

Abstract
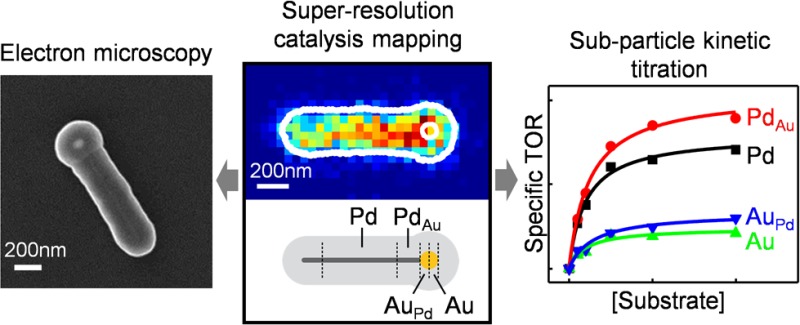
Compared with their monometallic counterparts, bimetallic nanoparticles often show enhanced catalytic activity associated with the bimetallic interface. Direct quantitation of catalytic activity at the bimetallic interface is important for understanding the enhancement mechanism, but challenging experimentally. Here using single-molecule super-resolution catalysis imaging in correlation with electron microscopy, we report the first quantitative visualization of enhanced bimetallic activity within single bimetallic nanoparticles. We focus on heteronuclear bimetallic PdAu nanoparticles that present a well-defined Pd–Au bimetallic interface in catalyzing a photodriven fluorogenic disproportionation reaction. Our approach also enables a direct comparison between the bimetallic and monometallic regions within the same nanoparticle. Theoretical calculations further provide insights into the electronic nature of N–O bond activation of the reactant (resazurin) adsorbed on bimetallic sites. Subparticle activity correlation between bimetallic enhancement and monometallic activity suggests that the favorable locations to construct bimetallic sites are those monometallic sites with higher activity, leading to a strategy for making effective bimetallic nanocatalysts. The results highlight the power of super-resolution catalysis imaging in gaining insights that could help improve nanocatalysts.
Short abstract
Super-resolution catalysis imaging in correlation with electron microscopy directly visualizes and quantifies catalytic enhancements at metal−metal interfaces within single bimetallic nanoparticles.
1. Introduction
Bimetallic nanoparticles are an important class of nanoscale catalysts in heterogeneous catalysis; they are widely used in industry due to the enhanced catalytic properties (i.e., activity, selectivity, and stability) compared with their monometallic counterparts.1−3 Many experimental and theoretical efforts have been spent in understanding the catalytic enhancements from the bimetallic effects.2,4,5 Two general mechanisms are often found to operate: (1) electronic (a.k.a. ligand) effect, in which the electronic properties of the catalytically active metal could be tuned by the other metal, leading to enhancement in activity;6 (2) geometric (a.k.a. ensemble) effect, where two metals together could form highly active surface sites made by an ensemble of atoms in some specific atomic arrangement.7,8 It is often challenging to deconvolute completely these two effects. Nevertheless the catalytic enhancement, in both cases, is a result of direct metal–metal interactions at the atomic scale, i.e., involving interfacing two types of metal components directly.
A majority of the scientific insights into bimetallic effects of nanoparticle catalysts came from studies at the ensemble level, where a large number of nanoparticles are measured simultaneously.2,4−14 By varying the composition of the bimetallic nanoparticles (typically alloy), the location, or the geometry of bimetallic sites, one could optimize the bimetallic catalysts for the largest activity enhancement. For such ensemble-averaged measurements, bimetallic nanocatalysts present particular challenges, however. First, these nanocatalysts are highly heterogeneous; their differences in catalytic activity are masked by the ensemble averaging. Second, even on a single bimetallic nanoparticle, there are both bimetallic and monometallic surface sites; the latter could contribute significantly to the measured activity. Spatially resolved activity measurements within a single catalyst particle are thus desirable. A recent tip-enhanced Raman spectroscopy (TERS) imaging study showed that, on a bimetallic PdAu surface, the Pd edge atoms better activate the reactant than the Pd terrace atoms on a Au(111) surface.15 Although with high spatial resolution (∼several nanometers), no quantitation of catalytic activity was available in such TERS imaging.
Single-molecule super-resolution fluorescence microscopy has emerged recently as an effective and complementary approach to study catalysis on nanoscale particles,16−29 including thermal catalysis,19−22,29 photocatalysis,23,24,28 electrocatalysis,25 and photoelectrocatalysis.26 Individual nanocatalysts can be interrogated at nanometer spatial resolution and single-turnover temporal resolution in real time under operando conditions, where the activity differences among different nanoparticles or even among different surface sites on the same nanoparticle can be directly quantified.
Here we report the first single-molecule super-resolution imaging of the bimetallic effects within single nanoparticle catalysts, in particular single heteronuclear PdAu nanoparticles in catalyzing a photodriven fluorogenic disproportionation reaction. The heteronuclear architecture allows for a clear definition of an interfacial region between the two metal components and a concurrent comparison with monometallic regions on the same particle. We directly visualize the enhanced activity at the bimetallic regions within individual PdAu nanoparticles. DFT calculations further provide insights into the mechanism of the enhanced activation of the reactant molecule at bimetallic vs monometallic surface sites. The correlation between the catalytic activity of bimetallic and monometallic regions within the same particle reveals the desirable locations to form bimetallic sites, which could potentially guide the design of effective bimetallic nanocatalysts.
2. Results and Discussions
2.1. PdAu Nanoparticles Can Catalyze the Photodriven Fluorogenic Disproportionation Reaction of Resazurin
We chose a heteronuclear PdAu bimetallic nanoparticle as a model bimetallic catalyst. Each PdAu nanoparticle consists of a pentatwinned crystalline Pd nanorod (∼500–4000 nm in length and 25.5 ± 2.8 nm in diameter) with a pseudospherical Au nanoparticle (∼60–140 nm in diameter) grown on one or both of its tips (Figure 1A,B and Figure S1).30 The heteronuclear architecture and the geometries of the two respective components enable ready identification of the interface between the two metals in each PdAu nanoparticle in scanning electron microscopy (SEM) characterizations. Elemental line profiling via energy dispersive spectroscopy confirms the heteronuclear nature with a clearly defined bimetallic junction between the Pd nanorod and the Au particle (Figure 1B, lower).
Figure 1.
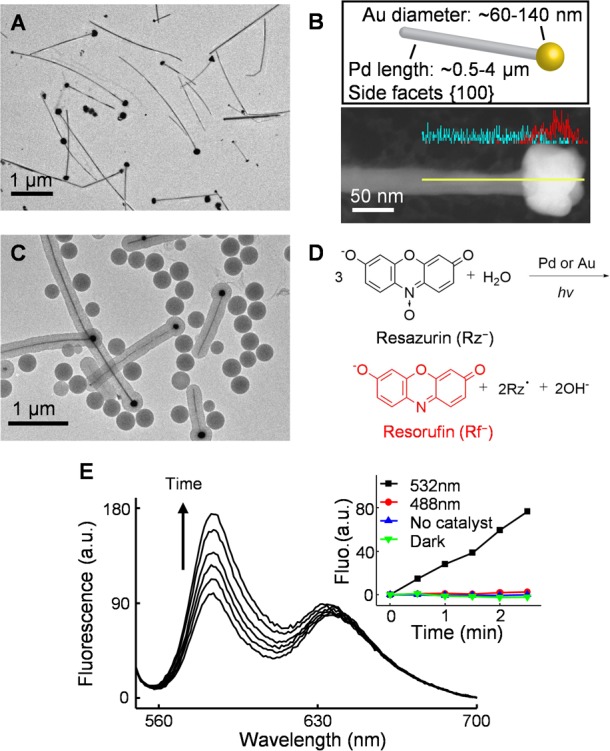
PdAu heteronuclear nanoparticles can catalyze the photodriven disproportionation reaction of resazurin to generate resorufin. (A) TEM image of heteronuclear bimetallic PdAu nanoparticles; structural measurements in Figure S1. (B) Upper: cartoon of PdAu nanoparticle. Lower: elemental line profile on a PdAu nanoparticle. Cyan, Pd; red, Au. Yellow line: the position of line scanning. (C) TEM image of mesoporous silica coated PdAu nanoparticles. (D) Proposed chemical equation of photodriven disproportionation reaction of resazurin (Rz–) to resorufin (Rf–) and a one-electron-oxidized radical species (Rz•) catalyzed by Pd or Au catalysts. (E) Fluorescence spectra of reaction solution containing 30.7 μM Rz– in 0.2 M pH 7.2 phosphate buffer in the presence of mesoporous silica coated PdAu nanoparticles under 50 mW 532 nm laser illumination; the increase of fluorescence at 583 nm indicates the formation of Rf–. Inset: the fluorescence intensity at 583 nm vs time profile of the reaction solution with catalysts under 50 mW 532 nm illumination (black), under 50 mW 488 nm illumination (red), without catalyst under 50 mW 532 nm excitation (blue), or with catalyst but no light illumination (green).
We further coated the PdAu nanoparticles with a mesoporous silica shell of 114 ± 11 nm thickness (Figure 1C), which allowed us to use UV-ozone to remove the organic ligands (e.g., polyvinylpyrrolidone (PVP)) on PdAu nanoparticles while maintaining their morphologies (especially the Pd–Au interface) and the accessibility of the metal surface to the reactant molecules via the mesopores.31,32 NaBH4 was then used to reduce the oxide layer on the metal to activate the surface for catalysis after UV-ozone treatment. In the subsequent imaging experiment (see section 2.2), the mesoporous silica shell also facilitates the fluorescence detection of the catalytic product molecules. These fluorescent product molecules desorb rapidly from the metal surface after catalytic formation, but temporarily adsorb onto the walls of the silica mesopores to be imaged, where the possible fluorescence quenching associated with direct detection on metal surfaces can also be circumvented (more discussions in Supporting Information section 2.2). (Note: for simplicity, we use “PdAu nanoparticles” to refer to these mesoporous silica coated nanoparticles in the writing below unless specified otherwise.)
Past studies have shown that the nonfluorescent molecule resazurin (Rz–), upon illumination with 532 nm light, can disproportionate to a one-electron reduced (Rz2–•) and a one-electron oxidized (Rz•) radical species,33 and the former one (Rz2–•) can further disproportionate to generate the highly fluorescent molecule resorufin (Rf–) (two-electron reduced from Rz–) and resazurin (Rz–).34Figure 1D gives the expected overall reaction equation. We have independently confirmed this photodriven disproportionation reaction, and further determined its 3:1 reaction stoichiometry for Rz:Rf (Supporting Information sections 3.1 and 3.2). We found that the PdAu nanoparticles, as well as their monometallic components, can catalyze this photodriven fluorogenic reaction (Figure 1E, and black line in inset, Supporting Information sections 3.1 and 3.2). Control experiments show that, without the PdAu nanoparticle catalyst or light, the reaction is insignificant (Figure 1E inset, blue and green lines). Moreover, 488 nm light, which Rz– does not absorb but which can excite the conduction band electrons of the metal (Figure S4E), cannot drive this reaction (Figure 1E inset, red line), indicating that the photoexcitation of the reactant Rz–, rather than that of the metal, is a key step in the disproportionation reaction.
2.2. Single-Molecule Super-Resolution Catalysis Imaging of Single PdAu Nanoparticles
The fluorogenic nature of the photodriven resazurin disproportionation allowed us to use single-molecule super-resolution fluorescence microscopy to image this reaction on individual PdAu nanoparticles, an approach we developed previously.31,32 In this approach, the particles were dispersed on a quartz slide within a microfluidic reactor. A continuous flow of a reactant solution and a constant 532 nm laser illumination led to a steady-state catalytic kinetics. Using wide-field total internal reflection fluorescence (TIRF) microscopy, we imaged the fluorescence of the product resorufin at the single-molecule level and localized their positions individually with ∼30–40 nm precision. The product molecules reside on the nanoparticle (i.e., adsorbed within the mesoporous silica shell) for ∼20 ms on average (i.e., ∼1 image frame) (Figure S7E) and do not show significant lateral diffusion (Supporting Information section 4.2.1) before desorbing and disappearing into the surrounding solution. We further confirmed the identity of the product resorufin by measuring the fluorescence spectrum on single PdAu nanoparticles (Figure S6B).
Figure 2A shows an exemplary map of thousands of product molecules imaged and localized on a single PdAu nanoparticle; its rod shape is apparent. Subsequent imaging through SEM independently confirmed the heteronuclear bimetallic morphology of each particle (Figure 2B), in which the Au particle is clearly visible at the tip of the Pd nanorod. Moreover, many product molecules are detected clearly in the mesoporous shell region, at distances of greater than ∼40 nm (our localization precision) away from the long-axis of the PdAu nanoparticle core; these detections directly support that the products adsorb onto silica-based sites within the mesopores.
Figure 2.
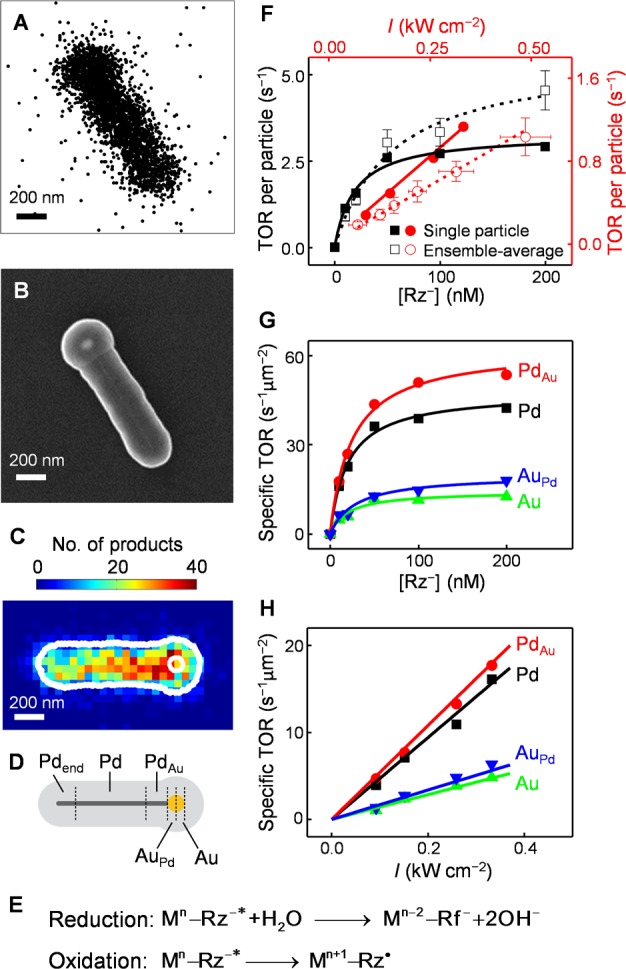
Super-resolution catalysis imaging and single-to-sub particle level analysis of reaction kinetics of PdAu nanoparticles. (A) Positions of ∼2900 products detected on a single mesoporous silica coated PdAu nanoparticle. (B) SEM image of the nanoparticle in A. (C) Two-dimensional histogram of product positions on the nanoparticle in panels A and B in 50 × 50 nm2 bins. The nanoparticle has been reoriented to align horizontally. Outer white line, structural contour; white circle, the contour of the Au particle; both determined from the SEM image in panel B. (D) Segmentation of a typical PdAu nanoparticle: regions of monometallic Pd (Pd), Au-doped Pd next to the Pd–Au interface (PdAu), monometallic Au (Au), and Pd-doped Au next to the Pd–Au interface (AuPd). The end segment (Pdend) is described in section 2.6. (E) Proposed kinetic mechanism of the catalytic disproportionation. Mn–Rz–*: a metal-surface-adsorbed excited-state resazurin; n is the number of electrons on the metal; the changes in electron counts on the metal particle are denoted. (F) Reactant concentration [Rz–] (black) and 532 nm laser power density I (red) dependences of the turnover rate (TOR) per PdAu nanoparticle. Solid symbols: the single particle in panels A–C. Open symbols: results averaged over 53 nanoparticles. Error bars: SEM for y and SD for x. Solid and dashed lines: global fits of eq 1. (G, H) [Rz–] and I dependences of specific turnover rates of the segments (Pd, PdAu, Au, AuPd) for the nanoparticle in panels A–C.
We titrated the reactant concentration [Rz–] and the incident 532 nm laser power density I, and examined how the catalytic activity of individual PdAu nanoparticles depends on them. Over the course of these titrations, the catalytic activity of the particles showed no significant deactivation for ∼6 h (Figure S12A). We further accounted for potential changes in fluorescence detection efficiency due to changes in background noises during these titrations (Supporting Information section 4.2.2).
With increasing [Rz–], the turnover rates of PdAu nanoparticles follow the classic Langmuir saturation kinetics for heterogeneous catalysis, both at the single-particle level and after ensemble averaging (Figure 2F), further supporting that the catalysis here is mediated on metal surfaces, in which the mass transport through the silica mesopores does not limit reaction kinetics (otherwise a linear dependence on [Rz–] is expected). With increasing I, the turnover rates of individual particles increase linearly (Figure 2F), in contrast to the quadratic power dependence of the uncatalyzed photodriven disproportionation reaction (Figure S4F). This linear power dependence suggests that the rate-limiting step of the catalytic reaction merely involves one photoexcited reactant molecule, resazurin (Rz–*).
Taking together all above results, we formulated a working kinetic mechanism of the catalytic disproportionation (Figure 2E; details in Supporting Information section 4.4). The reduction part of disproportionation is imaged directly in our experiments, in which a surface-adsorbed photoexcited resazurin (Mn–Rz–*, n is the number of electrons on the metal catalyst particle) obtained two electrons from the catalyst to generate resorufin (Rf–). For the oxidation part, which is not directly measured, we proposed that it is a surface-adsorbed excited-state resazurin that donates one electron to the metal catalyst generating a neutral radical (Rz•), giving the overall reaction as that in Figure 1D. This mechanism gives the catalytic turnover rate v as (derivation in Supporting Information section 4.4)
| 1 |
Here k is a catalytic rate constant and K is a reactant adsorption equilibrium constant on the metal surface. This equation predicts the saturating kinetics on [Rz–] and the linear dependence on I, as observed experimentally (Figure 2F). By globally fitting both [Rz–] and I dependences of v, we extracted k and K of PdAu nanoparticles at both the single-particle and the ensemble-averaged level.
2.3. Subparticle Analysis Reveals Enhanced Catalytic Activity around the Pd–Au Interface
The unique patterns of nanoparticles allowed us to overlay their super-resolution catalysis images onto their respective SEM images with ∼40 nm precision (Figure 2C; and Figure S10), on which the Pd–Au interface of the heteronuclear particle can be readily located in reference to the Au particle position. A higher catalytic activity (i.e., more reaction products) is apparent around the interfacial region (Figure 2C), suggesting a direct, and first-of-its-kind, visualization of bimetallic effect of catalytic enhancement.
We then dissected each heteronuclear PdAu nanoparticle into four different regions (Figure 2D): (1) a 100 nm long segment of the Pd nanorod adjacent to the interface, i.e., PdAu region, corresponding to a bimetallic component that is dominantly Pd with minor Au doping on the surface from the attached Au particle (Figure 1B, lower); (2) the rest of Pd nanorod away from the interface (excluding the other end of the Pd nanorod), representing the monometallic Pd component; (3) the Au hemisphere adjacent to the interface, i.e., AuPd region, corresponding to the other bimetallic component that is dominantly Au with minor Pd doping (Figure 1B, lower); and (4) the other hemisphere of Au, representing the monometallic Au component. The dimensions of all four regions are significantly larger than our spatial resolution (30–40 nm) to make these subparticle dissections physically meaningful. The specific catalytic rates of all four regions show saturation kinetics with increasing reactant concentration [Rz–] and a linear dependence on the light power density I (Figure 2G,H), as observed for the whole particle (Figure 2F). Fitting these results with eq 1 gave the catalytic rate constant k and the reactant adsorption equilibrium constant K of each region for each PdAu nanoparticle.
Pooling results from 53 PdAu nanoparticles, the catalytic rate constants k of the bimetallic PdAu and AuPd regions are ∼50% more active on average than their monometallic Pd and Au counterparts (Figure 3A and Figure S12B), directly reporting bimetallic catalytic enhancement. On the other hand, the reactant adsorption equilibrium constant K shows an opposite trend: the K of bimetallic regions are ∼30% smaller on average than their monometallic counterparts (Figure 3B). Furthermore, for each type of region, the k and K of individual PdAu nanoparticles are anticorrelated (Figure 3C,D). Therefore, the enhanced catalytic activity at bimetallic regions is associated with weakened reactant adsorption, rationalizable by transition state theory, in which a less stable reactant is closer in energy to the transition state resulting in a lower activation barrier.
Figure 3.
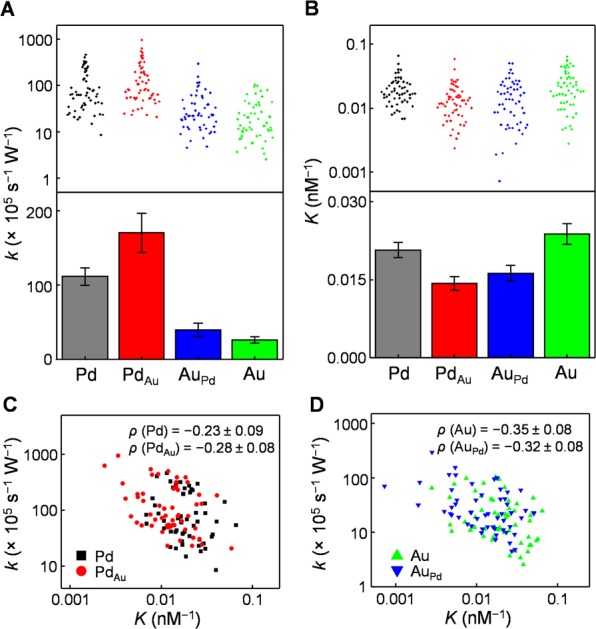
Subparticle catalytic properties of bimetallic vs monometallic regions. (A, B) The catalytic rate constant k (A) and reactant adsorption equilibrium constant K (B) of the bimetallic regions (PdAu, AuPd) vs monometallic regions (Pd, Au). Upper: results of individual nanoparticles; each dot is one PdAu nanoparticle. Lower: ensemble averages; error bars are SEM. (C, D) Scatter plots of k vs K for each type of region of individual PdAu nanoparticles; each dot is one region from one PdAu nanoparticle. ρ is Pearson’s cross correlation coefficient.
Control measurements and analyses show that (1) the mesoporous silica shells of the bimetallic and monometallic regions present equal accessibility to the reactant molecules (Figure S12E); (2) the fluorescence intensities and dwell times of the reaction products at the bimetallic regions are indistinguishable from those at the monometallic regions, and therefore the products are equally detected at different regions on the same nanoparticle (Figure S12D, F); and (3) the plasmonic enhancement effect is insignificant at the bimetallic regions (Supporting Information section 5). Altogether, these results support that the observed higher product detection rates at interfacial regions are indeed due to the bimetallic catalytic enhancement there.
2.4. Breaking the Metal–Metal Junction Abolishes the Enhanced Catalytic Activity at Bimetallic Regions
To further support that the enhanced catalytic activity is indeed related to the Pd–Au interface in the heteronuclear PdAu nanoparticle, we broke the Pd–Au junction by heating the PdAu nanoparticles at 450 °C for 1 h. A nanoscale gap appears between the Pd nanorod and the Au nanoparticle within the mesoporous silica shell (Figure 4A; more examples in Figure S3).
Figure 4.
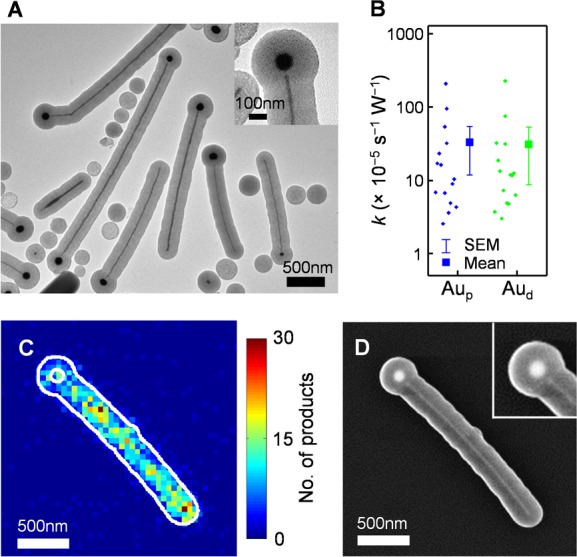
Super-resolution catalysis imaging of PdAu nanoparticles with nanoscale gaps (gap-PdAu nanoparticles). (A) TEM image of gap-PdAu nanoparticles coated with mesoporous silica. Inset: a zoom in. (B) Box plot of the catalytic rate constant k of the proximal (Aup) and distant (Aud) Au hemispheres with regard to the Pd nanorods in the gap-PdAu nanoparticles. (C) Two-dimensional histogram of ∼2500 product locations on a gap-PdAu nanoparticle. White line: structural contours of the outer shell and the spherical Au particle. (D) SEM image of the gap-PdAu nanoparticle in C. Inset: zoom in of the nanogap.
We then imaged these PdAu nanoparticles with nanoscale gaps (i.e., gap-PdAu nanoparticles) in catalyzing the same reaction (Figure 4C and Figure S14A), and with SEM, in which the gaps are visible (Figure 4D and Figure S14B). Strikingly, the enhanced catalytic activity originally around the Pd–Au interface vanished, supporting that the enhanced catalytic activity is specific to the heteronuclear bimetallic structure with direct Pd–Au contact.
Since the spherical Au particles are easily identifiable in the SEM images of these gap-PdAu nanoparticles, onto which the super-resolution catalysis images were mapped (Figure 4C,D), we compared the catalytic kinetics of the Au hemisphere proximal to the Pd nanorod (i.e., Aup) with the other hemisphere distant from the nanorod (i.e., Aud). (We could not reliably dissect out a Pd nanorod segment proximal to the Au particle because the nanorod end at the gap cannot be clearly located in the SEM image.) No significant differences in the catalytic rate constants were observed between the two hemispheres (Figure 4B) (nor in the reactant adsorption equilibrium constant K; Figure S14C), further supporting that the enhanced catalytic activity originally around the interface region is specific to the intact heteronuclear PdAu bimetallic structure.
It is worth noting that heat-breaking the Pd–Au junction should have left some Pd atoms on the Au particle, and therefore the proximal Au hemisphere should still behave like a AuPd region. However, the heating could have homogenized the structural and compositional differences between the two hemispheres. In any case, the abolishment of observable catalytic enhancement after breaking the junction supports that the enhancement is associated with the particular geometry with direct Pd–Au contact.
2.5. Electronic Structure Elucidation of Reactant Activation on Monometallic and Bimetallic Surfaces
2.5.1. Resazurin Interaction with Pd and PdAu Surfaces
We performed DFT calculations using the VASP package35 to gain more insights into the activation on Pd, Au, or bimetallic surface sites of resazurin’s N–O bond, which is cleaved to become resorufin (Figure 1D; calculation details in Supporting Information sections 6.1–6.3). Although the photodriven disproportionation of resazurin involves its excited state (Figure 2E), these ground-state DFT calculations could still inform on how resazurin’s N–O bond gets activated upon interaction with metal surfaces. We first examined resazurin interaction with a Pd(100) surface, the dominant facets on the sides of Pd nanorods.30 Note that the actual Pd nanorod side surface might not be the same as the perfect facet structure because there are always defects, edges, etc. and the sample preparation procedures such as NaBH4 reduction might also alter the surface structure. We chose Pd(100) (and Au(111) later) as a reasonable model surface for calculation purposes. The optimized adsorption geometry has resazurin lying flat on the surface (Figure 5A). The N–O fragment of resazurin is tilted away from the surface, in which the O atom sits approximately above the center of four Pd atoms and the N atom is above an edge bridging two Pd atoms. Importantly, compared with that in the free resazurin molecule, the N–O bond is elongated by 0.007 Å (Table S2), reflecting its weakening and thus activation for cleavage, consistent with Pd’s being a catalyst for the observed reaction.
Figure 5.

Electronic interactions of resazurin with Pd
and Au surfaces. (A,
B) Optimized adsorption geometries of resazurin on Pd(100) and Au(111).
The Pd or Au atoms to be substituted for evaluating bimetallic effects
are numbered. (C, D) COHP analyses for the interactions between the
N–O fragment of resazurin and the closest metal atoms on Pd(100)
and Au(111), respectively. The horizontal lines represent the Fermi
level (EF), referenced to the vacuum (E = 0). (E, F) The molecular orbitals of resazurin that
are dominantly πN–O (doubly occupied) and
πN–O* (unoccupied) in nature. (G, H) Schematics of the forward-donation
and back-donation on Pd(100). (I, J) Schematics of the forward-donation
and back-donation on Au(111). Only the N–O fragment of resazurin
is drawn. We use 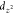 to represent d orbitals
of metal since they are pointed toward N–O. The solid black
circles and triangles mark the projected positions of O and N on the
metal surface, respectively.
to represent d orbitals
of metal since they are pointed toward N–O. The solid black
circles and triangles mark the projected positions of O and N on the
metal surface, respectively.
We performed crystal orbital Hamiltonian population (COHP) analysis36 to examine the interactions between the N–O fragment of resazurin and the Pd surface. COHP analysis allows for an energy-resolved visualization of bonding and antibonding orbital interactions between specific atoms (Figure 5C). The interactions mainly come from two types of Pd–O bonding interactions (Figure 5C, red shades below the Fermi level). One type involves N–O bonding orbitals (Figure 5C, blue line), located energetically below the Pd d band (at about −14 to −11.5 eV); this type of interaction is referred to as forward-donation.37 The other type involves N–O antibonding orbitals, located energetically within the Pd d band (at about −7.6 to −6.6 eV); this type is referred to as back-donation.37
We further performed DFT calculations using the Gaussian package38 to visualize the molecular orbitals of resazurin (Supporting Information section 6.6). Two N–O fragment based orbitals are identified to be likely involved in the forward- and back-donations between the N–O fragment and the Pd surface (Supporting Information section 6.6): one a doubly occupied π bonding orbital, denoted as πN–O (Figure 5E); the other an unoccupied π* orbital, denoted as πN–O* (Figure 5F). Both πN–O and πN–O have orbital lobes on the O atom pointing toward the Pd surface for bonding interactions.
Combining the adsorption geometry, the COHP analysis, and the resazurin MOs, we schematically depicted the forward-donation and back-donation interactions between Pd and the O atom of resazurin, labeled as πN–O + d and d + πN–O*, respectively (Figure 5G,H). Both types of interactions weaken the N–O bond of resazurin, consistent with the elongated N–O bond of resazurin adsorbed on the Pd(100) surface. But these two interactions have opposite charge transfer directions: the forward-donation takes away charge density from a N–O π bonding orbital, whereas the back-donation injects charge density into a N–O π* antibonding orbital; the net charge transfer is from Pd to the N–O fragment (Table S2), indicating that the back-donation is a more dominant bonding interaction here.
To computationally evaluate how bimetallic sites can possibly enhance the activation of resazurin’s N–O bond for cleavage, we substituted by Au the eight surface Pd atoms (numbered in Figure 5A) that directly interact with the carbon atoms of resazurin. The substitution was done one atom at a time to dissect the effect of each substitution in forming bimetallic Au@Pd(100) surface sites, on which resazurin adsorption was further geometry optimized. No significant change of the resazurin adsorption geometry is observed on these model bimetallic surfaces (Supporting Information section 6.3.1). Among the eight structures on these Au@Pd(100) surfaces, one of the most stable structures is a Au substitution at the Pd atom no. 2 (Figure 5A), which is far from the N–O fragment of the resazurin. Here the O of N–O is closer to the metal surface and N–O is further elongated compared to that on Pd(111) (Table S2), indicating a more weakened N–O bond and likely a bimetallic effect of enhancing the activation of the N–O bond for cleavage, even though the second metal (i.e., Au here) is not directly bonded to the N–O fragment. COHP analysis identified the same forward- and back-donation interactions between Pd and O atoms (Figure S22A). More important, both interactions increase in magnitude compared with those on monometallic Pd(100) (Table S4), consistent with the further weakening and thus enhanced activation of the N–O bond for cleavage on bimetallic PdAu surfaces.
2.5.2. Resazurin Interactions with Au and AuPd Surfaces
We performed similar calculations of resazurin adsorption on a Au(111) surface. Au(111) surface was chosen as a model because the capping ligands PVP and iodide, used in the synthesis, prefer Au(111) facet sites in growing Au nanoparticles.39,40 The optimized adsorption geometry has resazurin lying flat on the surface with 4 of its carbons and the O atom of the N–O fragment sitting on top of Au atoms (Figure 5B). Moreover, the N–O fragment is tilted toward the Au surface, suggesting strong interactions between Au and the N–O fragment; this is in contrast to that of resazurin–Pd(100) interactions where the N–O fragment is not directly on top of any Pd atom and is tilted away from the surface (Figure 5A). The N–O bond is also elongated (by 0.006 Å) on Au(111) compared with that in the free resazurin (Table S3), indicating a weakening of the N–O bond and thus activation toward cleavage.
Similarly, we did single Pd atom substitutions for the five Au atoms sitting directly below the resazurin atoms (Figure 5B) to evaluate the bimetallic effect. All five substitutions lead to shorter metal–O bonds (by ∼0.1 Å) compared with that on the Au(111) surface (Table S3), indicating even stronger interactions between the bimetallic surface and the N–O fragment. All of them also have a further elongated N–O bond (Table S3), indicating its enhanced activation via the bimetallic effect.
COHP analysis of resazurin on Au(111) identified forward- and back-donation interactions between Au and the O atom of the N–O fragment (Figure 5D), as in resazurin–Pd(100) interactions. The orbital overlap pattern here is different, as the O atom here sits directly on top of a Au atom (Figure 5I,J). The local net charge transfer is from Au to the N–O fragment, indicating that the back-donation is also more important here (Table S3). For the bimetallic surface Pd@Au(111), the COHP analysis also shows that both forward- and back-donation interactions increase between the metal and the N–O fragment (Table S4), leading to further activation of N–O.
Altogether, DFT calculations show that both substituting Pd by Au on Pd(100) and substituting Au by Pd on Au(111) could give a more lengthened N–O bond, from increases in both forward- and back-donation interactions, leading to the enhanced activation of the N–O bond for cleavage on the bimetallic surfaces.
2.6. Activity Correlation within Single Particle Reveals Optimal Locations for Bimetallic Sites
Our subparticle level activity quantitation also allowed us to examine how the activity enhancements of the bimetallic regions are related to the activity of the corresponding monometallic regions within the same particle (Figure 6A,B, black and red points); the latter would represent the original “monometallic” activity of the bimetallic region before the second metal was introduced. Strikingly, positive correlations are clearly observed. For the particles whose monometallic Pd region has higher catalytic rate constant k, their bimetallic activity enhancement ΔkBI (defined as k(PdAu) – k(Pd)) of the bimetallic PdAu region is also larger (Figure 6A). The same trend applies to the bimetallic AuPd region (Figure 6B). These trends indicate that when the particle, Pd or Au, is more active, the bimetallic enhancement is larger.
Figure 6.
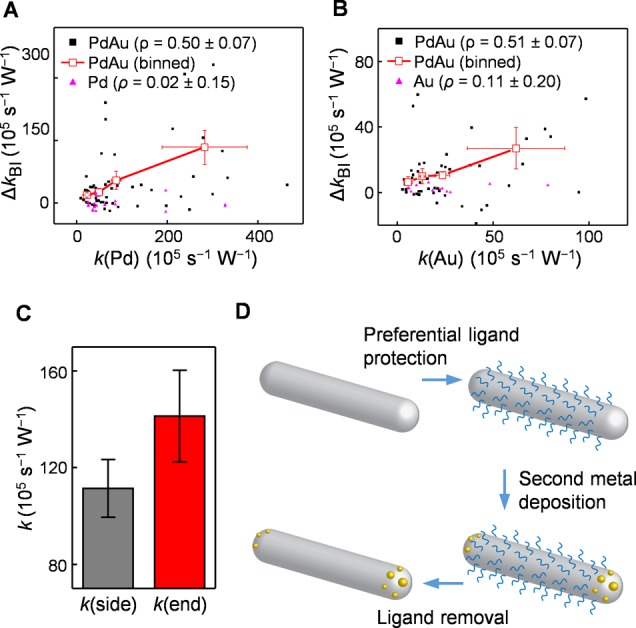
Rational strategy for generating more active bimetallic PdAu nanocatalysts. (A) Correlation between the catalytic activity enhancement of the bimetallic PdAu region and the monometallic Pd region on the same heteronuclear PdAu nanoparticle. Each black dot is a single PdAu nanoparticle. Hollow red squares are binned and averaged results. The pure single Pd nanorods (pink) serve as the control. ρ is Pearson’s correlation coefficient for the individual particles. (B) Same as panel A, but for the bimetallic AuPd vs the monometallic Au regions with pure Au nanoparticles (pink) as the control. (C) The catalytic rate constant k of the side and end regions of Pd nanorods (average from 46 nanoparticles). (D) Schematic of the proposed strategy to deposit the second metal onto the ends of nanorods of the first metal. Error bars are SEM for all y-axes and SD for all x-axes.
As a control, we also analyzed the corresponding subparticle regions of pure single Pd nanorods and single Au particles, which are minor components in the sample and imaged together with PdAu bimetallic nanoparticles. For each Pd nanorod, we compared the activity of the end regions adjacent to the two tips (which are at the same location as the bimetallic PdAu region in a PdAu nanoparticle) with the side activity (excluding the ends), which corresponds to the monometallic Pd region in a PdAu nanoparticle. For each Au nanoparticle, we arbitrarily dissected it into two hemispheres vertically and compared the left vs right hemisphere. Neither of the two controls shows any significant correlation (Figure 6A,B, pink points), indicating that possible intraparticle activity heterogeneity would not give the observed positive correlations between the bimetallic activity enhancement and the original monometallic activity.
These positive correlations suggest that, to obtain larger bimetallic activity enhancement, one should deposit the second metal onto originally more active monometallic surface sites of Pd or Au, constituting a possible rationalized strategy in making more active bimetallic nanoparticle catalysts. Implementing this strategy would require one to (1) identify what types of surface sites are more active on the initial monometallic particle and (2) selectively deposit the second metal onto the more active sites of the first metal.
Structurally anisotropic nanoparticles, such as 1-dimensional nanorods, offer opportunities to fulfill these two requirements. Regarding requirement 1, our previous study has identified that, for Au nanorods, their two ends are generally more active than their side facets in catalyzing an oxidative deacetylation reaction, possibly due to the higher activity of low coordination surface sites abundant at the ends.31 Here we found the same phenomenon on Pd nanorods: their free end segment Pdend (Figure 2D) in the heteronuclear PdAu bimetallic particle is more active than their side facets (Figure 6C). Regarding requirement 2, one could utilize appropriate capping ligands that could selectively bind to the side facets of the first metal nanorods, leaving the two ends preferentially for the deposition of the second metal (Figure 6D). (Note that, for generating just bimetallic PdAu sites, the deposition of Au would certainly need to be much less in quantity to not grow a big Au particle.) Subsequent removal of the capping ligands would result in a bimetallic system where the second metal is at the desired higher activity sites of the first metal. Interestingly, this scheme is what underlies the synthesis of the heteronuclear bimetallic PdAu nanorod–nanoparticle in the current study, in which the Au particle grew selectively at the ends of Pd nanorods and the side facets of Pd nanorods were preferentially blocked by the PVP and iodide ligands.30 Similar strategies have been reported in synthesizing Pd-tipped Au nanorods41 and Pd-edged Ag nanocubes42 for detecting catalytic products via surface-enhanced Raman scattering, and Pt-tipped Au nanorods for surface-plasmon-enhanced catalysis.43 With appropriate ligands, one could expect this synthesis strategy to be applicable to particles that are less structurally anisotropic (e.g., pseudospheres) for generating bimetallic sites at desired locations.
3. Conclusions
In summary, using single-molecule super-resolution catalysis imaging in correlation with SEM, we have directly visualized and quantified the bimetallic activity enhancement on heteronuclear PdAu nanoparticles at the subparticle level in catalyzing the photodriven disproportionation of resazurin. DFT calculations provide insights into the electronic nature of the activation of the resazurin N–O bond on the metal surfaces via synergistic forward- and back-donation interactions. The results further suggest that higher activity monometallic sites are the optimal locations to form bimetallic sites for larger activity enhancement, demonstrating the effectiveness of the super-resolution catalysis imaging approach in deriving knowledge for guiding the development of better nanocatalysts.
Acknowledgments
We thank Roald Hoffmann for access to computational facilities and comments, Tai-Yen Chen for providing Matlab codes, and Xiaochun Zhou and Hao Shen for assistance in microscopy setups. This research is supported mainly by the Army Research Office (W911NF-14-1-0377) and in part by the Department of Energy (DE-SC0004911), Army Research Office (W911NF-14-1-0620), and National Science Foundation (CBET1263736). This work made use of the Cornell Center for Materials Research Shared Facilities, which are supported through the NSF MRSEC program (DMR1120296), and of the Cornell NanoScale Facility, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), supported by the National Science Foundation (ECCS1542081). B.C. and the computational facilities used in this work at Cornell are supported by the Energy Frontier Research in Extreme Environments (EFree) Center, an Energy Frontier Research Center funded by the U.S. Department of Energy (DE-SC0001057).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00377.
Experimental details, additional results, and analyses (PDF)
Author Present Address
† J.B.S.: Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523, U.S.A. E.C.: Building 201, B-S-05, 3M Center, St. Paul, Minnesota 55144, U.S.A.
The authors declare no competing financial interest.
Supplementary Material
References
- Bartholomew C. H.; Farrauto R. J.. Fundamentals of industrial catalytic processes, 2nd ed.; Wiley: Hoboken, NJ, 2006. [Google Scholar]
- Gilroy K. D.; Ruditskiy A.; Peng H. C.; Qin D.; Xia Y. N. Bimetallic Nanocrystals: Syntheses, Properties, and Applications. Chem. Rev. 2016, 116, 10414–10472. 10.1021/acs.chemrev.6b00211. [DOI] [PubMed] [Google Scholar]
- Smith J. G.; Chakraborty I.; Jain P. K. In Situ Single-Nanoparticle Spectroscopy Study of Bimetallic Nanostructure Formation. Angew. Chem., Int. Ed. 2016, 55, 9979–9983. 10.1002/anie.201604710. [DOI] [PubMed] [Google Scholar]
- Norskov J. K.; Abild-Pedersen F.; Studt F.; Bligaard T. Density functional theory in surface chemistry and catalysis. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 937–943. 10.1073/pnas.1006652108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W. T.; Porosoff M. D.; Chen J. G. G. Review of Pt-Based Bimetallic Catalysis: From Model Surfaces to Supported Catalysts. Chem. Rev. 2012, 112, 5780–5817. 10.1021/cr300096b. [DOI] [PubMed] [Google Scholar]
- Bligaard T.; Norskov J. K. Ligand effects in heterogeneous catalysis and electrochemistry. Electrochim. Acta 2007, 52, 5512–5516. 10.1016/j.electacta.2007.02.041. [DOI] [Google Scholar]
- Maroun F.; Ozanam F.; Magnussen O. M.; Behm R. J. The role of atomic ensembles in the reactivity of bimetallic electrocatalysts. Science 2001, 293, 1811–1814. 10.1126/science.1061696. [DOI] [PubMed] [Google Scholar]
- Chen M. S.; Kumar D.; Yi C. W.; Goodman D. W. The promotional effect of gold in catalysis by palladium-gold. Science 2005, 310, 291–293. 10.1126/science.1115800. [DOI] [PubMed] [Google Scholar]
- Chen G. X.; Zhao Y.; Fu G.; Duchesne P. N.; Gu L.; Zheng Y. P.; Weng X. F.; Chen M. S.; Zhang P.; Pao C. W.; et al. Interfacial Effects in Iron-Nickel Hydroxide-Platinum Nanoparticles Enhance Catalytic Oxidation. Science 2014, 344, 495–499. 10.1126/science.1252553. [DOI] [PubMed] [Google Scholar]
- Guo X. G.; Fang G. Z.; Li G.; Ma H.; Fan H. J.; Yu L.; Ma C.; Wu X.; Deng D. H.; Wei M. M.; et al. Direct, Nonoxidative Conversion of Methane to Ethylene, Aromatics, and Hydrogen. Science 2014, 344, 616–619. 10.1126/science.1253150. [DOI] [PubMed] [Google Scholar]
- Kim D.; Resasco J.; Yu Y.; Asiri A. M.; Yang P. D. Synergistic geometric and electronic effects for electrochemical reduction of carbon dioxide using gold-copper bimetallic nanoparticles. Nat. Commun. 2014, 5, 4948. 10.1038/ncomms5948. [DOI] [PubMed] [Google Scholar]
- Zhang S. R.; Nguyen L.; Liang J. X.; Shan J. J.; Liu J. Y.; Frenkel A. I.; Patlolla A.; Huang W. X.; Li J.; Tao F. Catalysis on singly dispersed bimetallic sites. Nat. Commun. 2015, 6, 7938. 10.1038/ncomms8938. [DOI] [PubMed] [Google Scholar]
- Zhang H. J.; Watanabe T.; Okumura M.; Haruta M.; Toshima N. Catalytically highly active top gold atom on palladium nanocluster. Nat. Mater. 2012, 11, 49–52. 10.1038/nmat3143. [DOI] [PubMed] [Google Scholar]
- Zheng X.; Deng J.; Wang N.; Deng D.; Zhang W. H.; Bao X.; Li C. Podlike N-doped carbon nanotubes encapsulating FeNi alloy nanoparticles: high-performance counter electrode materials for dye-sensitized solar cells. Angew. Chem., Int. Ed. 2014, 53, 7023–7027. 10.1002/anie.201400388. [DOI] [PubMed] [Google Scholar]
- Zhong J. H.; Jin X.; Meng L. Y.; Wang X.; Su H. S.; Yang Z. L.; Williams C. T.; Ren B. Probing the electronic and catalytic properties of a bimetallic surface with 3 nm resolution. Nat. Nanotechnol. 2017, 12, 132–136. 10.1038/nnano.2016.241. [DOI] [PubMed] [Google Scholar]
- Janssen K. P. F.; De Cremer G.; Neely R. K.; Kubarev A. V.; Van Loon J.; Martens J. A.; De Vos D. E.; Roeffaers M. B. J.; Hofkens J. Single molecule methods for the study of catalysis: from enzymes to heterogeneous catalysts. Chem. Soc. Rev. 2014, 43, 990–1006. 10.1039/C3CS60245A. [DOI] [PubMed] [Google Scholar]
- Chen P.; Zhou X. C.; Andoy N. M.; Han K. S.; Choudhary E.; Zou N. M.; Chen G. Q.; Shen H. Spatiotemporal catalytic dynamics within single nanocatalysts revealed by single-molecule microscopy. Chem. Soc. Rev. 2014, 43, 1107–1117. 10.1039/C3CS60215J. [DOI] [PubMed] [Google Scholar]
- Chen T.; Dong B.; Chen K.; Zhao F.; Cheng X.; Ma C.; Lee S.; Zhang P.; Kang S. H.; Ha J. W.; et al. Optical Super-Resolution Imaging of Surface Reactions. Chem. Rev. 2017, 117, 7510–7537. 10.1021/acs.chemrev.6b00673. [DOI] [PubMed] [Google Scholar]
- Roeffaers M. B. J.; Sels B. F.; Uji-i H.; De Schryver F. C.; Jacobs P. A.; De Vos D. E.; Hofkens J. Spatially resolved observation of crystal-face-dependent catalysis by single turnover counting. Nature 2006, 439, 572–575. 10.1038/nature04502. [DOI] [PubMed] [Google Scholar]
- Xu W. L.; Kong J. S.; Yeh Y. T. E.; Chen P. Single-molecule nanocatalysis reveals heterogeneous reaction pathways and catalytic dynamics. Nat. Mater. 2008, 7, 992–996. 10.1038/nmat2319. [DOI] [PubMed] [Google Scholar]
- Decan M. R.; Impellizzeri S.; Marin M. L.; Scaiano J. C. Copper nanoparticle heterogeneous catalytic ’click’ cycloaddition confirmed by single-molecule spectroscopy. Nat. Commun. 2014, 5, 4612. 10.1038/ncomms5612. [DOI] [PubMed] [Google Scholar]
- Ristanovic Z.; Kerssens M. M.; Kubarev A. V.; Hendriks F. C.; Dedecker P.; Hofkens J.; Roeffaers M. B. J.; Weckhuysen B. M. High-Resolution Single-Molecule Fluorescence Imaging of Zeolite Aggregates within Real-Life Fluid Catalytic Cracking Particles. Angew. Chem., Int. Ed. 2015, 54, 1836–1840. 10.1002/anie.201410236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachikawa T.; Yamashita S.; Majima T. Evidence for Crystal-Face-Dependent TiO2 Photocatalysis from Single-Molecule Imaging and Kinetic Analysis. J. Am. Chem. Soc. 2011, 133, 7197–7204. 10.1021/ja201415j. [DOI] [PubMed] [Google Scholar]
- Zhang Y. W.; Lucas J. M.; Song P.; Beberwyck B.; Fu Q.; Xu W. L.; Alivisatos A. P. Superresolution fluorescence mapping of single-nanoparticle catalysts reveals spatiotemporal variations in surface reactivity. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 8959–8964. 10.1073/pnas.1502005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W. L.; Shen H.; Kim Y. J.; Zhou X. C.; Liu G. K.; Park J.; Chen P. Single-Molecule Electrocatalysis by Single-Walled Carbon Nanotubes. Nano Lett. 2009, 9, 3968–3973. 10.1021/nl900988f. [DOI] [PubMed] [Google Scholar]
- Sambur J. B.; Chen T. Y.; Choudhary E.; Chen G. Q.; Nissen E. J.; Thomas E. M.; Zou N. M.; Chen P. Sub-particle reaction and photocurrent mapping to optimize catalyst-modified photoanodes. Nature 2016, 530, 77–80. 10.1038/nature16534. [DOI] [PubMed] [Google Scholar]
- Cordes T.; Blum S. A. Opportunities and challenges in single-molecule and single-particle fluorescence microscopy for mechanistic studies of chemical reactions. Nat. Chem. 2013, 5, 993–999. 10.1038/nchem.1800. [DOI] [PubMed] [Google Scholar]
- Ha J. W.; Ruberu T. P.; Han R.; Dong B.; Vela J.; Fang N. Super-resolution mapping of photogenerated electron and hole separation in single metal-semiconductor nanocatalysts. J. Am. Chem. Soc. 2014, 136, 1398–1408. 10.1021/ja409011y. [DOI] [PubMed] [Google Scholar]
- Han R.; Ha J. W.; Xiao C.; Pei Y.; Qi Z.; Dong B.; Bormann N. L.; Huang W.; Fang N. Geometry-assisted three-dimensional superlocalization imaging of single-molecule catalysis on modular multilayer nanocatalysts. Angew. Chem., Int. Ed. 2014, 53, 12865–12869. 10.1002/anie.201407140. [DOI] [PubMed] [Google Scholar]
- Huang X. Q.; Zheng N. F. One-Pot, High-Yield Synthesis of 5-Fold Twinned Pd Nanowires and Nanorods. J. Am. Chem. Soc. 2009, 131, 4602–4603. 10.1021/ja9009343. [DOI] [PubMed] [Google Scholar]
- Zhou X. C.; Andoy N. M.; Liu G. K.; Choudhary E.; Han K. S.; Shen H.; Chen P. Quantitative super-resolution imaging uncovers reactivity patterns on single nanocatalysts. Nat. Nanotechnol. 2012, 7, 237–241. 10.1038/nnano.2012.18. [DOI] [PubMed] [Google Scholar]
- Andoy N. M.; Zhou X. C.; Choudhary E.; Shen H.; Liu G. K.; Chen P. Single-Molecule Catalysis Mapping Quantifies Site-Specific Activity and Uncovers Radial Activity Gradient on Single 2D Nanocrystals. J. Am. Chem. Soc. 2013, 135, 1845–1852. 10.1021/ja309948y. [DOI] [PubMed] [Google Scholar]
- Bueno C.; Villegas M. L.; Bertolotti S. G.; Previtali C. M.; Neumann M. G.; Encinas M. V. The excited-state interaction of resazurin and resorufin with amines in aqueous solutions. Photophysics and photochemical reaction. Photochem. Photobiol. 2002, 76, 385–390. 10.1562/0031-8655(2002)0760385TESIOR2.0.CO2. [DOI] [PubMed] [Google Scholar]
- Balcerzyk A.; Baldacchino G. Implementation of laser induced fluorescence in a pulse radiolysis experiment - a new way to analyze resazurin-like reduction mechanisms. Analyst 2014, 139, 1707–1712. 10.1039/c3an02000b. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmuller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54, 11169–11186. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Dronskowski R.; Blochl P. E. Crystal Orbital Hamilton Populations (Cohp) - Energy-Resolved Visualization of Chemical Bonding in Solids Based on Density-Functional Calculations. J. Phys. Chem. 1993, 97, 8617–8624. 10.1021/j100135a014. [DOI] [Google Scholar]
- Hoffmann R. A. Chemical and Theoretical Way to Look at Bonding on Surfaces. Rev. Mod. Phys. 1988, 60, 601–628. 10.1103/RevModPhys.60.601. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- Kim F.; Connor S.; Song H.; Kuykendall T.; Yang P. D. Platonic gold nanocrystals. Angew. Chem., Int. Ed. 2004, 43, 3673–3677. 10.1002/anie.200454216. [DOI] [PubMed] [Google Scholar]
- Langille M. R.; Personick M. L.; Zhang J.; Mirkin C. A. Defining Rules for the Shape Evolution of Gold Nanoparticles. J. Am. Chem. Soc. 2012, 134, 14542–14554. 10.1021/ja305245g. [DOI] [PubMed] [Google Scholar]
- Huang J. F.; Zhu Y. H.; Lin M.; Wang Q. X.; Zhao L.; Yang Y.; Yao K. X.; Han Y. Site-Specific. Growth of Au-Pd Alloy Horns on Au Nanorods: A Platform for Highly Sensitive Monitoring of Catalytic Reactions by Surface Enhancement Raman Spectroscopy. J. Am. Chem. Soc. 2013, 135, 8552–8561. 10.1021/ja4004602. [DOI] [PubMed] [Google Scholar]
- Li J. M.; Liu J. Y.; Yang Y.; Qin D. Bifunctional Ag@Pd-Ag Nanocubes for Highly Sensitive Monitoring of Catalytic Reactions by Surface-Enhanced Raman Spectroscopy. J. Am. Chem. Soc. 2015, 137, 7039–7042. 10.1021/jacs.5b03528. [DOI] [PubMed] [Google Scholar]
- Zheng Z. K.; Tachikawa T.; Majima T. Single-Particle Study of Pt-Modified Au Nanorods for Plasmon-Enhanced Hydrogen Generation in Visible to Near-Infrared Region. J. Am. Chem. Soc. 2014, 136, 6870–6873. 10.1021/ja502704n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.