

Abstract
Cirrhosis develops from liver fibrosis and is the severe pathological stage of all chronic liver injury. Cirrhosis caused by hepatitis B virus and hepatitis C virus infection is especially common. Liver fibrosis and cirrhosis involve excess production of extracellular matrix, which is closely related to liver sinusoidal endothelial cells (LSECs). Damaged LSECs can synthesize transforming growth factor-beta and platelet-derived growth factor, which activate hepatic stellate cells and facilitate the synthesis of extracellular matrix. Herein, we highlight the angiogenic cytokines of LSECs related to liver fibrosis and cirrhosis at different stages and focus on the formation and development of liver fibrosis and cirrhosis. Inhibition of LSEC angiogenesis and antiangiogenic therapy are described in detail. Targeting LSECs has high therapeutic potential for liver diseases. Further understanding of the mechanism of action will provide stronger evidence for the development of anti-LSEC drugs and new directions for diagnosis and treatment of liver diseases.
Keywords: Sinusoidal endothelial cells, Hepatitis, Fibrosis, Cirrhosis, Liver disease
Core tip: Liver sinusoidal endothelial cells (LSECs) comprise the highest proportion of nonparenchymal cells in the liver. Their fenestrae and basement membrane structure, and high endocytic clearance ability play an indispensable role in the physiology and pathology of the liver. LSECs mainly participate in the regulation of liver pathology, such as hepatitis, liver fibrosis, cirrhosis and liver regeneration, by exerting anti-inflammatory activity, endocytosis, secretion, synthesis of angiogenesis signaling molecules and maintaining the hepatic stellate cell phenotype. It is important to elucidate the mechanism of action of LSECs, which will provide important information for future targeted therapy and clinical diagnosis.
INTRODUCTION
Liver sinusoidal endothelial cells (LSECs) form the wall of the hepatic sinusoids and have unique morphology and function. These cells contain many fenestrae with uniform diameters of 100-150 nm, thus creating open channels for the exchange of substances between the blood and liver parenchyma[1]. However, the diameter and number of fenestrae are influenced by a variety of agents and liver disease. LSECs are reported to play a key role in triggering liver regeneration and contribute to hepatic complications, such as hepatitis, liver fibrosis and cirrhosis[2].
Hepatotropic viruses usually pass through the protective filter constructed by LSECs to gain access to the liver parenchyma. For example, hepatitis B virus (HBV) crosses the liver endothelium via transcytosis[3]. LSECs contribute to the clearance of HBV and hepatitis C virus (HCV) from the bloodstream and control HCV replication[4,5]. Meanwhile, LSECs produce large amounts of anti-inflammatory cytokines, such as transforming growth factor-beta (TGF-β)[6]. LSECs constitutively express major-histocompatibility complex I-restricted antigens and co-stimulatory molecules, which shift the hepatic immune balance toward tolerance[7,8]. Since many patients with chronic hepatitis often have liver fibrosis[9], it is important to know the changes in LSECs during development of liver diseases.
Liver fibrosis is characterized by excess deposition of extracellular matrix (ECM), which may lead to progression of chronic liver diseases like viral hepatitis[10]. Hyaluronic acid, a marker of ECM deposition, is synthesized by hepatic stellate cells (HSCs) and degraded by LSECs. So, liver fibrosis is closely related to LSECs[11]. When liver fibrosis happens, the LSECs lose their fenestrae and form a basement membrane, which is accompanied by production and release of soluble factors that affect the phenotype of neighboring cells[12]. For example, LSECs with defenestration cannot suppress activation of HSCs[13]. Early-stage fibrosis regresses to a near-normal level after successful treatment, but advanced fibrosis can cause irreversible damage. The capillarization of LSECs plays a key role in the pathogenesis and progression of this process[14].
In cirrhosis, LSECs frequently transform to a vascular type with a basement membrane, which interferes with the bidirectional exchange of molecules. Fibrosis, as a precursor of cirrhosis, is a common pathological process of all chronic liver diseases[15]. Angiogenesis contributes to progression of fibrosis to cirrhosis in patients with chronic liver diseases[16]. Many growth factors, such as vascular endothelial growth factor (VEGF), promote transformation of LSECs into the vascular type. Cellular crosstalk among LSECs, HSCs and hepatocytes plays an important role in the angiogenesis process during development of cirrhosis[17,18]. However, due to poor understanding of the molecular mechanisms leading to cirrhosis, there is still a lack of effective strategies to treat cirrhosis. So, it is important to know the pathogenesis of liver cirrhosis, and the change and function of LSECs and HSCs during the transformation of fibrosis to cirrhosis and liver regeneration.
In this review, we describe the morphological changes of LESCs in different liver diseases, such as hepatitis, liver fibrosis and cirrhosis. We discuss the proangiogenic markers and endothelial cell markers that can influence the angiogenic capability of LSECs. We list several agents that inhibit LESC-induced angiogenesis, such as tetramethylpyrazine and sorafenib plus gadolinium chloride (GdCl3).
LSECs IN NORMAL LIVER
The liver is composed of parenchymal and non-parenchymal cells. LSECs represent a small fraction of total liver cell volume, comprising about 70% of non-parenchymal cells. The LSEC surface is smooth and lacks filopodia, generally. The LSECs are held together through special intercellular junctions and there are no gaps existing between cells[19,20]. Transmission electron microscopy (TEM) shows that the thin attenuated cytoplasm of LSECs contains numerous fenestrae (Figure 1A). Scanning electron microscopy (SEM) shows high porosity of LSECs (Figure 1B)[21]. Atomic force microscopy (AFM) can retain the natural environment of LSECs[22]. AFM shows profound streaking artefacts (Figure 1C and D), which indicates that the AFM images on different kinds of liver sinusoidal cells interact closely with the cell surface. The LSEC cytoplasm contains vesicles and organelles with functions of uptake, transport and degradation of endocytosed material.
Figure 1.
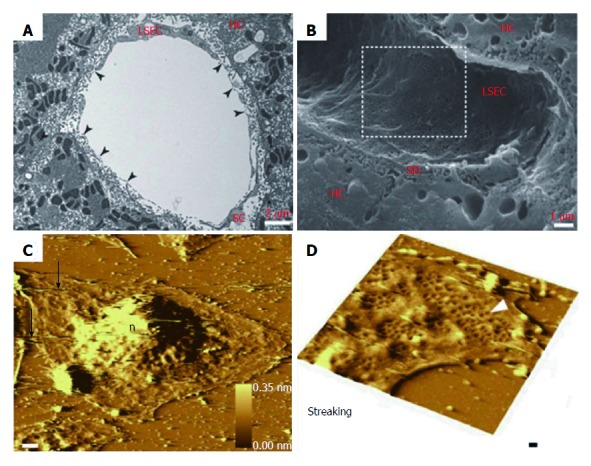
Morphological changes of liver sinusoidal endothelial cells. A: TEM image of rat liver sinusoid after transversal cut; B: SEM image of rat liver sinusoid; C: AFM image of LSECs with a 10 nm gold layer; D: A high scan range of AFM image of LSECs. A-B: ref 21 Copyright @1985 by the American Association for the Study of Liver Diseases; C-D: ref 22 Copyright @ 2012 Elsevier Ltd. AFM: Atomic force microscopy; LSECs: Liver sinusoidal endothelial cells; SEM: Scanning electron microscopy; TEM: Transmission electron microscopy.
The LSECs with fenestrae, absence of diaphragm and lack of basement membrane show high permeability, allowing exchanges of molecules but also active uptake and degradation of molecules[2]. The fenestrae, which comprise many small pores, are organized in clusters termed sieve plates. The number and size of fenestrae vary according to their localization in the liver. Larger but fewer fenestrae per sieve plate are located in the periportal region and smaller but more fenestrae per sieve plate are located in the centrilobular region, which is related to oxygen tension[23,24]. Furthermore, the size and number of fenestrae vary in different physiological and pathological conditions. For example, fasting individuals show fewer but larger fenestrae[25,26]. In normal liver, LSECs act as a selective sieve for soluble molecules, lipoproteins, metabolites, small chylomicron remnants, virus particles and other nanoparticles with a diameter below that of the fenestrae[27]. In addition, the cytoskeleton of LSECs, including its components such as actin, fibronectin, myosin and calmodulin, plays an important role in the modulation of fenestrae. In particular, actin filaments are in the vicinity of the fenestrae, and are involved in the contraction and dilatation of the fenestrae[28].
The maintenance of fenestrae of LSECs needs both paracrine and autocrine cell signaling[29]. VEGF, derived from adjacent hepatocytes and HSCs, is crucial in this regulation and acts through nitric oxide (NO)-dependent and -independent pathways. For example, VEGF stimulates NO release via endogenous NO synthase (eNOS) in the LSECs[30].
Meanwhile, the LSECs possess one of the highest endocytic capacities compared with other cells. Thus, LSECs can clear soluble macromolecules and small particles through endocytic receptors, resulting in LSECs having an immunological function. The endocytosis receptors of LSECs include scavenger receptors, mannose receptors and Fcγ receptor IIb2[31]. The scavenger receptors mediate endocytosis of polyanionic molecules, such as oxidized and acetylated low-density lipoproteins, and advanced glycation end products. The mannose receptors bind to glycoproteins and microbial glycans for further use in LSECs. Fcγ receptor IIb2 expressed by LSECs can mediate the clearance of small circulating immune complexes. Moreover, LSEC endocytosis also contributes to the transfer of molecules from the sinusoids to the space of Disse, a process named transcytosis[32]. In addition to expression of scavenger receptors, LSECs also express many pattern recognition receptors. For example, Toll-like receptors (TLRs) respond to low concentrations of lipopolysaccharide, which produces proinflammatory mediators such as interleukin (IL)-6[33]. Furthermore, cytokines, released by LSECs via TLRs, serve as antiviral effectors and contribute to liver targeting by hepatotropic viruses, such as HBV or HCV[4,34,35]. In summary, LSECs not only express scavenger functions in innate immune defense but also contribute to viral infection of the liver.
LSECs IN HEPATITIS
LSECs protect underlying tissues against external agents such as viruses, bacteria and parasites[36-37]. However, some viruses, such as HBV, have developed sophisticated ways to overcome the protective filter formed by LESCs and gain access to the underlying liver parenchyma. For example, HBV crosses the LSEC membrane via transcytosis[3]. In hepatitis, the number and size of fenestrae in LSECs decrease and a discontinuous basement membrane appears. These new characteristics become obvious with the aggravation of hepatitis[1]. Moreover, the scavenging LSECs play a key role in the initial uptake of viral pathogens into the liver. In order to observe the process, Breiner et al[3] combined duck HBV particles with fluorescent gold particles in test animals, as well as in primary liver cell culture. It is reported that molecules > 12 nm in diameter cannot pass through the LSECs to the hepatocytes[38]. The size of HBV-coated gold particles exceeds 50 nm, which cannot directly access the fenestrae of LSECs. The data support that viruses reach the hepatocytes through active transport across the liver endothelium. Furthermore, viral clearance in hepatitis only happens in hepatocytes so that viruses in LSECs could act as a reservoir for endogenous reinfection[39]. LSECs are unique antigen-presenting cells; for example, LESCs show antigen-specific immune tolerance in CD4+ and CD8+ cells. Thus, they may help viruses escape from the immune system[3].
The LSECs mediate hepatic immune tolerance toward self or foreign antigens by expression of anti-inflammatory mediators under physiological conditions, but they achieve proinflammatory functions upon viral infection[40]. In order to learn more about the role of LSECs in hepatic inflammation during acute viral hepatitis, Bleau et al[41] investigated the effects of LSECs infection on their proinflammatory profiles and aggravation of acute hepatitis using a mouse model of murine hepatitis virus type 3 (MHV3) infection. Three different types of MHV3 were injected intraperitoneally into mice: attenuated 51.6-MHV3, YAC-MHV3 and virulent MHV3. The inflammatory foci surrounded necrotic cells of virulent-MHV3-infected mice at 24 h after infection and disappeared at 72 h, while hepatocyte necrosis became extensive. The mice infected with 51.6-MHV3 showed delayed occurrence of inflammatory foci at 48 h after infection, while the mice infected with YAC-MHV3 had a few small inflammatory infiltrates and had no observable hepatic necrosis at 72 h after infection (Figure 2A and B). This demonstrated that the lower tropism of attenuated MHV3 variants for LSECs was associated with less severe damage and less viral replication in the liver. In vitro attenuated MHV3 strains induced less fibrinogen-like protein 2, a prothrombinase that promotes vascular thrombosis and hepatic inflammation, which is expressed by LSECs. Virulent MHV3 altered the production of anti-inflammatory cytokines expressed by LSECs; for example, lower induction of caveolin-1, which is a key component of LSEC fenestrae, and lower induction of IL-33, an alarmin secreted mainly by injured LSECs. These results indicate that virulent MHV3 strain promotes greater release of proinflammatory cytokines. Furthermore, specific activation of TLR2 signaling by MHV3 is related to the higher replication and proinflammatory activation in LSECs during acute hepatitis.
Figure 2.
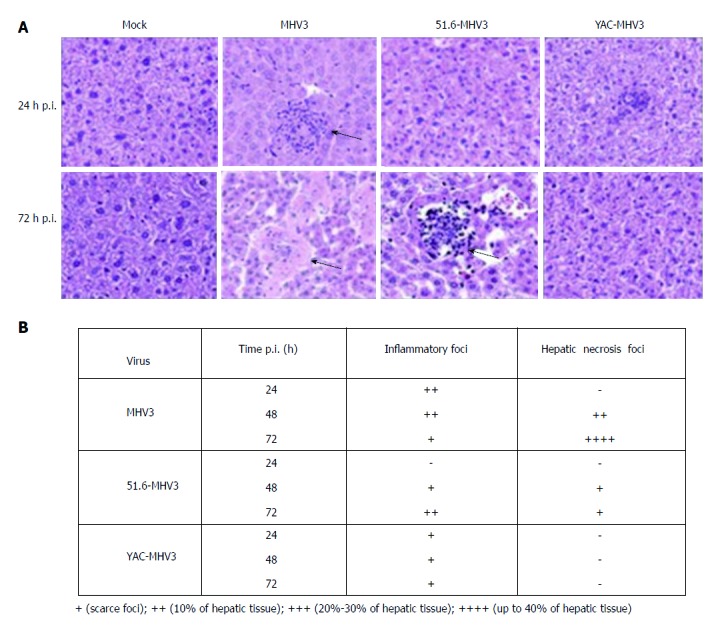
Pathological changes of hepatocytes after infection. A: Histopathological analysis was conducted on livers from mock-infected and virus-infected mice from each group at 24 and 72 h after infection; B: Summary of occurrence of necrotic and virus-infected mice at 24, 48 and 72 h after infection. A-B: ref 41 Copyright @ 2016 American Society for Microbiology.
Virus-specific T cells play an important role in the outcome and pathogenesis of viral infections in the liver[3,42]. Liu and co-workers[43] reported that the absence of LSEC lectin led to a high frequency of intrahepatic effector cytotoxic T lymphocytes (CTLs) in a mouse model of HBV infection. LSEC lectin is a cell surface molecule that belongs to the C-type lectin receptors. Compared with wild-type cells, LSEC lectin knockout cells show accelerated liver viral clearance due to a higher frequency of intrahepatic CTLs with higher antiviral activity. Moreover, LSEC lectin expression in the liver is induced by IL-10 or interferon-gamma. And, it also limits immunopathology at the cost of delaying viral clearance. All the results demonstrate that LSEC lectin might facilitate the reduction of liver inflammation.
LSECs IN LIVER FIBROSIS
Liver fibrosis results in disruption of the synthesis and degradation of ECM components, leading to excess connective tissue in the liver[44]. Hyaluronic acid is considered as a marker of ECM deposition. It is a glycosaminoglycan synthesized by HSCs and degraded by LSECs. Damaged LSECs lose their degradative capacity and secrete proinflammatory cytokines, such as TGF-β1, which can stimulate activation of HSCs to synthesize large amounts of ECM[2,11,45].
In the early stage of liver fibrosis, damaged LSECs begin to form capillaries[19]. Defenestration of LSECs is a distinctive structural change during capillarization, and the change can protect the liver from continuing damage by restricting toxins to a specific area. However, defenestration also promotes the formation of a continuous basement membrane of hepatic sinusoids, disrupting the bidirectional exchange of molecules between hepatocytes and hepatic blood sinuses[46]. Disruption of the exchange might induce ischemic atrophy of hepatocytes, which leads to increased fibrogenesis and compensatory hypertrophy of surrounding hepatocytes[14]. Moreover, LSECs with capillarization lose their capacity to inactivate HSCs, thus promoting fibrogenesis[47].
Defenestration of LSECs is regarded as the main pathological change of liver fibrosis. It is evident in LSECs by shrinking fenestrae and formation of a continuous basement membrane. Healthy LSECs produce a modest amount of macromolecules such as collagen type IV and fibronectin[48]. The healthy fenestrated LSECs and low density of ECM protein in the space of Disse facilitate transport of cargo from the blood vessel lumen to all kinds of liver cells. However, the quantity of ECM increases many fold in liver fibrosis, contributing to formation of a continuous basement membrane[12]. During the early stages of liver fibrosis, the basement membrane is made up of fibrillary collagen combined with excess collagen type IV and laminin[49]. Natarajan et al[44] reported the relationship between altered fenestral morphology and perisinusoidal matrix, and demonstrated that individual components of the ECM failed to maintain the fenestrae. In liver fibrosis, defenestration is related to accumulation of interstitial collagens in the space of Disse. After appearance of the continuous basement membrane in the liver endothelium, the changes in LSEC phenotype become almost irreversible[50]. So, it is important to inhibit liver fibrosis to control the excess production of ECM in sinusoidal vascular structures[51].
When studying the effects of Astragalus polysaccharide (APS), which is the primary effective component of the Chinese herbal medicine Astragalus membranaceus, on nanoscale mechanical properties of LSECs in rats, we found that as APS concentration increased, the value of Young’s modulus presented an increasing trend, and the surface topography demonstrated that APS was capable of increasing the total area of fenestrae. The observed changes in mechanical properties of LSECs may provide a new therapeutic strategy for mechanistic research of anti-hepatic fibrosis treatments in Chinese medicine[52].
Angiogenesis is defined as the formation of new microvasculature from pre-existing blood vessels and mature endothelial cells. Angiogenesis can occur in many physiological and pathological conditions, such as during liver regeneration and fibrosis[53]. Recent studies have reported that development of liver fibrosis is closely associated with angiogenesis. The inhibition of pathological angiogenesis can effectively alleviate liver fibrosis[10]. So, it is important to know which substances contribute to and which attenuate angiogenesis. Yan et al[54] investigated the biological roles of CD147 in LSECs and hepatocytes, and assessed its therapeutic value as a target molecule in a carbon tetrachloride-induced liver fibrosis mouse model. Bioinformatics and experimental data indicate that CD147 promotes liver fibrosis progression via VEGF-A/VEGF receptor-2 (VRGFR-2) signaling-mediated crosstalk between hepatocytes and LSECs. Increased expression of VEGFR-2 enhanced the angiogenic capability of LSECs, which induced intrahepatic angiogenesis, leading to liver fibrosis progression. Kantari-Mimoun et al[10] pointed out that genetic ablation of VEGF in myeloid cells or pharmacological inhibition of VEGFR-2 signaling could prevent angiogenic response and fibrosis progression. Meanwhile, they reported myeloid cell-derived VEGF as a critical regulator of ECM degradation by LSECs.
In order to attenuate LSEC-induced angiogenesis, Zhao and coworkers[55] evaluated the effect of tetramethylpyrazine (TMP) on angiogenesis and further elucidated the underlying mechanisms. They indicated that TMP alleviated liver fibrosis by hedgehog-dependent inhibition of angiogenesis in vitro and in vivo. In the mouse fibrotic liver experiment, SEM suggested that the number of fenestrae was decreased in fibrotic liver (Figure 3A and B) but was restored after treatment with TMP and imatinib (Figure 3C and D). Furthermore, expression of proangiogenic markers (VEGF-R2 and VEGF-A) and endothelial cell markers (CD31 and CD34) was up-regulated in fibrotic liver, but TMP and imatinib decreased these markers, indicating that the angiogenic niche involving LSECs was alleviated by treatment with TMP. In addition, in vitro experiments also consistently revealed that treatment with TMP inhibited LESC-induced angiogenesis. Furthermore, curcumin and nintedanib attenuated angiogenesis in liver fibrosis via inhibiting VEGF expression in HSCs rather than LSECs[13,56].
Figure 3.
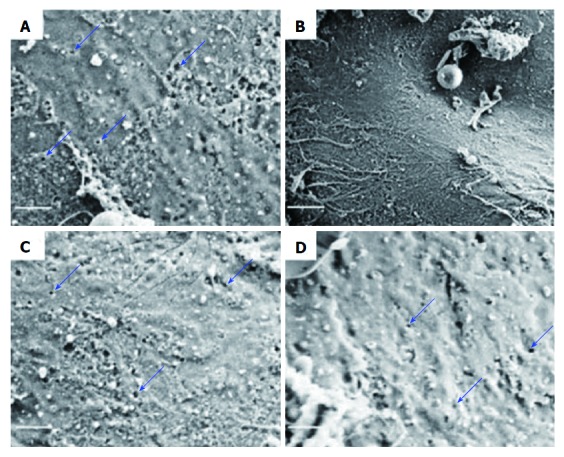
Scanning electron microscopy of liver sinusoidal endothelial cells sieve plates in CCl4-induced liver fibrosis. A: Control group; B: CCl4 group; C: TMP- (100 mg/kg) and CCl4-treated group; D: Imatinib- (10 mg/kg) and CCl4-treated group. Scale bar = 1 μm. A-D: Ref 54 Copyright @ 2017 International Union of Biochemistry and Molecular Biology. CCl4: Carbon tetrachloride; TMP: Tetramethylpyrazine.
However, traditional antiangiogenic therapy involves pharmacological inhibition of integrin αvβ3, which promotes fibrosis regression[57]. Liu et al[58] analyzed the effects of sorafenib plus GdCl3 on dimethylnitrosamine-induced liver fibrosis in rats and the interactions among LSECs, HSCs and Kupffer cells (KCs) using laser confocal microscopy. Compared with other antiangiogenic therapies, the sorafenib plus GdCl3 attenuated liver fibrosis by inhibiting the expression of angiogenesis-associated cell markers and cytokines, such as CD31 and VEGF. This suppressed the interactions of HSCs, LSECs and KCs, which were assessed by the colocation of α-smooth muscle actin, CD68 and von Willebrand factor. Furthermore, sorafenib plus GdCl3 significantly reduced liver function and hydroxyproline and suppressed collagen accumulation, which indicates that it could be a potential therapeutic strategy for treatment of liver fibrosis. To establish if the release of angiogenic factors from LSECs affect liver regeneration and fibrosis, Manavski et al[59] analyzed over-expression of Kruppel-like factor 2 (KLF2) in LSECs and found that KLF2 inhibited hepatocyte proliferation but had no effect on capillary density and liver fibrosis via induction of activin A.
Due to the special function of LSECs, the key step in liver regeneration is the replication of LSECs to expand the hepatic sinusoidal vascular network[60]. It is crucial for liver regeneration to replicate LSECs to connect with the existing vascular system. Ding et al[61] reported that natural and pharmacological ligands modulate endothelial sphingosine-1-phosphate receptor-1 (S1P1) to stimulate liver regeneration and inhibit fibrosis (Figure 4A). In mice lacking high-density lipoprotein (HDL)-S1P, the liver regenerative responses after partial hepatectomy were significantly inhibited, suggesting that HDL-S1P promotes functional recovery of liver mass after partial hepatectomy. Moreover, the distance between LSECs and hepatocytes was increased, and more perivascular deposition of ECM was observed (Figure 4B), which suggests that lack of HDL-S1P causes liver fibrosis. The data show that HDL-S1P may be a novel therapeutic target for liver fibrosis. All the above research points to the possibility of using antiangiogenic drugs for the treatment of liver fibrosis.
Figure 4.
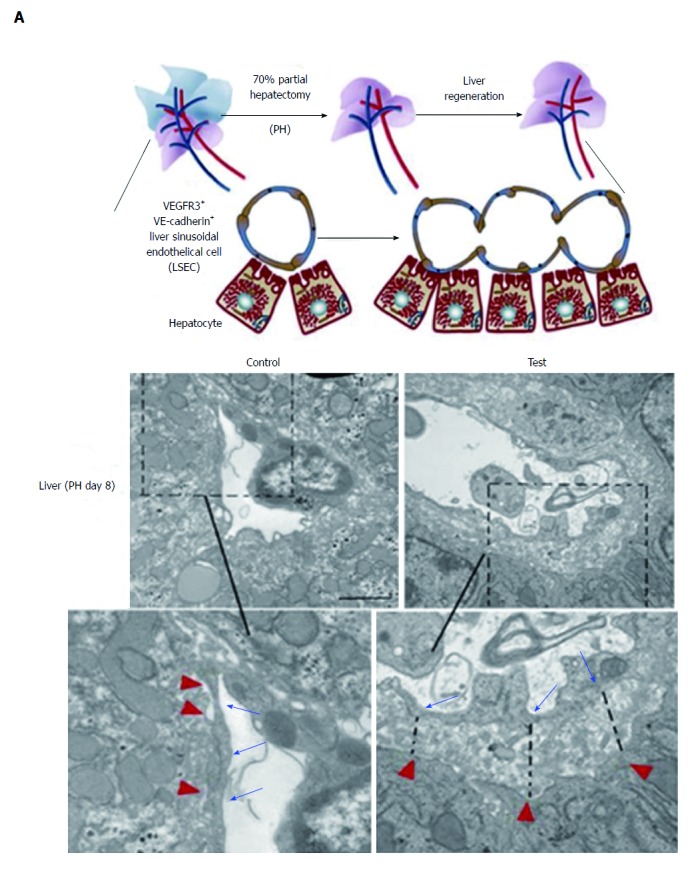
Relationship between liver sinusoidal endothelial cells and liver regeneration. A: Strategy to test liver regeneration in mice lacking HDL-S1P; B: Ultrastructure of LSECs in control and test groups after PH (dashed line represents increased LSEC-hepatocyte distance; inset shows higher level of perivascular matrix protein, black arrows and red arrowheads indicate the borders of LSECs and hepatocytes). Scale bar = 5 μm. A-B: ref 60 Copyright @ 2016 American Society for Clinical Investigation. HDL: High-density lipoprotein; LSECs: Liver sinusoidal endothelial cells; PH: Partial hepatectomy.
LSECs IN CIRRHOSIS
In cirrhosis, LSECs frequently transform fenestrae to a vascular type with the formation of a true basement membrane. The vascular type of LSECs interfere with the bidirectional exchange of molecules, resulting in decreased sinusoidal compliance with increased resistance to blood flow, which may contribute to development of portal hypertension in cirrhosis[17,18]. Gross hepatic structural disorders also associate with portal hypertension. The modified sinusoids are the main place of resistance to portal blood. The damaged LESCs became more sensitive to endogenous vasoconstrictors, which result in less expression of eNOS and less production of NO, and thus increased resistance for blood supply and oxygen delivery. And, the hypoxia leads more production of proangiogenic and profibrogenic factors. Moreover, impaired paracrine interaction between activated HSCs and LSECs and capillarization play the key role in improving the hepatic vascular resistance to portal blood flow. Meanwhile, they also add structural changes associated with diffuse fibrosis and regenerative nodules in liver cirrhosis[62].
Angiogenesis is an important factor that contributes to progression of fibrosis to cirrhosis in patients with chronic liver diseases. VEGF is an important regulator of angiogenesis through eNOS [16,63]. In experimental animal models of liver cirrhosis, HSCs express angiogenic cytokines and related receptors, such as VEGF and angiopoietin-1, which induce migration of HSCs, vessel formation and production of collagen, leading to liver disease progression[64]. Kaur and co-workers[65] focused on numbers and angiogenic functions of circulating endothelial progenitor cells (EPCs) and the interactions of EPCs with LSECs. Compared with controls, the co-culture of LSECs and EPCs from cirrhotic patients showed > 2-fold formation of tube-like closed network circular structures and considerable enhancement of two secretory factors, VEGF and platelet-derived growth factor (PDGF). Moreover, EPCs contribute to the pathogenesis of cirrhosis by stimulating resident LSECs via secretion of PDGF and VEGF. Medina et al[66] evaluated the cellular infiltrate phenotype and indicated VEGF expression in surrounding LSECs. To know more about hepatic sinusoidal angiogenesis during progression to cirrhosis, Xu et al[67] investigated the occurrence of autoantibodies to cell-surface-expressed molecules on LSECs in patients with cirrhosis. They tested the ability of these antibodies to transform LSECs to a vascular type and the reversibility of the transformation process in vitro. A high fraction of primary biliary cirrhosis patients had autoantibodies on LSECs compared to normal controls. These antibodies were capable of transforming LSECs into a vascular type. Furthermore, the antibodies induced cell surface expression of CD31, which may play an important role in transformation-inducing signals produced during cirrhosis. During the cirrhotic process, the LSECs with autoantibodies secreted collagen type IV and laminin, which is a component of basement membrane. In addition, patients with viral hepatitis and non-alcoholic and alcoholic steatohepatitis did not show the presence of these autoantibodies.
Yokomori et al[68] investigated the polymerase 1 and transcript release factor (PTRF) and serum deprivation protein response (SDPR) expression and found that these were up-regulated in small angiogenic LSECs with collagen deposition in the perisinusoidal space. Fixation approaches were designed to re-evaluate the precise ultrastructural localizations and changes of PTRF and SDPR expression on LSECs facing the sinusoidal blood flow. PTRF expression was localized primarily to caveola-like structures and vesicles in Child-Pugh class A cirrhotic liver tissues (Figure 5A and B). Several vacuolar components of LSECs, such as pinocytotic vesicles, lysosomes, Golgi apparatus and endosomes, were not observed. SDPR expression was mainly localized on caveola-like structures and to a lesser extent on vesicles of LSECs. Moreover, PTRF played an important role in regulating aspects of caveolin-1 in LSECs, and there was a direct association of PTRF and SDPR with the process of differentiation. Transformation of LSECs inducing hepatic sinusoidal capillarization was related to the progression of cirrhosis.
Figure 5.
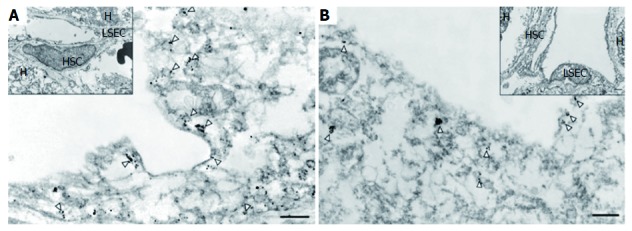
Ultrastructural localization of PTRF and SDPR in cirrhotic tissue. A: Immunogold particles suggesting PTRF expression was observed on caveola-like structures in capillarized, longitudinally cut LSECs; B: Immunogold particles suggesting SDPR expression on caveola-like structures in longitudinally cut LSECs (inset shows lower magnification; white arrowheads indicate caveola-like structures). A-B: ref 66 Copyright @ 2015 Elsevier Ltd. LSECs: Liver sinusoidal endothelial cells; PTRF: Polymerase 1 and transcript release factor; SDPR: Serum deprivation protein response.
In order to know more about the development of cirrhosis, Hollenbach and co-workers[69] analyzed the role of glyoxalase (Glo)-I in LSECs and HSCs from rats with early and advanced cirrhosis. Glo-I was the major detoxifying enzyme for methylglyoxal in cirrhosis. Compared with healthy cells, expression of Glo-I was reduced in early and advanced cirrhosis and methylglyoxal concentration was increased. Rats with advanced cirrhosis had a greater reduction in Glo-I expression than those with early cirrhosis. Protein and mRNA expression of Glo-I in LSECs was less than in HSCs. The reduced expression of Glo-I in cirrhosis leads to increased inflammation, which is more obvious in advanced cirrhosis.
Ex vivo expansion of autologous cells is essential for cell transplantation in patients with cirrhosis. Nakamura et al[70] investigated the efficacy of human ex vivo-expanded CD34+ cells for treatment of cirrhotic rat liver. Compared with nonexpanded CD34+ cells, the expanded cells had an increase in cell surface markers of vascular endothelial cadherin, VEGFR2 and Tie-2. The increased expression of proangiogenic growth factors and adhesion molecules increased hepatocyte and LSEC proliferative activity. Moreover, the transplanted expanded CD34+ cells enhanced the preventive efficacy of cell transplantation in a cirrhotic model.
CONCLUSION
Most of the recent studies have investigated LSECs at a single pathological stage of liver disease, but we have presented a comprehensive, continuous and dynamic observation of changes in the morphology and function of LSECs in hepatitis, liver fibrosis and cirrhosis in this review. By reviewing previous reports, the following conclusions can be drawn. (1) During hepatitis, the number and size of fenestrae in LSECs decrease and a discontinuous basement membrane appears, and the LSECs express anti-inflammatory cytokines and lectin to reduce the liver inflammation. Viruses such as HBV and HCV reach the hepatocytes through active transport across the LSECs; (2) During liver fibrosis, LSECs lose fenestrae and form a continuous basement membrane. The bidirectional exchange of molecules between hepatocytes and hepatic blood sinuses is disrupted due to the defenestration of LSECs. Meanwhile, damaged LSECs lose their ability to degrade ECM and secrete proinflammatory cytokines. Inhibition of LSEC-induced angiogenesis can reduce intrahepatic angiogenesis, thus alleviating liver fibrosis; and (3) during cirrhosis, LSECs form a vascular type with a true basement membrane, which contributes to the development of cirrhotic portal hypertension. Some secretions are important factors in the progression of angiogenesis, such as VEGF, PDGF and unique autoantibodies. At different stages of cirrhosis, the expression of detoxification enzymes in LSECs is decreased as cirrhosis deteriorates and this constitutes a novel target for antifibrotic therapy. Ex vivo expansion of autologous cells enhances the preventive efficacy of cell transplantation in cirrhosis.
As a result of LSECs playing important roles in different liver diseases, drug targeting therapy will be the focus in future research according to the pathological characteristics of LSECs at different stages of liver disease, which may be more effective at improving the fenestrae and basement membrane of LSECs. There are good prospects for new targeted therapy and clinical diagnosis in the area of LSECs for liver diseases.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Supported by the Young Elite Scientists Sponsorship Program by CAST, No. 2016QNRC001 and Beijing Natural Science Foundation, No. 7172187.
Conflict-of-interest statement: The authors declare no conflict of interests related to this article.
Peer-review started: July 24, 2017
First decision: August 28, 2017
Article in press: September 28, 2017
P- Reviewer: Doganay L, Garbuzenko DV, Zhu Y S- Editor: Qi Y L- Editor: Filipodia E- Editor: Huang Y
Contributor Information
Yao Ni, Department of Infection, Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing 100053, China.
Juan-Mei Li, Department of Infection, Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing 100053, China.
Ming-Kun Liu, Department of Infection, Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing 100053, China.
Ting-Ting Zhang, Department of Infection, Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing 100053, China.
Dong-Ping Wang, Department of Infection, Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing 100053, China.
Wen-Hui Zhou, Department of Infection, Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing 100053, China.
Ling-Zi Hu, Department of Infection, Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing 100053, China.
Wen-Liang Lv, Department of Infection, Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing 100053, China. lvwenliang@sohu.com.
References
- 1.Le Bail B, Bioulac-Sage P, Senuita R, Quinton A, Saric J, Balabaud C. Fine structure of hepatic sinusoids and sinusoidal cells in disease. J Electron Microsc Tech. 1990;14:257–282. doi: 10.1002/jemt.1060140307. [DOI] [PubMed] [Google Scholar]
- 2.Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D, Rautou PE. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J Hepatol. 2017;66:212–227. doi: 10.1016/j.jhep.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 3.Breiner KM, Schaller H, Knolle PA. Endothelial cell-mediated uptake of a hepatitis B virus: a new concept of liver targeting of hepatotropic microorganisms. Hepatology. 2001;34:803–808. doi: 10.1053/jhep.2001.27810. [DOI] [PubMed] [Google Scholar]
- 4.Pöhlmann S, Zhang J, Baribaud F, Chen Z, Leslie GJ, Lin G, Granelli-Piperno A, Doms RW, Rice CM, McKeating JA. Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J Virol. 2003;77:4070–4080. doi: 10.1128/JVI.77.7.4070-4080.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broering R, Wu J, Meng Z, Hilgard P, Lu M, Trippler M, Szczeponek A, Gerken G, Schlaak JF. Toll-like receptor-stimulated non-parenchymal liver cells can regulate hepatitis C virus replication. J Hepatol. 2008;48:914–922. doi: 10.1016/j.jhep.2008.01.028. [DOI] [PubMed] [Google Scholar]
- 6.Bissell DM, Wang SS, Jarnagin WR, Roll FJ. Cell-specific expression of transforming growth factor-βin rat liver evidence for autocrine regulation of hepatocyte proliferation. J Clin Invest. 1995;96:447–455. doi: 10.1172/JCI118055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Limmer A, Ohl J, Wingender G, Berg M, Jüngerkes F, Schumak B, Djandji D, Scholz K, Klevenz A, Hegenbarth S, et al. Cross-presentation of oral antigens by liver sinusoidal endothelial cells leads to CD8 T cell tolerance. Eur J Immunol. 2005;35:2970–2981. doi: 10.1002/eji.200526034. [DOI] [PubMed] [Google Scholar]
- 8.Knolle PA, Gerken G. Local control of the immune response in the liver. Immunol Rev. 2000;174:21–34. doi: 10.1034/j.1600-0528.2002.017408.x. [DOI] [PubMed] [Google Scholar]
- 9.Yamazaki T, Joshita S, Umemura T, Usami Y, Sugiura A, Fujimori N, Shibata S, Ichikawa Y, Komatsu M, Matsumoto A, et al. Association of Serum Autotaxin Levels with Liver Fibrosis in Patients with Chronic Hepatitis C. Sci Rep. 2017;7:46705. doi: 10.1038/srep46705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kantari-Mimoun C, Castells M, Klose R, Meinecke AK, Lemberger UJ, Rautou PE, Pinot-Roussel H, Badoual C, Schrödter K, Österreicher CH, et al. Resolution of liver fibrosis requires myeloid cell-driven sinusoidal angiogenesis. Hepatology. 2015;61:2042–2055. doi: 10.1002/hep.27635. [DOI] [PubMed] [Google Scholar]
- 11.Glińska-Suchocka K, Orłowska A, Spużak J, Jankowski M, Kubiak K. Suitability of using serum hialuronic acid concentrations in the diagnosis of canine liver fibrosis. Pol J Vet Sci. 2015;18:873–878. doi: 10.1515/pjvs-2015-0113. [DOI] [PubMed] [Google Scholar]
- 12.DeLeve LD. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology. 2015;61:1740–1746. doi: 10.1002/hep.27376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang F, Zhang Z, Chen L, Kong D, Zhang X, Lu C, Lu Y, Zheng S. Curcumin attenuates angiogenesis in liver fibrosis and inhibits angiogenic properties of hepatic stellate cells. J Cell Mol Med. 2014;18:1392–1406. doi: 10.1111/jcmm.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu M, Wang X, Zou Y, Zhong Y. Key role of liver sinusoidal endothelial cells in liver fibrosis. Biosci Trends. 2017;11:163–168. doi: 10.5582/bst.2017.01007. [DOI] [PubMed] [Google Scholar]
- 15.Zhou WC, Zhang QB, Qiao L. Pathogenesis of liver cirrhosis. World J Gastroenterol. 2014;20:7312–7324. doi: 10.3748/wjg.v20.i23.7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Medina J, Caveda L, Sanz-Cameno P, Arroyo AG, Martín-Vílchez S, Majano PL, García-Buey L, Sánchez-Madrid F, Moreno-Otero R. Hepatocyte growth factor activates endothelial proangiogenic mechanisms relevant in chronic hepatitis C-associated neoangiogenesis. J Hepatol. 2003;38:660–667. doi: 10.1016/s0168-8278(03)00053-9. [DOI] [PubMed] [Google Scholar]
- 17.Iwakiri Y, Groszmann RJ. Vascular endothelial dysfunction in cirrhosis. J Hepatol. 2007;46:927–934. doi: 10.1016/j.jhep.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 18.Iwakiri Y. Endothelial dysfunction in the regulation of cirrhosis and portal hypertension. Liver Int. 2012;32:199–213. doi: 10.1111/j.1478-3231.2011.02579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeLeve LD. Liver sinusoidal endothelial cells and liver regeneration. J Clin Invest. 2013;123:1861–1866. doi: 10.1172/JCI66025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elvevold K, Smedsrød B, Martinez I. The liver sinusoidal endothelial cell: a cell type of controversial and confusing identity. Am J Physiol Gastrointest Liver Physiol. 2008;294:G391–G400. doi: 10.1152/ajpgi.00167.2007. [DOI] [PubMed] [Google Scholar]
- 21.Sørensen KK, Simon-Santamaria J, McCuskey RS, Smedsrød B. Liver Sinusoidal Endothelial Cells. Compr Physiol. 2015;5:1751–1774. doi: 10.1002/cphy.c140078. [DOI] [PubMed] [Google Scholar]
- 22.Braet F, Wisse E. AFM imaging of fenestrated liver sinusoidal endothelial cells. Micron. 2012;43:1252–1258. doi: 10.1016/j.micron.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 23.Xie G, Wang L, Wang X, Wang L, DeLeve LD. Isolation of periportal, midlobular, and centrilobular rat liver sinusoidal endothelial cells enables study of zonated drug toxicity. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1204–G1210. doi: 10.1152/ajpgi.00302.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wisse E, De Zanger RB, Charels K, Van Der Smissen P, McCuskey RS. The liver sieve: considerations concerning the structure and function of endothelial fenestrae, the sinusoidal wall and the space of Disse. Hepatology. 1985;5:683–692. doi: 10.1002/hep.1840050427. [DOI] [PubMed] [Google Scholar]
- 25.O’Reilly JN, Cogger VC, Fraser R, Le Couteur DG. The effect of feeding and fasting on fenestrations in the liver sinusoidal endothelial cell. Pathology. 2010;42:255–258. doi: 10.3109/00313021003636469. [DOI] [PubMed] [Google Scholar]
- 26.Fraser R, Cogger VC, Dobbs B, Jamieson H, Warren A, Hilmer SN, Le Couteur DG. The liver sieve and atherosclerosis. Pathology. 2012;44:181–186. doi: 10.1097/PAT.0b013e328351bcc8. [DOI] [PubMed] [Google Scholar]
- 27.Géraud C, Evdokimov K, Straub BK, Peitsch WK, Demory A, Dörflinger Y, Schledzewski K, Schmieder A, Schemmer P, Augustin HG, et al. Unique cell type-specific junctional complexes in vascular endothelium of human and rat liver sinusoids. PLoS One. 2012;7:e34206. doi: 10.1371/journal.pone.0034206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokomori H. New insights into the dynamics of sinusoidal endothelial fenestrae in liver sinusoidal endothelial cells. Med Mol Morphol. 2008;41:1–4. doi: 10.1007/s00795-007-0390-7. [DOI] [PubMed] [Google Scholar]
- 29.DeLeve LD, Wang X, Hu L, McCuskey MK, McCuskey RS. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am J Physiol Gastrointest Liver Physiol. 2004;287:G757–G763. doi: 10.1152/ajpgi.00017.2004. [DOI] [PubMed] [Google Scholar]
- 30.Stutchfield BM, Forbes SJ. Liver sinusoidal endothelial cells in disease--and for therapy? J Hepatol. 2013;58:178–180. doi: 10.1016/j.jhep.2012.07.046. [DOI] [PubMed] [Google Scholar]
- 31.Wohlleber D, Knolle PA. The role of liver sinusoidal cells in local hepatic immune surveillance. Clinical & Translational immunology. 2016;5:e117. doi: 10.1038/cti.2016.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knolle PA, Wohlleber D. Immunological functions of liver sinusoidal endothelial cells. Cell Mol Immunol. 2016;13:347–353. doi: 10.1038/cmi.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knolle PA, Löser E, Protzer U, Duchmann R, Schmitt E, zum Büschenfelde KH, Rose-John S, Gerken G. Regulation of endotoxin-induced IL-6 production in liver sinusoidal endothelial cells and Kupffer cells by IL-10. Clin Exp Immunol. 1997;107:555–561. doi: 10.1046/j.1365-2249.1997.d01-959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu J, Lu M, Roggendorf M, Gerken G, Schlaak JF. TLR-mediated control of HBV replication by non-parenchymal liver cells. Zeitschrift Für Gastroenterologie. 2007;45:1–10. [Google Scholar]
- 35.Sørensen KK, McCourt P, Berg T, Crossley C, Le Couteur D, Wake K, Smedsrød B. The scavenger endothelial cell: a new player in homeostasis and immunity. Am J Physiol Regul Integr Comp Physiol. 2012;303:R1217–R1230. doi: 10.1152/ajpregu.00686.2011. [DOI] [PubMed] [Google Scholar]
- 36.Ganesan LP, Mohanty S, Kim J, Clark KR, Robinson JM, Anderson CL. Rapid and efficient clearance of blood-borne virus by liver sinusoidal endothelium. PLoS Pathog. 2011;7:e1002281. doi: 10.1371/journal.ppat.1002281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parent R, Durantel D, Lahlali T, Sallé A, Plissonnier ML, DaCosta D, Lesca G, Zoulim F, Marion MJ, Bartosch B. An immortalized human liver endothelial sinusoidal cell line for the study of the pathobiology of the liver endothelium. Biochem Biophys Res Commun. 2014;450:7–12. doi: 10.1016/j.bbrc.2014.05.038. [DOI] [PubMed] [Google Scholar]
- 38.Limmer A, Sacher T, Alferink J, Kretschmar M, Schönrich G, Nichterlein T, Arnold B, Hämmerling GJ. Failure to induce organ-specific autoimmunity by breaking of tolerance: importance of the microenvironment. Eur J Immunol. 1998;28:2395–2406. doi: 10.1002/(SICI)1521-4141(199808)28:08<2395::AID-IMMU2395>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 39.Guidotti LG, Borrow P, Brown A, McClary H, Koch R, Chisari FV. Noncytopathic clearance of lymphocytic choriomeningitis virus from the hepatocyte. J Exp Med. 1999;189:1555–1564. doi: 10.1084/jem.189.10.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamada Y, Sato M, Kida S, Akita M, Mizutani K, Fujii H, Sobajima T, Yoshida Y, Shinzaki S, Takamatsu S, et al. N-acetylglucosaminyltransferase V exacerbates concanavalin A-induced hepatitis in mice. Mol Med Rep. 2015;11:3573–3584. doi: 10.3892/mmr.2015.3168. [DOI] [PubMed] [Google Scholar]
- 41.Bleau C, Filliol A, Samson M, Lamontagne L. Mouse Hepatitis Virus Infection Induces a Toll-Like Receptor 2-Dependent Activation of Inflammatory Functions in Liver Sinusoidal Endothelial Cells during Acute Hepatitis. J Virol. 2016;90:9096–9113. doi: 10.1128/JVI.01069-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boonstra A, Woltman AM, Janssen HL. Immunology of hepatitis B and hepatitis C virus infections. Best Pract Res Clin Gastroenterol. 2008;22:1049–1061. doi: 10.1016/j.bpg.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 43.Liu B, Wang M, Wang X, Zhao D, Liu D, Liu J, Chen PJ, Yang D, He F, Tang L. Liver sinusoidal endothelial cell lectin inhibits CTL-dependent virus clearance in mouse models of viral hepatitis. J Immunol. 2013;190:4185–4195. doi: 10.4049/jimmunol.1203091. [DOI] [PubMed] [Google Scholar]
- 44.Natarajan V, Harris EN, Kidambi S. SECs (Sinusoidal Endothelial Cells), Liver Microenvironment, and Fibrosis. Biomed Res Int. 2017;2017:4097205. doi: 10.1155/2017/4097205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baranova A, Lal P, Birerdinc A, Younossi ZM. Non-invasive markers for hepatic fibrosis. BMC Gastroenterol. 2011;11:91. doi: 10.1186/1471-230X-11-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bartneck M, Warzecha KT, Tacke F. Therapeutic targeting of liver inflammation and fibrosis by nanomedicine. Hepatobiliary Surg Nutr. 2014;3:364–376. doi: 10.3978/j.issn.2304-3881.2014.11.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su TH, Kao JH, Liu CJ. Molecular mechanism and treatment of viral hepatitis-related liver fibrosis. Int J Mol Sci. 2014;15:10578–10604. doi: 10.3390/ijms150610578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Enomoto K, Nishikawa Y, Omori Y, Tokairin T, Yoshida M, Ohi N, Nishimura T, Yamamoto Y, Li Q. Cell biology and pathology of liver sinusoidal endothelial cells. Med Electron Microsc. 2004;37:208–215. doi: 10.1007/s00795-004-0261-4. [DOI] [PubMed] [Google Scholar]
- 49.Walsh KM, Fletcher A, MacSween RN, Morris AJ. Basement membrane peptides as markers of liver disease in chronic hepatitis C. J Hepatol. 2000;32:325–330. doi: 10.1016/s0168-8278(00)80079-3. [DOI] [PubMed] [Google Scholar]
- 50.Svistounov D, Zykova SN, Cogger VC, Warren A, Fraser R, Smedsrod B, McCuskey RS, Couteur DGL. 2011. Pseudocapillarization and the aging liver. Vascular Liver Disease. Springer New York; pp. 41–50. [Google Scholar]
- 51.Bocca C, Novo E, Miglietta A, Parola M. Angiogenesis and Fibrogenesis in Chronic Liver Diseases. Cell Mol Gastroenterol Hepatol. 2015;1:477–488. doi: 10.1016/j.jcmgh.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu WL, Li JM, Yang J, Xu CG, Zhang SS, Yan J, Zhang TT, Zhao HH. Effects of Astragalus polysaccharide on mechanical characterization of liver sinusoidal endothelial cells by atomic force microscopy at nanoscale. Chin J Integr Med. 2017 doi: 10.1007/s11655-017-2964-0. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 53.Kaur S, Anita K. Angiogenesis in liver regeneration and fibrosis: “a double-edged sword”. Hepatol Int. 2013;7:959–968. doi: 10.1007/s12072-013-9483-7. [DOI] [PubMed] [Google Scholar]
- 54.Yan Z, Qu K, Zhang J, Huang Q, Qu P, Xu X, Yuan P, Huang X, Shao Y, Liu C, et al. CD147 promotes liver fibrosis progression via VEGF-A/VEGFR2 signalling-mediated cross-talk between hepatocytes and sinusoidal endothelial cells. Clin Sci (Lond) 2015;129:699–710. doi: 10.1042/CS20140823. [DOI] [PubMed] [Google Scholar]
- 55.Zhao S, Zhang Z, Yao Z, Shao J, Chen A, Zhang F, Zheng S. Tetramethylpyrazine attenuates sinusoidal angiogenesis via inhibition of hedgehog signaling in liver fibrosis. IUBMB Life. 2017;69:115–127. doi: 10.1002/iub.1598. [DOI] [PubMed] [Google Scholar]
- 56.Öztürk Akcora B, Storm G, Prakash J, Bansal R. Tyrosine kinase inhibitor BIBF1120 ameliorates inflammation, angiogenesis and fibrosis in CCl4-induced liver fibrogenesis mouse model. Sci Rep. 2017;7:44545. doi: 10.1038/srep44545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hennenberg M, Trebicka J, Kohistani Z, Stark C, Nischalke HD, Krämer B, Körner C, Klein S, Granzow M, Fischer HP, et al. Hepatic and HSC-specific sorafenib effects in rats with established secondary biliary cirrhosis. Lab Invest. 2011;91:241–251. doi: 10.1038/labinvest.2010.148. [DOI] [PubMed] [Google Scholar]
- 58.Liu C, Yang Z, Wang L, Lu Y, Tang B, Miao H, Xu Q, Chen X. Combination of sorafenib and gadolinium chloride (GdCl3) attenuates dimethylnitrosamine(DMN)-induced liver fibrosis in rats. BMC Gastroenterol. 2015;15:159. doi: 10.1186/s12876-015-0380-5. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Manavski Y, Abel T, Hu J, Kleinlützum D, Buchholz CJ, Belz C, Augustin HG, Boon RA, Dimmeler S. Endothelial transcription factor KLF2 negatively regulates liver regeneration via induction of activin A. Proc Natl Acad Sci U S A. 2017;114:3993–3998. doi: 10.1073/pnas.1613392114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ding BS, Nolan DJ, Butler JM, James D, Babazadeh AO, Rosenwaks Z, Mittal V, Kobayashi H, Shido K, Lyden D, et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature. 2010;468:310–315. doi: 10.1038/nature09493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ding BS, Liu CH, Sun Y, Chen Y, Swendeman SL, Jung B, Chavez D, Cao Z, Christoffersen C, Nielsen LB, et al. HDL activation of endothelial sphingosine-1-phosphate receptor-1 (S1P1) promotes regeneration and suppresses fibrosis in the liver. JCI Insight. 2016;1:e87058. doi: 10.1172/jci.insight.87058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garbuzenko DV, Arefyev NO, Belov DV. Mechanisms of adaptation of the hepatic vasculature to the deteriorating conditions of blood circulation in liver cirrhosis. World J Hepatol. 2016;8:665–672. doi: 10.4254/wjh.v8.i16.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iwakiri Y, Shah V, Rockey DC. Vascular pathobiology in chronic liver disease and cirrhosis - current status and future directions. J Hepatol. 2014;61:912–924. doi: 10.1016/j.jhep.2014.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Novo E, Cannito S, Zamara E, Valfrè di Bonzo L, Caligiuri A, Cravanzola C, Compagnone A, Colombatto S, Marra F, Pinzani M, et al. Proangiogenic cytokines as hypoxia-dependent factors stimulating migration of human hepatic stellate cells. Am J Pathol. 2007;170:1942–1953. doi: 10.2353/ajpath.2007.060887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kaur S, Tripathi D, Dongre K, Garg V, Rooge S, Mukopadhyay A, Sakhuja P, Sarin SK. Increased number and function of endothelial progenitor cells stimulate angiogenesis by resident liver sinusoidal endothelial cells (SECs) in cirrhosis through paracrine factors. J Hepatol. 2012;57:1193–1198. doi: 10.1016/j.jhep.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 66.Medina J, Sanz-Cameno P, García-Buey L, Martín-Vílchez S, López-Cabrera M, Moreno-Otero R. Evidence of angiogenesis in primary biliary cirrhosis: an immunohistochemical descriptive study. J Hepatol. 2005;42:124–131. doi: 10.1016/j.jhep.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 67.Xu B, Broome U, Uzunel M, Nava S, Ge X, Kumagai-Braesch M, Hultenby K, Christensson B, Ericzon BG, Holgersson J, et al. Capillarization of hepatic sinusoid by liver endothelial cell-reactive autoantibodies in patients with cirrhosis and chronic hepatitis. Am J Pathol. 2003;163:1275–1289. doi: 10.1016/S0002-9440(10)63487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yokomori H, Ando W, Yoshimura K, Yamazaki H, Takahashi Y, Oda M. Increases in endothelial caveolin-1 and cavins correlate with cirrhosis progression. Micron. 2015;76:52–61. doi: 10.1016/j.micron.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 69.Hollenbach M, Thonig A, Pohl S, Ripoll C, Michel M, Zipprich A. Expression of glyoxalase-I is reduced in cirrhotic livers: A possible mechanism in the development of cirrhosis. PLoS One. 2017;12:e0171260. doi: 10.1371/journal.pone.0171260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakamura T, Koga H, Iwamoto H, Tsutsumi V, Imamura Y, Naitou M, Masuda A, Ikezono Y, Abe M, Wada F, et al. Ex vivo expansion of circulating CD34(+) cells enhances the regenerative effect on rat liver cirrhosis. Mol Ther Methods Clin Dev. 2016;3:16025. doi: 10.1038/mtm.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]