

Abstract
The stem cell factor (SCF) is a cytokine that specifically binds the tyrosine kinase receptor c-KIT. The SCF/c-KIT interaction leads to receptor dimerization, activation of kinase activity and initiation of several signal transduction pathways that control cell proliferation, apoptosis, differentiation and migration in several tissues. The activity of SCF/c-KIT system is linked with the phosphatidylinositol 3-kinase (PI3-K), the Src, the Janus kinase/signal transducers and activators of transcription (JAK/STAT), the phospholipase-C (PLC-γ) and the mitogen-activated protein kinase (MAPK) pathways. Moreover, it has been reported that cancer cases display an overactivation of c-KIT due to the presence of gain-of-function mutations or receptor overexpression, which renders c-KIT a tempting target for cancer treatment. In the case of male cancers the most documented activated pathways are the PI3-K and Src, both enhancing abnormal cell proliferation. It is also known that the Src activity in prostate cancer cases depends on the presence of tr-KIT, the cytoplasmic truncated variant of c-KIT that is specifically expressed in tumour tissues and, thus, a very interesting target for drug development. The present review provides an overview of the signalling pathways activated by SCF/c-KIT and discusses the potential application of c-KIT inhibitors for treatment of testicular and prostatic cancers.
Keywords: C-KIT, KIT ligand, Prostate cancer, SCF, Signalling, Testicular cancer
Introduction
The c-KIT is a tyrosine kinase receptor belonging to the type III receptor tyrosine kinase (RTK) family, which has the stem cell factor (SCF) as its specific ligand. The SCF/c-KIT interaction triggers several signal transduction pathways that regulate fundamental biological processes, such as apoptosis, cell proliferation, differentiation and migration (Ronnstrand 2004).
Despite the important function of SCF and c-KIT in healthy tissues, namely in the control of gametogenesis, melanogenesis, and haematopoiesis (Cardoso et al. 2014; Figueira et al. 2014) it has been shown that the SCF/c-KIT system is associated with the development and progression of human cancers (Cardoso et al. 2014). Gain-of-function mutations in the c-KIT receptor and/or c-KIT overexpression have been related to the onset and progression of several types of tumours (Ali and Ali 2007; Ashman and Griffith 2013; Capelli et al. 2016; Di Lorenzo et al. 2004; Mitchell et al. 2017), which have placed c-KIT on the road of the anti-cancer therapy. Currently, c-KIT inhibitors interfering with c-KIT signal transduction pathways are being used for treatment of leukemias and gastrointestinal tumours (Ashman and Griffith 2013), and under evaluation for other oncological diseases.
The testicular and prostatic cancers are the emblematic representatives of male gender malignancies, for which increasing incidence has been reported worldwide in the recent decades (Ferlay et al. 2013). Prostate cancer is a common concern of aging male and age is its most recognized risk factor, whereas testicular cancer is a disease of young and middle-aged men (Damaschke et al. 2013; Filippou et al. 2016; Hayes-Lattin and Nichols 2009). Nevertheless, these urological cancers are commonly influenced by a variety of genetic, epigenetic, and environmental factors (Wu et al. 2016), and for both prostate and testicular cancers, the improvement of treatment strategies and better disease management are clearly warranted. For prostate cancer, the situation is even more urgent considering the progression of disease to advanced stages, with a great complexity of androgen-dependent and -independent signalling mechanisms and for which limited therapeutic options exist (Farooqi and Sarkar 2015). The present review summarizes the current knowledge concerning the expression of c-KIT and the SCF/c-KIT activated pathways in testicular and prostate cancer cases, and discusses the perspectives of treatment of these oncological diseases targeting the c-KIT receptor.
C-KIT receptor in the context of RTKs family
The RTKs are important regulators of intracellular signal-transduction pathways playing a crucial role in the control of cell fate decisions. The human RTK superfamily consists of 58 proteins grouped into 20 sub-families (Fig. 1) that share structural features and some functional roles (Blume-Jensen and Hunter 2001). The families are the ErbB / EGFR, epidermal growth factor receptor; InsR, insulin receptor; PDGFR, platelet-derived growth factor receptor; VEGFR, vascular endothelial growth factor receptor; FGFR, fibroblast growth factor receptor; KLG/CCK, colon carcinoma kinase; NGFR, nerve growth factor receptor; HGFR, hepatocyte growth factor receptor; EphR, ephrin receptor; Axl, a Tyro3 PTK; Tie, tyrosine kinase receptor in endothelial cells; Ryk, receptor related to tyrosine kinases; DDR, discoidin domain receptor; Ret, rearranged during transfection; Ros, RPTK expressed in some epithelial cell types; LTK, leukocyte tyrosine kinase; Ror, receptor orphan; MuSK, muscle-specific kinase; LMR, Lemur and STYK1, serine, threonine, tyrosine kinase 1 (Blume-Jensen and Hunter 2001; Lemmon and Schlessinger 2010; Robinson et al. 2000). The EGFR, InsR and PDGFR families also are widely known as class I, class II and class III RTKs, respectively.
Fig. 1.

General structure of RTKs families. The extracellular, transmembrane and intracellular structural domains of the 20 families of human RTKs are represented schematically. Green cylinders represent the intracellular regions containing the kinase domains and purple rectangles show the transmembrane region. The main structural differences between all RTKs classes are located in the extracellular domain and confer ligand specificity. The classical EGFR, InsR and PDGFR families are also known as class I, class II and class III RTKs, respectively. c-KIT belongs to the class III, PDGFR family.  - L;
- L;  - cysteine-rich;
- cysteine-rich;  - fibronectin type III;
- fibronectin type III;  - immunoglobulin domain;
- immunoglobulin domain; 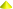 - acid box;
- acid box;  - leucine-rich;
- leucine-rich;  - sema;
- sema;  - psi;
- psi;  - ephrin binding domain;
- ephrin binding domain;  - EGF;
- EGF;  - WIF;
- WIF;  - discoidin;
- discoidin;  - cadherin;
- cadherin;  - YWTD propeller;
- YWTD propeller;  - mam domain;
- mam domain;  - Ldla;
- Ldla;  - fz;
- fz;  - kringle
- kringle
The general structure of RTKs (Fig. 1) encompasses the ligand binding domain in the glycosylated N-terminal extracellular region, a single transmembrane helix, and a cytoplasmic region that contains the tyrosine kinase domain, and C-terminal and juxtamembrane regulatory regions (Lemmon and Schlessinger 2010). The intracellular juxtamembrane region and the C-terminal tail differ in size and tyrosine content among family members, which creates differences in intracellular signalization. Nevertheless, RTKs differ mainly by the extracellular domains (Fig. 1) that display specific molecular features (immunoglobulin domains, cysteine, and leucine-rich domains, fibronectin type III domain, among others) in the distinct receptor families (Maruyama 2014; Segaliny et al. 2015).
The c-KIT belongs to the PDGFR family (Class III), that besides PDGFRα and PDGFRβ also includes the colony-stimulating factor 1 receptor (CSF1R) and the Fms-like tyrosine kinase 3 receptor (FLT3) (Verstraete and Savvides 2012).
Brief overview of the molecular biology of SCF/c-KIT system
C-KIT receptor
The c-KIT receptor was first described in 1986 as the transforming gene of the Hardy-Zuckerman 4 feline sarcoma virus and identified as the proto-oncogene v-KIT (Yarden et al. 1987). CD117, SCF receptor or KIT receptor are other common designations for c-KIT (Yarden et al. 1987). The main product of c-KIT gene is a single 5 kb transcript encoding a transmembrane glycoprotein with approximately 145–160 kDa that belongs to the type III RTK family (Yarden et al. 1987). This class of receptors is structurally characterized by the presence of three main functional regions (Mol et al. 2003) (Fig. 1): an intracellular domain, containing proximal and distal kinase domains separated by an interkinase domain, that is involved in signalling transduction; a transmembrane region constituted by a short hydrophobic chain of amino acids that anchors c-KIT at cell membrane; and an extracellular domain comprising five immunoglobulin-like domains, which participate in recognition of c-KIT ligand and receptor dimerization.
Distinct c-KIT protein variants have been identified over the years (Fig. 2). The use of an alternative 5′-donor splice site produces c-KIT isoforms that differ by the presence or absence of the tetrapeptide Gly-Asn-Asn-Lys (GNNK) in the juxtamembrane region of the extracellular domain (Caruana et al. 1999). Recently, it was demonstrated that the GNNK peptide is an important regulatory element for fine tuning receptor activation and downstream signalling since GNNK-negative c-KIT variants displayed increased tyrosine phosphorylation and activity (Phung et al. 2013). In other words, the juxtamembrane region by the presence of GNNK peptide acts as a negative regulator of c-KIT activity.
Fig. 2.

Structure of SCF/c-KIT proteins and downstream signalling pathways. Membrane-bound SCF (mSCF) contains an extracellular domain (red) that is responsible for recognizing and binding to c-KIT, a transmembrane domain (orange), and an intracellular domain (dark green). SCF also exists as a soluble protein (sSCF) secreted to the extracellular space. The five immunoglobulin-like domains (dark blue) in the extracellular domain of c-KIT are involved in ligand-binding and receptor dimerization. The transmembrane domain (violet) anchors c-KIT at the cytoplasm membrane and the intracellular region (green) is responsible for signal transduction. The receptor can be proteolytically cleaved and released from cell membrane, giving rise to a soluble c-KIT (s-KIT) consisting only of the extracellular domain. A truncated form of c-KIT (tr-KIT) residing at cytoplasm lacks the extracellular and transmembrane domains but retains part of the kinase domain. Binding of SCF homodimer to c-KIT induces receptor dimerization and auto-phosphorylation, and consequent activation of downstream signalling cascades. The cell survival PI3K/Akt pathway (dark blue) depends on the phosphorylation of p70 S6 kinase (p70S6K) via mammalian target of rapamycin (mTOR), which leads to increased expression of cyclin D3 and phosphorylation of retinoblastoma protein (Rb). SCF/c-KIT/PI3K/Akt signalling also contributes to cell survival by inactivation of the pro-apoptotic factor Bad. The Src pathway (pink) controls proliferation, cell-cycle progression, and migration by distinct actions of Akt, Fyn, and Lyn kinases. c-KIT activates PLC-γ (orange), that can also be activated by tr-KIT through SRC member Fyn, which induces cell proliferation. SCF/c-KIT signalling has also been linked with the activity of JAK/STAT pathway (purple) through the activation of JAK2 and consequent phosphorylation of STAT transcriptional regulators, which enhance proliferative activity. Activation of the MAPK pathway (light green) occurs upon binding of adaptor proteins, such as growth-factor receptor-bound protein-2 (Grb2) and guanine nucleotide exchange factor, son of sevenless (SOS), which determines the activation of the small G-protein RAS and the propagation of kinases signalling cascade
A mechanism of alternative promoter usage originates a 30–50 kDa truncated c-KIT protein (tr-KIT, Fig. 2) that lacks the extracellular domain and the transmembrane region (Rossi et al. 1992). tr-KIT is also devoid of the first part of the kinase domain (Fig. 2) and, thus, do not display kinase activity (Rossi et al. 1992). However, tr-KIT seems to retain signalling transduction capability by interacting with other RTKs, or with other receptor types (Sette et al. 1998) as a scaffold protein at the cytoplasm.
Proteolytic cleavage of c-KIT releases the receptor from the cell membrane given rise to a soluble isoform (Fig. 2) that only contains the extracellular domain. This protein variant binds SCF with the same affinity as the full-length c-KIT, and it was suggested that it might play a role modulating the bioactivity of ligand (Dahlen et al. 2001).
SCF
The c-KIT ligand, the cytokine SCF also known as steel factor or mast cell growth factor, is a potent growth factor firstly identified in 1990 (Williams et al. 1990). The SCF gene encodes a 45 kDa glycoprotein predominantly located at plasma membrane (Mansuroglu et al. 2009).
The SCF protein contains three distinct regions (Fig. 2): the intracellular domain, the hydrophobic transmembrane domain, and the extracellular domain responsible for recognizing and binding c-KIT (Langley et al. 1994). Besides the full-length membrane-bound SCF (mSCF), soluble forms of SCF have also been identified (Fig. 2). The proteolytic cleavage of an alternative spliced variant originates a soluble SCF (sSCF) that also binds and activates c-KIT. However, sSCF promotes transient activation and faster degradation of c-KIT whereas mSCF induces persistent activity and prolongs the life span of receptor (Miyazawa et al. 1995).
Generalities of c-KIT activation by SCF
SCF is a noncovalent homodimer composed of two protomers; an hydrophobic crevice with a charged region on the tail of each protomer functions as the potential receptor-binding site (Zhang et al. 2000). Thus, SCF binds simultaneously two molecules of c-KIT, inducing a conformational change that exposes a key dimerization site located in the fourth immunoglobulin-like domain of c-KIT (Lemmon et al. 1997). Receptor dimerization allows its autophosphorylation (Paulhe et al. 2009), and triggers the initiation of multiple signal transduction pathways (Ali and Ali 2007; Mol et al. 2003), namely, the phosphatidylinositol 3-kinase (PI3-K), the Src, the Janus kinase/signal transducers and activators of transcription (JAK/STAT), the phospholipase-C (PLC-γ) and the mitogen-activated protein kinase (MAPK). The physiological actions of c-KIT controlling cell survival, proliferation, differentiation, and migration depend on the activation of specific or overlapping pathways (Ronnstrand 2004) (Fig. 2), which endows the activity of SCF/c-KIT system of a great complexity. Disclosure of the c-KIT activated pathways in carcinogenesis will be a crucial step towards the development of c-KIT targeted therapies.
SCF/c-KIT signalling pathways
The PI3-K pathway
PI3-K heterodimer is one of the major pro-survival pathways influencing cell fate in a variety of tissues. The PI3-K regulatory subunit p85 contains two Src homology 2 (SH2) domains (Klippel et al. 1994) that are responsible for the interaction with c-KIT. Genetically modified mice lacking the p85 subunit of PI3-K displayed a dramatic reduction in the proliferative effects of SCF/c-KIT, which demonstrates the involvement of PI3-K downstream signal pathway (Fukao et al. 2002). The Tyr719 and Tyr821 residues in the interkinase domain of c-KIT are involved in PI3-K activation (Serve et al. 1994; Serve et al. 1995). PI3-K can also be indirectly activated by c-KIT through its binding to the tyrosine phosphorylated adaptor protein GAB2 (Nishida et al. 2002).
PI3-K activation in response to c-KIT is followed by the phosphorylation of downstream signalling molecules in the PI3-K cascade (Fig. 2), as is the case of cell survival regulator Akt (Nakai et al. 2005). Akt seems to mediate SCF/c-KIT/PI3-K signalling by phosphorylating the p70 S6 kinase (p70S6K) via mammalian target of rapamycin (mTOR) kinase (Feng et al. 2000). Phosphorylation of p70S6K leads to increased expression of cyclin D3 and phosphorylation of retinoblastoma (Rb) protein (Feng et al. 2000), allowing cell cycle progression and cell division. On the other hand, inhibition of mTOR by rapamycin prevented the SCF/c-KIT effects controlling cell survival and proliferation, cell adhesion, cytokine production and chemotaxis (Blume-Jensen et al. 1998; Feng et al. 2000).
The pro-apoptotic factor Bad is also a target of Akt upon SCF activation of the PI3-K pathway. Phosphorylation of Bad by Akt leads to its inactivation and, thus, contributes to cell survival (Blume-Jensen et al. 1998).
c-KIT mutations in the juxtamembrane and kinase domain (Table 1) have been described in human germ-cell tumours, the most common type of testicular cancer. All the mutations in the kinase domain constitutively activate the receptor (Table 1) and promote its association with PI3-K (Kemmer et al. 2004; Nakai et al. 2005; Tian et al. 1999). In addition, c-KIT activity in seminomas also phosphorylates Akt via PI3-K, thereby promoting the progression of germ-cell neoplasia (Nakai et al. 2005). Germ-cell tumours also appear to be enriched for expression of the GNNK-negative isoforms of c-KIT (Sakuma et al. 2003). These c-KIT variants lacking the GNNK tetrapeptide are strongly activated upon SCF binding and display pronounced signalling effects, namely, increased c-KIT phosphorylation and enhanced activation of MAP kinase protein ERK (Montero et al. 2008; Phung et al. 2013). Thus, blocking c-KIT activity would be a valuable strategy to treat testicular cancer, namely seminomas, since non-seminomas rarely express c-KIT (Izquierdo et al. 1995). At the present moment, the information concerning c-KIT inactivation in testicular cancer is very limited, but imatinib mesylate, the first tyrosine kinase inhibitor used as a chemotherapeutic drug, has shown beneficial effects. A patient with disseminated testicular seminoma that was refractory to salvage chemotherapy had complete remission after administration of imatinib for 3 months (400 mg/day) (Pedersini et al. 2007). However, all the c-KIT mutations identified in germ-cell tumours (Table 1) have been indicated as resistant to imatinib, which is apparently explained by the capability of imatinib inhibiting kinase activity only in its inactive conformation (Lennartsson and Ronnstrand 2012; Todd et al. 2013). Therefore, it would be important to screen patients for the presence of imatinib-sensitive mutations before embracing for this therapy. On the other hand, since the described oncogenic mutations of c-KIT (Table 1) have shown to be insensitive to imatinib but sensitive to the second-generation tyrosine kinase inhibitor dasatinib, which binds to the kinase domains in its active conformation (Lennartsson and Ronnstrand 2012), it is warrantable to test dasatinib for treatment of germ-cell tumours.
Table 1.
c-KIT mutations identified in germ-cell tumours
| Amino acid position | Amino acid change | Structural Region | Functional Effect* | References |
|---|---|---|---|---|
| 557 | W557R | Juxtamembrane domain | ? | (Coffey et al. 2008; Sakuma et al. 2003) |
| 814 | D814Y | Kinase domain | Enhanced tyrosine kinase activity | (Piao and Bernstein 1996; Schnabel et al. 2005) |
| 816 | D816H, D816V or D816Y | Kinase domain | Enhanced tyrosine kinase activity | (Biermann et al. 2007; Rapley et al. 2004; Willmore-Payne et al. 2006) |
| 820 | D820G | Kinase domain | Enhanced tyrosine kinase activity | (Rapley et al. 2004; Willmore-Payne et al. 2006) |
| 821 | S821F | Kinase domain | Enhanced tyrosine kinase activity | (Biermann et al. 2007) |
| 822 | N822 K | Kinase domain | Enhanced tyrosine kinase activity | (Biermann et al. 2007) |
| 823 | Y823D | Kinase domain | Enhanced tyrosine kinase activity | (Biermann et al. 2007; Willmore-Payne et al. 2006) |
*- all the identified mutations were reported to be insensitive to tyrosine kinase inhibitor imatinib;? – The exact consequence of c-KIT mutations in the juxtamembrane domain has not been demonstrated yet, but since this region is a negative regulator of c-KIT kinase activity (Phung et al. 2013) it is expected that they also could constitutively activate receptor
The Src pathway
Studies in different types of cells have demonstrated that the SCF/c-KIT system activates several Src family members (Fig. 2), such as Src, Lyn and Fyn (Linnekin et al. 1997a; Samayawardhena et al. 2007). Src kinases interact with Tyr568 and Tyr570 in the juxtamembrane domain of c-KIT, but only phosphorylation of Tyr568 seems to be required for the activation of Src family members (Price et al. 1997). Moreover, c-KIT isoforms negative for the GNNK sequence in the juxtamembrane domain display stronger activation of Src members than GNNK-positive isoforms (Voytyuk et al. 2003).
The activation of Src pathway is associated with the SCF/c-KIT actions promoting cell proliferation. It was demonstrated that SCF activates Lyn before the G1 to S phase transition (Mou and Linnekin 1999). Lyn activation by SCF triggered cell cycle progression by increasing the activity of cyclin-dependent kinase 2 (Cdk2) and phosphorylation of Rb protein. Moreover, Src inhibitors caused a strong reduction in Akt phosphorylation in response to c-KIT signalling (De Miguel et al. 2002; Farini et al. 2007), which demonstrated that the activation of Akt survival pathway can also be mediated by the Src pathway. The alternative activation of Akt pathway via PI3-K or Src family members seems to be cell specific (De Miguel et al. 2002).
c-KIT signal transduction triggering the Src pathway is also important in the regulation of cell migration. Experiments in cells with deficient forms of Lyn or the use of Src family kinase inhibitors provided evidence for the involvement of the Src pathway in SCF-dependent migratory response (Farini et al. 2007; O'Laughlin-Bunner et al. 2001).
The activity of Src family members has been closely associated with the action of tr-KIT. The tr-KIT is a stronger activator of Src kinases, comparatively with the full-length protein (Paronetto et al. 2004; Paronetto et al. 2003). Higher levels of Src activity were found in prostate cancer cells and tissues expressing the tr-KIT (Paronetto et al. 2004). It is known that Src activity leads to the phosphorylation of the RNA-binding protein Sam68, and that this event is linked to the neoplastic transformation of prostate cells (Derry et al. 2003). Interestingly, Sam68 phosphorylation is only detected in prostate tumours expressing the tr-Kit (Paronetto et al. 2004). Moreover, the expression of tr-KIT seems to be tumour specific and markedly increases with the progression of prostate cancer (Paronetto et al. 2004), which renders tr-KIT a pharmacological target.
The tyrosine kinase inhibitor imatinib blocks the activity of the full-length c-KIT but has no effect on the tr-KIT, which may explain the little efficacy this drug has been showing in prostate cancer treatment, as well as, the inconsistency between results of in vitro experiments and clinical findings (Corcoran and Costello 2005; Tiffany et al. 2004). Several reports have shown the growth inhibitory actions of imatinib alone or in combination with other cytotoxic drugs in distinct cell line models of prostate cancer, namely, in androgen-sensitive (LNCaP) and castration-resistant models (DU145 and PC3) (Huang et al. 2012; Pinto et al. 2011). However, these findings failed to be translated into the clinical setting with imatinib treatment showing only modest efficacy (Corcoran and Costello 2005; Lipton et al. 2010; Mathew et al. 2004; Nabhan et al. 2012; Tiffany et al. 2004). Moreover, we recently showed that imatinib significantly decreased the viability of DU145 cells whereas augmented the survival of PC3 cells, which was accompanied by a distinct expression pattern of apoptosis regulators (Cardoso et al. 2015). Also, an angiogenic factor, the vascular endothelial growth factor (VEGF) displayed a distinct response to imatinib in DU145 and PC3 cells. Imatinib treatment diminished the expression levels of VEGF in DU145 cells whereas an opposite effect was seen in PC3 (Cardoso et al. 2015). These opposed effects of imatinib in distinct cell lines may contribute explaining the lack of efficacy of this anti-cancer drug controlling prostate tumour’s growth and raise the concern about stimulation and progression of metastasis when applying imatinib treatment in prostate cancer patients. Ultimately, the expression levels of c-KIT and tr-KIT in DU145 and PC3 cells consubstantiate the distinct effects observed in response to imatinib, with PC3 cells displaying diminished expression of the full-length c-KIT and increased expression of tr-KIT (Cardoso et al. 2015). Overall, the current knowledge and the findings described above strongly stimulate future research aiming at discovering drugs that specifically block the tr-KIT.
The JAK/STAT pathway
The Janus kinases (JAKs) are cytoplasmic tyrosine kinases activated by ligand, which lead to phosphorylation and transcription of signal transducer and activation of transcription (STAT) proteins (Kerr et al. 2003). After being phosphorylated, STATs dimerize and are translocated to the nucleus, where they regulate the transcription of target genes (Kerr et al. 2003). Signal transduction by SCF and its receptor was shown to be related to the JAK/STAT pathway (Fig. 2) by the activation of JAK2 (Deberry et al. 1997; Linnekin et al. 1997b). SCF induced the association of c-KIT with JAK2 and the consequent phosphorylation of JAK2 (Weiler et al. 1996). The transient activation of JAK2 in response to SCF was associated with the downstream activation of STAT1 and STAT5 (Deberry et al. 1997; Ryan et al. 1997). Moreover, SCF also increased the expression of JAK2 and STAT1 (Imura et al. 2012). It was also observed an increase in differentiation but not in the self-renewal of stem cells in the testis of the fruit fly, Drosophila sp., in the absence of JAK/STAT signalling (Tulina and Matunis 2001). Thus, the activation of JAK2/STAT1 pathway seems to underpin the SCF/c-KIT actions regulating stem cell proliferation. Although the enhanced renewal of testicular stem cells has been linked with the development of germ-cell tumours (Chieffi 2014), the involvement of SCF/c-KIT and JAK/STAT signalling in testicular carcinogenesis remains to be clarified.
In the case of prostate cancer, the idea that a population of “cancer stem cells” implicated in the ontogeny of disease and therapeutic resistance also has been gaining consistency in the last years (Lin et al. 2016b; Yun et al. 2016). These cancer stem cells seem to be associated with the basal layer of prostate epithelium; a region reported to have high expression levels of c-KIT (Ceder et al. 2017; Leong et al. 2008). Moreover, it was demonstrated that tumorigenesis induced by prostate cancer stem cells is accompanied by the increased expression of c-KIT (Peng et al. 2015). Interestingly, several signalling pathways were shown to be deregulated in prostate cancer stem cells and accounting for its tumorigenic potential, which included the JAK-STAT pathway (Birnie et al. 2008; Lin et al. 2016a; Lin et al. 2016b).
The PLC-γ pathway
The phospholipase-Cγ (PLC-γ) pathway is one of the less characterized relatively to the activity of the SCF/c-KIT system. The activation of c-KIT by SCF leads to c-KIT autophosphorylation and its association with PLC-γ1 via the phosphorylated Tyr728 residue (Gommerman et al. 2000). Several reports demonstrated that the inhibition of PLC-γ disrupts the SCF effects in cells expressing c-KIT (Gommerman et al. 2000; Maddens et al. 2002). However, other study failed to detect PLC-γ activity induced by SCF/c-KIT, observing an activation of phospholipase D instead (Koike et al. 1993).
Interaction of tr-KIT with the PLC-γ pathway has also been described, though independently of SCF since this c-KIT variant is devoid of the extracellular domain (Rossi et al. 1992; Sette et al. 1998). It was demonstrated that tr-KIT in sperm is able to activate the PLC-γ1 in mouse oocytes (Schnabel et al. 2005; Sette et al. 1998). Moreover, it was proposed that the action of tr-KIT phosphorylating and activating PLC-γ1 is mediated by the interaction with the Src member Fyn. The physical association of tr-KIT with Fyn promotes the catalytic activity of Fyn and the consequent phosphorylation and activation of phospholipase (Sette et al. 2002).
Further studies are needed to establish a connection between the role of c-KIT activating PLC-γ and neoplastic conditions of male reproductive tract.
The MAPK pathway
Activation of the MAPK cascade occurs upon binding of SH2 adaptor proteins to the phosphorylated residues of RTKs, which determines the activation of small G-protein Ras. Phosphorylated Tyr703 and Tyr936 residues in the carboxyl-terminal tail of c-KIT were identified as binding sites for the adaptor proteins, growth factor receptor bound protein-2 (Grb2) and the guanine nucleotide exchange factor, son of sevenless (SOS) protein (Thommes et al. 1999). The Grb2/SOS complex has been shown to link RTKs to the Ras/MAPK pathway (Fig. 2). Cell signalling via c-KIT activates several MAPKs such as extracellular-signal-regulated kinase (ERK) 1/2, p38, c-Jun N-terminal kinase (JNK), and ERK5.
As expected, the MAPK pathway is linked with the proliferative activity of SCF upon activation of c-KIT. Inhibition of MEK/MAPK kinase effectively abolished the SCF-induced proliferation while the anti-apoptotic effect of SCF remained unchanged (Dolci et al. 2001). Moreover, membrane-bound and soluble isoforms of SCF seem to have specific MAPKs targets. The mSCF effectively induced cell proliferation through the ERK whereas the sSCF proliferative effects were achieved by the activity of p38 (Kapur et al. 2002).
SCF/c-KIT effects mediating cell migration also depend on the MAPK pathway via phosphorylation and activity of p38 (Kuang et al. 2008); MEK/MAPK inhibitors reduced the response to c-KIT by 30% (Farini et al. 2007).
Interestingly, it has been shown that the SCF/c-KIT activation of MAPK pathway is dependent on PI3-K signalling. The use of PI3-K inhibitors decreased the activation of RAF and ERK, but did not affect Ras activation (Wandzioch et al. 2004). The crosstalk between PI3-K/Akt and Ras/MAPK pathways has been described at multiple levels in distinct cell types endowing the cells with incredibly higher response patterns to the combinatorial variety of external stimuli (Aksamitiene et al. 2012). Moreover, it is known that abnormalities in MAPK signalling play a critical role in the development and progression of cancer (Dhillon et al. 2007). However, much is yet to be discovered about the relationship of SCF/c-KIT with MAPK pathway in male cancer, namely, in testicular and prostatic cancer.
Conclusion
The SCF/c-KIT system is involved in the control of basic biological processes, such as apoptosis, cell proliferation, differentiation, and migration. The deviation to the strict control of these cellular processes is closely related with the neoplastic transformation and tumours progression, thus, not surprisingly, deregulated actions of c-KIT have been associated with different types of human cancers.
Considerable research efforts have started to disclose the signalling pathways activated by c-KIT and how they, independently or in cross-talk, regulate cell fate and contribute to cell tissue homoeostasis. Moreover, it has been shown that due to the presence of gain-of-function mutations or overexpression of c-KIT, some of these pathways are overactivated in pathological conditions, as is the case of testicular and prostatic cancers. Therefore, the inhibition of c-KIT emerged as a promising strategy for treatment of testicular cancer, though being much less effective in the case of prostatic tumours. As discussed through the review, the differential response of human solid tumours to c-KIT inhibitors can be related with the existence of tissue-specific protein variants (e.g tr-KIT), and/or with c-KIT mutations. The characterization of c-KIT mutations sensitive to imatinib (or other second-generation tyrosine kinase inhibitors) in testicular and prostatic cancers deserves further investigation and should be a prerequisite before advancing in treatment. Moreover, a deeper understanding of c-KIT signalling mechanisms in cancer cells will be paramount to identify the crucial targets points for therapeutic intervention, which in the case of prostate cancer means the development of specific inhibitors for the tr-KIT.
Finally, human tumours are usually dependent on the activation of several survival and proliferation pathways. In this way, clarifying the interaction between the distinct signalling pathways driven by c-KIT (and its variants) in cancer cells might help to develop highly effective inhibitors acting simultaneously at different molecular targets. At this point, should also be included the microRNAs (miRNAs), a class of small endogenous RNAs with important actions in the regulation of gene expression and modulation of signalling pathways by its capability of enhancing or repressing the activity of downstream effectors. The role of miRNAs in prostate cancer was reviewed having been clear their oncogenic or tumour suppressor abilities, as well as the interaction with androgen receptor and other signalling pathways (Fayyaz and Farooqi 2013). This opens new avenues of research to ascertain the relationship between miRNAs and c-KIT signalling, which would be therapeutically relevant.
Acknowledgments
This work was supported by FEDER funds through the POCI - COMPETE 2020 - Operational Programme Competitiveness and Internationalisation in Axis I - Strengthening research, technological development and innovation (Project No. 007491) and National Funds by FCT-Foundation for Science and Technology (Project UID/Multi/00709/2013). Henrique Cardoso and Marília Figueira were funded by FCT fellowships (SFRH/BD/ 111351/2015 and SFRH/BD/104671/2014, respectively).
The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors.
Abbreviations
- ERK
Extracellular-signal-regulated kinase
- GNNK
Gly-Asn-Asn-Lys
- Grb2
Growth factor receptor bound protein-2
- JAK
Janus kinase
- JNK
c-Jun N-terminal kinase
- MAPK
Mitogen-activated protein kinase
- mSCF
membrane-bound SCF
- mTOR
mammalian target of rapamycin
- p70S6K
p70 S6 kinase
- PI3-K
Phosphatidylinositol 3-kinase
- PLC-γ
phospholipase-C
- Rb
Retinoblastoma
- SCF
Stem cell factor
- SH2
Src homology 2
- sSCF
soluble SCF
- STAT
Signal transducers and activators of transcription
- tr-KIT
truncated c-KIT protein
Footnotes
Henrique J. Cardoso and Marília I. Figueira contributed equally to this work.
References
- Aksamitiene E, Kiyatkin A, Kholodenko BN. Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: a fine balance. Biochem Soc Trans. 2012;40:139–146. doi: 10.1042/BST20110609. [DOI] [PubMed] [Google Scholar]
- Ali S, Ali S. Role of c-kit/SCF in cause and treatment of gastrointestinal stromal tumors (GIST) Gene. 2007;401:38–45. doi: 10.1016/j.gene.2007.06.017. [DOI] [PubMed] [Google Scholar]
- Ashman LK, Griffith R. Therapeutic targeting of c-KIT in cancer. Expert Opin Investig Drugs. 2013;22:103–115. doi: 10.1517/13543784.2013.740010. [DOI] [PubMed] [Google Scholar]
- Biermann K, Goke F, Nettersheim D, Eckert D, Zhou H, Kahl P, Gashaw I, Schorle H, Buttner R. C-KIT is frequently mutated in bilateral germ cell tumours and down-regulated during progression from intratubular germ cell neoplasia to seminoma. J Pathol. 2007;213:311–318. doi: 10.1002/path.2225. [DOI] [PubMed] [Google Scholar]
- Birnie R, Bryce SD, Roome C, Dussupt V, Droop A, Lang SH, Berry PA, Hyde CF, Lewis JL, Stower MJ, Maitland NJ, Collins AT. Gene expression profiling of human prostate cancer stem cells reveals a pro-inflammatory phenotype and the importance of extracellular matrix interactions. Genome Biol. 2008;9:R83. doi: 10.1186/gb-2008-9-5-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume-Jensen P, Hunter T (2001) Oncogenic kinase signalling. Nature 411:355–365 [DOI] [PubMed]
- Blume-Jensen P, Janknecht R, Hunter T. The kit receptor promotes cell survival via activation of PI 3-kinase and subsequent Akt-mediated phosphorylation of bad on Ser136. Curr Biol. 1998;8:779–782. doi: 10.1016/S0960-9822(98)70302-1. [DOI] [PubMed] [Google Scholar]
- Capelli L, Petracci E, Quagliuolo V, Saragoni L, Colombo P, Morgagni P, Calistri D, Tomezzoli A, Di Cosmo M, Roviello F, Vindigni C, Coniglio A, Villanacci V, Catarci M, Coppola L, Alfieri S, Ricci R, Capella C, Rausei S, Gulino D, Amadori D, Ulivi P. Gastric GISTs: analysis of c-kit, PDGFRA and BRAF mutations in relation to prognosis and clinical pathological characteristics of patients - a GIRCG study. European journal of surgical oncology: the journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology. 2016;42:1206–1214. doi: 10.1016/j.ejso.2016.05.022. [DOI] [PubMed] [Google Scholar]
- Cardoso HJ, Figueira MI, Correia S, Vaz CV, Socorro S. The SCF/c-KIT system in the male: survival strategies in fertility and cancer. Mol Reprod Dev. 2014;81:1064–1079. doi: 10.1002/mrd.22430. [DOI] [PubMed] [Google Scholar]
- Cardoso HJ, Vaz CV, Correia S, Figueira MI, Marques R, Maia CJ, Socorro S. Paradoxical and contradictory effects of imatinib in two cell line models of hormone-refractory prostate cancer. Prostate. 2015;75:923–935. doi: 10.1002/pros.22976. [DOI] [PubMed] [Google Scholar]
- Caruana G, Cambareri AC, Ashman LK. Isoforms of c-KIT differ in activation of signalling pathways and transformation of NIH3T3 fibroblasts. Oncogene. 1999;18:5573–5581. doi: 10.1038/sj.onc.1202939. [DOI] [PubMed] [Google Scholar]
- Ceder JA, Aalders TW, Schalken JA. Label retention and stem cell marker expression in the developing and adult prostate identifies basal and luminal epithelial stem cell subpopulations. Stem Cell Res Ther. 2017;8:95. doi: 10.1186/s13287-017-0544-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieffi P. Recent advances in molecular and cell biology of testicular germ-cell tumors. Int Rev Cell Mol Biol. 2014;312:79–100. doi: 10.1016/B978-0-12-800178-3.00003-8. [DOI] [PubMed] [Google Scholar]
- Coffey J, Linger R, Pugh J, Dudakia D, Sokal M, Easton DF, Timothy Bishop D, Stratton M, Huddart R, Rapley EA. Somatic KIT mutations occur predominantly in seminoma germ cell tumors and are not predictive of bilateral disease: report of 220 tumors and review of literature. Genes, chromosomes & cancer. 2008;47:34–42. doi: 10.1002/gcc.20503. [DOI] [PubMed] [Google Scholar]
- Corcoran NM, Costello AJ. Combined low-dose imatinib mesylate and paclitaxel lack synergy in an experimental model of extra-osseous hormone-refractory prostate cancer. BJU Int. 2005;96:640–646. doi: 10.1111/j.1464-410X.2005.05699.x. [DOI] [PubMed] [Google Scholar]
- Dahlen DD, Lin NL, Liu YC, Broudy VC. Soluble kit receptor blocks stem cell factor bioactivity in vitro. Leuk Res. 2001;25:413–421. doi: 10.1016/S0145-2126(00)00122-3. [DOI] [PubMed] [Google Scholar]
- Damaschke NA, Yang B, Bhusari S, Svaren JP, Jarrard DF. Epigenetic susceptibility factors for prostate cancer with aging. Prostate. 2013;73:1721–1730. doi: 10.1002/pros.22716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miguel MP, Cheng L, Holland EC, Federspiel MJ, Donovan PJ. Dissection of the c-kit signaling pathway in mouse primordial germ cells by retroviral-mediated gene transfer. Proc Natl Acad Sci U S A. 2002;99:10458–10463. doi: 10.1073/pnas.122249399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deberry C, Mou S, Linnekin D. Stat1 associates with c-kit and is activated in response to stem cell factor. Biochem J. 1997;327:73–80. doi: 10.1042/bj3270073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derry JJ, Prins GS, Ray V, Tyner AL. Altered localization and activity of the intracellular tyrosine kinase BRK/Sik in prostate tumor cells. Oncogene. 2003;22:4212–4220. doi: 10.1038/sj.onc.1206465. [DOI] [PubMed] [Google Scholar]
- Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- Di Lorenzo G, Autorino R, D'Armiento FP, Mignogna C, De Laurentiis M, De Sio M, D'Armiento M, Damiano R, Vecchio G, De Placido S. Expression of proto-oncogene c-kit in high risk prostate cancer. European journal of surgical oncology: the journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology. 2004;30:987–992. doi: 10.1016/j.ejso.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Dolci S, Pellegrini M, Di Agostino S, Geremia R, Rossi P. Signaling through extracellular signal-regulated kinase is required for spermatogonial proliferative response to stem cell factor. J Biol Chem. 2001;276:40225–40233. doi: 10.1074/jbc.M105143200. [DOI] [PubMed] [Google Scholar]
- Farini D, La Sala G, Tedesco M, De Felici M. Chemoattractant action and molecular signaling pathways of kit ligand on mouse primordial germ cells. Dev Biol. 2007;306:572–583. doi: 10.1016/j.ydbio.2007.03.031. [DOI] [PubMed] [Google Scholar]
- Farooqi AA, Sarkar FH. Overview on the complexity of androgen receptor-targeted therapy for prostate cancer. Cancer Cell Int. 2015;15:7. doi: 10.1186/s12935-014-0153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayyaz S, Farooqi AA. miRNA and TMPRSS2-ERG do not mind their own business in prostate cancer cells. Immunogenetics. 2013;65:315–332. doi: 10.1007/s00251-012-0677-2. [DOI] [PubMed] [Google Scholar]
- Feng LX, Ravindranath N, Dym M. Stem cell factor/c-kit up-regulates cyclin D3 and promotes cell cycle progression via the phosphoinositide 3-kinase/p70 S6 kinase pathway in spermatogonia. J Biol Chem. 2000;275:25572–25576. doi: 10.1074/jbc.M002218200. [DOI] [PubMed] [Google Scholar]
- Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D, Bray F. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49:1374–1403. doi: 10.1016/j.ejca.2012.12.027. [DOI] [PubMed] [Google Scholar]
- Figueira MI, Cardoso HJ, Correia S, Maia CJ, Socorro S. Hormonal regulation of c-KIT receptor and its ligand: implications for human infertility? Prog Histochem Cytochem. 2014;49:1–19. doi: 10.1016/j.proghi.2014.09.001. [DOI] [PubMed] [Google Scholar]
- Filippou P, Ferguson JE, 3rd, Nielsen ME. Epidemiology of prostate and testicular cancer. Semin Interv Radiol. 2016;33:182–185. doi: 10.1055/s-0036-1586146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukao T, Yamada T, Tanabe M, Terauchi Y, Ota T, Takayama T, Asano T, Takeuchi T, Kadowaki T, Hata Ji J, Koyasu S. Selective loss of gastrointestinal mast cells and impaired immunity in PI3K-deficient mice. Nat Immunol. 2002;3:295–304. doi: 10.1038/ni768. [DOI] [PubMed] [Google Scholar]
- Gommerman JL, Sittaro D, Klebasz NZ, Williams DA, Berger SA. Differential stimulation of c-kit mutants by membrane-bound and soluble steel factor correlates with leukemic potential. Blood. 2000;96:3734–3742. [PubMed] [Google Scholar]
- Hayes-Lattin B, Nichols CR. Testicular cancer: a prototypic tumor of young adults. Semin Oncol. 2009;36:432–438. doi: 10.1053/j.seminoncol.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DY, Chao Y, Tai MH, Yu YH, Lin WW. STI571 reduces TRAIL-induced apoptosis in colon cancer cells: c-Abl activation by the death receptor leads to stress kinase-dependent cell death. J Biomed Sci. 2012;19:35. doi: 10.1186/1423-0127-19-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imura M, Kojima Y, Kubota Y, Hamakawa T, Yasui T, Sasaki S, Hayashi Y, Kohri K. Regulation of cell proliferation through a KIT-mediated mechanism in benign prostatic hyperplasia. Prostate. 2012;72:1506–1513. doi: 10.1002/pros.22500. [DOI] [PubMed] [Google Scholar]
- Izquierdo MA, Van der Valk P, Van Ark-Otte J, Rubio G, Germa-Lluch JR, Ueda R, Scheper RJ, Takahashi T, Giaccone G. Differential expression of the c-kit proto-oncogene in germ cell tumours. J Pathol. 1995;177:253–258. doi: 10.1002/path.1711770307. [DOI] [PubMed] [Google Scholar]
- Kapur R, Chandra S, Cooper R, McCarthy J, Williams DA. Role of p38 and ERK MAP kinase in proliferation of erythroid progenitors in response to stimulation by soluble and membrane isoforms of stem cell factor. Blood. 2002;100:1287–1293. [PubMed] [Google Scholar]
- Kemmer K, Corless CL, Fletcher JA, McGreevey L, Haley A, Griffith D, Cummings OW, Wait C, Town A, Heinrich MC. KIT mutations are common in testicular seminomas. Am J Pathol. 2004;164:305–313. doi: 10.1016/S0002-9440(10)63120-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr IM, Costa-Pereira AP, Lillemeier BF, Strobl B. Of JAKs, STATs, blind watchmakers, jeeps and trains. FEBS Lett. 2003;546:1–5. doi: 10.1016/S0014-5793(03)00411-3. [DOI] [PubMed] [Google Scholar]
- Klippel A, Escobedo JA, Hirano M, Williams LT. The interaction of small domains between the subunits of phosphatidylinositol 3-kinase determines enzyme activity. Mol Cell Biol. 1994;14:2675–2685. doi: 10.1128/MCB.14.4.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike T, Hirai K, Morita Y, Nozawa Y. Stem cell factor-induced signal transduction in rat mast cells. Activation of phospholipase D but not phosphoinositide-specific phospholipase C in c-kit receptor stimulation J Immunol. 1993;151:359–366. [PubMed] [Google Scholar]
- Kuang D, Zhao X, Xiao G, Ni J, Feng Y, Wu R, Wang G. Stem cell factor/c-kit signaling mediated cardiac stem cell migration via activation of p38 MAPK. Basic Res Cardiol. 2008;103:265–273. doi: 10.1007/s00395-007-0690-z. [DOI] [PubMed] [Google Scholar]
- Langley KE, Mendiaz EA, Liu N, Narhi LO, Zeni L, Parseghian CM, Clogston CL, Leslie I, Pope JA, Lu HS, Zsebo KM, Boone TC. Properties of variant forms of human stem cell factor recombinantly expressed in Escherichia coli. Arch Biochem Biophys. 1994;311:55–61. doi: 10.1006/abbi.1994.1208. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J (2010) Cell signaling by receptor tyrosine kinases. Cell 141:1117–1134 [DOI] [PMC free article] [PubMed]
- Lemmon MA, Pinchasi D, Zhou M, Lax I, Schlessinger J. Kit receptor dimerization is driven by bivalent binding of stem cell factor. J Biol Chem. 1997;272:6311–6317. doi: 10.1074/jbc.272.10.6311. [DOI] [PubMed] [Google Scholar]
- Lennartsson J, Ronnstrand L. Stem cell factor receptor/c-kit: from basic science to clinical implications. Physiol Rev. 2012;92:1619–1649. doi: 10.1152/physrev.00046.2011. [DOI] [PubMed] [Google Scholar]
- Leong KG, Wang BE, Johnson L, Gao WQ. Generation of a prostate from a single adult stem cell. Nature. 2008;456:804–808. doi: 10.1038/nature07427. [DOI] [PubMed] [Google Scholar]
- Lin X, Aslam A, Attar R, Yaylim I, Qureshi MZ, Hasnain S, Qadir MI, and Farooqi AA (2016a) Signaling lansdscape of prostate cancer. Cellular and molecular biology (noisy-le-grand, France) 62:45-50 [PubMed]
- Lin X, Farooqi AA, Qureshi MZ, Romero MA, Tabassum S, Ismail M. Prostate cancer stem cells: viewing signaling cascades at a finer resolution. Arch Immunol Ther Exp. 2016;64:217–223. doi: 10.1007/s00005-016-0383-0. [DOI] [PubMed] [Google Scholar]
- Linnekin D, DeBerry CS, Mou S. Lyn associates with the juxtamembrane region of c-kit and is activated by stem cell factor in hematopoietic cell lines and normal progenitor cells. J Biol Chem. 1997;272:27450–27455. doi: 10.1074/jbc.272.43.27450. [DOI] [PubMed] [Google Scholar]
- Linnekin D, Mou S, Deberry CS, Weiler SR, Keller JR, Ruscetti FW, Longo DL. Stem cell factor, the JAK-STAT pathway and signal transduction. Leuk Lymphoma. 1997;27:439–444. doi: 10.3109/10428199709058310. [DOI] [PubMed] [Google Scholar]
- Lipton A, Campbell-Baird C, Harvey H, Kim C, Demers L, Costa L. Phase I trial of zoledronic acid + imatinib mesylate (Gleevec) in patients with bone metastases. Am J Clin Oncol. 2010;33:75–78. doi: 10.1097/COC.0b013e31819cccdc. [DOI] [PubMed] [Google Scholar]
- Maddens S, Charruyer A, Plo I, Dubreuil P, Berger S, Salles B, Laurent G, Jaffrezou JP. Kit signaling inhibits the sphingomyelin-ceramide pathway through PLC gamma 1: implication in stem cell factor radioprotective effect. Blood. 2002;100:1294–1301. [PubMed] [Google Scholar]
- Mansuroglu T, Ramadori P, Dudas J, Malik I, Hammerich K, Fuzesi L, Ramadori G. Expression of stem cell factor and its receptor c-kit during the development of intrahepatic cholangiocarcinoma. Lab Investig. 2009;89:562–574. doi: 10.1038/labinvest.2009.15. [DOI] [PubMed] [Google Scholar]
- Maruyama IN (2014) Mechanisms of activation of receptor tyrosine kinases: monomers or dimers. Cells 3:304–330 [DOI] [PMC free article] [PubMed]
- Mathew P, Thall PF, Jones D, Perez C, Bucana C, Troncoso P, Kim SJ, Fidler IJ, Logothetis C. Platelet-derived growth factor receptor inhibitor imatinib mesylate and docetaxel: a modular phase I trial in androgen-independent prostate cancer. J Clin Oncol. 2004;22:3323–3329. doi: 10.1200/JCO.2004.10.116. [DOI] [PubMed] [Google Scholar]
- Mitchell SG, Bunting ST, Saxe D, Olson T, and Keller FG (2017) A variant c-KIT mutation, D816H, fundamental to the sequential development of an ovarian mixed germ cell tumor and systemic mastocytosis with chronic myelomonocytic leukemia. Pediatric blood & cancer 64 [DOI] [PubMed]
- Miyazawa K, Williams DA, Gotoh A, Nishimaki J, Broxmeyer HE, Toyama K. Membrane-bound steel factor induces more persistent tyrosine kinase activation and longer life span of c-kit gene-encoded protein than its soluble form. Blood. 1995;85:641–649. [PubMed] [Google Scholar]
- Mol CD, Lim KB, Sridhar V, Zou H, Chien EY, Sang BC, Nowakowski J, Kassel DB, Cronin CN, McRee DE. Structure of a c-kit product complex reveals the basis for kinase transactivation. J Biol Chem. 2003;278:31461–31464. doi: 10.1074/jbc.C300186200. [DOI] [PubMed] [Google Scholar]
- Montero JC, Lopez-Perez R, San Miguel JF, Pandiella A. Expression of c-kit isoforms in multiple myeloma: differences in signaling and drug sensitivity. Haematologica. 2008;93:851–859. doi: 10.3324/haematol.12171. [DOI] [PubMed] [Google Scholar]
- Mou S, Linnekin D. Lyn is activated during late G1 of stem-cell-factor-induced cell cycle progression in haemopoietic cells. Biochem J. 1999;342(Pt 1):163–170. [PMC free article] [PubMed] [Google Scholar]
- Nabhan C, Villines D, Valdez TV, Tolzien K, Lestingi TM, Bitran JD, Christner SM, Egorin MJ, Beumer JH. Phase I study investigating the safety and feasibility of combining imatinib mesylate (Gleevec) with sorafenib in patients with refractory castration-resistant prostate cancer. Br J Cancer. 2012;107:592–597. doi: 10.1038/bjc.2012.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai Y, Nonomura N, Oka D, Shiba M, Arai Y, Nakayama M, Inoue H, Nishimura K, Aozasa K, Mizutani Y, Miki T, Okuyama A. KIT (c-kit oncogene product) pathway is constitutively activated in human testicular germ cell tumors. Biochem Biophys Res Commun. 2005;337:289–296. doi: 10.1016/j.bbrc.2005.09.042. [DOI] [PubMed] [Google Scholar]
- Nishida K, Wang L, Morii E, Park SJ, Narimatsu M, Itoh S, Yamasaki S, Fujishima M, Ishihara K, Hibi M, Kitamura Y, Hirano T. Requirement of Gab2 for mast cell development and KitL/c-kit signaling. Blood. 2002;99:1866–1869. doi: 10.1182/blood.V99.5.1866. [DOI] [PubMed] [Google Scholar]
- O'Laughlin-Bunner B, Radosevic N, Taylor ML, Shivakrupa DBC, Metcalfe DD, Zhou M, Lowell C, Linnekin D. Lyn is required for normal stem cell factor-induced proliferation and chemotaxis of primary hematopoietic cells. Blood. 2001;98:343–350. doi: 10.1182/blood.V98.2.343. [DOI] [PubMed] [Google Scholar]
- Paronetto MP, Venables JP, Elliott DJ, Geremia R, Rossi P, Sette C. Tr-kit promotes the formation of a multimolecular complex composed by Fyn, PLCgamma1 and Sam68. Oncogene. 2003;22:8707–8715. doi: 10.1038/sj.onc.1207016. [DOI] [PubMed] [Google Scholar]
- Paronetto MP, Farini D, Sammarco I, Maturo G, Vespasiani G, Geremia R, Rossi P, Sette C. Expression of a truncated form of the c-kit tyrosine kinase receptor and activation of Src kinase in human prostatic cancer. Am J Pathol. 2004;164:1243–1251. doi: 10.1016/S0002-9440(10)63212-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulhe F, Wehrle-Haller M, Jacquier MC, Imhof BA, Tabone-Eglinger S, Wehrle-Haller B. Dimerization of kit-ligand and efficient cell-surface presentation requires a conserved Ser-Gly-Gly-Tyr motif in its transmembrane domain. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2009;23:3037–3048. doi: 10.1096/fj.09-129577. [DOI] [PubMed] [Google Scholar]
- Pedersini R, Vattemi E, Mazzoleni G, Graiff C. Complete response after treatment with imatinib in pretreated disseminated testicular seminoma with overexpression of c-KIT. Lancet Oncol. 2007;8:1039–1040. doi: 10.1016/S1470-2045(07)70344-3. [DOI] [PubMed] [Google Scholar]
- Peng Y, Chen Q, Gu M, Chen Y, Zhang M, Zhou J, Wang H, Gao Y, Li W, Wang Z, Cai Z. Human stromal cells in the peripheral zone of the prostate promote tumorigenesis of prostatic cancer stem cells through up-regulation of C-kit expression. J Cancer. 2015;6:776–785. doi: 10.7150/jca.9961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phung B, Steingrimsson E, Ronnstrand L. Differential activity of c-KIT splice forms is controlled by extracellular peptide insert length. Cell Signal. 2013;25:2231–2238. doi: 10.1016/j.cellsig.2013.07.011. [DOI] [PubMed] [Google Scholar]
- Piao X, Bernstein A. A point mutation in the catalytic domain of c-kit induces growth factor independence, tumorigenicity, and differentiation of mast cells. Blood. 1996;87:3117–3123. [PubMed] [Google Scholar]
- Pinto AC, Angelo S, Moreira JN, Simoes S. Schedule treatment design and quantitative in vitro evaluation of chemotherapeutic combinations for metastatic prostate cancer therapy. Cancer Chemother Pharmacol. 2011;67:275–284. doi: 10.1007/s00280-010-1315-z. [DOI] [PubMed] [Google Scholar]
- Price DJ, Rivnay B, Fu Y, Jiang S, Avraham S, Avraham H. Direct association of Csk homologous kinase (CHK) with the diphosphorylated site Tyr568/570 of the activated c-KIT in megakaryocytes. J Biol Chem. 1997;272:5915–5920. doi: 10.1074/jbc.272.9.5915. [DOI] [PubMed] [Google Scholar]
- Rapley EA, Hockley S, Warren W, Johnson L, Huddart R, Crockford G, Forman D, Leahy MG, Oliver DT, Tucker K, Friedlander M, Phillips KA, Hogg D, Jewett MA, Lohynska R, Daugaard G, Richard S, Heidenreich A, Geczi L, Bodrogi I, Olah E, Ormiston WJ, Daly PA, Looijenga LH, Guilford P, Aass N, Fossa SD, Heimdal K, Tjulandin SA, Liubchenko L, Stoll H, Weber W, Einhorn L, Weber BL, McMaster M, Greene MH, Bishop DT, Easton D, Stratton MR. Somatic mutations of KIT in familial testicular germ cell tumours. Br J Cancer. 2004;90:2397–2401. doi: 10.1038/sj.bjc.6601880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DR, Wu YM, Lin SF (2000) The protein tyrosine kinase family of the human genome Oncogene 19:5548–5557 [DOI] [PubMed]
- Ronnstrand L. Signal transduction via the stem cell factor receptor/c-kit. Cell Mol Life Sci. 2004;61:2535–2548. doi: 10.1007/s00018-004-4189-6. [DOI] [PubMed] [Google Scholar]
- Rossi P, Marziali G, Albanesi C, Charlesworth A, Geremia R, Sorrentino V. A novel c-kit transcript, potentially encoding a truncated receptor, originates within a kit gene intron in mouse spermatids. Dev Biol. 1992;152:203–207. doi: 10.1016/0012-1606(92)90172-D. [DOI] [PubMed] [Google Scholar]
- Ryan JJ, Huang H, McReynolds LJ, Shelburne C, Hu-Li J, Huff TF, Paul WE. Stem cell factor activates STAT-5 DNA binding in IL-3-derived bone marrow mast cells. Exp Hematol. 1997;25:357–362. [PubMed] [Google Scholar]
- Sakuma Y, Sakurai S, Oguni S, Hironaka M, Saito K. Alterations of the c-kit gene in testicular germ cell tumors. Cancer Sci. 2003;94:486–491. doi: 10.1111/j.1349-7006.2003.tb01470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samayawardhena LA, Kapur R, Craig AW. Involvement of Fyn kinase in kit and integrin-mediated Rac activation, cytoskeletal reorganization, and chemotaxis of mast cells. Blood. 2007;109:3679–3686. doi: 10.1182/blood-2006-11-057315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel D, Ramirez L, Gertsenstein M, Nagy A, Lomeli H. Ectopic expression of KitD814Y in spermatids of transgenic mice, interferes with sperm morphogenesis. Developmental dynamics: an official publication of the American Association of Anatomists. 2005;233:29–40. doi: 10.1002/dvdy.20292. [DOI] [PubMed] [Google Scholar]
- Segaliny AI, Tellez-Gabriel M, Heymann MF, Heymann D (2015) Receptor tyrosine kinases: Characterisation, mechanism of action and therapeutic interests for bone cancers. Journal of Bone Oncology 4:1–12 [DOI] [PMC free article] [PubMed]
- Serve H, Hsu YC, Besmer P. Tyrosine residue 719 of the c-kit receptor is essential for binding of the P85 subunit of phosphatidylinositol (PI) 3-kinase and for c-kit-associated PI 3-kinase activity in COS-1 cells. J Biol Chem. 1994;269:6026–6030. [PubMed] [Google Scholar]
- Serve H, Yee NS, Stella G, Sepp-Lorenzino L, Tan JC, Besmer P. Differential roles of PI3-kinase and kit tyrosine 821 in kit receptor-mediated proliferation, survival and cell adhesion in mast cells. EMBO J. 1995;14:473–483. doi: 10.1002/j.1460-2075.1995.tb07023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sette C, Bevilacqua A, Geremia R, Rossi P. Involvement of phospholipase Cgamma1 in mouse egg activation induced by a truncated form of the C-kit tyrosine kinase present in spermatozoa. J Cell Biol. 1998;142:1063–1074. doi: 10.1083/jcb.142.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sette C, Paronetto MP, Barchi M, Bevilacqua A, Geremia R, Rossi P. Tr-kit-induced resumption of the cell cycle in mouse eggs requires activation of a Src-like kinase. EMBO J. 2002;21:5386–5395. doi: 10.1093/emboj/cdf553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thommes K, Lennartsson J, Carlberg M, Ronnstrand L. Identification of Tyr-703 and Tyr-936 as the primary association sites for Grb2 and Grb7 in the c-kit/stem cell factor receptor. Biochem J. 1999;341(Pt 1):211–216. doi: 10.1042/bj3410211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Q, Frierson HF, Jr, Krystal GW, Moskaluk CA. Activating c-kit gene mutations in human germ cell tumors. Am J Pathol. 1999;154:1643–1647. doi: 10.1016/S0002-9440(10)65419-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffany NM, Wersinger EM, Garzotto M, Beer TM. Imatinib mesylate and zoledronic acid in androgen-independent prostate cancer. Urology. 2004;63:934–939. doi: 10.1016/j.urology.2003.12.022. [DOI] [PubMed] [Google Scholar]
- Todd JR, Becker TM, Kefford RF, Rizos H. Secondary c-kit mutations confer acquired resistance to RTK inhibitors in c-kit mutant melanoma cells. Pigment Cell Melanoma Res. 2013;26:518–526. doi: 10.1111/pcmr.12107. [DOI] [PubMed] [Google Scholar]
- Tulina N, Matunis E. Control of stem cell self-renewal in drosophila spermatogenesis by JAK-STAT signaling. Science. 2001;294:2546–2549. doi: 10.1126/science.1066700. [DOI] [PubMed] [Google Scholar]
- Verstraete K, Savvides SN (2012) Extracellular assembly and activation principles of oncogenic class III receptor tyrosine kinases. Nat Rev Cancer 12:753–766 [DOI] [PubMed]
- Voytyuk O, Lennartsson J, Mogi A, Caruana G, Courtneidge S, Ashman LK, Ronnstrand L. Src family kinases are involved in the differential signaling from two splice forms of c-kit. J Biol Chem. 2003;278:9159–9166. doi: 10.1074/jbc.M211726200. [DOI] [PubMed] [Google Scholar]
- Wandzioch E, Edling CE, Palmer RH, Carlsson L, Hallberg B. Activation of the MAP kinase pathway by c-kit is PI-3 kinase dependent in hematopoietic progenitor/stem cell lines. Blood. 2004;104:51–57. doi: 10.1182/blood-2003-07-2554. [DOI] [PubMed] [Google Scholar]
- Weiler SR, Mou S, DeBerry CS, Keller JR, Ruscetti FW, Ferris DK, Longo DL, Linnekin D. JAK2 is associated with the c-kit proto-oncogene product and is phosphorylated in response to stem cell factor. Blood. 1996;87:3688–3693. [PubMed] [Google Scholar]
- Williams DE, Eisenman J, Baird A, Rauch C, Van Ness K, March CJ, Park LS, Martin U, Mochizuki DY, Boswell HS, Burgess GS, Cosma D, Stewart DL. Identification of a ligand for the c-kit proto-oncogene. Cell. 1990;63:167–174. doi: 10.1016/0092-8674(90)90297-R. [DOI] [PubMed] [Google Scholar]
- Willmore-Payne C, Holden JA, Chadwick BE, Layfield LJ. Detection of c-kit exons 11- and 17-activating mutations in testicular seminomas by high-resolution melting amplicon analysis. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2006;19:1164–1169. doi: 10.1038/modpathol.3800623. [DOI] [PubMed] [Google Scholar]
- Wu P, Cao Z, Wu S. New Progress of Epigenetic Biomarkers in Urological Cancer. 2016;2016:9864047. doi: 10.1155/2016/9864047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden Y, Kuang WJ, Yang-Feng T, Coussens L, Munemitsu S, Dull TJ, Chen E, Schlessinger J, Francke U, Ullrich A. Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987;6:3341–3351. doi: 10.1002/j.1460-2075.1987.tb02655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun E-J, Lo UG, Hsieh J-T. The evolving landscape of prostate cancer stem cell: therapeutic implications and future challenges. Asian Journal of Urology. 2016;3:203–210. doi: 10.1016/j.ajur.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhang R, Joachimiak A, Schlessinger J, Kong XP. Crystal structure of human stem cell factor: implication for stem cell factor receptor dimerization and activation. Proc Natl Acad Sci U S A. 2000;97:7732–7737. doi: 10.1073/pnas.97.14.7732. [DOI] [PMC free article] [PubMed] [Google Scholar]