for 30 s, and 68°C for 3 min, followed by 35 cycles of 94°C for 20 s, 58°C for 30 s, and 68°C for 3 min, with a final extension at 68°C for 10 min.
To assess the FluA/B RT-PCR amplification products, 3 μl of the amplicons was loaded into a 1% agarose gel for electrophoresis until all bands were clearly separated. DNA was stained with ethidium bromide that was precast in the gel.
Next-generation sequencing.
The products of the various influenza A and B virus amplicons were sequenced on an Ion Torrent PGM (Life technologies) and a MiSeq (Illumina) using standard protocols appropriate for each sequencing platform. Influenza virus sequencing and analysis at the J. Craig Venter Institute (JCVI) were performed as previously described (14). The procedures at the CDC were also previously described (16). Sequencing using the Ion Torrent PGM at WHOCC-Melbourne was performed according to the manufacturer's protocol: PCR amplicons were fragmented to 200 bp using the Ion Xpress Plus fragment library kit (Life technologies) and then ligated to Ion Xpress barcode adapters 1 to 96 (Life Technologies). Up to 96 individual amplicons with adapters were pooled and cleaned with AMPure XP reagent (Agencourt). Pooled libraries were quantified with the Ion library quantitation kit (Life Technologies), 10 pM of final library concentration was used to prepare template-positive Ion Sphere particles on the Ion OneTouch 2 instrument (Life Technologies), and Ion 316 Chip version 2 was used for sequencing on the Ion Torrent PGM.
Influenza virus analysis pipeline.
We developed a sequence analysis pipeline, FluLINE, using several open-source programs to identify the closest viral strain genome, generate the consensus genome, and detect variance within the sample viral population. The main steps of the pipeline are: (i) quality control and filtering of the sequencing reads; (ii) a search of each read against the NCBI database to find the nearest sequence; (iii) clustering and identification of the viral species; (iv) generation of consensus genomic sequences iteratively; (v) mapping of the reads to the final consensus genome; and (vi) identification of single nucleotide variants (SNVs) and visualization of the coverage of the genome (Fig. 5). FluLINE is available on https://umasangumathi.github.io/FluLINE/.
FastQC (26) was used to assess the quality of the NGS reads, such as quality of base calling and read length distribution, and to identify adapter sequences in reads. Cutadapt (27) was used to trim adapter sequences and filter out low-quality reads. A minimum base quality cutoff of 20 and minimum read length of 50 bp were chosen to ensure good-quality reads for consensus genome generation.
To identify coinfection, each read was searched for the best matching nucleotide sequence in the NCBI nucleotide database using the BLAST toolkit (28). The reads were clustered based on the species or strain using the metagenomic analysis software MEGAN (29) and generated phylogenetic clustering at the viral strain level to investigate the potential for coinfection by multiple viral species or strains of virus and to identify the closest viral genome in NCBI.
With the closest viral genome as a guide, the consensus genome of the sample was generated by using the ‘bam2cons_iter.sh’ script from the ViPR pipeline (https://github.com/CSB5/vipr). The script uses BWA (30) to iteratively map reads to the reference genome until a working consensus is generated based on the maximum frequency of nucleotide at each position. BLAST is then run again using the working consensus as the query, and the iterative consensus building procedure is run again with this optimal reference as the root to produce the final consensus sequence for the sample.
Bowtie2 (31) with local fast alignment option was used to map the reads to the consensus reference genome for mutations and indels detection, as suggested by Caboche et al. (32). Finally, LoFreq2 (33) was used to detect the SNVs present in the sample for the regions with a minimum of 100× genome coverage. The visualization of genome coverage for the viral gene segments of interest was plotted using the Circos software (34).
ACKNOWLEDGMENTS
The Melbourne WHO Collaborating Centre for Reference and Research on Influenza is supported by the Australian Government Department of Health and Ageing.
The opinions expressed in this article are our own and do not reflect the views of the Centers for Disease Control and Prevention, the Department of Health and Human Services, or the U.S. Government.
We thank all of the National Influenza Centres and other contributing laboratories who sent influenza virus-positive samples to the WHO Collaborating Centres for influenza virus reference and research.
REFERENCES
- 1.WHO. 2009. Influenza (seasonal). Fact sheet 211. World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs211/en/. [Google Scholar]
- 2.Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. 2003. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289:179–186. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- 3.Molinari NA, Ortega-Sanchez IR, Messonnier ML, Thompson WW, Wortley PM, Weintraub E, Bridges CB. 2007. The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine 25:5086–5096. doi: 10.1016/j.vaccine.2007.03.046. [DOI] [PubMed] [Google Scholar]
- 4.Wright PF, Neumann G, Kawaoka Y. 2013. Orthomyxoviruses, p 1186–1243. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6 ed, vol 1 Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 5.Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. 2001. Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol 146:2275–2289. doi: 10.1007/s007050170002. [DOI] [PubMed] [Google Scholar]
- 6.Chan CH, Lin KL, Chan Y, Wang YL, Chi YT, Tu HL, Shieh HK, Liu WT. 2006. Amplification of the entire genome of influenza A virus H1N1 and H3N2 subtypes by reverse-transcription polymerase chain reaction. J Virol Methods 136:38–43. doi: 10.1016/j.jviromet.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 7.Jindal N, Chander Y, de Abin M, Sreevatsan S, Stallknecht D, Halvorson DA, Goyal SM. 2009. Amplification of four genes of influenza A viruses using a degenerate primer set in a one step RT-PCR method. J Virol Methods 160:163–166. doi: 10.1016/j.jviromet.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Höper D, Hoffmann B, Beer M. 2009. Simple, sensitive, and swift sequencing of complete H5N1 avian influenza virus genomes. J Clin Microbiol 47:674–679. doi: 10.1128/JCM.01028-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Höper D, Hoffmann B, Beer M. 2011. A comprehensive deep sequencing strategy for full-length genomes of influenza A. PLoS One 6:e19075. doi: 10.1371/journal.pone.0019075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng YM, Spirason N, Iannello P, Jelley L, Lau H, Barr IG. 2015. A simplified Sanger sequencing method for full genome sequencing of multiple subtypes of human influenza A viruses. J Clin Virol 68:43–48. doi: 10.1016/j.jcv.2015.04.019. [DOI] [PubMed] [Google Scholar]
- 11.Djikeng A, Halpin R, Kuzmickas R, Depasse J, Feldblyum J, Sengamalay N, Afonso C, Zhang X, Anderson NG, Ghedin E, Spiro DJ. 2008. Viral genome sequencing by random priming methods. BMC Genomics 9:5. doi: 10.1186/1471-2164-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rutvisuttinunt W, Chinnawirotpisan P, Simasathien S, Shrestha SK, Yoon IK, Klungthong C, Fernandez S. 2013. Simultaneous and complete genome sequencing of influenza A and B with high coverage by Illumina MiSeq platform. J Virol Methods 193:394–404. doi: 10.1016/j.jviromet.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Zhou B, Donnelly ME, Scholes DT, St. George K, Hatta M, Kawaoka Y, Wentworth DE. 2009. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and swine origin human influenza a viruses. J Virol 83:10309–10313. doi: 10.1128/JVI.01109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou B, Lin X, Wang W, Halpin RA, Bera J, Stockwell TB, Barr IG, Wentworth DE. 2014. Universal influenza B virus genomic amplification facilitates sequencing, diagnostics, and reverse genetics. J Clin Microbiol 52:1330–1337. doi: 10.1128/JCM.03265-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bao YM, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D. 2008. The influenza virus resource at the National Center for Biotechnology Information. J Virol 82:596–601. doi: 10.1128/JVI.02005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shepard SS, Meno S, Bahl J, Wilson MM, Barnes J, Neuhaus E. 2016. Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler. BMC Genomics 17:708. doi: 10.1186/s12864-016-3030-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bright RA, Shay DK, Shu B, Cox NJ, Klimov AI. 2006. Adamantane resistance among influenza A viruses isolated early during the 2005-2006 influenza season in the United States. JAMA 295:891–894. doi: 10.1001/jama.295.8.joc60020. [DOI] [PubMed] [Google Scholar]
- 18.Deyde VM, Xu X, Bright RA, Shaw M, Smith CB, Zhang Y, Shu Y, Gubareva LV, Cox NJ, Klimov AI. 2007. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J Infect Dis 196:249–257. doi: 10.1086/518936. [DOI] [PubMed] [Google Scholar]
- 19.Hurt AC. 2014. The epidemiology and spread of drug resistant human influenza viruses. Curr Opin Virol 8:22–29. doi: 10.1016/j.coviro.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 20.Jhung MA, Epperson S, Biggerstaff M, Allen D, Balish A, Barnes N, Beaudoin A, Berman L, Bidol S, Blanton L, Blythe D, Brammer L, D'Mello T, Danila R, Davis W, de Fijter S, Diorio M, Durand LO, Emery S, Fowler B, Garten R, Grant Y, Greenbaum A, Gubareva L, Havers F, Haupt T, House J, Ibrahim S, Jiang V, Jain S, Jernigan D, Kazmierczak J, Klimov A, Lindstrom S, Longenberger A, Lucas P, Lynfield R, McMorrow M, Moll M, Morin C, Ostroff S, Page SL, Park SY, Peters S, Quinn C, Reed C, Richards S, Scheftel J, Simwale O, Shu B, et al. . 2013. Outbreak of variant influenza A(H3N2) virus in the United States. Clin Infect Dis 57:1703–1712. doi: 10.1093/cid/cit649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindstrom S, Garten R, Balish A, Shu B, Emery S, Berman L, Barnes N, Sleeman K, Gubareva L, Villanueva J, Klimov A. 2012. Human infections with novel reassortant influenza A(H3N2)v viruses, United States, 2011. Emerg Infect Dis 18:834–837. doi: 10.3201/eid1805.111922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Wesenbeeck L, D'Haese D, Tolboom J, Meeuws H, Dwyer DE, Holmes M, Ison MG, Katz K, McGeer A, Sadoff J, Weverling GJ, Stuyver L. 2015. A downward trend of the ratio of influenza RNA copy number to infectious viral titer in hospitalized influenza A-infected patients. Open Forum Infect Dis 2:ofv166. doi: 10.1093/ofid/ofv166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suess T, Remschmidt C, Schink SB, Schweiger B, Heider A, Milde J, Nitsche A, Schroeder K, Doellinger J, Braun C, Haas W, Krause G, Buchholz U. 2012. Comparison of shedding characteristics of seasonal influenza virus (sub)types and influenza A(H1N1)pdm09; Germany, 2007–2011. PLoS One 7:e51653. doi: 10.1371/journal.pone.0051653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saira K, Lin X, DePasse JV, Halpin R, Twaddle A, Stockwell T, Angus B, Cozzi-Lepri A, Delfino M, Dugan V, Dwyer DE, Freiberg M, Horban A, Losso M, Lynfield R, Wentworth DN, Holmes EC, Davey R, Wentworth DE, Ghedin E, INSIGHT FLU002 Study Group, INSIGHT FLU003 Study Group . 2013. Sequence analysis of in vivo defective interfering-like RNA of influenza A H1N1 pandemic virus. J Virol 87:8064–8074. doi: 10.1128/JVI.00240-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao J, Liu J, Vemula SV, Lin C, Tan J, Ragupathy V, Wang X, Mbondji-Wonje C, Ye Z, Landry ML, Hewlett I. 2016. Sensitive detection and simultaneous discrimination of influenza A and B viruses in nasopharyngeal swabs in a single assay using next-generation sequenced-based diagnotics. PLoS One 11:e0163175. doi: 10.1371/journal.pone.0163175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andrews S. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 27.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 28.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huson DH, Auch AF, Qi J, Schuster SC. 2007. MEGAN analysis of metagenomic data. Genome Res 17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caboche S, Audebert C, Lemoine Y, Hot D. 2014. Comparison of mapping algorithms used in high-throughput sequencing: application to Ion Torrent data. BMC Genomics 15:264. doi: 10.1186/1471-2164-15-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilm A, Aw PPK, Bertrand D, Yeo GH, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N. 2012. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 40:11189–11201. doi: 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crooks GE, Hon G, Chandonia J-M, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]