

 This work is licensed under a
This work is licensed under a Abstract
Background
The occurrence of parathyroid carcinoma in multiple endocrine neoplasia type I (MENI) is rare and the 15 cases of malignant parathyroid tumor reported so far have been associated with MENI in individuals and not with multiple members within a family.
Methods
We report on a 61-year-old male, operated for a 7.3 cm parathyroid carcinoma infiltrating the esophagus. In his brother, a 4.6 cm parathyroid carcinoma was diagnosed histologically, while in the daughter, neck ultrasonography revealed 2 extrathyroidal nodules, yet to be excised.
Results
Screening of the MEN1 gene identified a known germline heterozygous missense mutation (c.1252G>A; p.D418N) in exon 9, in all affected subjects.
Conclusions
The occurrence of parathyroid carcinoma in more than one affected member of a single MEN1 family represents the first reported familial case. This suggests that additional constitutional genetic mutations may contribute to the variation in malignant potential and clinical behavior of parathyroid tumors in MEN1.
Keywords: MEN1, parathyroid carcinoma, multiple endocrine neoplasia, familial
Introduction
The multiple endocrine neoplasia type 1 syndrome (MEN1, MIM131100) is primarily characterized by parathyroid, gastro-entero-pancreatic and pituitary tumors (1, 2). Less frequently, it is associated with adrenocortical (3), thyroid, carcinoid tumors, lipomas and leiomyomas (1). A few cases of melanoma have been reported that may be associated with the MEN1 syndrome (4, 5, 6). Clinical association of familial primary hyperparathyroidism (PHPT) with lesions in at least two of the three ‘P’ glands (pancreas, pituitary, parathyroid) is diagnostic for MEN1 (7). Lesions of the parathyroid gland, adenoma or hyperplasia are almost always benign. The syndrome that is inherited in an autosomal dominant fashion is due to inactivating mutations of the MEN1 gene, encoding menin, an intracellular protein that interacts with transcription factors involved in cell cycle regulation and proliferation (8). Fifteen well-documented cases of parathyroid carcinoma (PC) and one atypical adenoma in the MEN1 syndrome have been reported (9, 10, 11), and in seven of these, an MEN1 mutation was identified.
PC accounts for <1% of all PHPT cases and is detected as a sporadic case or as part of the hyperparathyroidism and jaw-tumor syndrome (HPT-JT, MIM145001), characterized by ossifying fibromas of the jaw and mandible, renal and uterine lesions (12). The HPT-JT syndrome is caused by inactivating mutations of the tumor suppressor CDC73 gene encoding parafibromin, a ubiquitously expressed protein involved in RNA polymerase II transcription and chromatin remodeling (13). A number of investigators have suggested that menin and parafibromin are involved in common molecular pathways and share similar functions (14, 15, 16).
Here, we report an unusual association of MEN1 gene mutation with PC in two siblings from the same MEN1 family, representing the first reported familial occurrence.
Clinical case
A 61-year-old male (Fig. 1A, II-4) was admitted in April 2013 to our Health Care Centre for follow-up of a pathologically diagnosed PC (size 7.3 cm) showing capsular invasion and infiltration into the esophagus. At surgery, a hyperplastic parathyroid gland was removed. The patient had a history of nephrolithiasis with repeated renal colic, and bilateral adrenal lesions were present by computed tomography (CT). At admission, laboratory values were unremarkable. Assessment of first-degree relatives (Fig. 1A) revealed the presence of hypercalcemia and hypercalciuria with high levels of PTH in a brother (II-6) and in the proband’s daughter (III-1) (Table 1). In the brother, imaging revealed an extrathyroidal lesion (4.6 cm) at the upper left lobe that at surgery was histopathologically identified as PC. He had suffered from renal stones and had been operated on for an in situ melanoma and lipoma, and during the follow-up, bilateral adrenal lesions had been discovered (Table 1). In the daughter, two extrathyroidal nodules (1.4 and 1 cm) were detected at neck ultrasonography and confirmed by 99mTc-Sestamibi scan, but are yet to be excised. Biochemical evaluation of other relatives, one brother and sister, and paternal nephew (Fig. 1A: II-1, II-3, and III-2) was negative.
Figure 1.
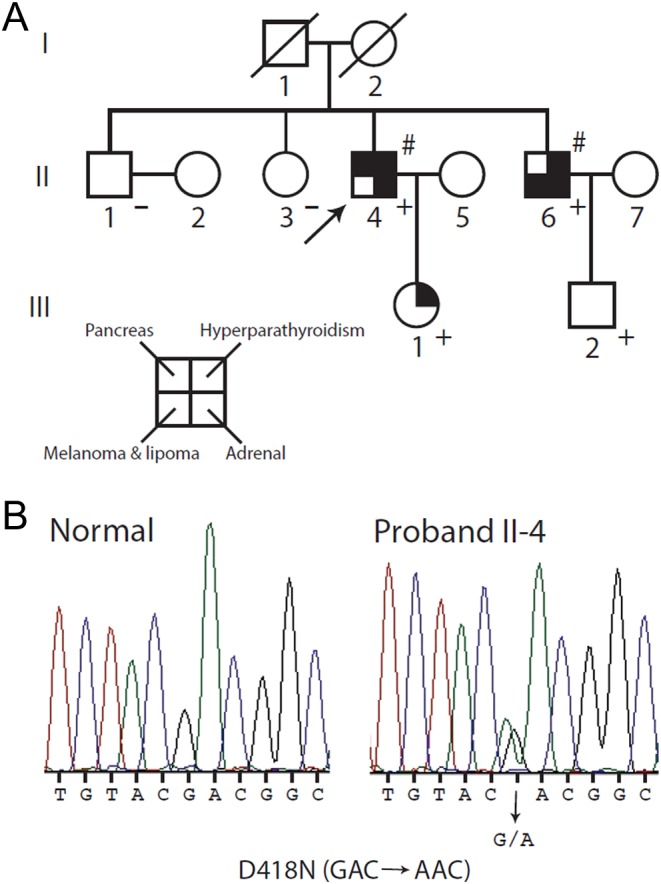
Pedigree of the family under study. (A) Clinical status is indicated by open symbols (unaffected or unknown) and filled symbols (affected). Filled quadrants indicate a diagnosis as indicated in the inset legend. Histological diagnosis of carcinoma is indicated (#). A diagonal slash mark through a symbol indicates deceased. Proband is indicated by the arrow. The presence (+) or absence (−) of a mutation in tested family members is shown. (B) Detection of a mutation in the MEN1 gene. Direct sequence analysis of the exon 9 genomic DNA amplicon of proband II-4 (right) revealed a heterozygous G to A transition encoding the missense D418N mutation compared with an unrelated normal individual (left).
Table 1.
Clinical and molecular features of the proband and relatives.
| Sex | Age | Ca2+ib | PTHc | PHPT | Kidney stones | Pituitary | Pancreas | Parathyroid | Size (cm) | Adrenal | Other | MEN1 | CDC73 | IHC | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Menin | Parafibromin | |||||||||||||||
| II:4a | M | 62 | NA | NA | Yes | Yes | No | Multiple PNET | Carcinoma, hyperplasia | 7.3 | Yes | Prostate | p.D418N | Neg | NP | NP |
| II:6 | M | 55 | 1.48 | 286 | Yes | Yes | No | No | Carcinoma | 4.6 | Yes | Melanoma lipoma schwannoma | p.D418N | Neg | Pos | Pos |
| III:1 | F | 26 | 1.34 | 77 | Yes | Yes | NA | NA | NA | 1.4/1 | No | No | p.D418N | NP | / | / |
| III:2 | M | 21 | 1.30 | 43 | No | No | No | No | No | / | No | No | p.D418N | NP | / | / |
a, proband; b, normal range: 1.12–1.32 mmol/L; c, normal range: 10–65 pg/mL.
IHC, immunohistochemistry; NA, not available; neg, negative; NP, Not Performed/Not Applicable; PHPT, primary hyperparathyroidism; PNET, pancreatic neuroendocrine tumor; pos, positive; PTH, parathyroid hormone.
After the identification of the MEN1 mutation, gastrointestinal echoendoscopy of the proband revealed multiple pancreatic neuroendocrine tumors (PNETs), <1 cm in diameter, while pituitary MRI was negative. In the brother (II-6), a right lesion close to the pharynx was identified during follow-up and removed: histopathological diagnosis was an ancient schwannoma.
Materials and methods
Informed consent was obtained from each patient or subject, and the study was conducted according to a protocol that was approved by the IRCCS Casa Sollievo della Sofferenza Hospital, Research Ethics Board.
Genetic screening
DNA was extracted from peripheral blood and formalin-fixed-paraffin-embedded (FFPE) tissue, and MEN1 and CDC73 gene coding sequences were PCR amplified and directly sequenced (12, 17). Loss of heterozygosity (LOH) at the MEN1 locus (chr 11q13.1) was searched for as described previously (17). Lipoma tumoral tissue was not available.
SNP array
A search for CDC73 and MEN1 gene deletions was performed using the CytoScan HD array platform (Affymetrix) as described (18).
Immunohistochemistry
Four micron-thick tissue sections of FFPE parathyroid tumor tissues were rehydrated in pH 7.5 buffer and processed for staining. Antigen retrieval was performed by heating the slides in 0.01 M citrate buffer (pH 6.0) in a bath for 20 min at 97°C. After blocking endogenous peroxidase activity in 0.3% hydrogen peroxide and methanol solution for 15 min, tissue sections were stained either with primary monoclonal anti-menin antibody (clone B-9, sc-374371, 1:200 dilution) or anti-parafibromin antibody (clone 2H1, sc-33638, 1:200 dilution) (Santa Cruz Biotechnology) for 20 min.
Slides were then incubated with a commercially available detection kit (EnVision FLEX+, Dako) following the manufacturer’s instructions, developing peroxidase activity with 3-3′-diaminobenzidine. Finally, slides were counterstained with hematoxylin, dehydrated and mounted. Specificity of reactions was assessed by (i) replacing the primary antibody with an unrelated mouse immunoglobulin at a comparable dilution or (ii) using normal serum alone. Positive and negative controls were included.
Results
Molecular screening
No coding mutation (germline or somatic) or deletion of the CDC73 gene (1q31.2) was found. No deletion of the MEN1 gene (11q13.1) was found. A known MEN1 mutation in exon 9 (c.1252G>A – p.D418N) was identified in the proband (Fig. 1B; II-4) and confirmed in not only the affected brother and daughter (II-6 and III-1), but also in the normocalcemic nephew (III-2). The mutation was not found in the unaffected sister and brother (II-1 and II-3). No LOH at the MEN1 locus was detected in the tumor tissue of the affected brother (II-6).
Pathology and immunohistochemistry (IHC)
Hematoxylin and eosin (H&E)-stained sections of the parathyroid lesion of the brother (II-6) were reviewed according to World Health Organization classification criteria (19). Clear-cut capsular penetration with infiltration of the surrounding extra-glandular adipose tissue was observed. Necrosis was not seen (Fig. 2). By IHC, diffuse positivity of menin nuclear expression was observed (Fig. 3). Likewise, positivity for parafibromin nuclear expression was seen on separate tissue sections (data not shown).
Figure 2.
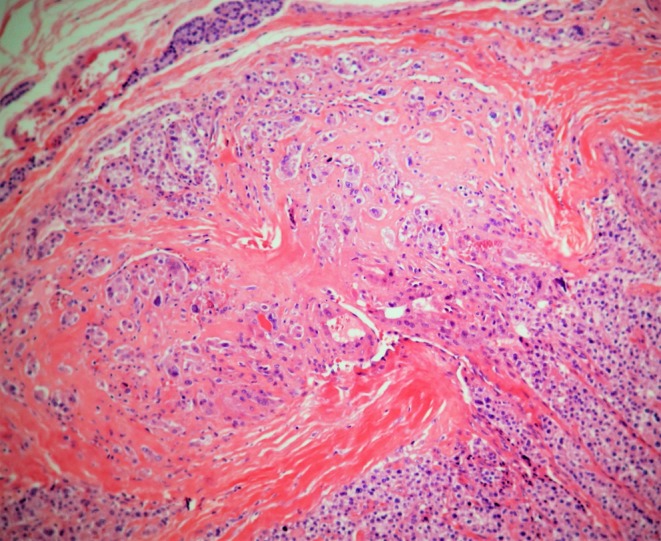
(H&E X10) Fungus-like capsular infiltration by neoplastic cells of parathyroid carcinoma.
Figure 3.
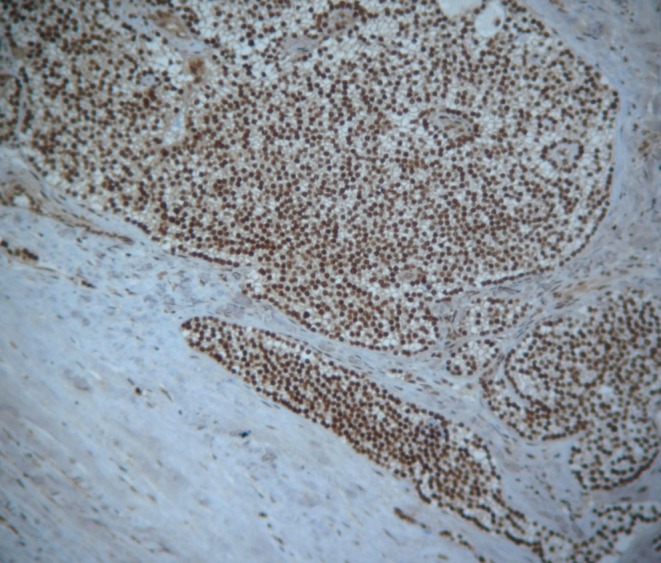
Diffuse positivity of menin protein in parathyroid carcinoma tissue excised from the proband’s brother.
Discussion
The first manifestation of MEN1 syndrome is hypercalcemia and PHPT in up to 95% of cases. The parathyroid lesion is almost always a benign adenoma or hyperplasia. Gastrinomas, non-functioning islet adenomas and insulinomas are frequently present (up to 40%) together with anterior pituitary tumors (up to 30%) (7, 20). MEN1 is due to inactivating mutations of menin, a widely expressed intracellular protein interacting with factors or complexes that modulate gene transcription (reviewed in 21).
Thus far, fifteen cases of PC associated with the MEN1 syndrome have been reported, with an overall prevalence of 0.5% (3/590 MEN1 subjects recruited at the Mayo Clinic and at the MD Anderson Cancer Center, 9–11). In seven of those 15 cases, an MEN1 mutation was identified. For all the 15 index cases, in which familial PHPT was established, there was no report of a malignant parathyroid lesion in other affected family members (9–11 and references therein). On the one hand, younger age at diagnosis (e.g., through screening) and subtotal parathyroidectomy might lower the possibility of malignant transformation in parathyroid tissue in some of these cases. On the other hand, these data suggest that the MEN1 gene is not necessarily a major genetic driver per se of malignancy for a parathyroid lesion. Rather, the malignant progression of the parathyroid lesion in those cases could be due to additional unknown genetic events occurring at the somatic level, as frequently reported in many cancers (22).
The p.D418N mutation affects menin binding with the suppressor of variegation 3–9 homolog family, SUV39H1, protein, whose complex is involved in the methylation of H3K9 (23). It has never been found previously associated with malignant MEN1 tumors, and specifically PC (21 and references therein). Taking into account the prevalence of MEN1 in the general population (0.0033%, 24), given the low frequency of PC in MEN1 syndrome (0.5%, 9 and 10), the occurrence of parathyroid carcinoma affecting two siblings of an MEN1 family is unlikely a random event (P concurrence = 0.0033 × 0.5 × 0.5 = 0.00082/ 1000000 = 8.2 × 10−10). Instead, one might argue that this mutation is being expressed on different genetic backgrounds in our family. The presence of two carcinoma cases in the same family suggests the key genetic event was pre-somatic. Our family may well represent the first reported MEN1 familial case with PC. Additional studies––whole-exome sequencing and gene expression profiling––are ongoing, in order to identify any putative genetic factors that might underlie the aggressive nature of this familial occurrence.
One further point concerns the atypical clinical presentation of the family. The concurrent presence of: (i) parathyroid disease in 3 affected members and gross carcinoma (7.3 and 4.6 cm) in 2 siblings; (ii) familial PHPT with PC and (iii) renal colic, might suggest HPT-JT. However, as no mutations were found in the CDC73 gene, the MEN1 syndrome was considered, supported by the involvement of adrenal lesions, melanoma and lipoma. This case is instructive in that it highlights the importance of implementing a stepwise screening protocol for all the genes showing some association with familial PHPT, especially in families with atypical clinical presentation and independently of age. This would aid the identification of unaffected carriers, such as the 19-year-old nephew of the proband in our family, thus allowing closer clinical follow-up.
In conclusion, the occurrence of PC in more than one affected member of a single MEN1 family suggests that additional constitutional genetic mutations may contribute to the variation in malignant potential and clinical behavior of parathyroid tumors in MEN1.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research was supported by the Ricerca Corrente and Finalizzata-GR-2011-02351489 funding granted by the Italian Ministry of Health (to V G), by the ‘5 × 1000’ voluntary contributions and a Canadian Institutes of Health Research Operating Grant (to G N H).
Acknowledgements
The authors are indebted to the family members for their participation and to Luigi Bisceglia for insightful scientific discussion.
References
- 1.Trump D, Farren B, Wooding C, Pang JT, Besser GM, Buchanan KD, Edwards CR, Heath DA, Jackson CE, Jansen S, et al Clinical studies of multiple endocrine neoplasia type 1 (MEN1). QJM 1996. 89 653–669. ( 10.1093/qjmed/89.9.653) [DOI] [PubMed] [Google Scholar]
- 2.Benson L, Ljunghall S, Akerstrom G, Oberg K. Hyperparathyroidism presenting as the first lesion in multiple endocrine neoplasia type 1. American Journal of Medicine 1987. 82 731–737. ( 10.1016/0002-9343(87)90008-8) [DOI] [PubMed] [Google Scholar]
- 3.Skogseid B, Larsson C, Lindgren PG, Kvanta E, Rastad J, Theodorsson E, Wide L, Wilander E, Oberg K. Clinical and genetic features of adrenocortical lesions in multiple endocrine neoplasia type 1. Journal of Clinical Endocrinology and Metabolism 1992. 75 76–81. ( 10.1210/jcem.75.1.1352309) [DOI] [PubMed] [Google Scholar]
- 4.Nord B, Platz A, Smoczynski K, Kytölä S, Robertson G, Calender A, Murat A, Weintraub D, Burgess J, Edwards M, et al Malignant melanoma in patients with multiple endocrine neoplasia type 1 and involvement of the MEN1 gene in sporadic melanoma. International Journal of Cancer 2000. 874 463–467. ( 10.1002/1097-0215(20000815)87:4<463::AID-IJC1>3.0.CO;2-8) [DOI] [PubMed] [Google Scholar]
- 5.Fang M, Xia F, Mahalingam M, Virbasius CM, Wajapeyee N, Green MR. MEN1 is a melanoma tumor suppressor that preserves genomic integrity by stimulating transcription of genes that promote homologous recombination-directed DNA repair. Molecular and Cellular Biology 2013. 33 2635–2647. ( 10.1128/MCB.00167-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baldauf C, Vortmeyer AO, Koch CA, Sticherling M. Combination of multiple skin malignancies with multiple endocrine neoplasia type 1: coincidental or pathogenetically related? Dermatology 2009. 219 365–367. ( 10.1159/000193058) [DOI] [PubMed] [Google Scholar]
- 7.Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, Melmed S, Sakurai A, Tonelli F, Brandi ML. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). Journal of Clinical Endocrinology and Metabolism 2012. 97 2990–3011. ( 10.1210/jc.2012-1230) [DOI] [PubMed] [Google Scholar]
- 8.Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, et al Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997. 276 404–406. ( 10.1126/science.276.5311.404) [DOI] [PubMed] [Google Scholar]
- 9.Singh Ospina N, Sebo TJ, Thompson GB, Clarke BL, Young WF., Jr Prevalence of parathyroid carcinoma in 348 patients with multiple endocrine neoplasia type 1 – case report and review of the literature. Clinical Endocrinology 2014. 84 244–249. ( 10.1111/cen.12714) [DOI] [PubMed] [Google Scholar]
- 10.Christakis I, Busaidy NL, Cote GJ, Williams MD, Hyde SM, Silva Figueroa AM, Kwatampora LJ, Clarke CN, Qiu W, Lee JE, et al Parathyroid carcinoma and atypical parathyroid neoplasms in MEN1 patients; A clinico-pathologic challenge. The MD Anderson case series and review of the literature. International Journal of Surgery 2016. 31 10–16. ( 10.1016/j.ijsu.2016.05.035) [DOI] [PubMed] [Google Scholar]
- 11.Cinque L, Sparaneo A, Cetani F, Coco M, Clemente C, Chetta M, Balsamo T, Battista C, Sanpaolo E, Pardi E, et al Novel association of MEN1 gene mutations with parathyroid carcinoma. Oncology Letters 2017. 14 23–30. ( 10.21641/los.14.1.22) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, Simonds WF, Gillanders EM, Kennedy AM, Chen JD, et al HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nature Genetics 2002. 32 676–680. ( 10.1038/ng1048) [DOI] [PubMed] [Google Scholar]
- 13.Newey PJ, Bowl MR, Cranston T, Thakker RV. Cell division cycle protein 73 homolog (CDC73) mutations in the hyperparathyroidism-jaw tumor syndrome (HPT-JT) and parathyroid tumors. Human Mutation 2010. 31 295–307. ( 10.1002/humu.21188) [DOI] [PubMed] [Google Scholar]
- 14.Agarwal SK, Kennedy PA, Scacheri PC, Novotny EA, Hickman AB, Cerrato A, Rice TS, Moore JB, Rao S, Ji Y, et al Menin molecular interactions: insights into normal functions and tumorigenesis. Hormone and Metabolic Research 2005. 37 369–374. ( 10.1055/s-2005-870139) [DOI] [PubMed] [Google Scholar]
- 15.Chen G, A J, Wang M, Farley S, Lee LY, Lee LC, Sawicki MP. Menin promotes the Wnt signaling pathway in pancreatic endocrine cells. Molecular Cancer Research 2008. 6 1894–1907. ( 10.1158/1541-7786.MCR-07-2206) [DOI] [PubMed] [Google Scholar]
- 16.Rozenblatt-Rosen O, Hughes CM, Nannepaga SJ, Shanmugam KS, Copeland TD, Guszczynski T, Resau JH, Meyerson M. The parafibromin tumor suppressor protein is part of a human Paf1 complex. Molecular and Cellular Biology 2005. 25 612–620. ( 10.1128/MCB.25.2.612-620.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cetani F, Pardi E, Giovannetti A, Cerrai P, Borsari S, Vignali E, Picone A, Cianferotti L, Miccoli P, Pinchera A, et al Six novel MEN1 gene mutations in sporadic parathyroid tumors. Human Mutation 2000. 16 44 ( 10.1002/1098-1004(200011)16:5<445::AID-HUMU12>3.0.CO;2-6) [DOI] [PubMed] [Google Scholar]
- 18.Palumbo O, Fischetto R, Palumbo P, Nicastro F, Papadia F, Zelante L, Carella M. De novo microduplication of CHL1 in a patient with non-syndromic developmental phenotypes. Molecular Cytogenetics 2015. 8 66 ( 10.1186/s13039-015-0170-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pathology and Genetics of Tumours of Endocrine Organs, 3rd Edition. IARC WHO Classification of Tumours. Lyon, France: IARC Press, 2004. [Google Scholar]
- 20.Thakker RV. Multiple endocrine neoplasia: syndromes of the twentieth century. Journal of Clinical Endocrinology and Metabolism 1998. 83 2617–2620. ( 10.1210/jc.83.8.2617) [DOI] [PubMed] [Google Scholar]
- 21.Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Human Mutation 2008. 29 22–32. ( 10.1002/humu.20605) [DOI] [PubMed] [Google Scholar]
- 22.Roberts SA, Gordenin DA. Hypermutation in human cancer genomes: footprints and mechanisms. Nature Reviews: Cancer 2014. 14 786–800. ( 10.1038/nrc3816) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang YJ, Song TY, Park J, Lee J, Lim J, Jang H, Kim YN, Yang JH, Song Y, Choi A, et al Menin mediates epigenetic regulation via histone H3 lysine 9 methylation. Cell Death and Disease 2013. 4 e583 ( 10.1038/cddis.2013.98) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lairmore TC, Piersall LD, DeBenedetti MK, Dilley WG, Mutch MG, Whelan AJ, Zehnbauer B. Clinical genetic testing and early surgical intervention in patients with multiple endocrine neoplasia type 1 (MEN 1). Annals of Surgery 2004. 5 637–645. ( 10.1097/01.sla.0000124383.98416.8d) [DOI] [PMC free article] [PubMed] [Google Scholar]