

Abstract
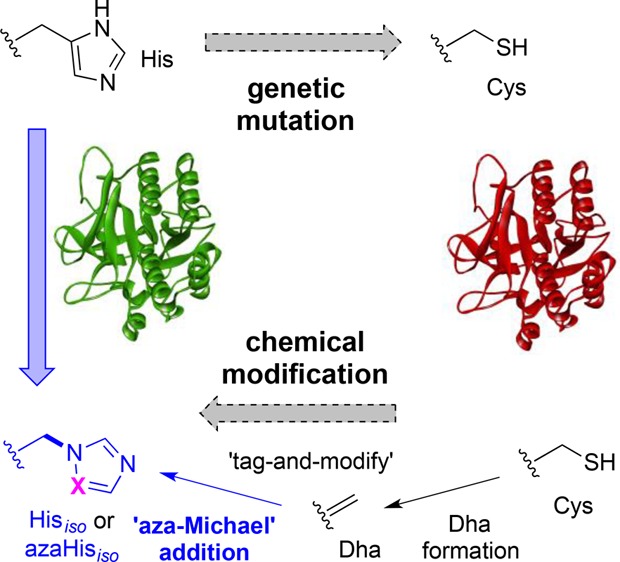
Biomimicry valuably allows the understanding of the essential chemical components required to recapitulate biological function, yet direct strategies for evaluating the roles of amino acids in proteins can be limited by access to suitable, subtly-altered unnatural variants. Here we describe a strategy for dissecting the role of histidine residues in enzyme active sites using unprecedented, chemical, post-translational side-chain-β,γ C–N bond formation. Installation of dehydroalanine (as a “tag”) allowed the testing of nitrogen conjugate nucleophiles in “aza-Michael”-1,4-additions (to “modify”). This allowed the creation of a regioisomer of His (iso-His, Hisiso) linked instead through its pros-Nπ atom rather than naturally linked via C4, as well as an aza-altered variant aza-Hisiso. The site-selective generation of these unnatural amino acids was successfully applied to probe the contributing roles (e.g., size, H-bonding) of His residues toward activity in the model enzymes subtilisin protease from Bacillus lentus and Mycobacterium tuberculosis pantothenate synthetase.
Short abstract
Chemical mutagenesis can expand genetic methods. Now, chemical, post-translational side-chain-β,γ C−N bond formation probes activities of new histidine regioisomers and analogues (Hisiso, aza-Hisiso).
Introduction
Covalent protein chemistry provides well-appreciated opportunities in protein labeling1−3 and the attachment of cargoes.4 It also has great potential in allowing the creation of subtle changes and in the creation of precise alterations and functional mimics in proteins, beyond the limits of traditional biology, that in turn can allow deeper understanding of protein mechanism.5,6 Among one of the most intriguing strategies is a notion of alteration to test recapitulation of (e.g., enzymatic) activity through the replacement of a “lost” functional group with a variant (sometimes known as chemical “rescue”).7,8 This has been applied in various ways including, for example, the rescue by a noncovalently associated prosthetic9,10 (that may have relevance to more general activation by small molecules11,12); or by the “uncaging” of encoded “caged” unnatural amino acids.13−15 As a form of “covalent chemical rescue”, chemical modifications of amino acid residues in enzyme active sites have primarily explored cysteine (Cys) aminoethylation to mimic lysine (Lys) (e.g., ribulosebisphosphate carboxylase/oxygenase,16 aspartate aminotransferase,17 leader peptidase,18 aldolase,19 topoisomerase,20 sugar lyase,21 and pantothenate synthetase22) with other, more rare, examples of attempted mimicry of arginine (Arg),23 glutamate (Glu),24 and even histidine (His).25−27 However, most of these mimics differ in critical parameters such as size, constitution, and/or side-chain length. To our knowledge, no strategies for mimicking amino acids as their direct regioisomers have been possible to date; these would have the potential to act as precisely different isosteres allowing careful dissection of contributing functions. Here, we create and test the effectiveness of the first such regioisomer of His in proteins, using a variant that is linked regioisomerically through its pros-Nπ atom (iso-His, Hisiso) rather than naturally linked via C4 (Figure 1a) and its aza-analogue (aza-Hisiso).
Figure 1.

(a) Concept of protein activity probing via post-translational mutagenesis to regioisomeric Hisiso or aza-Hisiso in a protein (e.g., enzyme active site). (b) Proposed “tag-and-modify” approach to the installation of Hisiso or aza-Hisiso based on dehydroalanine (Dha) formation followed by novel β,γ-C,N aza-Michael addition.
Results
We chose to explore the effect of chemical mutagenesis on enzyme activity through conversion via cysteine (Cys) to the His-mimic residue iso-histidine (Hisiso, Figure 1). His can be a sensitive catalytic residue in several enzymes,28 and Cys would provide a dramatically different side chain in a logical mutational analysis pathway: e.g., His → Cys → Hisiso (Figure 1a). We reasoned that Hisiso (and variants) could in principle be introduced site-selectively using a “tag-and-modify” approach29 (Figure 1b) based on the installation of dehydroalanine (Dha) (potentially from cysteine (Cys)30) followed by conjugate Michael-type addition. Until now, addition of sulfur nucleophiles to Dha (“thio-Michael-type” additions)31 has been the dominant use of this unnatural amino acid residue in proteins, including use in “chemical rescue”.21 However, other potential nucleophiles, such as amines, could be considered (“aza-Michael-type” additions, Figure 1b), thereby opening the door to this new form of protein modification.32 We report here a validation of this approach on intact proteins through the first examples of β,γ-C,N bond formation.33−35 Prior examples of aza-Michael additions, attempted only on extended acrylamido/vinylsulfonamido motifs, are rare36 and would not allow the precise mutagenesis via β,γ-C,N bond formation intended here. While aza-Michael reactions have typically required essentially protein-incompatible conditions (harsh catalysis37 and/or the use of organic solvents38,39), the noted beneficial effect of aqueous media upon rate40 and the success of vinylsulfonamides36 suggested some potential.
We tested the proposed β,γ-C,N “aza-Michael” addition of imidazole to Dha on intact proteins using the subtilisin from Bacillus lentus(41) (SBL, EC 3.4.21.62). SBL is a suitably robust model protein for this approach; it has no native Cys allowing the positioning of Dha via the chemical conversion of a Cys residue introduced through mutagenesis. Thus, the single cysteine mutant SBL-S156C (Ser156 → Cys156) was used to generate SBL-Dha156 using 2,5-dibromohexanediamide (DBHDA).30 Site 156 sits at the base of the S1 (Schechter–Berger nomenclature42) pocket of the active site. Gratifyingly, full conversion to the desired SBL-S156Hisiso, demonstrating the first examples of β,γ-C,N bond formation on proteins, was observed after incubation of SBL-S156Dha with imidazole for 5 h at 37 °C (Figure S1). Importantly, determination of the activity of SBL-Hisiso156 revealed that the conditions of installation were compatible with maintaining protein function. Prior studies have allowed chemical installation of bulky charged groups43 or classical mutagenesis to His at this site;44,45 through the determination of kinetic parameters (SBL-Hisiso156, kcat = 122 ± 6 s–1; KM = 0.71 ± 0.06 mM–1; SBL-wt, kcat = 153 ± 4 s–1; KM = 0.73 ± 0.05 mM–1), it was confirmed that this site, where Hisiso is essentially isosteric to His without H-bond donation, has a minimal role in activity determination, consistent with an outward facing disposition.46
Next, M. tuberculosis pantothenate synthetase (PanC; EC 6.3.2.1)47 was selected as a model protein to probe the proposed His-to-Hisiso activity “modulation” (Figure 1a). PanC catalyzes the ATP-dependent condensation of d-pantoate and β-alanine forming pantothenate (Figure 2a).47 Biosynthesis of pantothenate (vitamin B5) is essential for the growth of bacteria, yeast, and plants and, in particular, for virulence of M. tuberculosis; it is thus a potential therapeutic target.48 In the catalytic site of PanC, histidines His44 and His47 were identified as having a proposed49 critical role in ATP binding (Figure 2b); prior mutation of His → Ala in either site led to decrease in activity.22 These therefore presented potentially sensitive sites at which to test regioisomeric Hisiso mutation. Notably, both sites are buried (cf. the more surface-exposed SBL-156 site) and would provide a more stringent test of the Hisiso formation strategy.
Figure 2.
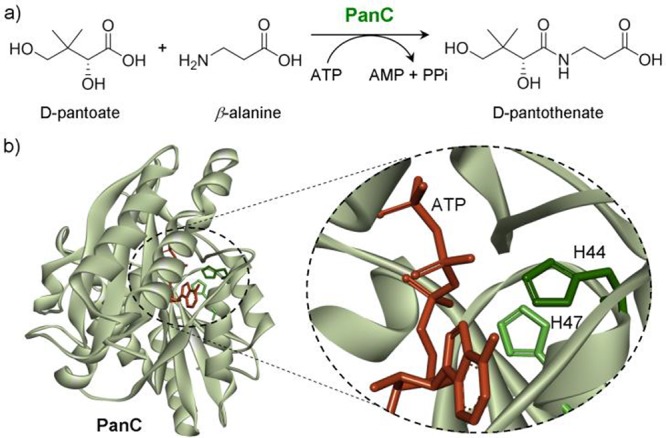
(a) The condensation of d-pantoate with β-alanine catalyzed by pantothenate synthetase (PanC). (b) PanC active site image highlighting His47 and His44 and their proposed interaction with the α and β phosphates of ATP, respectively, during catalysis [derived from PDB 1N2E (ref (48))].
Dha was introduced to position 44 or 47 of PanC protein by site-directed mutagenesis first to Cys44 or Cys47 to give PanC-H44C or PanC-H47C. We then explored chemical conversion to Dha through bis-alkylation/elimination reagent. Wild type PanC-wt has no native cysteine, which, as for SBL, we envisaged would enable full control of the site of Dha formation. Consistent with literature reports,22 PanC proved a challenging substrate protein, strikingly susceptible to unfolding and precipitation: this necessitated a novel strategy for Dha generation. Typical Dha-forming reaction with DBHDA30 (Figure 3, R1 = R2 = CONH2) or the use of temperatures >25 °C over prolonged periods with any reagents proved detrimental to PanC (as judged by CD spectroscopy (Figure S5)). It was therefore not possible to directly chemically introduce Dha into this sensitive protein using standard methods.
Figure 3.

Creation of Hisiso in PanC at sites (a) 44 and (c) 47 using a novel “interrupted” elimination strategy to generate Dha: step 1 monoalkylation and purification in this interrupted state prevented loss of this sensitive protein; then steps 2 and 3 were simply performed by incubation to give Dha. (b). Conditions: (i) site-directed mutagenesis; (ii) 50 equiv of dibromide, 25 °C, aqueous buffer (NaPi 50 mM, pH 8.0); DBHDA, R1 = R2 = CONH2; MDBP, R1 = COOMe, R2 = H, then gel filtration, then incubation 25 °C; (iii) imidazole (aqueous), pH 9–10.
Methyl 2,5-dibromopentanoate (MDBP)50 (Figure 3, R1 = COOMe, R2 = H), however, proved both sufficiently reactive with PanC and advantageously soluble in aqueous buffer so as to allow rapid monoalkylation. This allowed us to consider the exploitation of a rate-limiting second alkylation step to enable isolation of intermediates not typically accessible with DBHDA. This, in turn, allowed us to develop a novel, “interrupted” alkylation–elimination method (Figure 3b) as a solution to the problems of accessing Dha: first, rapid monoalkylation (prior to purification away from conditions detrimental to protein stability), followed by, second, simple prolonged incubation to allow sulfonium formation and elimination to Dha. Thus, with only 50 equiv of MDBP complete monoalkylation (step 1, Figure 3b) of PanC-H44C was observed after 45 min at 25 °C (Figure S6). Removal of excess MDBP by gel filtration at this monoalkylated state successfully ablated protein precipitation in later steps and then allowed incubation at 25 °C for 16 h with concomitant full conversion to PanC-H44Dha via intramolecular alkylation–elimination (steps 2 and 3, Figure 3b, and Figure S7). In this way, even these highly sensitive proteins proved tractable substrates. Moreover, an essentially identical “interrupted” approach also allowed the site variant PanC-H47Dha, in which site 47 is part of a helix and thus more buried, to be generated (Figure 3c). Thus, PanC-H47C was fully monoalkylated after 16 h incubation with MDBP (50 equiv), purified, then incubated, and hence, gratifyingly, fully converted after excess reagent removal and incubation (48 h at 25 °C, Figure 3c and Figure S8) to PanC-H47Dha.
Next, the reactivities of these relatively buried sites (Dha44 and Dha47) were tested. First, “thio-Michael-type” addition of β-mercaptoethanol to PanC-H44Dha allowed complete conversion to the desired adduct (Figure S9), thereby confirming both accessibility and electrophilic reactivity of Dha44 in PanC. Then, β,γ-C,N “aza-Michael” addition of imidazole to PanC-H44Dha and PanC-H47Dha was initially tested at 25 °C due to the noted instability (Figure S5). Pleasingly, initial examples of aza-Michael reaction at pH 9–10 were observed to PanC-H44Hiso and PanC-H47Hiso; however these required high excesses of imidazole and long reaction times (3 days) and typically resulted in only partial or variable conversions (50 to >95%, Figure S10). Despite instability over long periods, shorter reaction times at 37 °C proved successful (Figure 3a,b). It also proved possible to install an aza-analogue to create PanC-H44azaHiso, simply through variation of the nucleophile to 1,2,4-triazole (>80% conversion, Figure S14).51 The precise site locations of installation of Hisiso and aza-Hisiso in PanC-H44Hiso, PanC-H44azaHiso, and PanC-H47Hiso (as well as stability to enzymatic digestion) were fully confirmed by peptide mapping using proteolytic digest followed by LC–MS/MS analysis (Figures S11, S12, and S14). Notably, attempted use of a variety of substituted imidazoles under essentially identical reaction conditions (Figure S13) failed, suggesting that only something isosteric to His (such as Hisiso or aza-Hisiso) could be accommodated at either site and highlighting the highly hindered nature of these sites. Interestingly, lower conversions were observed (Figure S15) for unoptimized test reactions with other N-heterocycles, suggesting that electronics (with potential influence upon nucleophilicity and/or basicity) may also play an important role in reactivity in such reactions.
Having successfully installed Hisiso, even into these buried sites, we next tested its effect upon the enzyme activity of PanC. Kinetic parameters for PanC variants were determined by coupled spectrophotometric assay (Table 1 and Figure S16–18).52 Regioisomeric Hisiso, which lacks hydrogen bond donor character, allows examination of hydrogen bonding; His44’s primary role has been suggested as such a hydrogen bond donor. The measurement of these steady state parameters in the Bi-Uni-Uni-Bi Ping-Pong mechanism suggested for PanC22 requires some caution with direct mechanistic interpretation, but kcat reports directly on ADP formation allowing interrogation of the first half-reaction, the reversible formation of adenyl-pantoate·ADP. Mutation of His44 to Cys44 or Dha44 reduced this activity to 1–2%, as judged by kcat, to levels consistent with prior analyses for Ala44.22 Through the creation of PanC-H44Hiso we were able to probe the effect of altered function through this regioisomeric, “chemical mutation”. Thus, while Hisiso is a poor substitute for His in PanC, its insertion apparently creates some activity (as judged by kcat), ∼5% of that of wt. Notably, insertion of the aza variant in PanC-H44azaHiso gives only the lower levels of activity seen for Ala, Cys, or Dha.
Table 1. . Steady-State Kinetic Parameters of Wild-Type and “Chemical Mutants” of M. tuberculosis PanCa.
|
d-pantoate |
β-alanine |
|||||
|---|---|---|---|---|---|---|
| entry | PanC | kcat [s–1] | KM [mM] | kcat/KM [s–1·M–1] | KM [mM] | kcat/KM [s–1·M–1] |
| 1 | wild-type | 1.6 ± 0.1 | 1.3 ± 0.1 | 1219 | 0.61 ± 0.01 | 2598 |
| 2 | H44C | 0.011 ± 0.001 | 0.5 ± 0.1 | 22 | 0.03 ± 0.02 | 368 |
| 3 | H44Dha | 0.020 ± 0.003 | 1.1 ± 0.3 | 19 | 0.106 ± 0.008 | 193 |
| 4 | H44Hiso | 0.08 ± 0.01 | ndb | ndb | 0.16 ± 0.09 | 500 |
| 5 | H44azaHiso | 0.0123 ± 0.002 | ndb | ndb | 0.12 ± 0.01 | 102 |
25 °C, 100 mM Hepes (pH 7.8), 10 mM MgCl2, 10 mM ATP, [d-pantoate] or [β-alanine] is 0.01–5 mM while the other is saturated and constant at 5 mM. Coupled assay with 1 mM phosphoenolpyruvate, 200 μM NADH, myokinase, pyruvate kinase, and lactate dehydrogenase (18 U/mL each).
nd = not determined due to an inability to determine a KM using concentration range <5 mM, and the associated β-alanine. KM is thus a KMapp; see Supporting Information.
To probe the potential structural implications of the intermediate activity that was observed only for Hisiso further, we conducted a series of molecular dynamics simulations (Figure 4 and Figures S21–25), based on the available X-ray structure.48 Three cases were considered: the wt enzyme and the His44Hisiso and His44Cys mutants (Figure 4). Both neutral and protonated forms were considered for His44Hisiso. Stable Michaelis complexes were readily obtained for each complex after 40 ns; no substantial changes were seen upon increasing the simulation time. The simulations of the wt enzyme show that the side chain of His44 forms a tele-N–H···O-ATP hydrogen bond with the β-phosphate (Figure 4, top left). This interaction is still present in the His44Hisiso mutant if Hisiso44 is protonated (Figure 4, top right). However, only a small population of the protonated form would be expected to be present at the working pH, and the predominant form (neutral Hisiso) preferentially adopts a rotated conformation that displays no interaction with the β-phosphate. In this case Hisiso moves out toward the solvent (Figure 4, bottom right). Together these observations are consistent with the intermediate kcat of PanC-Hisiso44:53 the retained 5% activity might be attributed to a small population of protonated Hisiso and/or the facilitation of ADP release caused by the conformational rearrangement that Hisiso44 undergoes upon deprotonation. This population would be expected to be altered in the aza variant aza-Hisiso44, a mutant that showed only lower activity. Mutation of His44 to Cys (Figure 4, bottom left) also suppresses the hydrogen bond, since Cys is further away (almost 8 Å) and prefers to interact with other residues, consistent with the lowest kcat. Notably, suppression of the hydrogen bond in all cases changes the orientation of the β-phosphate of ATP, moving it away from the histidine that aids its activation (Figure S26), suggesting that this conformational change could be a structural determinant in the lower activities of the Hisiso and Cys mutants.
Figure 4.
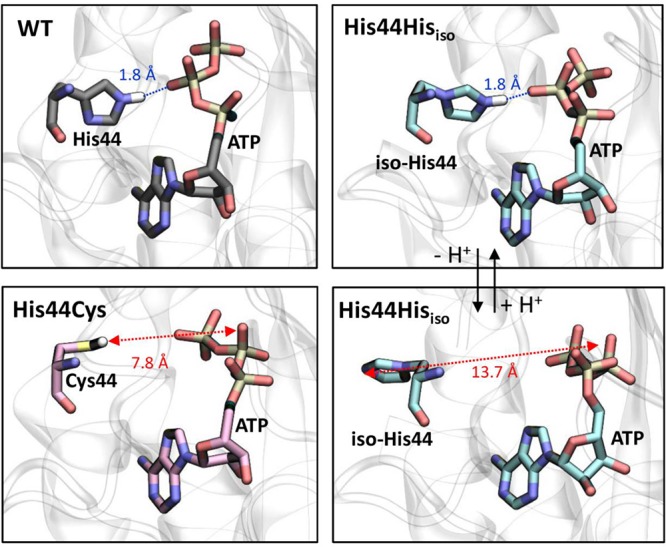
Representative structures obtained from molecular dynamics simulations (40 ns) of the wt enzyme (top left) and the mutants of the His44 residue to Cys (bottom left), neutral iso-His (bottom right), and protonated iso-His (top right).
Discussion
We demonstrate for the first time the creation of a regioisomeric pros-N-linked imidazole ring in the chemical mutant Hisiso as a mimic histidine for the C4-linked imidazole in His in two enzymes, as well as a corresponding aza variant (aza-Hisiso). This ability to perform aza-Michael-type chemical, post-translational mutation not only allows mechanistic hypothesis testing, as we have shown here, but may have broader utility in exploring other His-mediated modulation of activities such as nucleophilicity, basicity, metal-binding, and even the pH range in which a protein might operate. As such, iso-His highlights the broader value of regioisomeric mutants and might now be considered a new, conservative mutation that may be installed even into active sites in sensitive proteins, as we show here. As such, both this post-translational strategy and the new Hisiso and aza-Hisiso mutations disclosed here complement other methods for exploring the role of histidine in proteins.54−56 For example, prior cotranslational biosynthetic methods using residue-specific replacement54 or amber-codon suppression55,56 have allowed incorporation of certain nonisomeric analogues but all can struggle with other analogues, dependent on the method and associated translational and biosynthetic tolerances. More generally, post-translational, aza-Michael-type chemistry at Dha may provide a usefully general mode of protein conjugation for the attachment of other cargoes, probes, or labels.32
Acknowledgments
We thank the EU Horizon 2020 program under the Marie Sklodowska-Curie (700124, J.D.), the NSF (J.M.C.), and BBSRC (P.G.I.) for funding. C.R. is supported by MINECO (CTQ2014-55174-P) and AGAUR (2014SGR-987). L.R. thanks the University of Barcelona for a predoctoral fellowship (APIF). We gratefully acknowledge the computational resources at Minotauro and the technical support provided by BSC-CNS. We thank Prof J. Blanchard for supplying the gene for wt-PanC in plasmid and Dr. Tiago Rodrigues for initial structural analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00341.
Figures S1–26 and details of protein expression and purification, LC–MS and MS/MS analyses, enzyme activity assays, chemical mutations, structural analyses, sequences, and computational methods (PDF)
Author Present Address
‡ G.J.L.B.: Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, U.K., and Instituto de Medicina Molecular, Faculdade de Medicina da Universidade de Lisboa, Av. Prof. Egas Moniz, 1649-028 Lisboa, Portugal. J.M.C.: School of Chemical and Physical Sciences, Flinders University, Sturt Road, Bedford Park, South Australia, 5042, Australia.
The authors declare no competing financial interest.
Supplementary Material
References
- Lang K.; Chin J. W. Cellular Incorporation of Unnatural Amino Acids and Bioorthogonal Labeling of Proteins. Chem. Rev. 2014, 114, 4764. 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- Xue L.; Karpenko I. A.; Hiblot J.; Johnsson K. Imaging and manipulating proteins in live cells through covalent labeling. Nat. Chem. Biol. 2015, 11, 917. 10.1038/nchembio.1959. [DOI] [PubMed] [Google Scholar]
- Boutureira O.; Bernardes G. J. L. Advances in Chemical Protein Modification. Chem. Rev. 2015, 115, 2174. 10.1021/cr500399p. [DOI] [PubMed] [Google Scholar]
- Agarwal P.; Bertozzi C. R. Site-Specific Antibody–Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjugate Chem. 2015, 26, 176. 10.1021/bc5004982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicer C. D.; Davis B. G. Selective Chemical Protein Modification. Nat. Commun. 2014, 5, 4740. 10.1038/ncomms5740. [DOI] [PubMed] [Google Scholar]
- Krall N.; da Cruz F. P.; Boutureira O.; Bernardes G. J. L. Site-selective protein-modification chemistry for basic biology and drug development. Nat. Chem. 2016, 8, 103. 10.1038/nchem.2393. [DOI] [PubMed] [Google Scholar]
- Qiao Y.; Molina H.; Pandey A.; Zhang J.; Cole P. A. Chemical Rescue of a Mutant Enzyme in Living Cells. Science 2006, 311 (3), 1293. 10.1126/science.1122224. [DOI] [PubMed] [Google Scholar]
- Peracchi A. How (and Why) to Revive a Dead Enzyme: The Power of Chemical Rescue. Curr. Chem. Biol. 2008, 2 (1), 32. 10.2174/187231308783334162. [DOI] [Google Scholar]
- Carter P.; Wells J. A. Engineering enzyme specificity by substrate-assisted catalysis. Science 1987, 237 (4813), 394. 10.1126/science.3299704. [DOI] [PubMed] [Google Scholar]
- Toney M. D.; Kirsch J. F. Direct Bronsted Analysis of the Restoration of Activity to a Mutant Enzyme by exogenous amines. Science 1989, 243, 1485. 10.1126/science.2538921. [DOI] [PubMed] [Google Scholar]
- Bishop A. C.; Chen V. L. Brought to life: targeted activation of enzyme function with small molecules. J. Chemical Biology 2009, 2 (1), 1. 10.1007/s12154-008-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorn J. A.; Wells J. A. Turning enzymes ON with small molecules. Nat. Chem. Biol. 2010, 6 (3), 179. 10.1038/nchembio.318. [DOI] [PubMed] [Google Scholar]
- Mendel D.; Ellman J. A.; Schultz P. G. Construction of a Light-Activated Protein by Unnatural Amino Acid Mutagenesis. J. Am. Chem. Soc. 1991, 113, 2758. 10.1021/ja00007a063. [DOI] [Google Scholar]
- Baker A. S.; Deiters A. Optical Control of Protein Function through Unnatural Amino Acid Mutagenesis and Other Optogenetic Approaches. ACS Chem. Biol. 2014, 9 (7), 1398. 10.1021/cb500176x. [DOI] [PubMed] [Google Scholar]
- Li J.; Yu J.; Zhao J.; Wang J.; Zheng S.; Lin S.; Chen L.; Yang M.; Jia S.; Zhang X.; et al. Palladium-triggered deprotection chemistry for protein activation in living cells. Nat. Chem. 2014, 6 (4), 352. 10.1038/nchem.1887. [DOI] [PubMed] [Google Scholar]
- Smith H. B.; Hartman F. C. Restoration of activity to catalytically deficient mutants of ribulosebisphosphate carboxylase/oxygenase by aminoethylation. J. Biol. Chem. 1988, 263 (10), 4921. [PubMed] [Google Scholar]
- Gloss L. M.; Kirsch J. F. Decreasing the Basicity of the Active Site Base, Lys-258, of Escherichia coli Aspartate Aminotransferase by Replacement with.gamma.-thialysine. Biochemistry 1995, 34 (12), 3990. 10.1021/bi00012a017. [DOI] [PubMed] [Google Scholar]
- Paetzel M.; Strynadka N. C. J.; Tschantz W. R.; Casareno R.; Bullinger P. R.; Dalbey R. E. Use of Site-directed Chemical Modification to Study an Essential Lysine in Escherichia coli Leader Peptidase. J. Biol. Chem. 1997, 272 (15), 9994. 10.1074/jbc.272.15.9994. [DOI] [PubMed] [Google Scholar]
- Hopkins C. E.; O’Connor P. B.; Allen K. N.; Costello C. E.; Tolan D. R. Chemical-modification rescue assessed by mass spectrometry demonstrates that γ-thia-lysine yields the same activity as lysine in aldolase. Protein Sci. 2002, 11 (7), 1591. 10.1110/ps.3900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovleva L.; Shuman S. Chemical Mutagenesis of Vaccinia DNA Topoisomerase Lysine 167 Provides Insights to the Catalysis of DNA Transesterification. Biochemistry 2013, 52 (5), 984. 10.1021/bi301643h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timms N.; Windle C. L.; Polyakova A.; Ault J. R.; Trinh C. H.; Pearson A. R.; Nelson A.; Berry A. Structural Insights into the Recovery of Aldolase Activity in N-Acetylneuraminic Acid Lyase by Replacement of the Catalytically Active Lysine with γ-Thialysine by Using a Chemical Mutagenesis Strategy. ChemBioChem 2013, 14 (4), 474. 10.1002/cbic.201200714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng R.; Dam T. K.; Brewer C. F.; Blanchard J. S. Active site residues in Mycobacterium tuberculosis pantothenate synthetase required in the formation and stabilization of the adenylate intermediate. Biochemistry 2004, 43 (22), 7171. 10.1021/bi049676n. [DOI] [PubMed] [Google Scholar]
- Dhalla A. M.; Li B.; Alibhai M. F.; Yost K. J.; Hemmingsen J. M.; Atkins W. M.; Schineller J.; Villafranca J. J. Regeneration of catalytic activity of glutamine synthetase mutants by chemical activation: Exploration of the role of arginines 339 and 359 in activity. Protein Sci. 1994, 3 (3), 476. 10.1002/pro.5560030313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson S. L.; Wakarchuk W. W.; Withers S. G. Effects of both shortening and lengthening the active site nucleophile of Bacillus circulans xylanase on catalytic activity. Biochemistry 1996, 35, 10110. 10.1021/bi960586v. [DOI] [PubMed] [Google Scholar]
- Earnhardt J. N.; Wright S. K.; Qian M.; Tu C.; Laipis P. J.; Viola R. E.; Silverman D. N. Introduction of Histidine Analogs Leads to Enhanced Proton Transfer in Carbonic Anhydrase V. Arch. Biochem. Biophys. 1999, 361 (2), 264. 10.1006/abbi.1998.0984. [DOI] [PubMed] [Google Scholar]
- Colleluori D. M.; Reczkowski R. S.; Emig F. A.; Cama E.; Cox J. D.; Scolnick L. R.; Compher K.; Jude K.; Han S.; Viola R. E.; et al. Probing the role of the hyper-reactive histidine residue of arginase. Arch. Biochem. Biophys. 2005, 444, 15. 10.1016/j.abb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Watt E. D.; Rivalta I.; Whittier S. K.; Batista V. S.; Loria J. P. Reengineering Rate-Limiting, Millisecond Enzyme Motions by Introduction of an Unnatural Amino Acid. Biophys. J. 2011, 101 (2), 411. 10.1016/j.bpj.2011.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polgar L. The catalytic triad of serine peptidases. Cell. Mol. Life Sci. 2005, 62 (19–20), 2161. 10.1007/s00018-005-5160-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker J. M.; Bernardes G. J. L.; Davis B. G. A “Tag-and-Modify” Approach to Site-Selective Protein Modification. Acc. Chem. Res. 2011, 44 (9), 730. 10.1021/ar200056q. [DOI] [PubMed] [Google Scholar]
- Chalker J. M.; Gunnoo S. B.; Boutureira O.; Gerstberger S. C.; Fernandez-Gonzalez M.; Bernardes G. J. L.; Griffin L.; Hailu H.; Schofield C. J.; Davis B. G. Methods for converting cysteine to dehydroalanine on peptides and proteins. Chem. Sci. 2011, 2 (9), 1666. 10.1039/c1sc00185j. [DOI] [Google Scholar]
- Bernardes G. J.; Chalker J. M.; Errey J. C.; Davis B. G. Facile conversion of cysteine and alkyl cysteines to dehydroalanine on protein surfaces: versatile and switchable access to functionalized proteins. J. Am. Chem. Soc. 2008, 130 (15), 5052. 10.1021/ja800800p. [DOI] [PubMed] [Google Scholar]
- Freedy A. M.; Matos M. J.; Boutureira O.; Corzana F.; Guerreiro A.; Somovilla V. J.; Rodrigues T.; Nicholls K.; Xie B.; Jiménez-Osés G.; Brindle K. M.; Neves A. A.; Bernardes G. J. L.. Chemoselective Installation of Amine Bonds on Proteins Through Aza-Michael Ligation.Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker J. M.Reaction Engineering for Protein Modification: Tools for Chemistry and Biology; D.Phil. thesis; University of Oxford, 2011. [Google Scholar]
- Bernardes G. J. L.Building Synthetic Proteins; D.Phil. thesis; University of Oxford: 2008. [Google Scholar]
- Branigan E.Introducing Spin Labels into Proteins to Determine Their Solution Conformation by Pulsed EPR Methods; Ph.D. thesis;University of St Andrews: 2013. [Google Scholar]
- Furman J. L.; Kang M.; Choi S.; Cao Y.; Wold E. D.; Sun S. B.; Smider V. V.; Schultz P. G.; Kim C. H. A Genetically Encoded aza-Michael Acceptor for Covalent Cross-Linking of Protein–Receptor Complexes. J. Am. Chem. Soc. 2014, 136 (23), 8411. 10.1021/ja502851h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez M.; Pleixats R. FeCl3-catalyzed conjugate addition of secondary amines, imidazole and pyrazole to methyl 2-acetamidoacrylate. Preparation of β-dialkylamino-α-alanine and β-(N-heteroaryi)-α-alanine derivatives. Tetrahedron 1995, 51 (30), 8355. 10.1016/0040-4020(95)00446-F. [DOI] [Google Scholar]
- Ferreira P. M. T.; Maia H. L. S.; Monteiro L. S.; Sacramento J.; Sebastiao J. Synthesis of b-substituted alanines via Michael addition of nucleophiles to dehydroalanine derivatives. J. Chem. Soc., Perkin Trans. 2000, 1, 3317. 10.1039/b003353g. [DOI] [Google Scholar]
- Ferreira P. M. T.; Maia H. L. S.; Monteiro L. S.; Sacramento J. Michael addition of thiols, carbon nucleophiles and amines to dehydroamino acid and dehydropeptide derivatives. J. Chem. Soc., Perkin Trans. 2001, 1, 3167. 10.1039/b106487h. [DOI] [Google Scholar]
- Naidu B. N.; Sorenson M. E.; Connolly T. P.; Ueda Y. Michael Addition of Amines and Thiols to Dehydroalanine Amides: A Remarkable Rate Acceleration in Water. J. Org. Chem. 2003, 68 (26), 10098. 10.1021/jo034762z. [DOI] [PubMed] [Google Scholar]
- DeSantis G.; Berglund P.; Stabile M. R.; Gold M.; Jones J. B. Site-directed mutagenesis combined with chemical modification as a strategy for altering the specificity of the S1 and S1’ pockets of subtilisin Bacillus lentus. Biochemistry 1998, 37 (17), 5968. 10.1021/bi9727951. [DOI] [PubMed] [Google Scholar]
- Schechter I.; Berger A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27 (2), 157. 10.1016/S0006-291X(67)80055-X. [DOI] [PubMed] [Google Scholar]
- Davis B. G.; Khumtaveeporn K.; Bott R. R.; Jones J. B. Altering the specificity of subtilisin Bacillus lentus through the introduction of positive charge at single amino acid sites. Bioorg. Med. Chem. 1999, 7 (11), 2303. 10.1016/S0968-0896(99)00168-6. [DOI] [PubMed] [Google Scholar]
- Minning S.; Draborg H.; Roggen E. L.; Soni N. K.; Berg N. W.; Lyngstrand S. T.; US patent 8,389,262 ed., 2013.
- Babe L. M.; Estell D. A.; Goedegebuur F.; Bott R. R.; Kolkman M.; Mulder H.; Schmidt B. F.; Augustyn K.; Atwal M.; Marquez A.; PCT WO2015038792, 2015.
- Kuhn P.; Knapp M.; Soltis S. M.; Ganshaw G.; Thoene M.; Bott R. The 0.78 Å Structure of a Serine Protease: Bacillus lentus Subtilisin. Biochemistry 1998, 37 (39), 13446. 10.1021/bi9813983. [DOI] [PubMed] [Google Scholar]
- Zheng R.; Blanchard J. S. Steady-State and Pre-Steady-State Kinetic Analysis of Mycobacterium tuberculosis Pantothenate Synthetase. Biochemistry 2001, 40 (43), 12904. 10.1021/bi011522+. [DOI] [PubMed] [Google Scholar]
- Wang S.; Eisenberg D. Crystal structures of a pantothenate synthetase from M. tuberculosis and its complexes with substrates and a reaction intermediate. Protein Sci. 2003, 12 (5), 1097. 10.1110/ps.0241803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Eisenberg D. Crystal Structure of the Pantothenate Synthetase from Mycobacterium tuberculosis, Snapshots of the Enzyme in Action. Biochemistry 2006, 45 (6), 1554. 10.1021/bi051873e. [DOI] [PubMed] [Google Scholar]
- Morrison P. M.; Foley P. J.; Warriner S. L.; Webb M. E. Chemical generation and modification of peptides containing multiple dehydroalanines. Chem. Commun. 2015, 51 (70), 13470. 10.1039/C5CC05469A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- We thank a referee for this excellent suggestion.
- Kiianitsa K.; Solinger J. A.; Heyer W. D. NADH-coupled microplate photometric assay for kinetic studies of ATP-hydrolyzing enzymes with low and high specific activities. Anal. Biochem. 2003, 321 (2), 266. 10.1016/S0003-2697(03)00461-5. [DOI] [PubMed] [Google Scholar]
- Molecular mechanics revealed too that, as might be expected, any d-isomer (Figure S21) formed from the aza-Michael addition would face directly away from ATP. It may therefore be that any measurement of kcat determined here is an underestimate from a population of l-isomer PanC-Hisiso in a mixture with much less active d-isomer PanC-Hisiso
- Ikeda Y.; Kawahara S. i.; Taki M.; Kuno A.; Hasegawa T.; Taira K. Synthesis of a novel histidine analogue and its efficient incorporation into a protein in vivo. Protein Eng., Des. Sel. 2003, 16 (9), 699. 10.1093/protein/gzg084. [DOI] [PubMed] [Google Scholar]
- Xiao H.; Peters F. B.; Yang P. Y.; Reed S.; Chittuluru J. R.; Schultz P. G. Genetic incorporation of histidine derivatives using an engineered pyrrolysyl-tRNA synthetase. ACS Chem. Biol. 2014, 9 (5), 1092. 10.1021/cb500032c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.; Wang Y. S.; Liu W. R. Probing the Catalytic Charge-Relay System in Alanine Racemase with Genetically Encoded Histidine Mimetics. ACS Chem. Biol. 2016, 11 (12), 3305. 10.1021/acschembio.6b00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.