

Abstract

APOBEC3G (A3G) is a restriction factor that provides innate immunity against HIV-1 in the absence of viral infectivity factor (Vif) protein. However, structural information about A3G, which can aid in unraveling the mechanisms that govern its interactions and define its antiviral activity, remains unknown. Here, we built a computer model of a full-length A3G using docking approaches and molecular dynamics simulations, based on the available X-ray and NMR structural data for the two protein domains. The model revealed a large-scale dynamics of the A3G monomer, as the two A3G domains can assume compact forms or extended dumbbell type forms with domains visibly separated from each other. To validate the A3G model, we performed time-lapse high-speed atomic force microscopy (HS-AFM) experiments enabling us to get images of a fully hydrated A3G and to directly visualize its dynamics. HS-AFM confirmed that A3G exists in two forms, a globular form (∼84% of the time) and a dumbbell form (∼16% of the time), and can dynamically switch from one form to the other. The obtained HS-AFM results are in line with the computer modeling, which demonstrates a similar distribution between two forms. Furthermore, our simulations capture the complete process of A3G switching from the DNA-bound state to the closed state. The revealed dynamic nature of monomeric A3G could aid in target recognition including scanning for cytosine locations along the DNA strand and in interactions with viral RNA during packaging into HIV-1 particles.
Short abstract
APOBEC3G structure and dynamics are revealed in simulations and validated by high-speed AFM experiments: it exists in and switches between two forms, compact globular and extended dumbbell forms.
1. Introduction
APOBEC proteins are cellular cytidine deaminases with important roles in mammalian innate immune responses.1−3 Among them, major attention is given to APOBEC3G (A3G), which restricts the replication of HIV-1, hepatitis B virus, retrotransposons, and other DNA-based parasites.4−6 Inhibition of HIV-1 replication can occur in two ways: by deamination of viral ssDNA during reverse transcription1,7,8 and by a deaminase-independent mechanism in which a roadblock is created during the synthesis of complementary DNA.9 Both of these mechanisms require the incorporation of A3G into the viral particle, which is driven by A3G interactions with RNA.10 Advancing A3G stability and its incorporation into virions could be a potent strategy for the improvement of new antiviral therapies.11 However, HIV-1 developed a counteracting viral infectivity factor (Vif) protein that can act against the antiviral activity of A3G.12 A3G gets degraded when bound to Vif, which forms complexes with E3 ubiquitin ligase complex proteins Cullin5, Elongin B/C, and CBF-β.13−15 Therefore, another strategy for HIV restriction is to design antiviral A3G-based treatments that will prevent A3G binding to Vif. All these properties of A3G suggest that elucidating molecular details of A3G structure and understanding A3G interactions with its binding partners, especially those involved in the enzymatic activity (deamination of nucleic acids), are needed for the development of efficient A3G based HIV restrictions.
A3G is a two-domain protein: its N-terminal domain (NTD) interacts with nucleic acids and Vif, and its C-terminal catalytic domain (CTD) carries out the deamination activity.16−18 A first step toward detailed understanding of A3G functional activity is determining its atomic structure. Yet, high-resolution atomic structure of a full-length A3G remains undetermined, due to oligomerization and precipitation of A3G at concentrations required for crystallization.19,20 However, structures of individual mutated CTD and NTD have been determined by NMR spectroscopy and X-ray crystallography,20−23 and the shape of the full-length A3G protein was revealed by small-angle X-ray experiments and advanced envelope restoration methods.24,25 Structures of other APOBEC3 subfamily members have also been reported.26−33
In the present paper, we built the atomic scale model of the full-size A3G via computational modeling and docking of A3G C-terminal and N-terminal domains based on available structures of CTD and NTD of A3G,34,35,23 followed by microsecond-long molecular dynamics (MD) simulations. The simulations revealed a highly dynamic feature of A3G monomer that can lead to extended conformations in which two domains are separated by distances as large as 4.5 nm. To validate the major features of the A3G model, we probed the dynamics of A3G monomer with time-lapse high-speed (HS) AFM imaging. The experimental results are concordant with the computational model. Additional structural and dynamic properties of A3G are revealed, and they are discussed in the light of its functional activities.
2. Results
To date, crystallization and structure determination of a full-size wild-type A3G has been unsuccessful, due to A3G oligomerization and precipitation at the concentrations necessary in crystallization experiments. In the present study, we used molecular modeling to develop computational models for the full-size A3G structures and assess their dynamics. To test the computational models, we compare shapes and dynamics of these models and of the full-size A3G monomers examined in AFM experiments. Nanoscale parameters of experimentally measured and computationally modeled A3G monomers are compared, as described below.
2.1. Computational Models of Monomeric A3G
2.1.1. Ensemble of Structures of A3G Monomer
A computational model of a complete wild-type A3G monomer was obtained by docking the existing structures of NTD and CTD of A3G into a single protein structure, as described in Materials and Methods. In summary, six independent pairs of NTD and CTD structures were docked using the HADDOCK server,36−38 with a restraint that two terminal residues of CTD and NTD need to be involved in mutual interaction (i.e., they are to form a covalent peptide bond). Figure 1 shows an ensemble of complete modeled A3G structures, aligned with respect to either CTD (a) or NTD (b). Several features of the obtained ensemble are to be noted. First, two domains have several preferential positions with respect to each other, leaving significant surface areas of the two domains always exposed. The preferential positions of two domains are partly determined by conformations of the polypeptide chain linker between CTD and NTD (A3G residues 196–203), and partly through favored noncovalent interactions between CTD and NTD surface residues.
Figure 1.

Ensemble of structures of A3G monomer, determined in molecular docking calculations. (a, b) Overlaid A3G structures aligned with respect to the CTD (red) or NTD (blue). (c) Two representative structures of A3G monomer, determined by clustering of the complete ensemble. CTD (red) of the two structures are aligned. (d, e) DNA/RNA-interacting residues (cyan spheres) or Vif-interacting residues (yellow spheres), shown for the two representative structures displayed in panel c.
Analysis of protein shapes in the computed ensemble of A3G structures determined that A3G monomer exists predominantly in two forms: globular and dumbbell forms (Figure 2). These forms are characterized by a parameter d3, defined as the distance between centers of masses of NTD and CTD (defined in Figure S1). To characterize the whole ensemble of docked structures, we computed d3 distances for all the modeled A3G structures. Figure 2 shows the histogram distribution of d3 distances for the whole A3G ensemble, where d3 values range from 3.2 to 4.6 nm. While most of the A3G structures are found to exist in the compact globular shape (here, taken to be d3 < 4.2 nm), there is a significant population that assumes a dumbbell-like form (here, taken to be d3 > 4.2 nm). The overall population of dumbbell conformations in the ensemble of docked structures is ∼25%, determined using the threshold of d3 = 4.2 nm (Figure 2). However, due to the limitations of the docking procedure in which direct interactions between CTD and NTD are favored, the obtained percentage of the dumbbell structures with d3 > 4.5 nm is deemed to be a lower estimate for such structures. Because the docking procedure favors structures where CTD and NTD interact with each other rather than being fully immersed in the solvent, only 3% of all structures in the docked ensemble have d3 > 4.5 nm.
Figure 2.

Histogram distribution of distances between NTD and CTD, d3, for the complete docking ensemble of A3G structures. The histogram is separated into structures that acquire globular form (d3 < 4.2 nm; black rectangles) and dumbbell form d3 > 4.2 (gray rectangles). Insets show examples of A3G in globular and dumbbell forms. A3G is shown in surface representation, where NTD is in blue, and CTD is in red.
To examine the nature of interactions that hold together CTD and NTD, we determined the residues of these domains that are in contact within the docked ensemble, as analyzed in Figure 3. The figure shows that many types of interactions, including charged, polar, and hydrophobic interactions, all contribute to the contact between NTD and CTD. Both in the docked ensemble and in MD simulations, the specific residues present at the interface of two domains vary, but the chemical nature of the residues is similar, as evidenced by low standard deviations in the calculated contact areas. Furthermore, a contact map in Figure S2 summarizes CTD and NTD amino acid residues that are in direct contact for A3G in representative globular and dumbbell forms.
Figure 3.

Interactions at the CTD-NTD interface in the A3G ensemble obtained by docking. (a) CTD and NTD residues that are in contact within a representative A3G conformation. (b) Contact areas between amino acids at interfaces of individual domains, calculated separately for dumbbell and globular A3G protein structures in docked ensembles. The contact areas of residues involved in charge–charge, nonpolar–nonpolar, and polar–polar interactions are plotted in separate categories.
2.1.2. Dynamics and Conformational Changes of A3G Monomers in MD Simulations
In order to explore the dynamics and stability of the A3G monomer, two A3G structures representative of the whole ensemble, shown in Figure 1c, were examined in 1 μs long MD simulations. These structures were obtained through clustering of the complete A3G ensemble, and broadly represent globular and dumbbell conformations of the A3G monomer. RMSDs of two representative A3G structures, shown in Figure 4a, plateau during the first 200 ns of MD simulations, indicating the stability of these structures in aqueous solution. However, the globular structure undergoes a large conformational change first after 200 ns and then after 340 ns of MD simulation, where the NTD and CTD readjust with respect to each other, as shown in Figure 4c,d and Movies S1 and S2. The conformational changes of the globular structure are also evident in the RMSD plots calculated for the 100 ns pieces of the partitioned 1 μs MD trajectory (Figure S3). During this conformational change of the globular A3G, the distance between NTD and CTD, d3, increases from ∼3.2 nm to ∼4.0 nm. This conformational change captures the dynamic nature of our A3G model, and samples the transition between its globular and more extended dumbbell-like forms.
Figure 4.

Stability of A3G structural models in MD simulations. (a) RMSDs of two A3G structural models, visually representative of a globular conformation (red) and a dumbbell conformation (blue). (b) Distances d3 between CTDs and NTDs of the two A3G structural models. Red and blue plots represent the results for globular and dumbbell conformations, respectively. (c, d) Conformational transition of the globular form of a full A3G during a 1 μs MD trajectory. The initial structure of A3G is shown in light blue, the final structure of A3G is shown in green, and the flexible linker, which repositions during the simulation course and correlates with the reorientation of two A3G domains, is shown as a red tube. For comparison, panels c and d show both opaque and transparent structures of initial and final A3G states, aligned with respect to the NTD domain of A3G. Zn2+ ions are present in the simulations, but are not shown for convenience. Movies S1 and S2 display the dynamics of the transition shown in panels c and d.
The simulations reveal that the transition between globular and dumbbell-like forms of A3G is facilitated by a flexible linker that connects NTD and CTD of A3G. While many residues at the interface of CTD and NTD have flexible coil conformations, including the flexible coil residues 195 to 219, our simulations show that residues 196–203 form a flexible linker that can reorganize and lead to a large conformational change of A3G on the time scale of ∼0.34 μs (Figure 4c,d). In this conformational change, two A3G domains change their orientations with respect to each other, but largely preserve their secondary and tertiary structures (Figures S4 and S5). During the globular to dumbbell transition, the number of residues on NTD and CTD that are in direct contact with each other is significantly reduced (Figure S6).
Conformational analyses of individual NTD and CTD in A3G models, presented in Figures S4, S5, and S7, show that these domains preserve their overall structures on 1 μs time scales, as RMSD values for CTD and NTD do not exceed 0.5 and 0.3 nm, respectively. In CTD, a moderate readjustment of helix5 is observed. However, the NTD undergoes a transition in its DNA binding pocket. In computed structures, the NTD is initially prepared with its DNA binding pocket in the DNA-bound conformation (here called an open state),35 yet without the DNA substrate. The open state of the DNA binding pocket, formed by W94, Y124, and neighboring residues, is shown in Figure 5a. Within several nanoseconds, DNA binding pockets switch from open states to transition states in MD simulations of both A3G models (Movies S3 and S5). The transition states in which W94 is no longer available for DNA binding, due to interactions with neighboring protein residues, occurred in ∼180 ns (dumbbell model) and in <30 ns (globular model). In simulations of the globular A3G model, the DNA binding pocket remained in the transition state after 1 μs. However, in simulations of the dumbbell A3G model, the DNA binding pocket fully transitioned to a closed state34 after 0.53 μs, as shown in Figure 5c,d and in Movie S4. In the closed state (Figure 5c), the DNA binding residue W94 is no longer available for DNA binding. The simulations of the dumbbell A3G model showed that the neighboring residues F21, R24, W34, S95, and R122 show major conformational changes during the open to closed state switching, in agreement with observations of the reference (35).
Figure 5.

Conformational changes of the DNA binding pocket of A3G-NTD in MD simulations. (a–c) Snapshots of the open, transition, and closed states of the DNA binding pocket of NTD. The open state is obtained from the crystal structure (pdb id: 5k83; bound DNA is shown in pink), and the other states are obtained from MD simulations of A3G in the dumbbell conformation. (d, e) Time series of the distance between side chains of W94 and Y124 amino acids, shown for dumbbell and globular conformations of A3G. The distance is calculated between centers of mass of the side chains of two amino acids. Movies S3, S4, S5, and S6 display the dynamics of the DNA-binding site during MD simulations of A3G in dumbbell and globular forms.
DNA/RNA-interacting residues (R24, W94, W127, R213, R215, R313, R320, R374, R376) and Vif-interacting residues (Y19, I26, L27, W34, V58, Y59, Y124, F126, W127, D128, P129, D130) of representative A3G structures are shown in Figures 1d and 1e, respectively. In both structures, most of these A3G residues involved in binding to substrates remain available for recognizing and binding to A3G binding partners. In both MD simulations and in the whole docked ensemble, these functionally important residues remain largely exposed to the solvent, as shown in Figures S8 and S9.
2.2. Dynamics of A3G Monomer in Solution Visualized by Time-Lapse HS-AFM
To characterize the dynamics of A3G monomer, time-lapse experiments of fully hydrated samples were obtained with the high-speed AFM (HS-AFM). To make sure that we followed the dynamics of the A3G monomer, the data were collected for A3G monomer dissociated from the complex between A3G dimer and RNA. The data were acquired by continuous scanning in liquid over the selected area with the rate 398 ms/frame, and all frames were assembled as movies. One such movie is shown in Movie S7, and a few selected frames illustrating the dynamics of A3G are shown in Figure 6a.
Figure 6.

A3G can exist in compact globular and extended dumbbell conformations. (a) Selected frames from Movie S7. The size of the images is 40 nm. The scanning rate corresponds to 398 ms per frame. (b) A representative AFM image of the globular-shaped A3G monomer. Inset i provides the definition of d1 and d2. Inset ii shows the plot of fluctuations of the d1/d2 ratio of A3G monomer calculated for ∼250 frames captured. The mean value of the d1/d2 ratio is equal to 1.3 ± 0.3. (c) A representative image of the dumbbell-shaped A3G monomer. Inset iii presents the cross section of the dumbbell structure as measured by AFM. Inset iv shows the histogram of the measured d3 distance between two maxima, obtained from 3 separate movies. The maximum corresponds to 4.5 ± 1.0 nm.
Frame 1 shows A3G in a slightly elongated globular shape, which is getting more extended in frame 15 and then returns to the initial shape in frame 22. To characterize the globular shape of the A3G, shown in Figure 6b, we calculated the ratio of two orthogonal parameters, d1 and d2, defined in inset i of Figure 6b. The measurements of d1/d2 ratio were done for over 250 frames from three separate movies, and the data are presented in Figure 6b, inset ii. The graph from Figure 6b demonstrates that most of the time A3G has compact, globular shape that fluctuates between slightly elongated (d1/d2 ∼ 1.1) and ellipsoid (d1/d2 ∼ 1.7).
In the remaining 16% of cases, the A3G monomer adopts a clear dumbbell shape, as shown in Figure 6c, and this value is roughly in line with the occurrence of the dumbbell structure in the simulation. Frames 25 and 39 from Movie S7 demonstrate the dumbbell structure of A3G with clear separation between domains, which later changes back to the globular structure, as shown in frame 49. To characterize the dumbbell shape of A3G, parameter d3 is used, defined as the distance between maxima of the peaks of the height cross section of the A3G dumbbell (Figure 6c, inset iii). Analysis of the dynamics of the dumbbell shape of A3G was performed based on the data obtained from three separate movies, resulting in a histogram of d3 distribution shown in Figure 6c (inset iv). The data set was approximated by a Gaussian with maxima at 4.5 nm, indicating that distance between CTD and NTD of A3G fluctuates.
To graphically illustrate the dynamics of A3G, the horizontal diameter d1 was measured for both globular and dumbbell structures of A3G. The data obtained from one of the movies are presented in the Figure S10. The time trajectory of the d1 value of the protein against the frame number is shown in Figure S10A. Green and red symbols correspond to globular and dummbell shapes of A3G, respectively. Figure S10B shows the size distributions of globular (black bars) and dumbbell (red bars) structures assembled as a histogram. Together, data in Figure 6 and Figure S10 demonstrate that A3G monomer is structurally dynamic, switching between compact globular and extended dumbbell conformations.
3. Discussion
In the present study, we provided a model of a full monomeric structure of A3G and validated this model using HS AFM data. The most striking and unique feature of monomeric A3G observed in the present study is its broad dynamics. A3G can be in a rather compact structure in which two domains are close to each other or they are separated far with little to no intermolecular interaction between domains. These two major conclusions are fully supported by high-speed AFM imaging in which the dynamics of A3G was directly visualized. Not only are overall sizes of A3G in both conformations consistent with the model but also the partition of A3G between the two states is in good coincidence. Computational modeling revealed a number of important properties of A3G that are discussed below.
Within an ensemble of docked structures, NTD and CTD are held together by a variety of contacts, as shown for a representative A3G structure in Figure 3. Yet, A3G needs to be able to perform its functions, which occur via binding to nucleic acids. The solvent exposure of the DNA-, RNA-, and Vif- binding residues are shown in Figures S8 and S9. In most of the obtained A3G structures, including the structures within the whole docked ensemble and the representative A3G structures obtained by clustering, the A3G residues that bind nucleic acids and Vif substrates are exposed to solvent, and thus accessible for binding to these substrates.
Long-range electrostatic interactions are likely important for guiding A3G binding to RNA and DNA substrates. Therefore, the electrostatic properties of NTD and CTD should influence the A3G–nucleic acid binding process. The electrostatic properties of the A3G surface are shown in Figure 7: NTD surface has largely positive electrostatic potential, whereas the CTD surface has largely negative potential, except around the Zn-binding site, where the nucleic acids should be deaminated. Since nucleic acid substrates need to search and bind to the A3G and find the deaminase sites, dynamics of A3G domains with respect to each other are likely important for the search process, which eventually should lead to accommodation of bound nucleic acids into the enzymatic Zn-binding site.
Figure 7.
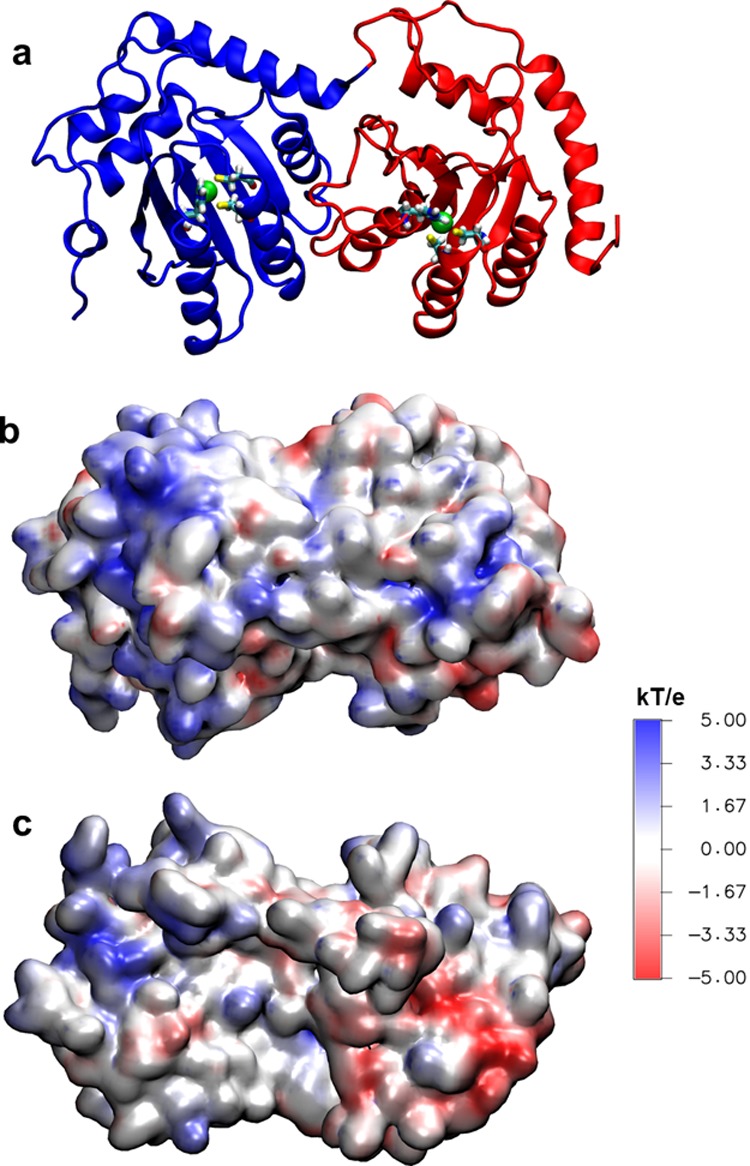
(a) Energy minimized A3G structure in dumbbell conformation, used to run the MD simulations, with highlighted zinc active sites. NTD is shown in blue, CTD is shown in red, and zinc ions are shown in green. (b, c) A3G, in two opposite orientations, colored according to the electrostatic potential. The right domain is CTD, and the left domain is the NTD. A3G structure in panel c is rotated by 180° around its long axis, with respect to the A3G structure shown in panel b.
The dynamics of NTD and CTD with respect to each other are facilitated by a flexible linker that connects them. While there are many residues with flexible coil conformations that connect CTD and NTD, our simulations identified that A3G residues 196–203 form a coiled linker that can reorganize and lead to a large conformational change of A3G on the time scale of MD simulations, where two A3G domains change their orientations but preserve the secondary and tertiary structures (Figure 4 and Movies S1 and S2). The conformational change in MD simulations occurred in 0.34 μs, showing that the dynamics of the linker and the domains can be very quick with respect to the time scales tracked in experiments (milliseconds to seconds).
The NTD in simulated A3G models undergo transitions from open state to closed state during simulations (Movies S3, S4, S5, and S6). This transition is facilitated primarily by Y124 residue via stacking interactions with W94, suggesting a role of a “molecular switch” for Y124 residue as proposed in previous experimental studies.35 This transition also results in conformational changes for residues that are neighboring to W94 residue, which are proposed to make direct or indirect contacts with DNA when binding to A3G. The interface between CTD and NTD of our globular model is significantly different from the proposed dimer CD1–CD1 model,35 CD2–CD2 model,21 and CD2–CD2 model39 dimers whereas our dumbbell model has an interface similar to the head-to-tail model of the CD2–CD2 dimer.39 The NTD and CTD interface in the dumbbell model is well maintained during the MD simulations.
A3G structures and dynamics identified above should have functional importance. A3G function involves binding to ssDNA, processive scanning,40−42 target recognition, and stochastic deamination of ssDNA.42 Furthermore, FRET results43 reported two modes of A3G-ssDNA interactions: (1) a mode in which A3G quickly binds to and quickly dissociates from ssDNA; (2) a mode that involves a longer binding event, followed by rapid scanning of DNA by A3G (captured over ∼25 s time scale). When sliding on and scanning the ssDNA, A3G could behave as a rigid body or it could slide by internal reorganization of its domains. Rigid body sliding would need to involve simultaneous breaking of many bonds, which should have a large energy barrier, followed by a simultaneous creation of many new bonds between A3G and ssDNA. Instead, it could be easier for A3G to slide by overcoming the energy barrier associated with gradual reorganization of its individual domains on the ssDNA substrate, where bond breaking (or forming) between A3G and ssDNA can occur more gradually.
Overall, the observed inherent flexibility of the A3G monomer in the form of interdomain dynamics may facilitate target both binding to ssDNA (when A3G diffuses close to the target) and quick sliding on and scanning of the target ssDNA. Finally, the adjustment of A3G into the binding pose on ssDNA that leads to the deamination of ssDNA may require significant conformational changes of A3G. The interdomain dynamics could also contribute to A3G search for the catalytic binding pose, especially as the nucleic acid binding residues are located on different A3G domains and at solvent-exposed surfaces that can be spatially distant (Figure 1d,e). Furthermore, the interdomain dynamics could be facilitating the process of viral RNA packaging into HIV virion, which is aided by interactions with A3G.
Our observations provide new understanding of A3G monomer, the basic unit that possesses both catalytic and antiviral activities, and forms the basis for future studies of A3G interactions with its binding partners, and design of novel deamination-based antiviral therapies.
4. Materials and Methods
4.1. Atomic Models
To prepare a complete wild-type A3G monomer, structures of its C-terminal domain (pdb id: 2KBO(34)) and N-terminal domain (pdb id: 5K83(35)) were used. While CTD structure had a wild-type sequence, NTD crystal structure contained 57 mutations from the wild-type sequence (UniProtKB code: Q9HC16). To prepare a wild-type NTD, its mutations were mutated to their corresponding wild-type residues within VMD.44 A missing loop (residues 139–142) on NTD was added with VMD, by alignment to another solved NTD structure (pdb id: 2MZZ).23
4.2. Molecular Docking
Molecular docking calculations were performed with six pairs of independent structures of NTD (residues 1–195, previously relaxed in molecular dynamics simulations described below) and CTD (residues 196–384) domains. The six pairs of structures differed in orientations of terminal residues that should form a covalent bond in the complete A3G, namely, residues 195 and 196. Prepared pairs of NTD and CTD structures were docked using the HADDOCK server36−38 in three steps: rigid-body docking, semiflexible docking, and water refinement. During the docking procedure in HADDOCK, residues 195 of NTD and 196 of CTD were defined as active residues involved in the interaction. NTD–CTD docking was otherwise performed with the default parameters of other server settings in HADDOCK. Since HADDOCK program ignores the ions, the Zn2+ ions were not included during the docking process. The selected docking procedure resulted in an ensemble of 8500 complete A3G monomer structures. The representative A3G structures were obtained through cluster analysis of the docked ensemble with the g_cluster tool in GROMACS.45 Complete full-length A3G systems were modeled by placing Zn2+ ions into active site pockets, and ensuring that these ions maintain their appropriate active site interactions, as observed in crystal structures of individual NTD and CTD, which have been used for docking.
4.3. Molecular Dynamics Simulations
MD simulations of A3G systems, described with the CHARMM36 force field,46,47 were performed with NAMD2.11 code.48 The particle mesh Ewald (PME) method49 was used for the evaluation of long-range Coulomb interactions. The time step was set to 2.0 fs; all bonds involving hydrogen were constrained with the SHAKE algorithm. All simulations were performed in the isobaric–isothermal (NPT) ensemble, at a constant temperature of 310 K, with a friction constant of 1.0 ps–1, and at a constant pressure of 1 bar. Short-range and long-range interactions were evaluated every 2 and 4 fs, respectively. The MD simulations involved typically altogether ∼40,000 atoms (wild-type NTD, water, ions) and ∼63,500 atoms (A3G, water, ions). All systems prepared were minimized for 2000 steps. Then, ions and water molecules were equilibrated for 2 ns around proteins, which were restrained using harmonic forces with a spring constant of 1 kcal/(molÅ2). Unrestrained NTD was simulated for 100 ns (Figure S11), and the complete unrestrained models of A3G were simulated for 1 μs.
4.4. Size Analysis of Modeled A3G Ensemble
To compare sizes of A3G in the docked ensemble and in the images obtained in HS-AFM experiments, we defined three spatial parameters, d1, d2, and d3. Parameters d1 and d2 are visually estimated sizes of A3G along two orthogonal dimensions (x, y), when the long dimension of A3G is lined up with the x axis, and the short dimension of A3G is lined up with the y axis, as shown in Figure S1a. Parameter d3 is the distance between centers of mass of NTD and CTD, as defined in Figure S1b.
4.5. High Speed AFM Experiments
4.5.1. RNA Hybrid Substrate Preparation
Due to a high flexibility and low contrast of RNA in AFM images, we prepared a special RNA hybrid substrate. The detailed procedure for assembly of such a substrate is described in refs (50) and (51). Briefly, the RNA hybrid substrate consists of 69nt RNA spliced onto the end of 145bp dsDNA fragment. The RNA part is a template for binding with A3G and dsDNA functions as an imaging tag. Such a construct allows unambiguous identification and characterization of the RNA– A3G complex, which appeared at the end of the dsDNA part.
4.5.2. Preparation of A3G Complexes with RNA Substrate
Full wild-type human A3G protein purified as described in ref (52) was mixed with RNA hybrid at 4:1 protein to substrate ratio in reaction buffer containing 50 mM HEPES, pH 7.5, 100 mM NaCl, 5 mM MgCl2, 1 mM DTT and incubated for 15 min at 37 °C as indicated in ref (51).
4.5.3. Sample Preparation and Time-Lapse HS AFM Imaging
The samples were prepared according to ref (51). Namely, 2 nM A3G–RNA complex in the buffer, containing 50 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 1 mM DTT, was deposited on the APS treated mica surface52,53 for 2 min, rinsed with the buffer, and imaged continuously without drying the sample. To make sure that we followed the dynamics of the A3G monomer, the data were collected for A3G dissociated from the complex, which was initially formed between A3G dimer and RNA. HS-AFM instrument (RIBM, Japan; design of T. Ando54,55) was used for continuous scanning over a 200 nm × 200 nm area with the rate 398 ms/frame. AFM probes obtained by an electron beam deposition procedure on short cantilevers with spring constant between 0.1 and 0.2 N/m and resonance frequency of 400–1000 kHz (BL-AC10DS-A2, Olympus, Japan) were used.
4.5.4. Data analysis
To characterize the size and shape of A3G protein, three parameters were calculated: d1, d2, and d3. For each parameter, the value was obtained from the cross section feature of the FemtoScan Online software (Advance Technologies Center, Moscow, Russia) as described in ref (56). The value for the d1/d2 ratio was calculated from two orthogonal cross sections of the particle from the AFM image as seen in inset i in Figure 6b. The cross section of the dumbbell shape of A3G, d3, was calculated from the distance between two distinct maxima on the plot as seen in Figure 6c, inset iii.
Acknowledgments
This work is supported by a grant from the National Institutes of Health (NIH-R01GM11800601) to Y.L.L. and by the startup funding from the University of Texas at El Paso (S.G., L.V.). The authors gratefully acknowledge computer time provided by the Texas Advanced Computing Center (TACC). R.S.H. is an Investigator of the Howard Hughes Medical Institute.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00346.
Author Contributions
‡ S.G., Y.P., and L.S.S. made equal contributions.
The authors declare no competing financial interest.
Supplementary Material
References
- Harris R. S.; et al. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. 10.1016/S0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- Strebel K.; Luban J.; Jeang K. T. Human cellular restriction factors that target HIV-1 replication. BMC Med. 2009, 7, 48. 10.1186/1741-7015-7-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRue R. S.; et al. Guidelines for naming nonprimate APOBEC3 genes and proteins. J. Virol. 2009, 83, 494–497. 10.1128/JVI.01976-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter J. D.; Bennett R. P.; Smith H. C. The apobec protein family: United by structure, divergent in function. Trends Biochem. Sci. 2016, 41, 578–594. 10.1016/j.tibs.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias J.; Koyama F. T.; Kinomoto M.; Tokunaga K. Retroelements versus APOBEC3 family members: No great escape from the magnificent seven. Front. Microbiol. 2012, 3, 275. 10.3389/fmicb.2012.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris R. S.; Dudley J. P. APOBECs and virus restriction. Virology 2015, 479–480, 131–145. 10.1016/j.virol.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeat B.; et al. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- Zhang H.; et al. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 2003, 424, 94–98. 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaurasiya K. R. Oligomerization transforms human APOBEC3G from an efficient enzyme to a slowly dissociating nucleic acid-binding protein. Nat. Chem. 2014, 6, 28–33. 10.1038/nchem.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York A.; Kutluay S. B.; Errando M.; Bieniasz P. D. The RNA Binding specificity of human APOBEC3 proteins resembles that of HIV-1 nucleocapsid. PLoS Pathog. 2016, 12, e1005833. 10.1371/journal.ppat.1005833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene W. C. The brightening future of HIV therapeutics. Nat. Immunol. 2004, 5, 867–871. 10.1038/ni0904-867. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Baig T.; Love R. P.; Chelico L. Suppression of APOBEC3-mediated restriction of HIV-1 by Vif. Front. Microbiol. 2014, 5, 450. 10.3389/fmicb.2014.00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopak K.; de Noronha K.; Yonemoto W.; Greene W. C. HIV-1 vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 2003, 12, 591–601. 10.1016/S1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- Yu X.; et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 2003, 302, 1056–1060. 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- Jager S.; et al. Vif hijacks CBF-β to degrade APOBEC3G and promote HIV-1 infection. Nature 2012, 481, 371–375. 10.1038/nature10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goila-Gaur R.; Strebel K. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology 2008, 5, 51. 10.1186/1742-4690-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura S.; Ode H.; Iwatani Y. Structural features of antiviral apobec3 proteins are linked to their functional activities. Front. Microbiol. 2011, 2, 258. 10.3389/fmicb.2011.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haché G.; Liddament M. T.; Harris R. S. The retroviral hypermutation specificity of APOBEC3F and APOBEC3G is governed by the C-terminal DNA cytosine deaminase domain. J. Biol. Chem. 2005, 280, 10920–10924. 10.1074/jbc.M500382200. [DOI] [PubMed] [Google Scholar]
- Jarmuz A.; et al. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 2002, 79, 285–296. 10.1006/geno.2002.6718. [DOI] [PubMed] [Google Scholar]
- Harjes E.; et al. An extended structure of the APOBEC3G catalytic domain suggests a unique holoenzyme model. J. Mol. Biol. 2009, 389, 819–832. 10.1016/j.jmb.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shandilya S. M.; et al. Crystal structure of the APOBEC3G catalytic domain reveals potential oligomerization interfaces. Structure 2010, 18, 28–38. 10.1016/j.str.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K. M.; et al. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature 2008, 452, 116–119. 10.1038/nature06638. [DOI] [PubMed] [Google Scholar]
- Kouno T.; et al. Structure of the Vif-binding domain of the antiviral enzyme APOBEC3G. Nat. Struct. Mol. Biol. 2015, 22, 485–491. 10.1038/nsmb.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter J. D.; Krucinska J.; Raina J.; Smith H. C.; Wedekind J. E. A hydrodynamic analysis of APOBEC3G reveals a monomer-dimer-tetramer self-association that has implications for anti-HIV function. Biochemistry 2009, 48, 10685–10687. 10.1021/bi901642c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedekind J. E.; et al. Nanostructures of APOBEC3G support a hierarchical assembly model of high molecular mass ribonucleoprotein particles from dimeric subunits. J. Biol. Chem. 2006, 281, 38122–38126. 10.1074/jbc.C600253200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byeon I. J. L.; et al. NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat. Commun. 2013, 4, 1890. 10.1038/ncomms2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra M.; et al. Structural determinants of human APOBEC3A enzymatic and nucleic acid binding properties. Nucleic Acids Res. 2014, 42, 1095–1110. 10.1093/nar/gkt945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura S.; et al. The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat. Struct. Mol. Biol. 2012, 19, 1005–1010. 10.1038/nsmb.2378. [DOI] [PubMed] [Google Scholar]
- Siu K. K.; Sultana A.; Azimi F. C.; Lee J. E. Structural determinants of HIV-1 Vif susceptibility and DNA binding in APOBEC3F. Nat. Commun. 2013, 4, 2593. 10.1038/ncomms3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn M. F.; et al. Crystal structure of the DNA cytosine deaminase APOBEC3F: The catalytically active and HIV-1 Vif-binding domain. Structure 2013, 21, 1042–1050. 10.1016/j.str.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaban N. M.; Shi K.; Li M.; Aihara H.; Harris R. S. 1.92 Angstrom zinc-free APOBEC3F catalytic domain crystal structure. J. Mol. Biol. 2016, 428, 2307–2316. 10.1016/j.jmb.2016.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K.; Carpenter M. A.; Kurahashi K.; Harris R. S.; Aihara H. Crystal structure of the DNA deaminase APOBEC3B catalytic domain. J. Biol. Chem. 2015, 290, 28120–28130. 10.1074/jbc.M115.679951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K.; et al. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 2017, 24, 131–139. 10.1038/nsmb.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa A.; et al. Structure, interaction and real-time monitoring of the enzymatic reaction of wildtype APOBEC3G. EMBO J. 2009, 28, 440–451. 10.1038/emboj.2008.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X.; Li S. X.; Yang H.; Chen X. S. Crystal structures of APOBEC3G N-domain alone and its complex with DNA. Nat. Commun. 2016, 7, 12193. 10.1038/ncomms12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries S. J.; et al. HADDOCK versus HADDOCK: New features and performance of HADDOCK2.0 on the CAPRI targets. Proteins: Struct., Funct., Genet. 2007, 69, 726–733. 10.1002/prot.21723. [DOI] [PubMed] [Google Scholar]
- Schmitz C.; et al. Protein-protein docking with HADDOCK. In NMR of Biomolecules; Wiley-VCH Verlag GmbH & Co. KGaA: 2012; pp 520–535. [Google Scholar]
- van Zundert G. C. P.; et al. The HADDOCK2.2 web server: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- Lu X.; et al. Crystal structure of DNA cytidine deaminase ABOBEC3G catalytic deamination domain suggests a binding mode of full-length enzyme to single-stranded DNA. J. Biol. Chem. 2015, 290, 4010–4021. 10.1074/jbc.M114.624262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelico L.; Sacho E. J.; Erie D. A.; Goodman M. F. A model for oligomeric regulation of APOBEC3G cytosine deaminase-dependent restriction of HIV. J. Biol. Chem. 2008, 283, 13780–13791. 10.1074/jbc.M801004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelico L.; Prochnow C.; Erie D. A.; Chen X. S.; Goodman M. F. Structural model for deoxycytidine deamination mechanisms of the HIV-1 inactivation enzyme APOBEC3G. J. Biol. Chem. 2010, 285, 16195–16205. 10.1074/jbc.M110.107987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelico L.; Pham P.; Calabrese P.; Goodman M. F. APOBEC3G DNA deaminase acts processively 3′ --> 5′ on single-stranded DNA. Nat. Struct. Mol. Biol. 2006, 13, 392–399. 10.1038/nsmb1086. [DOI] [PubMed] [Google Scholar]
- Senavirathne G.; et al. Single-stranded DNA scanning and deamination by APOBEC3G cytidine deaminase at single molecule resolution. J. Biol. Chem. 2012, 287, 15826–15835. 10.1074/jbc.M112.342790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Van Der Spoel D.; et al. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Best R. B.; et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ, and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; MacKerell A. D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden; York T. D.; Pedersen L. Particle mesh Ewald: An n·log(n) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Shlyakhtenko L. S.; et al. Atomic force microscopy studies provide direct evidence for dimerization of the HIV restriction factor APOBEC3G. J. Biol. Chem. 2011, 286, 3387–3395. 10.1074/jbc.M110.195685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y.; et al. Nanoscale characterization of interaction of APOBEC3G with RNA. Biochemistry 2017, 56, 1473–1481. 10.1021/acs.biochem.6b01189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyakhtenko L. S.; et al. Atomic force microscopy studies provide direct evidence for dimerization of the HIV restriction factor APOBEC3G. J. Biol. Chem. 2011, 286, 3387–3395. 10.1074/jbc.M110.195685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyakhtenko L. S.; Gall A. A.; Lyubchenko Y. L. Mica functionalization for imaging of DNA and protein-DNA complexes with atomic force microscopy. Methods Mol. Biol. 2012, 931, 295–312. 10.1007/978-1-62703-056-4_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando T.; Uchihashi T.; Kodera N. High-speed AFM and applications to biomolecular systems. Annu. Rev. Biophys. 2013, 42, 393–414. 10.1146/annurev-biophys-083012-130324. [DOI] [PubMed] [Google Scholar]
- Shlyakhtenko L. S.; et al. Atomic force microscopy studies of APOBEC3G oligomerization and dynamics. J. Struct. Biol. 2013, 184, 217–225. 10.1016/j.jsb.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyakhtenko L. S.; et al. Molecular mechanism underlying RAG1/RAG2 synaptic complex formation. J. Biol. Chem. 2009, 284, 20956–20965. 10.1074/jbc.M109.028977. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.