

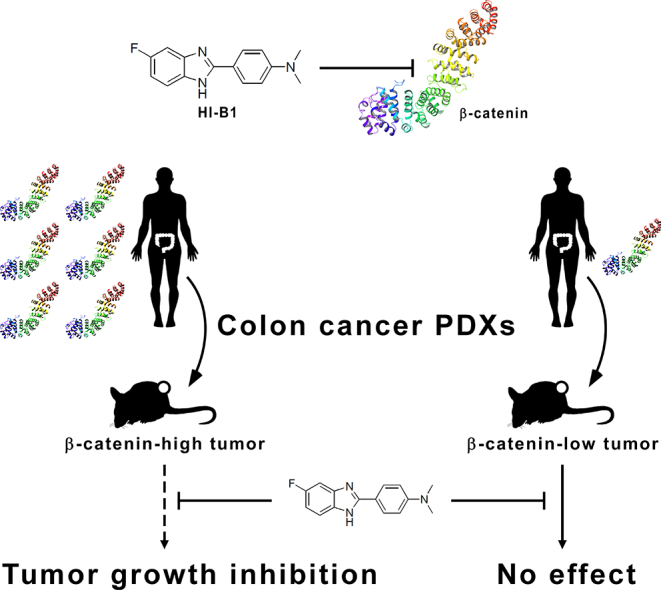
Abstract
Colorectal cancer is associated with aberrant activation of the Wnt pathway. β-Catenin plays essential roles in the Wnt pathway by interacting with T-cell factor 4 (TCF4) to transcribe oncogenes. We synthesized a small molecule, referred to as HI-B1, and evaluated signaling changes and biological consequences induced by the compound. HI-B1 inhibited β-catenin/TCF4 luciferase activity and preferentially caused apoptosis of cancer cells in which the survival is dependent on β-catenin. The formation of the β-catenin/TCF4 complex was disrupted by HI-B1 due to the direct interaction of HI-B1 with β-catenin. Colon cancer patient-derived xenograft (PDX) studies showed that a tumor with higher levels of β-catenin expression was more sensitive to HI-B1 treatment, compared to a tumor with lower expression levels of β-catenin. The different sensitivities of PDX tumors to HI-B1 were dependent on the β-catenin expression level and potentially could be further exploited for biomarker development and therapeutic applications against colon cancer.
Keywords: Colorectal cancer, β-catenin, HI-B1, Small molecule inhibitor, Patient-derived xenograft, Precision medicine
Graphical Abstract
Highlights
-
•
HI-B1 is a synthesized compound identified as a β-catenin inhibitor suppressing the β-catenin-TCF4 protein interaction.
-
•
HI-B1 preferentially causes apoptosis in β-catenin-dependent cancer cells.
-
•
A colon cancer PDX mouse model with a high level of β-catenin is sensitive to HI-B1.
β-catenin is an important protein that facilitates colon cancer. Shin et al. synthesized and identified HI-B1 as a direct β-catenin inhibitor. HI-B1 disrupted formation of the β-catenin-TCF4 protein complex. HI-B1 preferentially caused apoptosis of cancer cells in which the survival is dependent on β-catenin. In a comparison of two colon cancer PDX models with different β-catenin levels, they showed that β-catenin-high PDX is more sensitive to HI-B1 treatment than β-catenin-low PDX. HI-B1 could thus be further developed as a colon cancer drug, and β-catenin expression levels might be a predictive biomarker for colon cancer therapy using β-catenin inhibitors.
1. Introduction
Colon cancer is the third most common cancer diagnosed in both men and women in the United State (Siegel et al., 2016). Although the incidence rate of colon cancer is decreasing due to the introduction of colonoscopy, and the removal of precancerous lesions, incidence and death rates are still increasing among people younger than 50 years old (Siegel et al., 2016). Chemotherapy of colon cancer has been heavily dependent on 5-fluorouracil (5-FU), a genotoxic drug that blocks DNA synthesis and replication. Since target-based intervention was introduced in cancer therapy, epidermal growth factor receptor (EGFR) inhibitors combined with 5-FU-based treatment have increased the overall survival rate of colorectal cancer patients with K-ras wild-type gene expression (Bokemeyer et al., 2009; Douillard et al., 2010). However, the elucidation of additional important targets, and the development of specific inhibitors are still challenging goals.
β-catenin is a central component of the Wnt signaling pathway, and aberrant expression or prolonged activation of β-catenin is frequently associated with various diseases, including cancer (Polakis, 2012; Clevers and Nusse, 2012; Kahn, 2014). The role of β-catenin and Wnt signaling in carcinogenesis has been studied extensively in cancer, especially in colorectal cancer. The expression level and the activity of β-catenin is tightly regulated by its upstream regulator, the destruction complex, which includes the tumor suppressor adenomatous polyposis coli (APC), glycogen synthase kinase 3β (GSK3β), casein kinase 1α (CK1α) and the scaffold protein AXIN (Gumbiner, 1997). When β-catenin is translocated from the cytosol to the nucleus, it binds with T-cell factor 4 (TCF4), to transcribe target genes such as axin2 (Leung et al., 2002), cyclin D1 (Shtutman et al., 1999) and c-myc (He et al., 1998).
The importance of the Wnt pathway in tumorigenesis has made it a promising target for drug development (Kahn, 2014). Over the past decade, the down-regulation of Wnt signaling in cancer cells was achieved by small molecules (Anastas and Moon, 2013). Compounds that target upstream of β-catenin include tankyrase inhibitors IWR-1 (Chen et al., 2009) and XAV939 (Huang et al., 2009) and casein kinase 1α activator pyrvinium (Thorne et al., 2010). These inhibitors facilitate β-catenin degradation by enhancing the activity of the β-catenin destruction complex. Direct inhibition of β-catenin by PKF115-584 (Sukhdeo et al., 2007; Lepourcelet et al., 2004) and CGP049090 (Lepourcelet et al., 2004) reduces target gene expression without affecting the protein expression level of β-catenin. Methyl 3-([(4-methylphenyl)sulfonyl]amino)benzoate (MSAB) was recently reported to target β-catenin and induce ubiquitination (Hwang et al., 2016). MSAB selectively killed Wnt-dependent cancer cells (Hwang et al., 2016). BC2059 (Fiskus et al., 2015), LF3 (Fang et al., 2016), PKF118-310 (Lepourcelet et al., 2004), PKF118-744 (Lepourcelet et al., 2004), PKF222-815 (Lepourcelet et al., 2004), ZTM000990 (Lepourcelet et al., 2004), iCRT3 (Gonsalves et al., 2011), iCRT5 (Gonsalves et al., 2011), iCRT14 (Gonsalves et al., 2011), ZINC02092166 (Catrow et al., 2015), and henryin (Li et al., 2013) also directly inhibit β-catenin (Supplementary Fig. 1). Despite these efforts, the effectiveness of Wnt/β-catenin inhibitors in clinical trials is yet to be determined, and strategies to identify patients who will respond to the inhibitors are still largely elusive (Kahn, 2014).
The patient-derived xenograft (PDX) model comprises a surgically dissected clinical tumor sample that is implanted into immuno-deficient mice (Byrne et al., 2017). Unlike established cell lines that are cultured in vitro for many passages, the PDX tumor is believed to recapitulate tumor heterogeneity and, thus, better reflects the features of the original human cancer (Aparicio et al., 2015; Hidalgo et al., 2014). In particular, the PDX model has become a valuable tool to test small molecules with anti-cancer activities in drug discovery and biomarker development (Aparicio et al., 2015; Cho et al., 2016). Although the PDX model can be perceived as time-consuming to establish and might be highly variable, drug responses obtained from PDX mice are highly consistent with responses observed in human patients (Byrne et al., 2017). For example, the overall response rates of EGFR antibodies in PDX colorectal cancer studies were similar to those found in the clinic (Bertotti et al., 2011; Cunningham et al., 2004). A comparative analysis of EGFR antibody sensitivities in PDX models (Bardelli et al., 2013) and patient (Kawazoe et al., 2015) populations in different studies revealed that the response rate in PDX can reflect the clinical outcome (Byrne et al., 2017).
Herein, we synthesized a small molecule, referred to as HI-B1, and report that the small molecule shows an inhibitory effect against β-catenin/TCF4 luciferase activity in colon cancer cells. HI-B1 preferentially causes apoptosis of cancer cells in which the survival is dependent on β-catenin. The inhibition of the β-catenin/TCF4 pathway by HI-B1 resulted in apoptosis, and binding assays show that β-catenin is a direct target protein of HI-B1. HI-B1 disrupts the interaction between β-catenin and TCF4 in vitro and ex vivo. A PDX colon cancer study showed that HI-B1 inhibits the growth of tumors with a high expression level of β-catenin, but was not effective against tumors with a low level of β-catenin. We anticipate that these preclinical results will provide the groundwork for the translational research of β-catenin inhibitors, and the development of biomarkers to match patients with specific inhibitors in the era of precision oncology.
2. Materials and Methods
2.1. Reagents
All antibodies were commercially available, including β-catenin, β-actin, cyclin D1, α-tubulin and GST from Santa Cruz Biotechnology (Santa Cruz, CA), c-Myc and TCF4 from Cell Signaling Technology (Beverly, MA), lamin B from Calbiochem (San Diego, CA). The luciferase assay substrate and the Cell Titer 96 Aqueous One Solution Reagent [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS)] kit were from Promega (Madison, WI).
2.2. Synthesis of 4-(5-Fluoro-1H-Benzo[d]Imidazol-2-yl)-N, N-Dimethylaniline (HI-B1)
To a solution of 4-(Dimethylamino) benzaldehyde (2.98 g, 20 mmol) in ethanol (40 ml), Sodium dithionite (3.48 g, 20 mmol) in water (20 ml) was added. The resulting mixture was stirred at room temperature for 20 min and then 4-Fluoro-1,2-phenylenediamine (2.52 g, 20 mmol) was added portion wise. The resulting reaction mixture was refluxed for about 14 h. The solution was then diluted with water. The obtained brown precipitate was filtered and purified by recrystallization from ethanol to give 2.5 g of 4-(5-fluoro-1H-benzo[d]imidazol-2-yl)-N, N-dimethylaniline. Nuclear magnetic resonance (NMR) spectra were determined in DMSO using a Varian instrument (1H, 400 MHz). 1H NMR (400 MHz, DMSO-d6) δ 7.97 (d, J = 8.8 Hz, 2H), 7.50 (dd, J = 8.4, 4.8 Hz, 1H), 7.32 (dd, J = 9.1, 2.4 Hz, 1H), 7.02 (td, J = 8.6, 2.4 Hz, 1H), 6.85 (d, J = 8.8 Hz, 2H), 3.01 (s, 6H). Electrospray ionisation mass spectrometry (ESI-MS) calculated (M + H) + 256.1172, found 256.1294.
2.3. Cell Culture
All cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA) except for LIM1215 from Sigma-Aldrich (St. Louis, MO). Cells were comprehensively authenticated by PCR using human-specific primers and morphology checks through microscopes before being frozen as recommended (Almeida et al., 2016), and free of mycoplasma from Hoechest 33258 staining (Chen, 1977; Marx, 2014). Each vial of frozen cells was thawed and used in experiments for a maximum of 8 weeks. Cells were cultured in RPMI 1640 (DLD1, LIM1215, H838) or MEM (CACO2, LS174T, WiDr, CCD-18Co) or McCoy's 5A (HCT116, HT29) or DMEM (293 T) containing penicillin (100 units/ml), streptomycin (100 μg/ml), and 10% fetal bovine serum (FBS; Gemini Bio-Products, Calabasas, CA). The NIH3T3 cell line was cultured in DMEM with 10% calf serum (CS; Gemini Bio-Products). All cells were incubated at 37 °C with 5% CO2.
2.4. Luciferase Assay
Transient transfection was performed using jetPEI (Promega). Assays to detect firefly luciferase and Renilla activities were performed according to the manufacturer's manual (Promega). Briefly, cells were seeded onto 10-cm plates and co-transfected with 400 ng of the Renilla luciferase internal control gene and 4 μg of the TOPFlash luciferase reporter construct containing three tandem Tcf consensus binding sites upstream of luciferase cDNA, or the FOPFlash luciferase reporter construct, a plasmid with mutated Tcf binding sites. After 16 h of transfection, cells were trypsinized and seeded onto 48-well plates, and then treated with respective compounds for 24 h. Luciferase and Renilla activities were measured using their respective substrates.
2.5. MTS Assay
Cells were seeded (1 × 103 cells/well) in 96-well plates, incubated overnight and then treated with different doses of HI-B1. After incubation for 2 days, 20 μl of CellTiter96 Aqueous One Solution (Promega) were added and then cells were incubated for 1 h at 37 °C in a 5% CO2 incubator. Absorbance was measured at 492 nm.
2.6. Lentivirus Production and Infection
The shRNA of β-catenin clones (sh-ctnnb1 #1: TRCN0000003843, AGGTGCTATCTGTCTGCTCTA; #3: TRCN0000003845, GCTTGGAATGAGACTGCTGAT; #5: TRCN0000010824, CCATTGTTTGTGCAGCTGCTT) were purchased from University of Minnesota Genomics Center (Minneapolis, MN). The lentiviruses were generated from 293T packaging cells. The cell culture supernatant fraction containing lentivirus was harvested from 293T cells at 24 or 48 h after transfection. The cells were infected with the lentiviruses in the presence of 10 μg/ml of polybrene (Sigma Aldrich, St. Louis, MO). After 24 h of infection, the cells were treated with puromycin for selection, and then pooled for subsequent analysis.
2.7. Flow Cytometry for Analysis of Apoptosis
For analysis of apoptosis, cells (1.5 × 105 cells per well) were seeded into six-well plates and cultured overnight, then exposed to 50 μM HI-B1 for 48 h. Cells were trypsinized, and washed twice with cold PBS. They were then resuspended with PBS and incubated for 5 min at room temperature with annexin V-FITC plus propidium iodide. Cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA).
2.8. Anchorage-independent Cell Transformation Assay
Cells (8 × 103/well) suspended in 10% Basal Medium Eagle (Sigma Aldrich) were added to 0.3% agar with different doses of HI-B1 in a top layer over a base layer of 0.5% agar with the same concentration of HI-B1. The cultures were maintained at 37 °C in a 5% CO2 incubator for 1 wk. and then colonies were counted under a microscope using the Image-Pro Plus software (v.6.1) program (Media Cybernetics, Rockville, MD).
2.9. 7-Day Proliferation Assay
CACO2 cells were seeded into 6-well plates (2 × 105 cells/well) and cultured with or without different doses of HI-B1. After 7 days of incubation, cells were fixed with 4% paraformaldehyde (in PBS), and then stained with 0.5% crystal violet (in water) (Sun et al., 2014).
2.10. Foci Formation Assay
Transformation of NIH3T3 cells was performing following standard protocols. Cells were transiently transfected with combinations of pcDNA3-H-RasG12V (100 ng), pcDNA3-β-catenin (1 μg) and pcDNA3-mock (as compensation to achieve equal amounts of DNA) plasmids and incubated overnight. Then the cells were cultured in 5% CS-DMEM with or without HI-B1 for 2 wk. The media were changed every other day. Foci were fixed and stained with 0.5% crystal violet (Clark et al., 1995).
2.11. Western Blot Analysis
Cells were disrupted on ice for 30 min in cell lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM sodium vanadate, and 1 mM PMSF [phenylmethylsulfonyl fluoride]) with protease inhibitor cocktail (Roche, Switzerland). The supernatant fraction was harvested after centrifugation at 13,000 rpm for 10 min. Protein concentrations were determined with the Bio-Rad protein assay reagent (Richmond, CA). For nuclear and cytosol fractions, NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, Waltham, MA) were used according to the manufacturer's protocol. The protein lysates were separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes in 20 mM Tris-HCl (pH 8.0), containing 150 mM glycine and 15% (v/v) methanol. Membranes were blocked with 5% non-fat dry milk in 1 × TBS containing 0.05% Tween 20 (TBS-T) and incubated with primary antibodies at 4 °C overnight. Protein bands were visualized by the LAS4000 Biomolecular Imager (GE Healthcare, United Kingdom) and Western Lightning Plus ECL (Perkin Elmer, Waltham, MA) after incubation with appropriate secondary antibodies.
2.12. Quantitative PCR (qRT-PCR)
Total RNA from cells was extracted using RNeasy Plus Mini Kit (Qiagen, Valencia, CA) following the manufacturer's instructions. The reverse transcription reaction was performed with the amfiRivert cDNA Synthesis Platinum Master Mix (GenDepot, Barker, TX). Expression of the indicated genes was assessed with a 7500 real time PCR system (Applied Biosystems, Carlsbad, CA) using the Power SYBR Green Master Mix (Life Technology, Grand Island, NY). Reaction plates were incubated in a 96-well thermal cycling plate at 95 °C for 10 min and then underwent 40 cycles of 15 s at 95 °C and 1 min at 59 °C. All reactions were performed in triplicate. Relative quantitation (RQ) was calculated using the 2−ΔCt method, where ΔCt symbolizes the change in Ct between the sample and reference mRNA. The following primers were used to detect expression: GAPDH, 5′-AGCCACATCGCTCAGACAC-3′ (forward), 5′-GCCCAATACGACCAAATCC-3′ (reverse); cyclin D1, 5′-AGCTCCTGTGCTGCGAAGTGGAAAC-3′ (forward), 5′-AGTGTTCAATGAAATCGTGCGGGGT-3′ (reverse); AXIN2, 5′-CTGGCTCCAGAAGATCACAAAG-3′ (forward) and 5′-ATCTCCTCAAACACCGCTCCA-3′ (reverse); β-catenin, 5′-ACCTTTCCCATCATCGTGAG-3′ (forward), 5′-AATCCACTGGTGAACCAAGC-3′ (reverse).
2.13. Protein Purification
A construct comprising residues 126–686 of human β-catenin (AB451264) was sub-cloned into pET-46 Ek/LIC vector (EMD Millipore). The recombinant His-fusion β-catenin was expressed in E. coli BL21 (DE3) and purified by affinity chromatography using nickel-nitrilotriacetic acid-agarose (HisPur™ Ni-NTA, Thermo Scientific). The protein was eluted from the resin with 250 mM imidazole, 150 mM NaCl, 20 mM Tris, pH 8.0, and purified further on a gel-filtration HiLoad 16/60 Superdex-200 column (GE Healthcare) equilibrated with buffer (150 mM NaCl, 20 mM CAPS, pH 10.5).
2.14. Ex Vivo Pull-Down Assays
Cell lysates (500 μg) were incubated with HI-B1-Sepharose 4B (or Sepharose 4B only as a control) beads (50 μl 50% slurry) in reaction buffer (50 mM Tris, pH 7.5, 5 mM EDTA, 150 mM NaCl, 1 mM DTT, 0.01% NP-40, and 2 mg/ml bovine serum albumin). After incubation with gentle rocking overnight at 4 °C, the beads were washed 3 times with buffer (50 mM Tris, pH 7.5, 5 mM EDTA, 150 mM NaCl, 1 mM DTT, and 0.01% NP-40) and the binding proteins were visualized by Western blotting.
2.15. Co-Immunoprecipitation Assay
Cells were treated with the indicated concentrations of HI-B1 for 24 h and then disrupted with lysis buffer. Immunoprecipitation was performed by incubating overnight with anti-β-catenin. Protein G Agarose beads (50 μl) (GenDepot, Barker, TX) were added and the solution was incubated for 6 h at 4 °C. The beads were washed 3 times with lysis buffer. Bound proteins were harvested and visualized by Western blotting.
2.16. In Silico Molecular Docking
The Glide module from Schrödinger Suite 2011 was used for prediction of a small molecule-protein binding mode. A crystal structure of a human β-catenin structure (PDB ID:1JPW) was downloaded from Protein Data Bank. The binding pocket was selected around the K312 and K435 residues, which are reported to be important in the β-catenin/TCF4 binding mode. The XP (extra precision) docking was repeated > 30 times, and the most frequent outcomes of β-catenin in chemical-protein interaction were selected as possible binding residues.
2.17. Animal Experiments
All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committees of the University of Minnesota (Minneapolis, MN) and Zhengzhou University (Zhengzhou, Henan, China).
2.18. Patient-derived Xenografts (PDX)
Fresh tumor tissue fragments were collected from colon cancer patients and transferred at 4 °C in FBS-free RPMI 1640 medium with antibiotics. Tumor tissues were trimmed into 3–5 mm sizes within 2 h after surgical resection, and implanted subcutaneously in 6 to 8 week-old female SCID mice (Vital River Laboratories Co., Ltd., Beijing, China). Once mass formation reached 1500 mm3, mice of this first generation of xenografts (named P1) were sacrificed and the tumors were passaged and expanded for 3 more generations (P2 and P3). When P4 tumors reached an average volume of 50 mm3, mice were divided into different groups (n = 8/group) and treated with vehicle or HI-B1 50 mg/kg, respectively, 3 times a week by oral gavage. HI-B1 was dissolved in 5% DMSO and 5% PEG containing PBS. Mice were sacrificed at the end of the experiment and tumors extracted. Tumor volume was calculated using the following formula: tumor volume (mm3) = (length × width × height × 0.5). These studies were approved by the Ethics Committee of Zhengzhou University. Patients whose tumors were used in these studies were completely informed and provided consent.
2.19. APCmin Mouse Model
C57BL⁄6J-ApcMin/+ mice (APCmin mice, RRID: IMSR_JAX:002020) were purchased from the Jackson Laboratory (Bar Harbor, ME), bred and genotyped according to the manufacturer's protocol. Male 6-week-old mice were treated with vehicle or 2 or 10 mg/kg/day of HI-B1 by oral gavage for 10 weeks (n = 6/group). At the end of the experimental period, the intestine and colon were removed, opened longitudinally, and fixed flat between filter paper sheets in 10% buffered formalin overnight. These sections were stained with a 0.2% methylene blue (Sigma-Aldrich) solution, and the mucosal surfaces were assessed for counting polyps with a stereoscopic microscope. One centimetre-long sections were prepared from the same position of ileum across the groups, and mRNA was extracted for further real-time PCR analysis.
2.20. Immunohistochemistry
Colon cancer patients-derived xenograft samples were fixed in 10% formalin overnight, embedded in paraffin, and sectioned at 5 μm of thickness. The sections were stained with haematoxylin and eosin (H&E) according to standard protocols. Immunohistochemistry staining for β-catenin and c-Myc was performed using an ABC complex kit (PK-6100, Vector Laboratories, Burlingame, CA). Harris's haematoxylin was used for counter staining. The staining intensity was quantified by calculating the integrated optical density (IOD) using the Image Pro-Plus 6.1 software program (Media Cybernetics) (Li et al., 2015).
2.21. Statistical Analysis
All quantitative results are expressed as mean values ± standard deviation. Statistically significant differences were obtained by one-way ANOVA. A p-value of < 0.05 was considered to be statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001).
3. Results
3.1. HI-B1 Preferentially Attenuates Growth and Induces Apoptosis of Cancer Cells in Which β-Catenin is Essential for Survival
HI-B1 (patent number: US9616047) was rationally designed by cyclization of resveratrol (Fig. 1a). Based on the effect of resveratrol against the Wnt/β-catenin pathway (Chen et al., 2012), we sought to change its structure and make small molecules with strong inhibitory effects on the pathway. HI-B1 inhibited β-catenin/TCF4 luciferase activity in a dose-dependent manner in two different colon cancer cell lines (Fig. 1b). Because the inhibition of Wnt/β-catenin activity can reportedly kill cancer cells (Lepourcelet et al., 2004; Sukhdeo et al., 2007; Chen et al., 2009; Grossmann et al., 2012; Hao et al., 2013; Li et al., 2013; Fang et al., 2016; Jang et al., 2015), the effect of HI-B1 on cell growth and apoptosis was investigated. HI-B1 treatment suppressed growth of several types of colon cancer cells (DLD1, CACO2 and HCT116), but did not affect the H838 lung cancer cell line (Fig. 1c). As previously reported (Licchesi et al., 2008), H838 expressed a low level of β-catenin compared to other cancer cell lines (Fig. 1d). Based on these findings, the specificity of HI-B1 on the β-catenin pathway was further examined. By using short hairpin RNA that target β-catenin, we were able to find cell lines in which the survival is dependent on β-catenin. Knockdown of β-catenin resulted in apoptosis in DLD1, CACO2 and HCT116 cells, whereas H838 and CCD-18Co (normal colon epithelial cell line) cells, were only marginally affected by the knockdown (Fig. 1e). HI-B1 treatment also preferentially resulted in more apoptosis in DLD1, CACO2 and HCT116 cell lines, compared to H838 and CCD-18Co cells (Fig. 1f).
Fig. 1.

HI-B1 is a small molecule that preferentially kills cancer cells in which β-catenin is critical for survival.
(a) Chemical structure of HI-B1.
(b) DLD1 (left panel) and CACO2 (right panel) cells were transfected with TOPFlash, which contains TCF4 binding sequences, or FOPFlash (as a negative control) and then treated with different doses of HI-B1 for 24 h. Firefly luciferase activity was normalized with Renilla luciferase activity.
(c) Cells were treated with HI-B1 at different doses for 48 h and then viability was assessed by MTS assay.
(d) Lysates of nine different cell lines were prepared and β-catenin expression was detected by western blots.
(e) Cells were incubated for 48 h after the shRNA infection and antibiotic selection. Apoptosis was measured by annexin V/propidium iodide staining and flow cytometry.
(f) DLD1, CACO2, HCT116, H838 and CCD-18Co cell lines were treated with HI-B1. Apoptosis was measured after 48 h of treatment with flow cytometry.
All values of graphs present mean ± standard deviation (SD) (in triplicate). Significant differences were calculated by one-way ANOVA compared with DMSO-treated group or sh-mock group (*P < 0.05, **P < 0.01, ***P < 0.001).
3.2. HI-B1 Inhibits Anchorage-independent Growth of Colon Cancer Cells by Reducing β-Catenin Target Gene Expression
β-catenin is a primary driver in colon cell transformation (Kolligs et al., 2002), therefore the effects of HI-B1 on cancer cells were examined using soft agar, a measure of cell transformation and anchorage-independent growth of cancer cells. Results indicated that HI-B1 treatment significantly inhibited anchorage-independent growth of DLD1 and HT-29 colon cancer cells (Fig. 2a). The growth of CACO2, another colon cancer cell line, was also suppressed by HI-B1 in a 7-day proliferation assay (Fig. 2b). To further confirm the effect of HI-B1 on the β-catenin pathway, an H-ras/β-catenin-induced foci formation assay was conducted. According to previous studies (Espada et al., 2009; Damalas et al., 2001), constitutively active Ras facilitates nuclear translocation of β-catenin to induce cell transformation. In agreement with these findings (Damalas et al., 2001), foci formation of NIH3T3 cells was substantially induced by co-transfection of H-ras and β-catenin, but not by single gene transfection (Fig. 2c). Notably, HI-B1 attenuated H-ras/β-catenin-induced foci formation. These results reveal that HI-B1 inhibits anchorage-independent growth and foci formation, where β-catenin plays an important role.
Fig. 2.

HI-B1 inhibits colorectal cancer cell growth by suppressing β-catenin activity.
(a) Cells were seeded into 0.3%-agar-containing medium with various concentrations of HI-B1 and the numbers of colonies was counted at Day 7.
(b) Cells were seeded on 6-well plates with various concentrations of HI-B1 and then stained with crystal violet solution at Day 7.
(c) Foci formation was induced by co-transfection of β-catenin and H-ras (H12V) in NIH3T3. After transfection, cells were treated with HI-B1 and stained with crystal violet solution (Day 14). The medium was changed every other day.
(d and e) After 24 h of HI-B1 treatment, mRNA (d) and protein (e) were harvested from the cells and relative amount was measured by qRT-PCR and Western blotting, respectively.
(f) The cytosol and the nucleus portions of the cells were fractionated at 24 h of HI-B1 treatment. Lamin B and α-tubulin were used as a cytosol and a nucleus marker, respectively.
All values of graphs present mean ± SD (in triplicate). Significant differences were calculated by one-way ANOVA compared with DMSO-treated group (*P < 0.05, **P < 0.01, ***P < 0.001).
We next focused on how HI-B1 affected the Wnt/β-catenin signaling pathway. Quantitative real-time PCR results showed that HI-B1 treatment decreased mRNA expression of cyclin D1 and axin2 in DLD1 and CACO2 cell lines (Fig. 2d). HI-B1 also down-regulated cyclin D1 and c-Myc protein levels (Fig. 2e). Of particular note, the β-catenin protein level was not changed by HI-B1, either at the protein or mRNA level. This suggests that HI-B1 could inhibit the nuclear translocation of β-catenin (Jamieson et al., 2014) or directly bind to β-catenin and suppress its activity (Kahn, 2014). To study this issue further, HI-B1 treated and untreated cytosolic and nuclear fractions were examined, and results showed that nuclear translocation of β-catenin was not altered by the compound, which suggested a direct-binding mechanism of action (Fig. 2f).
3.3. HI-B1 Directly Binds With β-Catenin, but not TCF4, to Disrupt the β-Catenin/TCF4 Interaction
To determine whether HI-B1 binds to β-catenin directly, HI-B1 was conjugated to sepharose 4B-beads and incubated with cell lysates. β-Catenin was detected by Western blot and results showed that HI-B1 directly binds with β-catenin (Fig. 3a). Notably, TCF4, which is a binding partner of β-catenin, and with which it forms a complex for inducing gene transcription, was not bound to HI-B1. The interaction between β-catenin and TCF4 has been implicated as a core element of the Wnt pathway in colorectal cancer cell growth (Segditsas and Tomlinson, 2006). The binding of HI-B1 with β-catenin disrupted the interaction between β-catenin and TCF4 in vitro (Fig. 3b). His-tagged β-catenin and GST-tagged TCF4 were incubated with or without HI-B1, and His-β-catenin was pulled down with nickel beads. The amount of TCF4 co-immunoprecipitated with β-catenin was decreased in HI-B1 treated groups. The effect of HI-B1 against β-catenin/TCF4 complex formation was also confirmed in DLD1 and CACO2 cell lines (Fig. 3c). These results demonstrate that HI-B1 directly binds with β-catenin and disrupts the β-catenin/TCF4 interaction.
Fig. 3.

HI-B1 directly targets β-catenin and disrupts β-catenin/TCF4 complex formation.
(a) HI-B1 directly binds with β-catenin. HI-B1 was conjugated with Sepharose 4B beads and incubated with cell lysates. Protein co-immunoprecipitated with the beads was analyzed by Western blotting.
(b) HI-B1 disrupts the formation of the β-catenin/TCF4 complex in vitro. His-tagged β-catenin (126–686) was immunoprecipitated with nickel beads and the amount of TCF4 was detected by Western blotting.
(c) DLD1 and CACO2 cell lysates were harvested after 24 h of HI-B1 treatment. β-Catenin was immunoprecipitated and binding proteins was detected by Western blotting.
(d) Molecular docking predicts the interaction of HI-B1 and β-catenin. A nitrogen atom of HI-B1 forms a hydrogen bond with β-catenin (upper panel) from Extra precision (XP) docking in silico (lower panel).
(e and f) HI-B1 and HI-B1-NC (a nitrogen atom was substituted with a carbon) were treated to DLD1 cells (n = 3). Proliferation was measured by MTS assay at 48 h (e), and β-catenin/TCF4 luciferase activity was measured by luminometer at 24 h (f). Significant difference with DMSO-treated group by One-way ANOVA (*P < 0.05, **P < 0.01, ***P < 0.001).
We next conducted in silico docking, to predict the important binding residues of HI-B1. A nitrogen atom was found to be responsible for a hydrogen bond with β-catenin (Fig. 3d). HI-B1-NC, a derivative of HI-B1 in which the nitrogen atom (N) is substituted with a carbon atom (C), was synthesized, and its biological activities were compared with HI-B1. The N to C substitution completely attenuated the effect of HI-B1 against DLD1 colon cancer cell growth (Fig. 3e). The inhibitory effect on β-catenin/TCF4 luciferase activity was also eliminated by the substitution (Fig. 3f). Overall, the nitrogen atom of HI-B1was found to be important for its direct binding with β-catenin.
3.4. HI-B1 Suppresses Polyp Formations in APCmin Model and Decreases the Growth of Patient-derived Xenograft (PDX) Tumors With a High Expression Level of β-Catenin
To verify the efficacy of HI-B1 in vivo, a PDX colon cancer model was utilized. Tumor fragments were from two different patients (JG5 and JG14) and immunohistochemistry analysis of the two PDX tumors indicated a difference in β-catenin expression. JG5 had a high level of β-catenin expression, compared to JG14 (Fig. 4a). The effect of HI-B1 was examined on each of the two colon cancer PDX models to determine whether the expression level of β-catenin could be a predictive marker for sensitivity to a β-catenin inhibitor. Interestingly, HI-B1 treatment reduced the weight and volume of the JG5 PDX tumor, but had no effect on the JG14 PDX tumor (Fig. 4b, c). Although the efficacy of HI-B1 against only two PDX tumors with differential levels of β-catenin expression was examined, these findings suggest that tumors with higher levels of β-catenin might be more sensitive to β-catenin inhibitor treatment. The decrease in c-myc expression by HI-B1 was confirmed by immunohistochemistry (Fig. 4d). Further studies testing HI-B1 in more PDX tumors will be needed.
Fig. 4.

HI-B1 inhibits β-catenin-driven tumorigenesis in vivo.
(a) β-Catenin expression levels in colon cancer PDX samples were examined by immunohistochemistry (IHC) and its integrated optical density (IOD) was quantified using Image-Pro Plus software (v.6.1) program (Media Cybernetics, Bethesda, MD). Scale bar: 50 μm.
(b and c) HI-B1 was administered per os (p.o) (orally) three times a week at a dose of 50 mg/kg (n = 8/group) to JG5 (b) and JG14 (c) tumors. Tumor size was measured three times a week and the weight was measured at the end of experiments. (One-way ANOVA and Dunnett's test; *P < 0.05, significant difference compared to the vehicle group.)
(d) In the JG5 PDX tumor, c-Myc Expression level change by HI-B1 treatment was measured by IHC (same as Fig. 4A). Scale bar: 50 μm.
(e) HI-B1 reduced polyp formation driven by aberrant activation of β-catenin in APCmin mouse model. The indicated doses of HI-B1 were administered p.o. for 10 weeks, and mice were sacrificed for analysis of intestinal carcinogenesis (n = 6/group).
(f and g) Small intestine tissue was prepared from the same position of ileum from mice, and then mRNA expression levels of c-myc (f) and cyclin D1 (g) were quantified by qRT-PCR. (One-way ANOVA; *P < 0.05, **P < 0.01, significant difference compared to the vehicle group.)
To further confirm the in vivo efficacy of HI-B1, the APCMin mouse model was utilized. This mouse model exhibits aberrant activation of β-catenin, and HI-B1 treatment reduced the number of polyps (Fig. 4e). In addition, the mRNA expression levels of c-myc and cyclin D1 (Fig. 4f, g) were also decreased. Overall, these results indicate that HI-B1 inhibits β-catenin-driven tumorigenesis in vivo.
4. Discussion
Most cases of sporadic colon cancer are caused by genetic mutations of several Wnt pathway proteins, including APC, axin and β-catenin, resulting in stabilization of β-catenin and the formation of the β-catenin/TCF4 complex (Clevers and Nusse, 2012; Korinek et al., 1997). Although a number of inhibitors against the Wnt pathway and β-catenin have already been identified (Catrow et al., 2015; Fang et al., 2016; Fiskus et al., 2015; Gonsalves et al., 2011; Lepourcelet et al., 2004; Li et al., 2013), none of them have been approved by the FDA for the treatment of colon cancer (Kahn, 2014). Thus, the development of specific β-catenin inhibitors is still valuable. In this study, we identified HI-B1 as an inhibitor of β-catenin/TCF4 luciferase activity (Fig. 1b), which could be promising for developing drugs that target the β-catenin pathway. The molecular mechanisms of HI-B1 were investigated, and we found that it directly binds β-catenin, and disrupts the β-catenin/TCF4 interaction (Fig. 3b, c). Based on these promising in vitro results, we examined its efficacy in vivo. We found that a PDX tumor expressing high levels of β-catenin was more sensitive to β-catenin inhibitor treatment, as compared to another PDX tumor expressing lower levels of β-catenin (Fig. 4b, c). Thus, we showed that a β-catenin inhibitor could attenuate the growth of a colon cancer patient-derived tumor, and that sensitivity to the inhibitor might be dependent on the expression level of β-catenin.
HI-B1 has a unique structure, as compared to other previously reported β-catenin/TCF4 inhibitors (Supplementary Fig. 1), and we examined the therapeutic potential of this small molecule. The relatively small molecular weight (255.3) of HI-B1 provides opportunities to further develop HI-B1 analogues. Using several biological approaches, we showed that HI-B1 specifically inhibited the β-catenin pathway. Importantly, the efficacy of the HI-B1 in a preclinical cancer model, the colon cancer PDX model, was verified. Our data suggest that HI-B1 is a β-catenin/TCF4 interaction inhibitor, and can be used for the drug development pipeline.
Our study provides a rationale for examining the β-catenin levels in colon cancer patients before Wnt/β-catenin inhibitors are prescribed. The significant differences observed in β-catenin expression levels between different colon cancer patients (Fig. 4a) were rather unexpected, regarding the well-known importance of β-catenin in colorectal cancer (Kahn, 2014). Similar to HER2 (human epidermal growth factor receptor 2)-targeted drugs, such as lapatinib and trastuzumab, which are recommended only to HER2-positive breast cancer patients (Arteaga et al., 2011), HI-B1 was only effective in a PDX model where β-catenin is highly expressed (Fig. 4b, c). These results suggest that the β-catenin expression level might be correlated with the efficacy of Wnt/β-catenin inhibitors against colon cancer. Such information could be further exploited for biomarker development, and therapeutic applications against colon cancer, especially in the ongoing clinical trials of Wnt pathway inhibitors.
The following is the supplementary data related to this article.
Chemical structures of HI-BI and β-catenin inhibitors previously reported. MSAB: Methyl 3-([(4-methylphenyl)sulfonyl]amino)benzoate.
Funding Sources
The research was supported by The Hormel Foundation and National Institutes of Health (R01-CA187027). S.H.S. was supported by a Bioinformatics and Computational Biology fellowship and a Doctoral Dissertation Fellowship from the University of Minnesota.
Conflicts of Interest
None of the authors have any financial conflict of interest that might be construed to influence the results or interpretation of this manuscript. Funders played no role in study design, data collection, data analysis, interpretation or writing of this report.
Author Contributions
S.H.S and Z.D. designed overall experiments. S.H.S. performed most of experiments. D.Y.L. helped with immunoprecipitation and APCmin mouse model. K.R. designed and synthesized HI-B1 and HI-B1-NC. M.M. purified and provided recombinant β-catenin. F.L, T.W, and M.S. performed PDX studies supervised by M.L and K.L. H.C. helped with research design and in silico docking. K.B.B. and J.R. helped with experiments and data analysis. S.H.S. and A.M.B wrote the manuscript. Z.D. supervised overall progress of the project.
Acknowledgments
Acknowledgements
We thank Todd Schuster for helping with flow cytometry, Ruihua Bai for helping with the evaluation of IHC slides, Dhilli Rao Gorja for helping with NMR, and Tia Rai and Nicki Brickman-Van Erkel for critical reading, and assisting with the manuscript submission.
References
- Almeida J.L., Cole K.D., Plant A.L. Standards for cell line authentication and beyond. PLoS Biol. 2016;14 doi: 10.1371/journal.pbio.1002476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastas J.N., Moon R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- Aparicio S., Hidalgo M., Kung A.L. Examining the utility of patient-derived xenograft mouse models. Nat. Rev. Cancer. 2015;15:311–316. doi: 10.1038/nrc3944. [DOI] [PubMed] [Google Scholar]
- Arteaga C.L., Sliwkowski M.X., Osborne C.K., Perez E.A., Puglisi F., Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat. Rev. Clin. Oncol. 2011;9:16–32. doi: 10.1038/nrclinonc.2011.177. [DOI] [PubMed] [Google Scholar]
- Bardelli A., Corso S., Bertotti A., Hobor S., Valtorta E., Siravegna G., Sartore-Bianchi A., Scala E., Cassingena A., Zecchin D., Apicella M., Migliardi G., Galimi F., Lauricella C., Zanon C., Perera T., Veronese S., Corti G., Amatu A., Gambacorta M., Diaz L.A., Jr., Sausen M., Velculescu V.E., Comoglio P., Trusolino L., Di Nicolantonio F., Giordano S., Siena S. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3:658–673. doi: 10.1158/2159-8290.CD-12-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertotti A., Migliardi G., Galimi F., Sassi F., Torti D., Isella C., Cora D., Di Nicolantonio F., Buscarino M., Petti C., Ribero D., Russolillo N., Muratore A., Massucco P., Pisacane A., Molinaro L., Valtorta E., Sartore-Bianchi A., Risio M., Capussotti L., Gambacorta M., Siena S., Medico E., Sapino A., Marsoni S., Comoglio P.M., Bardelli A., Trusolino L. A molecularly annotated platform of patient-derived xenografts ("xenopatients") identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1:508–523. doi: 10.1158/2159-8290.CD-11-0109. [DOI] [PubMed] [Google Scholar]
- Bokemeyer C., Bondarenko I., Makhson A., Hartmann J.T., Aparicio J., De Braud F., Donea S., Ludwig H., Schuch G., Stroh C., Loos A.H., Zubel A., Koralewski P. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J. Clin. Oncol. 2009;27:663–671. doi: 10.1200/JCO.2008.20.8397. [DOI] [PubMed] [Google Scholar]
- Byrne A.T., Alferez D.G., Amant F., Annibali D., Arribas J., Biankin A.V., Bruna A., Budinska E., Caldas C., Chang D.K., Clarke R.B., Clevers H., Coukos G., Dangles-Marie V., Eckhardt S.G., Gonzalez-Suarez E., Hermans E., Hidalgo M., Jarzabek M.A., De Jong S., Jonkers J., Kemper K., Lanfrancone L., Maelandsmo G.M., Marangoni E., Marine J.C., Medico E., Norum J.H., Palmer H.G., Peeper D.S., Pelicci P.G., Piris-Gimenez A., Roman-Roman S., Rueda O.M., Seoane J., Serra V., Soucek L., Vanhecke D., Villanueva A., Vinolo E., Bertotti A., Trusolino L. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer. 2017;17:254–268. doi: 10.1038/nrc.2016.140. [DOI] [PubMed] [Google Scholar]
- Catrow J.L., Zhang Y., Zhang M., Ji H. Discovery of selective small-molecule inhibitors for the beta-catenin/T-cell factor protein-protein interaction through the optimization of the acyl hydrazone moiety. J. Med. Chem. 2015;58:4678–4692. doi: 10.1021/acs.jmedchem.5b00223. [DOI] [PubMed] [Google Scholar]
- Chen T.R. In situ detection of mycoplasma contamination in cell cultures by fluorescent Hoechst 33258 stain. Exp. Cell Res. 1977;104:255–262. doi: 10.1016/0014-4827(77)90089-1. [DOI] [PubMed] [Google Scholar]
- Chen B., Dodge M.E., Tang W., Lu J., Ma Z., Fan C.W., Wei S., Hao W., Kilgore J., Williams N.S., Roth M.G., Amatruda J.F., Chen C., Lum L. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009;5:100–107. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.J., Hsu L.S., Shia Y.T., Lin M.W., Lin C.M. The beta-catenin/Tcf complex as a novel target of resveratrol in the Wnt/beta-catenin signaling pathway. Biochem. Pharmacol. 2012;84:1143–1153. doi: 10.1016/j.bcp.2012.08.011. [DOI] [PubMed] [Google Scholar]
- Cho S.Y., Kang W., Han J.Y., Min S., Kang J., Lee A., Kwon J.Y., Lee C., Park H. An integrative approach to precision cancer medicine using patient-derived xenografts. Mol. Cell. 2016;39:77–86. doi: 10.14348/molcells.2016.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark G.J., Cox A.D., Graham S.M., Der C.J. Biological assays for Ras transformation. Methods Enzymol. 1995;255:395–412. doi: 10.1016/s0076-6879(95)55042-9. [DOI] [PubMed] [Google Scholar]
- Clevers H., Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Cunningham D., Humblet Y., Siena S., Khayat D., Bleiberg H., Santoro A., Bets D., Mueser M., Harstrick A., Verslype C., Chau I., Van Cutsem E. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 2004;351:337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- Damalas A., Kahan S., Shtutman M., Ben-Ze'ev A., Oren M. Deregulated beta-catenin induces a p53- and ARF-dependent growth arrest and cooperates with Ras in transformation. EMBO J. 2001;20:4912–4922. doi: 10.1093/emboj/20.17.4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douillard J.Y., Siena S., Cassidy J., Tabernero J., Burkes R., Barugel M., Humblet Y., Bodoky G., Cunningham D., Jassem J., Rivera F., Kocakova I., Ruff P., Blasinska-Morawiec M., Smakal M., Canon J.L., Rother M., Oliner K.S., Wolf M., Gansert J. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J. Clin. Oncol. 2010;28:4697–4705. doi: 10.1200/JCO.2009.27.4860. [DOI] [PubMed] [Google Scholar]
- Espada J., Galaz S., Sanz-Rodriguez F., Blazquez-Castro A., Stockert J.C., Bagazgoitia L., Jaen P., Gonzalez S., Cano A., Juarranz A. Oncogenic H-Ras and PI3K signaling can inhibit E-cadherin-dependent apoptosis and promote cell survival after photodynamic therapy in mouse keratinocytes. J. Cell. Physiol. 2009;219:84–93. doi: 10.1002/jcp.21652. [DOI] [PubMed] [Google Scholar]
- Fang L., Zhu Q., Neuenschwander M., Specker E., Wulf-Goldenberg A., Weis W.I., Von Kries J.P., Birchmeier W. A small-molecule antagonist of the beta-catenin/TCF4 interaction blocks the self-renewal of cancer stem cells and suppresses tumorigenesis. Cancer Res. 2016;76:891–901. doi: 10.1158/0008-5472.CAN-15-1519. [DOI] [PubMed] [Google Scholar]
- Fiskus W., Sharma S., Saha S., Shah B., Devaraj S.G., Sun B., Horrigan S., Leveque C., Zu Y., Iyer S., Bhalla K.N. Pre-clinical efficacy of combined therapy with novel beta-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia. 2015;29:1267–1278. doi: 10.1038/leu.2014.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsalves F.C., Klein K., Carson B.B., Katz S., Ekas L.A., Evans S., Nagourney R., Cardozo T., Brown A.M., Dasgupta R. An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 2011;108:5954–5963. doi: 10.1073/pnas.1017496108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann T.N., Yeh J.T., Bowman B.R., Chu Q., Moellering R.E., Verdine G.L. Inhibition of oncogenic Wnt signaling through direct targeting of beta-catenin. Proc. Natl. Acad. Sci. U. S. A. 2012;109:17942–17947. doi: 10.1073/pnas.1208396109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner B.M. Carcinogenesis: a balance between beta-catenin and APC. Curr. Biol. 1997;7:R443–6. doi: 10.1016/s0960-9822(06)00214-4. [DOI] [PubMed] [Google Scholar]
- Hao J., Ao A., Zhou L., Murphy C.K., Frist A.Y., Keel J.J., Thorne C.A., Kim K., Lee E., Hong C.C. Selective small molecule targeting beta-catenin function discovered by in vivo chemical genetic screen. Cell Rep. 2013;4:898–904. doi: 10.1016/j.celrep.2013.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He T.C., Sparks A.B., Rago C., Hermeking H., Zawel L., Da Costa L.T., Morin P.J., Vogelstein B., Kinzler K.W. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Hidalgo M., Amant F., Biankin A.V., Budinska E., Byrne A.T., Caldas C., Clarke R.B., De Jong S., Jonkers J., Maelandsmo G.M., Roman-Roman S., Seoane J., Trusolino L., Villanueva A. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4:998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S.M., Mishina Y.M., Liu S., Cheung A., Stegmeier F., Michaud G.A., Charlat O., Wiellette E., Zhang Y., Wiessner S., Hild M., Shi X., Wilson C.J., Mickanin C., Myer V., Fazal A., Tomlinson R., Serluca F., Shao W., Cheng H., Shultz M., Rau C., Schirle M., Schlegl J., Ghidelli S., Fawell S., Lu C., Curtis D., Kirschner M.W., Lengauer C., Finan P.M., Tallarico J.A., Bouwmeester T., Porter J.A., Bauer A., Cong F. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- Hwang S.Y., Deng X., Byun S., Lee C., Lee S.J., Suh H., Zhang J., Kang Q., Zhang T., Westover K.D., Mandinova A., Lee S.W. Direct targeting of beta-catenin by a small molecule stimulates proteasomal degradation and suppresses oncogenic Wnt/beta-catenin signaling. Cell Rep. 2016;16:28–36. doi: 10.1016/j.celrep.2016.05.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson C., Sharma M., Henderson B.R. Targeting the beta-catenin nuclear transport pathway in cancer. Semin. Cancer Biol. 2014;27:20–29. doi: 10.1016/j.semcancer.2014.04.012. [DOI] [PubMed] [Google Scholar]
- Jang G.B., Hong I.S., Kim R.J., Lee S.Y., Park S.J., Lee E.S., Park J.H., Yun C.H., Chung J.U., Lee K.J., Lee H.Y., Nam J.S. Wnt/beta-catenin small-molecule inhibitor CWP232228 preferentially inhibits the growth of breast cancer stem-like cells. Cancer Res. 2015;75:1691–1702. doi: 10.1158/0008-5472.CAN-14-2041. [DOI] [PubMed] [Google Scholar]
- Kahn M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014;13:513–532. doi: 10.1038/nrd4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawazoe A., Shitara K., Fukuoka S., Kuboki Y., Bando H., Okamoto W., Kojima T., Fuse N., Yamanaka T., Doi T., Ohtsu A., Yoshino T. A retrospective observational study of clinicopathological features of KRAS, NRAS, BRAF and PIK3CA mutations in Japanese patients with metastatic colorectal cancer. BMC Cancer. 2015;15:258. doi: 10.1186/s12885-015-1276-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolligs F.T., Bommer G., Goke B. Wnt/beta-catenin/Tcf signaling: a critical pathway in gastrointestinal tumorigenesis. Digestion. 2002;66:131–144. doi: 10.1159/000066755. [DOI] [PubMed] [Google Scholar]
- Korinek V., Barker N., Morin P.J., Van Wichen D., De Weger R., Kinzler K.W., Vogelstein B., Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC −/− colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Lepourcelet M., Chen Y.N., France D.S., Wang H., Crews P., Petersen F., Bruseo C., Wood A.W., Shivdasani R.A. Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer Cell. 2004;5:91–102. doi: 10.1016/s1535-6108(03)00334-9. [DOI] [PubMed] [Google Scholar]
- Leung J.Y., Kolligs F.T., Wu R., Zhai Y., Kuick R., Hanash S., Cho K.R., Fearon E.R. Activation of AXIN2 expression by beta-catenin-T cell factor. A feedback repressor pathway regulating Wnt signaling. J. Biol. Chem. 2002;277:21657–21665. doi: 10.1074/jbc.M200139200. [DOI] [PubMed] [Google Scholar]
- Li X., Pu J., Jiang S., Su J., Kong L., Mao B., Sun H., Li Y. Henryin, an ent-kaurane diterpenoid, inhibits Wnt signaling through interference with beta-catenin/TCF4 interaction in colorectal cancer cells. PLoS One. 2013;8 doi: 10.1371/journal.pone.0068525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Zhu F., Boardman L.A., Wang L., Oi N., Liu K., Li X., Fu Y., Limburg P.J., Bode A.M., Dong Z. Aspirin prevents colorectal cancer by normalizing EGFR expression. EBioMedicine. 2015;2:447–455. doi: 10.1016/j.ebiom.2015.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licchesi J.D., Westra W.H., Hooker C.M., Machida E.O., Baylin S.B., Herman J.G. Epigenetic alteration of Wnt pathway antagonists in progressive glandular neoplasia of the lung. Carcinogenesis. 2008;29:895–904. doi: 10.1093/carcin/bgn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx V. Cell-line authentication demystified. Nat. Methods. 2014;11:483–488. doi: 10.1038/nmeth.2932. [DOI] [PubMed] [Google Scholar]
- Polakis P. Drugging Wnt signalling in cancer. EMBO J. 2012;31:2737–2746. doi: 10.1038/emboj.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segditsas S., Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25:7531–7537. doi: 10.1038/sj.onc.1210059. [DOI] [PubMed] [Google Scholar]
- Shtutman M., Zhurinsky J., Simcha I., Albanese C., D'amico M., Pestell R., Ben-Ze'ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. U. S. A. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2016. CA Cancer J. Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- Sukhdeo K., Mani M., Zhang Y., Dutta J., Yasui H., Rooney M.D., Carrasco D.E., Zheng M., He H., Tai Y.T., Mitsiades C., Anderson K.C., Carrasco D.R. Targeting the beta-catenin/Tcf transcriptional complex in the treatment of multiple myeloma. Proc. Natl. Acad. Sci. U. S. A. 2007;104:7516–7521. doi: 10.1073/pnas.0610299104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C., Hobor S., Bertotti A., Zecchin D., Huang S., Galimi F., Cottino F., Prahallad A., Grernrum W., Tzani A., Schlicker A., Wessels L.F., Smit E.F., Thunnissen E., Halonen P., Lieftink C., Beijersbergen R.L., Di Nicolantonio F., Bardelli A., Trusolino L., Bernards R. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 2014;7:86–93. doi: 10.1016/j.celrep.2014.02.045. [DOI] [PubMed] [Google Scholar]
- Thorne C.A., Hanson A.J., Schneider J., Tahinci E., Orton D., Cselenyi C.S., Jernigan K.K., Meyers K.C., Hang B.I., Waterson A.G., Kim K., Melancon B., Ghidu V.P., Sulikowski G.A., Lafleur B., Salic A., Lee L.A., Miller D.M., 3rd, Lee E. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1alpha. Nat. Chem. Biol. 2010;6:829–836. doi: 10.1038/nchembio.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Chemical structures of HI-BI and β-catenin inhibitors previously reported. MSAB: Methyl 3-([(4-methylphenyl)sulfonyl]amino)benzoate.