

 This work is licensed under a
This work is licensed under a Abstract
Aryl hydrocarbon receptor-interacting protein (AIP) gene mutations (AIPmut) are the most frequent germline mutations found in apparently sporadic pituitary adenomas (SPA). Our aim was to evaluate the frequency of AIPmut among young Brazilian patients with SPA. We performed an observational cohort study between 2013 and 2016 in a single referral center. AIPmut screening was carried out in 132 SPA patients with macroadenomas diagnosed up to 40 years or in adenomas of any size diagnosed until 18 years of age. Twelve tumor samples were also analyzed. Leukocyte DNA and tumor tissue DNA were sequenced for the entire AIP-coding region for evaluation of mutations. Eleven (8.3%) of the 132 patients had AIPmut, comprising 9/74 (12%) somatotropinomas, 1/38 (2.6%) prolactinoma, 1/10 (10%) corticotropinoma and no non-functioning adenomas. In pediatric patients (≤18 years), AIPmut frequency was 13.3% (2/15). Out of the 5 patients with gigantism, two had AIPmut, both truncating mutations. The Y268* mutation was described in Brazilian patients and the K273Rfs*30 mutation is a novel mutation in our patient. No somatic AIP mutations were found in the 12 tumor samples. A tumor sample from an acromegaly patient harboring the A299V AIPmut showed loss of heterozygosity. In conclusion, AIPmut frequency in SPA Brazilian patients is similar to other populations. Our study identified two mutations exclusively found in Brazilians and also shows, for the first time, loss of heterozygosity in tumor DNA from an acromegaly patient harboring the A299V AIPmut. Our findings corroborate previous observations that AIPmut screening should be performed in young patients with SPA.
Keywords: AIP, germline mutations, sporadic pituitary adenomas, tumor suppressor gene
Introduction
Although most pituitary adenomas occur sporadically, with only 5% of all cases being related to inherited syndromes (1), the mechanisms underlying pituitary tumorigenesis in a non-familial setting are poorly understood. Somatic mutations and other genetic and/or epigenetic abnormalities have been related to SPA, but a minor subgroup of these adenomas can have a germline mutation in a predisposing gene with no known familial history of pituitary adenoma (2). Germline aryl hydrocarbon receptor-interacting protein (AIP) gene mutations (AIPmut) were first described by Vierimaa and coworkers in 2006 (3). This study has found AIPmut in seemingly sporadic acromegaly patients and in familial isolated pituitary adenomas (FIPA) (3), which is characterized by the presence of pituitary adenomas in two or more members of the same family in the absence of other syndromic clinical features.
AIP appears to act as a tumor suppressor gene (TSG) (3). It is a cytoplasmic protein and a co-chaperone of heat-shock protein 90 (HSP90), and several studies demonstrated the involvement of AIP in various nuclear receptor signaling pathways, such as in estrogen receptor α (ERA) and glucocorticoid receptor (GR) signaling pathways (4, 5, 6). However, the exact molecular mechanisms by which AIPmut promotes pituitary adenomas are unclear. There is evidence that a failure to inhibit cyclic adenosine monophosphate (cAMP) synthesis underlies the development of pituitary adenomas in AIPmut patients (7). The observation of loss of heterozygosity (LOH) at the chromosome 11q13 in pituitary adenomas containing AIPmut provides another argument for the role of these genetic mutations in pituitary tumorigenesis (3, 8). Functional evaluation of AIPmut has shown reduced ability to inhibit cell proliferation and disruption of the protein–protein interaction between AIP and phosphodiesterase-4A5 (PDE4A5) (9). In addition, the observation that most pathological mutations lead to a truncated protein, mostly affecting its C-terminal part, which is involved in interactions with other proteins, or conformational changes that lead to altered protein stability (10), reinforces the role of AIP as a TSG (11).
A number of studies have investigated the prevalence and the clinical characteristics of patients with all types of apparently SPA and AIPmut (12, 13, 14, 15, 16), and so far, it is established that AIPmut are the most frequent germline mutations found in SPA (17). The seemingly low prevalence of AIPmut in apparently sporadic cases is probably due to low penetrance (20%) (14, 18), as de novo mutations have only been described in 2 patients (19, 20). Patients harboring AIPmut are predominantly male (61%), are young at the time of diagnosis (78% aged <30 years) and tend to have macroadenomas (88%) with extrasellar extension making curative surgery less likely (21). In case of acromegaly, AIPmut patients have a poor response to medical treatment (14, 21). Therefore, recognition of AIPmut positive pituitary adenomas is of clinical importance and family member screening can provide early diagnosis of affected patients not yet diagnosed leading to higher chance of disease control.
The findings of previous studies that investigated the prevalence of germline AIPmut in patients with SPA suggest that screening should be focused on young patients (diagnosed before the age of 30–40 years) with macroadenomas or in patients with any size of tumors diagnosed before age 18 years (22). The studies that applied those criteria have found a prevalence ranging from 2.8 to 11.7% (13, 14, 16, 23, 24, 25). Most studies were performed in European populations, and only a few were multicentric (14, 16, 26). Although prevalence of AIPmut seems to be similar across different ethnicities, new studies can show variations in AIPmut profiles and bring more data from different populations. Thus, we analyzed patients with SPA, with diagnosis up to 40 years, for the presence of AIPmut in our tertiary referral center in Brazil.
Materials and methods
Subjects
Consecutive patients with SPA from a single referral center were prospectively enrolled from July 2013 to February 2016. This tertiary referral center is part of a University Hospital established in Rio de Janeiro, which is linked to the single health system of Brazil, receiving referrals from all the State. Inclusion criteria were evidence of macroadenoma (maximal diameter ≥10 mm on pituitary MRI) diagnosed up to 40 years. Patients with diagnosis until 18 years of age (pediatric patients) were included both with micro or macroadenoma. Clinical, laboratory and family history from all subjects was undertaken to exclude familial pituitary adenomas either isolated (FIPA and X-linked acro gigantism) or as a component of other genetic syndromes (e.g. multiple endocrine neoplasia types 1 (MEN1) and 4 (MEN4), Carney complex, familial pheochromocytoma/paraganglioma/pituitary adenoma syndrome) (27). Genomic analyses for the screening of these genetic syndromes were not performed. All subjects gave written informed consent. The Ethics Committee of the Medical School and the Hospital Universitário Clementino Fraga Filho of the Universidade Federal do Rio de Janeiro (HUCFF-UFRJ) approved the study. Genetic counseling was provided for family members of AIPmut-positive cases, and clinical testing and follow-up were offered, whenever possible.
Pituitary tumor samples
Formalin-fixed and paraffin-embedded tissue was available from patients who underwent surgery in our center. Histological sections were stained with H&E and submitted to immunohistochemical reactions for pituitary hormones (GH – dilution 1:5000, PRL – dilution 1:5000, ACTH – dilution 1:4000, FSH – dilution 1:3000, TSH – dilution 1:2000, LH – dilution 1:4000), all of them polyclonal rabbit antibody/cell marque. In addition, GH-positive tumors were immunostained with CAM 5.2 (monoclonal mouse antibody cytokeratin (CAM5.2)/Cell Marque, dilution 1:2000) to differentiate sparsely from densely granulated tumors.
Nine frozen and three paraffin-embedded tissue samples of the enrolled patients were available for genetic screening for somatic AIP gene mutations. In cases where AIPmut were identified in leukocyte DNA and tumor DNA was available, search for LOH was performed through AIP sequencing.
Genomic analyses of AIP
Mutation screening of AIP was done using genomic DNA isolated from peripheral blood leukocytes and from frozen or paraffin-embedded tumor tissues, using the Gentra PureGene Blood Kit (Qiagen), AllPrep DNA/RNA/miRNA Universal Kit (Qiagen) and QIAamp DNA FFPE Tissue Kit (Qiagen), respectively, following the manufacturer’s instructions.
The entire AIP-coding region (exons 1–6) as well as flanking intronic sequences were amplified and sequenced with AIP PCR/sanger sequencing primer pairs (Thermo Fisher Scientific). The promoter region was not analyzed. PCRs were performed on Applied Biosystems ProFlex PCR System (Applied Biosystems). PCR products clean up were performed with ExoSAP-IT (USB Corporation, Cleveland, OH, USA). DNA sequencing was performed using Big Dye Terminator 3.1 Cycle Sequencing kit and an automated capillary sequencer (ABI 3130xl Genetic Analyzer, Applied Biosystems). Electropherogram-derived sequences were compared with NCBI references for the AIP gene (NG_008969.1 RefSeq-Gene and NM_003977.3 transcript) using Benchling (http://benchling.com, Benchling Inc, San Francisco, CA, USA). All genetic alterations were confirmed by a repeated analysis.
AIP sequence variants were compared with human single-nucleotide polymorphism (SNP) databases (dbSNP, http://www.ncbi.nlm.nih.gov/SNP/snp_summary.cgi), ExAC database (http://exac.broadinstitute.org) and also against AIP mutation data from genetically diverse populations (28). Only the variants that met the mutation criterion, defined as a minor allele frequencies (MAF) <1%, were considered for further analysis (intronic variants outside the splicing site area were not analyzed). PolyPhen2 (http://genetics.bwh.harvard.edu) and Alamut Software, version 2.2e (Interactive Biosoftware, Rouen, France) were used to evaluate the pathogenicity of missense mutations on AIP structure. Mutations were classified as pathogenic, likely pathogenic, variants of uncertain significance (VUS), likely benign or benign, according to the Standards and Guidelines for the Interpretation of Sequence Variants (29).
Patients with somatotropinomas and pediatric patients with any pituitary adenoma in whom AIP sequencing did not find a mutation were screened for large deletions of the AIP using multiplex ligation-dependent probe amplification (Salsa MLPA probemix P244-B1 AIP-MEN1, MRC-Holland, Amsterdam, The Netherlands), whenever suitable quality DNA was available.
Statistical analyses
Normal distribution was tested by the Kolmogorov–Smirnov and Shapiro–Wilk tests. The Mann–Whitney U test and the χ 2 test were used for statistical analysis. Data are given as median (range). P values below 0.05 were considered as significant.
Results
Clinical characteristics of the study cohort
A total of 132 patients with sporadic pituitary macroadenomas diagnosed up to 40 years, and with micro or macroadenomas diagnosed until 18 years of age were included. Of these patients, 74 (56%) had acromegaly or gigantism, 38 (28.8%) had prolactinoma, 10 (7.6%) had non-functioning pituitary adenoma (NFPA) and 10 (7.6%) had Cushing’s disease. The median age at diagnosis was 28 (9–40) years, 15 (11.3%) had diagnosis during childhood or adolescence (age ≤18 years), 84 (63.6%) were female and the median tumor diameter at diagnosis was 22 (6–81) mm. Characteristics of each group at diagnosis are given in Table 1. Female predominance was seen in all groups.
Table 1.
Clinical, radiological and pathological data of the study cohort.
| Type of pituitary tumor and distribution (Dx until and after 18 years) | Females (%) | Median age (min–max) | Median tumor diameter mm (min–max) | Giant adenomas (%) | Available tumors |
|---|---|---|---|---|---|
| Somatotropinoma | 43 (58) | 29 (18–40) | 24 (11–61) | 7 (9.4) | 8 |
| n = 74; 56.0% | |||||
| ≤18 years = 1 | 1 (100) | 18 | 20 | 0 | 0 |
| >18 years = 73 | 42 (58) | 29 (20–40) | 25 (11–61) | 7 | 8 |
| Prolactinoma | 27 (71) | 23 (11–40) | 19 (10–81) | 12 (31.6) | 2 |
| n = 38; 28.8% | |||||
| ≤18 years = 8 | 5 (63) | 17 (11–18) | 19 (14–60) | 3 | 0 |
| >18 years = 30 | 22 (73) | 25 (19–40) | 19.5 (10–81) | 9 | 2 |
| NFPA | 6 (60) | 30.5 (11–37) | 27 (12–50) | 1 (10) | 0 |
| n = 10; 7.6% | |||||
| ≤18 years = 2 | 0 | 12.5 (11–14) | 22* | 0 | 0 |
| >18 years = 8 | 6 (75) | 32.5 (22–37) | 28.5 (12–50) | 1 | 0 |
| Cushing’s disease | 8 (80) | 21.5 (9–39) | 12 (6–35) | 0 | 2 |
| n = 10; 7.6% | |||||
| ≤18 years = 4 | 2 (50) | 14 (9–16) | 10 (6–11) | 0 | 1 |
| >18 years = 6 | 6 (100) | 25 (21–39) | 15.5 (12–35) | 0 | 1 |
| Total | 84 (63) | 28 (9–40) | 22 (6–81) | 20 (14.5) | 12 |
| n = 132 |
*Only one patient had tumor diameter available.
Dx, diagnosis; NFPA, non-functioning pituitary adenoma.
Patients with AIPmut
Germline AIPmut were observed in 11 (8.3%) of the 132 patients. Among these 11 patients, we found 8 different AIPmut (3 pathogenic mutations, 3 VUS and 2 likely benign mutations) (Table 2).
Table 2.
Clinical, radiological and genetic characteristics of patients with AIP germline mutations.
| Dx | Sex | Age at Dx (years) | Adenoma size at Dx (mm) | Mut | Protein change | Protein location | Functional study | Sig | PolyPhen2 (in silico) | Alamut (in silico) (SIFT/Mut Taster) | MAF | Type of mut | dbSNP reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acro | M | 33 | 12 | c.47G > A | R16H | N-terminal | Incomplete loss of interaction with PDE4A5 (11, 35)Similar half-life to WT AIP (10) | Likely benign | Probably damaging (0.966) | Deleterious (0.01)/disease causing (p.0.9) | 0.0020 | Mis | rs145047094 |
| Acro | F | 34 | 25 | c.382C > T | R128C | Between FKBP PPIase and TPR1 domains | NA | VUS | Benign (0.228) | Deleterious (0.04)/polymorphism (p.1.0) | 0.0001 | Mis | rs140530307 |
| Acro | M | 34 | Macro | c.382C > T | R128C | Between FKBP PPIase and TPR1 domains | NA | VUS | Benign (0.228) | Deleterious (0.04)/polymorphism (p.1.0) | 0.0001 | Mis | rs140530307 |
| Giant | M | 27 | 17 | c.804C > A | Y268* | TPR3 domain/predicted protein lacking the final 63 aa | Rapid degradation of truncated AIP (10) | Pathogenic | NA | Non | rs121908356 | ||
| Giant* | F | 22 | 22 | c.816delC | K273Rfs*30 | TPR3 domain | NA | Pathogenic | NA | Frame | In process | ||
| Acro | M | 40 | 19 | c.896C > T | A299V | C-terminal α-helix | Incomplete loss of interaction with PDE4A5 (11, 35)Short half-life compared to WT AIP (10) | VUS | Possibly damaging (0.934) | Deleterious (0.04)/disease causing (p.0.9) | 0.0004 | Mis | rs148986773 |
| CD | M | 15 | 10 | c.896C > T | A299V | C-terminal α-helix | Incomplete loss of interaction with PDE4A5 (11, 35)Short half-life compared to WT AIP (10) | VUS | Possibly damaging (0.934) | Deleterious (0.04)/disease causing (p.0.9) | 0.0004 | Mis | rs148986773 |
| PRL | M | 18 | 60 | c.911G > A | R304Q | C-terminal α-helix | No significant reduction in β-galactosidase activity for the R304Q AIP mutant (11, 35)Similar half-life to WTAIP (10) | Pathogenic | Benign (0.047) | Deleterious (0.04)/disease causing (p.0.8) | 0.0015 | Mis | rs104894190 |
| Acro | F | 37 | 31 | c.*14C > A | Not in protein (3′UTR) | NA | VUS | 0.0005 | rs142567224 | ||||
| Acro | F | 33 | 40 | c.*14C > A | Not in protein (3′UTR) | NA | VUS | 0.0005 | rs142567224 | ||||
| Acro | M | 38 | 25 | c.*64G > A | Not in protein (3′UTR) | NA | Likely benign | 0.0062 | rs115346238 | ||||
| Dx | Sex | Age at Dx (years) | Adenoma size at Dx (mm) | Mut | Protein change | Protein location | Functional study | Sig | PolyPhen2 (in silico) | Alamut (in silico) (SIFT/mut Taster) | MAF | Type of mut | dbSNP reference |
| Acro | M | 33 | 12 | c.47G > A | R16H | N-terminal | Incomplete loss of interaction with PDE4A5 (11, 35)Similar half-life to WT AIP (10) | Likely benign | Probably damaging (0.966) | Deleterious (0.01)/disease causing (p.0.9) | 0.0020 | Mis | rs145047094 |
| Acro | F | 34 | 25 | c.382C > T | R128C | Between FKBP PPIase and TPR1 domains | NA | VUS | Benign (0.228) | Deleterious (0.04)/polymorphism (p.1.0) | 0.0001 | Mis | rs140530307 |
| Acro | M | 34 | Macro | c.382C > T | R128C | Between FKBP PPIase and TPR1 domains | NA | VUS | Benign (0.228) | Deleterious (0.04)/polymorphism (p.1.0) | 0.0001 | Mis | rs140530307 |
| Giant | M | 27 | 17 | c.804C > A | Y268* | TPR3 domain/predicted protein lacking the final 63 aa | Rapid degradation of truncated AIP (10) | Pathogenic | NA | Non | rs121908356 | ||
| Giant* | F | 22 | 22 | c.816delC | K273Rfs*30 | TPR3 domain | NA | Pathogenic | NA | Frame | In process | ||
| Acro | M | 40 | 19 | c.896C > T | A299V | C-terminal α-helix | Incomplete loss of interaction with PDE4A5 (11, 35)Short half-life compared to WT AIP (10) | VUS | Possibly damaging (0.934) | Deleterious (0.04)/disease causing (p.0.9) | 0.0004 | Mis | rs148986773 |
| CD | M | 15 | 10 | c.896C > T | A299V | C-terminal α-helix | Incomplete loss of interaction with PDE4A5 (11, 35)Short half-life compared to WT AIP (10) | VUS | Possibly damaging (0.934) | Deleterious (0.04)/disease causing (p.0.9) | 0.0004 | Mis | rs148986773 |
| PRL | M | 18 | 60 | c.911G > A | R304Q | C-terminal α-helix | No significant reduction in β-galactosidase activity for the R304Q AIP mutant (11, 35)Similar half-life to WTAIP (10) | Pathogenic | Benign (0.047) | Deleterious (0.04)/disease causing (p.0.8) | 0.0015 | Mis | rs104894190 |
| Acro | F | 37 | 31 | c.*14C > A | Not in protein (3′UTR) | NA | VUS | 0.0005 | rs142567224 | ||||
| Acro | F | 33 | 40 | c.*14C > A | Not in protein (3′UTR) | NA | VUS | 0.0005 | rs142567224 | ||||
| Acro | M | 38 | 25 | c.*64G > A | Not in protein (3′UTR) | NA | Likelybenig n | 0.0062 | rs115346238 |
*Previously reported by Hernandez-Ramirez et al. (14).
Aa, amino acid; Acro, acromegaly; CD, Cushing’s disease; Dx, diagnosis; F, female; FKBP PPIase domain, FK506-binding protein peptidyl-prolyl cis-trans isomerase domain; frame, frameshift; giant, gigantism; M, male; Macro, macroadenoma; MAF, minor allele frequency (ExAC database was used for all AIPmut, except for the AIPmut c.*64G > A, where dbSNP database was used); Mis, missense; Mut, mutation; NA, not available; non, nonsense; PRL, prolactinoma; PDE4A5, phosphodiesterase-4A5; Sig, significance; TPR, tetratricopeptide repeat; VUS, variant of uncertain significance; UTR, untranslated region; WT, wild type.
Among the 74 patients with somatotropinomas, 9 (12.2%) presented AIPmut. From this group, 2 out of 5 patients with gigantism had a pathogenic truncating AIPmut (Y268* and K273Rfs*30) and 7 out of 69 (10.1%) patients with acromegaly had an AIP VUS or likely benign mutations (Table 2). Dosage analysis by MLPA was possible from 59 of the 65 patients with somatotropinomas without AIPmut, including the 3 patients with gigantism, and did not reveal any large deletions.
The male patient with gigantism harboring the nonsense AIPmut Y268* (c.804C > A), had the diagnosis at 27 years with a history of accelerated growth since the age of 13 years (height at diagnosis 217 cm), arthralgia and a 17 mm macroadenoma. He refused surgery, and treatment with first-generation somatostatin analogue (SA) did not result in normalization of his GH and IGF-1 levels. Parental DNA from his mother and 2 sisters were available for AIPmut screening, and the same mutation was found in one of the sisters who is clinically unaffected, although a proper evaluation with pituitary hormones and pituitary MRI was not performed due to her refusal (Fig. 1A).
Figure 1.

Pedigrees of the families of the probands. The scheme shows the three family trees (A, B and C) of the probands (black squares with an arrow). Male family members are represented by squares, females by circles.
The frameshifit AIPmut K273Rfs*30 (c.816delC) (14) was detected in a 22-year-old female patient who presented with a phenotype of gigantism, height of 181 cm and a 22 mm macroadenoma. She underwent a pituitary surgery, and histopathology of the tumor confirmed to be a somatotropinoma. She was started on clinical treatment with first-generation SA and cabergoline (CAB) with poor response, and then was started on pegvisomant. Unfortunately, genetic screening of her family is not available.
The missense likely benign AIPmut R16H (c.47G > A) was found in a male acromegaly patient with diagnosis at the age of 33 years with a macroadenoma of 12 mm. Pituitary surgery was curative and pathology demonstrated a sparsely granulated (SG) somatotropinoma. The other AIP likely benign mutation c.*64G > A, located at the 3′ untranslated region (3′UTR), was found in a male acromegaly patient (Table 2), diagnosed at the age of 38 years with a tumor of 25 mm, in a pre-operative evaluation for rhinoplasty. He underwent two pituitary surgeries, and the pathology revealed a SG somatotropinoma. Due to resistance to combined first-generation SA and CAB therapy, the patient was started on pasireotide LAR, which resulted in disease control.
The missense AIP VUS A299V (c.896C > T) was present in a male patient with acromegaly diagnosed at the age of 40 years with a tumor of 19 mm. A non-curative pituitary surgery was performed and the pathology showed a SG somatotropinoma. Medical treatment with first-generation SA was started with poor response. The other AIP VUS were found only in acromegaly patients, including the missense R128C (c.382C > T) at exon 3 and the 3′UTR c.*14C > A (Table 2). The R128C AIPmut was found in one male and one female patient, both diagnosed at the age of 34 years with macroadenomas. Both had non-curative surgeries. The female patient showed resistance to first-generation SA therapy, even after radiation therapy and to CAB association and is now under control with SA and pegvisomant. The male patient had radiotherapy and is under control with first-generation SA treatment. The c.*14C > A AIPmut was found in two female patients with acromegaly. The first one was diagnosed at the age of 37 with a tumor of 31 mm operated transsphenoidally. She had resistance to combined treatment of first-generation SA and CAB, but achieved control with the combination of SA and pegvisomant. The second one was diagnosed at the age of 33 years with a giant tumor. She was submitted to a pituitary surgery and the pathology showed a SG somatotropinoma (Fig. 2). Medical treatment with first-generation SA was started, and the patient did not achieve disease control.
Figure 2.

Sparsely granulated somatotropinoma from the patient with the AIPmut c.*14C > A. Pituitary adenoma stained with Hematoxilin & Eosin, consisting of eosinophilic cells (A and B) and immunopositive for GH (C), which are sparsely granulated (dot staining) with CAM 5.2 (D). There are blood cells and cholesterol clefts among the epithelial cells (A).
In the 38 patients with prolactinomas, 1 (2.6%) had the pathogenic AIPmut R304Q (c.911G > A). This male patient was diagnosed in the age of 18 years with a giant adenoma (60 mm) and very high prolactin (PRL) levels (20,000 ng/mL). A pituitary surgery was performed, but PRL remained elevated, and he showed resistance to high doses of CAB (3.5 mg/week). Tumor sample was not available for LOH analysis, but genetic screening of his family identified his father and paternal uncle as AIPmut carriers, since clinical evaluation, pituitary hormones dosage and pituitary MRI were normal for both relatives (Fig. 1B).
Among the 10 patients with corticotropinomas, 1 (10%) had the missense AIP VUS A299V (c.896C > T). Cushing’s disease was diagnosed in this male patient at the age of 15 years. He had a 10 mm pituitary adenoma that was surgically resected four times, with immunohistochemistry positive for adrenocorticotrophic hormone (ACTH) and a Ki-67 index of 5%. As the patient was not cured, a bilateral adrenalectomy was performed for disease control, and he developed Nelson’s syndrome 6 months after surgery. The same mutation was found in his clinically unaffected mother, who has presented normal levels of pituitary hormones and a normal pituitary MRI (Fig. 1C). This patient’s tissue sample has been tested negative for somatic ubiquitin-specific protease 8 (USP8) gene (30).
No mutations were detected among patients with NFPA. There was no difference regarding age, gender and tumor size at diagnosis between patients harboring or not AIPmut (P = 0.27, P = 0.053 and P = 0.94, respectively).
AIPmut in pituitary tumor samples and LOH analysis
Of the 12 genomic DNA tumor samples from our cohort, 9 were from patients who had no mutation on AIP sequencing of the peripheral blood DNA (6 somatotropinomas, 2 prolactinomas and 1 Cushing’s disease). These samples did not show somatic AIP mutations. Among the 3 patients that have shown an AIPmut on the peripheral blood leukocyte DNA analysis, LOH was investigated through AIP sequencing. LOH was found in the tumor sample of the acromegaly patient harboring the A299V (c.896C > T) VUS, with loss of the wild-type allele (Fig. 3A and B). No LOH was identified in the tumor sample from the patient with Cushing’s disease harboring the A299V VUS and the patient with acromegaly harboring the c.*14C > A AIPmut.
Figure 3.
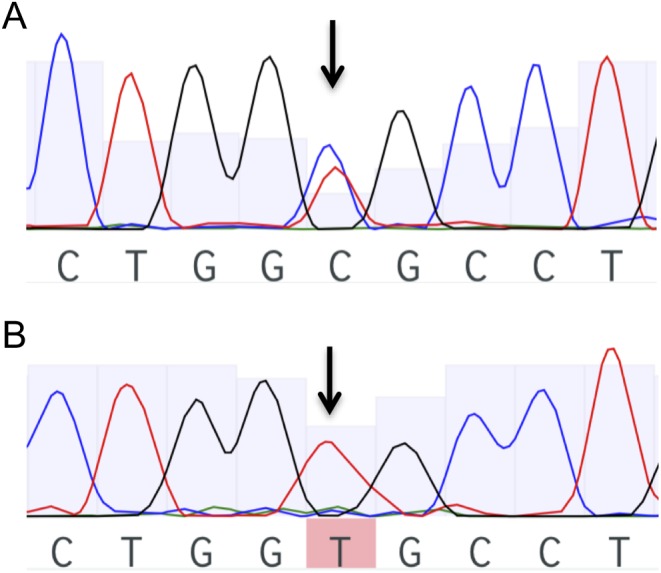
Sequencing electropherograms showing AIPmut c.896C > T (A299V) in exon 6. Black arrows show the position of the nucleotide change. (A) Blood leukocyte genomic DNA from acromegaly patient. (B) Tumor genomic DNA from acromegaly patient with loss of heterozygosity (LOH).
AIPmut in pediatric patients with pituitary adenomas
Considering only pediatric patients, AIPmut were identified in 2 of 15 patients (13.3%) diagnosed until 18 years of age. The pathogenic AIPmut R304Q was found in one patient with prolactinoma and the AIP VUS A299V was found in one patient with Cushing’s disease. Dosage analysis by MLPA was possible from 12 of the 13 pediatric patients without AIPmut and did not reveal any large deletions.
Discussion
Our finding of AIPmut in 8.3% of our cohort and in 13.3% among pediatric patients is consistent with the findings of previous studies in other populations (13, 14, 16, 24). Moreover, our study identified two AIPmut exclusively found in Brazilian patients and also shows, for the first time, LOH in the tumor DNA from an acromegaly patient harboring the A299V AIPmut. Although the study has included all types of pituitary adenomas matching the inclusion criteria, the acromegaly group is overrepresented, probably because we are a state referral center for the treatment of acromegaly, and the NFPA group has a limited number of patients; therefore, we cannot draw a definitive conclusion on the prevalence of AIP mutations in this type of adenoma.
Although AIPmut frequency in Brazil has shown to be similar to other populations, there are some interesting aspects of our study. Among the eight different AIPmut found, two out of three pathogenic mutations were never described in other populations. The first one is the nonsense AIPmut Y268*, which has been found in a Brazilian family with acromegaly (31), in a Brazilian patient with familial macroprolactinoma diagnosed in his twenties (18) and in the patient with gigantism presented here and elsewhere (14). This mutation results in a premature TAA-stop signal at codon 268 (31) (Table 2), and a missense mutation at this residue has previously been reported (AIPmut Y268C) (18). Another patient from our center who was diagnosed with acromegaly at 49 years, tested positive for the same AIPmut in another study (32). However, no family relationship between them was found, but a founder effect cannot be excluded. Family members of both patients were screened for the AIPmut Y268* and carriers’ relatives were found. The second one is the novel germline frameshift AIPmut K273Rfs*30 predicted to lead to a truncated protein (Table 2). This mutation was only described in our patient with gigantism (current study and (14)). Both mutations are located on exon 6, which is the most affected exon of the AIP gene.
The third pathogenic mutation found is the missense mutation R304Q (c.911G > A) (Table 2), first described by Georgitsi and coworkers (33) in a seemingly sporadic case of Cushing’s disease. Our study identified this mutation in an 18-year-old patient with a giant prolactinoma resistant to CAB treatment, with his unaffected father and uncle carrying the same mutation. The pathogenic role of R304Q has been strongly supported by clinical data, since it has been identified in several independent FIPA families as well as in sporadic patients including cases of acromegaly, Cushing’s disease and prolactinoma (9, 11, 12, 13, 16, 24, 25, 33). The c.911G > A is part of a CpG island mutational hotspot (34) and the missense mutation could possibly affect the interaction of AIP with AhR (33), but functional studies did not show significant reduction in β-galactosidase activity for the R304Q AIP mutant (11, 35) (Table 2). Moreover, the MAF of AIPmut R304Q, provided by ExAC, is very high when compared to other pathogenic or VUS AIPmut (Table 2). Therefore, the higher MAF together with discrepancies between experimental conditions and clinical data could lead to a review of the classification of the AIPmut R304Q to a likely pathogenic mutation or even to a VUS.
The AIP VUS A299V (Table 2) has been found in patients with acromegaly, prolactinoma and NFPA, both in sporadic and familial cohorts (11, 14, 15, 20, 33). None of these studies showed tumor LOH related to this VUS, but there are also no data of this VUS being found in any of the large general population databases. We identified the AIP VUS A299V, in a young patient with Cushing’s disease and in a patient with acromegaly resistant to treatment with SA. Both tumor samples were available, and LOH was found in the somatotropinoma, with retention of the mutated allele, which is in accordance with the Knudson’s two-hit hypothesis (36). Therefore, this is the first time that the LOH of the AIP VUS A299V is found in a somatotropinoma supporting the possibility that it might play a role in pituitary adenoma pathogenesis. In contrast, the corticotropinoma did not show LOH. This may be explained by contamination of the tumor sample with normal pituitary tissue, especially because it was taken from a second surgery of this originally 10 mm adenoma. Another possibility is that the Cushing’s disease patient may have a different (i.e. not loss of 11q13 chromosomal material) second hit for the development of the pituitary tumor, such as downregulation of gene expression through promoter methylation (37) or via microRNAs (38).
One of our important findings is that we have not identified somatic mutations in the tumor samples studied. This is in accordance with previous data in the literature (9, 26, 33, 39) and with the Catalogue Of Somatic Mutations In Cancer (http://cancer.sanger.ac.uk) suggesting that somatic AIPmut does not seem to contribute in the pathogenesis of the SPA.
The likely benign AIPmut R16H (Table 2), first described by Daly and coworkers (40) in a FIPA family, was identified in an acromegaly patient diagnosed at the age of 33 years with a macroadenoma. At first, the AIPmut R16H was considered a VUS, but although this mutation has been found in familial and sporadic patients, no LOH was identified in tumor samples, and it has been found in some control subjects, besides it has a high MAF (33, 40, 41, 42). Therefore, it is questioned whether the R16H is pathogenic, and it is hypothesized that it is a rare polymorphism (33, 42, 43).
Regarding the AIPmut R128C (c.382C > T), we found it in two acromegaly patients, and it has been previously described in two prolactinoma patients (15). In silico analyses predict both a benign and a deleterious mutation (Table 2), and there is lack of functional studies, restricting the conclusions about its pathogenicity. Therefore, we classified it as a VUS. Moreover, another mutation at this site (R128H) has been described in an acromegaly patient (44), suggesting that there might be a pathogenic role for the AIPmut R128C. The last two AIPmut (c.*14C > A and c.*64G > A) are located at the 3′ UTR; therefore, there is no amino acid change in AIP structure (Table 2). However, the 3′ UTR of AIP is a known target for microRNAs (miRNAs), which are small noncoding RNAs that inhibit posttranscriptional expression of target mRNAs by binding to target sequences (45). Therefore, changes in this area could change the affinity of a specific miRNAs to its target. The two most well-studied miRNAs that bind to the AIP 3′ UTR are the miR-34a and the miR-107 (14, 32, 45), but their binding sites do not overlap with these two new variants. The c.*64G > A has a high MAF and is classified as likely benign at dbSNP website (Table 2). The c.*14C > A, on the other hand, has a low MAF provided by ExAC (Table 2) and was detected in two patients of our cohort, although no LOH have been found in the tumor sample of one of the patients harboring this AIPmut. Therefore, due to these conflicting data, we classified the AIPmut c.*14C > A as a VUS.
Large genomic deletions of the AIP gene in mutation-negative patients can be detected by MLPA. The majority of studies that have used MLPA did not find large deletions (12, 13, 16, 19, 24, 46, 47, 48, 49, 50). The finding of deletions in blood DNA was restricted to 4 studies, 2 of them including only FIPA patients (11, 51), and other 2 studies, one including only GH-secreting adenomas, both sporadic or FIPA, that found deletions in 2 giant patients, one of them with FIPA (25), and the other one including all types of pituitary adenomas both sporadic or FIPA, that found deletion in 1 giant patient (14). In our study, MLPA analysis was restricted to pediatric and acromegaly patients with quality DNA available, with no detection of large deletions. Our finding is in agreement with previous studies of SPA (12, 13, 16, 24, 46, 47). Therefore, MLPA analysis may be restricted to FIPA cases, for sporadic pituitary adenoma patients diagnosed in childhood or adolescence, or even in patients with a phenotype highly suggestive of AIPmut, that were tested negative for AIPmut in sequencing analyses.
In conclusion, our AIPmut screening performed for the first time in a Brazilian population corroborates the low frequency of germline AIPmut in SPA, as previously reported in other ethnic populations. Moreover, we found two AIPmut that are only described in Brazilian patients. We also show that special populations like patients with gigantism and patients diagnosed in childhood present a higher prevalence of AIPmut, and therefore, should be considered for screening. This allows early identification of affected carriers, when the proband is identified. Finally, we described for the first time the presence of LOH in a somatotropinoma from an acromegaly patient harboring an A299V AIPmut, previously classified as VUS. Finally, the diversity of AIPmut found among all the studies points to the need for more functional studies for a better understanding of the role of AIP in the pituitary tumorigenesis.
Declaration of interest
M R G has received consulting fees from Novartis, Ipsen and Ionis, speaker fees from Novartis, Pfizer and Ipsen and research grants from Novartis, Ipsen and Pfizer. P B A is employed by Diagnósticos da América SA, but the company had no interference in the development of the study, and this affiliation does not alter policies on sharing data and materials.
Funding
The laboratory work was funded by a grant from Fundação de Amparo a Pesquisa do Estado do Rio de Janeiro – FAPERJ E-26/010.001967/2014 (to M R G) and from unrestricted research grants from Novartis and Ipsen (to M R G). The funder Diagnósticos da América SA provided support in the form of salaries for author P B A, but did not have any additional role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Acknowledgements
The authors would like to thank patients for accepting to take part of this study and all the neuroendocrinology team from HUCFF-UFRJ for the constant and valuable support.
References
- 1.Gadelha MR, Trivellin G, Hernández Ramírez LC, Korbonits M. Genetics of pituitary adenomas. Frontiers of Hormone Research 2013. 41 111–140. [DOI] [PubMed] [Google Scholar]
- 2.Melmed S. Pathogenesis of pituitary tumors. Nature Reviews Endocrinology 2011. 7 257–266. ( 10.1038/nrendo.2011.40) [DOI] [PubMed] [Google Scholar]
- 3.Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, Raitila A, Tuppurainen K, Ebeling TM, Salmela PI, Paschke R, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science 2006. 312 1228–1230. ( 10.1126/science.1126100) [DOI] [PubMed] [Google Scholar]
- 4.Trivellin G, Korbonits M. AIP and its interacting partners. Journal of Endocrinology 2011. 210 137–155. ( 10.1530/JOE-11-0054) [DOI] [PubMed] [Google Scholar]
- 5.Swedenborg E, Pongratz I. AhR and ARNT modulate ER signaling. Toxicology 2010. 268 132–138. ( 10.1016/j.tox.2009.09.007) [DOI] [PubMed] [Google Scholar]
- 6.Laenger A, Lang-Rollin I, Kozany C, Zschocke J, Zimmermann N, Rüegg J, Holsboer F, Hausch F, Rein T. XAP2 inhibits glucocorticoid receptor activity in mammalian cells. FEBS Letters 2009. 583 1493–1498. ( 10.1016/j.febslet.2009.03.072) [DOI] [PubMed] [Google Scholar]
- 7.Tuominen I, Heliövaara E, Raitila A, Rautiainen MR, Mehine M, Katainen R, Donner I, Aittomäki V, Lehtonen HJ, Ahlsten M, et al. AIP inactivation leads to pituitary tumorigenesis through defective Galphai-cAMP signaling. Oncogene 2015. 34 1174–1184. ( 10.1038/onc.2014.50) [DOI] [PubMed] [Google Scholar]
- 8.Gadelha MR. Loss of heterozygosity on chromosome 11q13 in two families with acromegaly/gigantism is independent of mutations of the multiple endocrine neoplasia type I gene. Journal of Clinical Endocrinology and Metabolism 1999. 84 249–256. [DOI] [PubMed] [Google Scholar]
- 9.Leontiou CA, Gueorguiev M, van der puy J, Quinton R, Lolli F, Hassan S, Chahal HS, Igreja SC, Jordan S, Rowe J, et al. The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. Journal of Clinical Endocrinology and Metabolism 2008. 93 2390–2401. ( 10.1210/jc.2007-2611) [DOI] [PubMed] [Google Scholar]
- 10.Hernandez-Ramirez LC, Martucci F, Morgan RM, Trivellin G, Tilley D, Ramos-Guajardo N, Iacovazzo D, D’Acquisto F, Prodromou C, Korbonits M. Rapid proteasomal degradation of mutant proteins is the primary mechanism leading to tumorigenesis in patients with missense AIP mutations. Journal of Clinical Endocrinology and Metabolism 2016. 101 3144–3154. ( 10.1210/jc.2016-1307) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Igreja S, Chahal HS, King P, Bolger GB, Srirangalingam U, Guasti L, Chapple JP, Trivellin G, Gueorguiev M, Guegan K, et al. Characterization of aryl hydrocarbon receptor interacting protein (AIP) mutations in familial isolated pituitary adenoma families. Human Mutation 2010. 31 950–960. ( 10.1002/humu.21292) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cazabat L, Bouligand J, Salenave S, Bernier M, Gaillard S, Parker F, Young J, Guiochon-Mantel A, Chanson P. Germline AIP mutations in apparently sporadic pituitary adenomas: prevalence in a prospective single-center cohort of 443 patients. Journal of Clinical Endocrinology and Metabolism 2012. 97 E663–E670. ( 10.1210/jc.2011-2291) [DOI] [PubMed] [Google Scholar]
- 13.Preda V, Korbonits M, Cudlip S, Karavitaki N, Grossman AB. Low rate of germline AIP mutations in patients with apparently sporadic pituitary adenomas before the age of 40: a single-centre adult cohort. European Journal of Endocrinology 2014. 171 659–666. ( 10.1530/EJE-14-0426) [DOI] [PubMed] [Google Scholar]
- 14.Hernandez-Ramirez LC, Gabrovska P, Dénes J, Stals K, Trivellin G, Tilley D, Ferrau F, Evanson J, Ellard S, Grossman AB, et al. Landscape of familial isolated and young-onset pituitary adenomas: prospective diagnosis in AIP mutation carriers. Journal of Clinical Endocrinology and Metabolism 2015. 100 E1242–E1254. ( 10.1210/jc.2015-1869) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lecoq AL, Bouligand J, Hage M, Cazabat L, Salenave S, Linglart A, Young J, Guiochon-Mantel A, Chanson P, Kamenický P. Very low frequency of germline GPR101 genetic variation and no biallelic defects with AIP in a large cohort of patients with sporadic pituitary adenomas. European Journal of Endocrinology 2016. 174 523–530. ( 10.1530/EJE-15-1044) [DOI] [PubMed] [Google Scholar]
- 16.Tichomirowa MA, Barlier A, Daly AF, Jaffrain-Rea ML, Ronchi C, Yaneva M, Urban JD, Petrossians P, Elenkova A, Tabarin A, et al. High prevalence of AIP gene mutations following focused screening in young patients with sporadic pituitary macroadenomas. European Journal of Endocrinology 2011. 165 509–515. ( 10.1530/EJE-11-0304) [DOI] [PubMed] [Google Scholar]
- 17.Lecoq AL, Kamenický P, Guiochon-Mantel A, Chanson P. Genetic mutations in sporadic pituitary adenomas – what to screen for? Nature Reviews Endocrinology 2015. 11 43–54. ( 10.1038/nrendo.2014.181) [DOI] [PubMed] [Google Scholar]
- 18.Beckers A, Aaltonen LA, Daly AF, Karhu A. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocrine Reviews 2013. 34 239–277. ( 10.1210/er.2012-1013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stratakis CA, Tichomirowa MA, Boikos S, Azevedo MF, Lodish M, Martari M, Verma S, Daly AF, Raygada M, Keil MF, et al. The role of germline AIP, MEN1, PRKAR1A, CDKN1B and CDKN2C mutations in causing pituitary adenomas in a large cohort of children, adolescents, and patients with genetic syndromes. Clinical Genetics 2010. 78 457–463. ( 10.1111/j.1399-0004.2010.01406.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramirez-Renteria C, Hernández-Ramírez LC, Portocarrero-Ortiz L, Vargas G, Melgar V, Espinosa E, Espinosa-de-Los-Monteros AL, Sosa E, González B, Zúñiga S, et al. AIP mutations in young patients with acromegaly and the Tampico Giant: the Mexican experience. Endocrine 2016. 53 402–411. [DOI] [PubMed] [Google Scholar]
- 21.Daly AF, Beckers A. Familial isolated pituitary adenomas (FIPA) and mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocrinology Metabolism Clinics of North America 2015. 44 19–25. ( 10.1016/j.ecl.2014.10.002) [DOI] [PubMed] [Google Scholar]
- 22.Korbonits M, Storr H, Kumar AV. Familial pituitary adenomas – who should be tested for AIP mutations? Clinical Endocrinology 2012. 77 351–356. ( 10.1111/j.1365-2265.2012.04445.x) [DOI] [PubMed] [Google Scholar]
- 23.Georgitsi M, De Menis E, Cannavò S, Mäkinen MJ, Tuppurainen K, Pauletto P, Curtò L, Weil RJ, Paschke R, Zielinski G, et al. Aryl hydrocarbon receptor interacting protein (AIP) gene mutation analysis in children and adolescents with sporadic pituitary adenomas. Clinical Endocrinology 2008. 69 621–627. ( 10.1111/j.1365-2265.2008.03266.x) [DOI] [PubMed] [Google Scholar]
- 24.Cuny T, Pertuit M, Sahnoun-Fathallah M, Daly A, Occhi G, Odou MF, Tabarin A, Nunes ML, Delemer B, Rohmer V, et al. Genetic analysis in young patients with sporadic pituitary macroadenomas: besides AIP don’t forget MEN1 genetic analysis. European Journal of Endocrinology 2013. 168 533–541. ( 10.1530/EJE-12-0763) [DOI] [PubMed] [Google Scholar]
- 25.Schofl C, Honegger J, Droste M, Grussendorf M, Finke R, Plöckinger U, Berg C, Willenberg HS, Lammert A, Klingmüller D, et al. Frequency of AIP gene mutations in young patients with acromegaly: a registry-based study. Journal of Clinical Endocrinology and Metabolism 2014. 99 E2789–E2793. ( 10.1210/jc.2014-2094) [DOI] [PubMed] [Google Scholar]
- 26.Barlier A, Vanbellinghen JF, Daly AF, Silvy M, Jaffrain-Rea ML, Trouillas J, Tamagno G, Cazabat L, Bours V, Brue T, et al. Mutations in the aryl hydrocarbon receptor interacting protein gene are not highly prevalent among subjects with sporadic pituitary adenomas. Journal of Clinical Endocrinology and Metabolism 2007. 92 1952–1955. ( 10.1210/jc.2006-2702) [DOI] [PubMed] [Google Scholar]
- 27.Minnetti M, Grossman A. Somatic and germline mutations in NETs: implications for their diagnosis and management. Best Practice and Research: Clinical Endocrinology and Metabolism 2016. 30 115–127. ( 10.1016/j.beem.2015.09.007) [DOI] [PubMed] [Google Scholar]
- 28.Craig Rowlands J, Urban JD, Wikoff DS, Budinsky RA. An evaluation of single nucleotide polymorphisms in the human aryl hydrocarbon receptor-interacting protein (AIP) gene. Drug Metabolism and Pharmacokinetics 2011. 26 431–439. ( 10.2133/dmpk.DMPK-11-SC-013) [DOI] [PubMed] [Google Scholar]
- 29.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015. 17 405–424. ( 10.1038/gim.2015.30) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perez-Rivas LG, Theodoropoulou M, Ferraù F, Nusser C, Kawaguchi K, Stratakis CA, Faucz FR, Wildemberg LE, Assié G, Beschorner R, et al. The gene of the ubiquitin-specific protease 8 is frequently mutated in adenomas causing Cushing’s disease. Journal of Clinical Endocrinology and Metabolism 2015. 100 E997–E1004. ( 10.1210/jc.2015-1453) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toledo RA, Lourenço DM, Liberman B, Jr, Cunha-Neto MB, Cavalcanti MG, Moyses CB, Toledo SP, Dahia PL. Germline mutation in the aryl hydrocarbon receptor interacting protein gene in familial somatotropinoma. Journal of Clinical Endocrinology and Metabolism 2007. 92 1934–1937. ( 10.1210/jc.2006-2394) [DOI] [PubMed] [Google Scholar]
- 32.Denes J, Kasuki L, Trivellin G, Colli LM, Takiya CM, Stiles CE, Barry S, de Castro M, Gadelha MR, Korbonits M. Regulation of aryl hydrocarbon receptor interacting protein (AIP) protein expression by MiR-34a in sporadic somatotropinomas. PLoS ONE 2015. 10 e0117107 ( 10.1371/journal.pone.0117107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Georgitsi M, Raitila A, Karhu A, Tuppurainen K, Mäkinen MJ, Vierimaa O, Paschke R, Saeger W, van der Luijt RB, Sane T, et al. Molecular diagnosis of pituitary adenoma predisposition caused by aryl hydrocarbon receptor-interacting protein gene mutations. PNAS 2007. 104 4101–4105. ( 10.1073/pnas.0700004104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chahal HS, Chapple JP, Frohman LA, Grossman AB, Korbonits M. Clinical, genetic and molecular characterization of patients with familial isolated pituitary adenomas (FIPA). Trends in Endocrinology and Metabolism 2010. 21 419–427. ( 10.1016/j.tem.2010.02.007) [DOI] [PubMed] [Google Scholar]
- 35.Bolger GB, Bizzi MF, Pinheiro SV, Trivellin G, Smoot L, Accavitti MA, Korbonits M, Ribeiro-Oliveira A., Jr cAMP-specific PDE4 phosphodiesterases and AIP in the pathogenesis of pituitary tumors. Endocrine-Related Cancer 2016. 23 419–431. ( 10.1530/ERC-15-0205) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knudson AG., Jr. Mutation and cancer: statistical study of retinoblastoma. PNAS 1971. 68 820–823. ( 10.1073/pnas.68.4.820) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramalho-Carvalho J, Pires M, Lisboa S, Graça I, Rocha P, Barros-Silva JD, Savva-Bordalo J, Maurício J, Resende M, Teixeira MR, et al. Altered expression of MGMT in high-grade gliomas results from the combined effect of epigenetic and genetic aberrations. PLoS ONE 2013. 8 e58206 ( 10.1371/journal.pone.0058206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luzi E, Marini F, Giusti F, Galli G, Cavalli L, Brandi ML. The negative feedback-loop between the oncomir Mir-24-1 and menin modulates the Men1 tumorigenesis by mimicking the ‘Knudson’s second hit’. PLoS ONE 2012. 7 e39767 ( 10.1371/journal.pone.0039767) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iwata T, Yamada S, Mizusawa N, Golam HM, Sano T, Yoshimoto K. The aryl hydrocarbon receptor-interacting protein gene is rarely mutated in sporadic GH-secreting adenomas. Clinical Endocrinology 2007. 66 499–502. [DOI] [PubMed] [Google Scholar]
- 40.Daly AF, Vanbellinghen JF, Khoo SK, Jaffrain-Rea ML, Naves LA, Guitelman MA, Murat A, Emy P, Gimenez-Roqueplo AP, Tamburrano G, et al. Aryl hydrocarbon receptor-interacting protein gene mutations in familial isolated pituitary adenomas: analysis in 73 families. Journal of Clinical Endocrinology and Metabolism 2007. 92 1891–1896. ( 10.1210/jc.2006-2513) [DOI] [PubMed] [Google Scholar]
- 41.Cazabat L, Libè R, Perlemoine K, René-Corail F, Burnichon N, Gimenez-Roqueplo AP, Dupasquier-Fediaevsky L, Bertagna X, Clauser E, Chanson P, et al. Germline inactivating mutations of the aryl hydrocarbon receptor-interacting protein gene in a large cohort of sporadic acromegaly: mutations are found in a subset of young patients with macroadenomas. European Journal of Endocrinology 2007. 157 1–8. ( 10.1530/EJE-07-0181) [DOI] [PubMed] [Google Scholar]
- 42.Zatelli MC, Torre ML, Rossi R, Ragonese M, Trimarchi F, degli Uberti E, Cannavò S. Should aip gene screening be recommended in family members of FIPA patients with R16H variant? Pituitary 2013. 16 238–244. ( 10.1007/s11102-012-0409-5) [DOI] [PubMed] [Google Scholar]
- 43.Ozfirat Z, Korbonits M. AIP gene and familial isolated pituitary adenomas. Molecular and Cellular Endocrinology 2010. 326 71–79. ( 10.1016/j.mce.2010.05.001) [DOI] [PubMed] [Google Scholar]
- 44.Jaffrain-Rea ML, Angelini M, Gargano D, Tichomirowa MA, Daly AF, Vanbellinghen JF, D’Innocenzo E, Barlier A, Giangaspero F, Esposito V, et al. Expression of aryl hydrocarbon receptor (AHR) and AHR-interacting protein in pituitary adenomas: pathological and clinical implications. Endocrine-Related Cancer 2009. 16 1029–1043. ( 10.1677/ERC-09-0094) [DOI] [PubMed] [Google Scholar]
- 45.Trivellin G, Butz H, Delhove J, Igreja S, Chahal HS, Zivkovic V, McKay T, Patócs A, Grossman AB, Korbonits M. MicroRNA miR-107 is overexpressed in pituitary adenomas and inhibits the expression of aryl hydrocarbon receptor-interacting protein in vitro. American Journal of Physiology: Endocrinology and Metabolism 2012. 303 E708–E719. ( 10.1152/ajpendo.00546.2011) [DOI] [PubMed] [Google Scholar]
- 46.Daly AF, Tichomirowa MA, Petrossians P, Heliövaara E, Jaffrain-Rea ML, Barlier A, Naves LA, Ebeling T, Karhu A, Raappana A, et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. Journal of Clinical Endocrinology and Metabolism 2010. 95 E373–E383. ( 10.1210/jc.2009-2556) [DOI] [PubMed] [Google Scholar]
- 47.Occhi G, Trivellin G, Ceccato F, De Lazzari P, Giorgi G, Demattè S, Grimaldi F, Castello R, Davì MV, Arnaldi G, et al. Prevalence of AIP mutations in a large series of sporadic Italian acromegalic patients and evaluation of CDKN1B status in acromegalic patients with multiple endocrine neoplasia. European Journal of Endocrinology 2010. 163 369–376. ( 10.1530/EJE-10-0327) [DOI] [PubMed] [Google Scholar]
- 48.Pinho LK, Vieira Neto L, Wildemberg LE, Moraes AB, Takiya CM, Frohman LA, Korbonits M, Gadelha MR. Familial isolated pituitary adenomas experience at a single center: clinical importance of AIP mutation screening. Arquivos Brasileiros de Endocrinologia e Metabologia 2010. 54 698–704. ( 10.1590/S0004-27302010000800006) [DOI] [PubMed] [Google Scholar]
- 49.Cai F, Zhang YD, Zhao X, Yang YK, Ma SH, Dai CX, Liu XH, Yao Y, Feng M, Wei JJ, et al. Screening for AIP gene mutations in a Han Chinese pituitary adenoma cohort followed by LOH analysis. European Journal of Endocrinology 2013. 169 867–884. ( 10.1530/EJE-13-0442) [DOI] [PubMed] [Google Scholar]
- 50.Oriola J, Lucas T, Halperin I, Mora M, Perales MJ, Alvarez-Escolá C, Paz de MN, Díaz Soto G, Salinas I, Julián MT, et al. Germline mutations of AIP gene in somatotropinomas resistant to somatostatin analogues. European Journal of Endocrinology 2013. 168 9–13. ( 10.1530/EJE-12-0457) [DOI] [PubMed] [Google Scholar]
- 51.Georgitsi M, Heliövaara E, Paschke R, Kumar AV, Tischkowitz M, Vierimaa O, Salmela P, Sane T, De Menis E, Cannavò S, et al. Large genomic deletions in AIP in pituitary adenoma predisposition. Journal of Clinical Endocrinology and Metabolism 2008. 93 4146–4151. ( 10.1210/jc.2008-1003) [DOI] [PubMed] [Google Scholar]