

Abstract
The MEK1 kinase directly phosphorylates ERK2, after the activation loop of MEK1 is itself phosphorylated by Raf. Studies over the past decade have revealed a large number of disease-related mutations in the MEK1 gene that lead to tumorigenesis and abnormal development. Several of these mutations result in MEK1 constitutive activity, but how they affect MEK1 regulation and function remains largely unknown. Here, we address these questions focusing on two pathogenic variants of the Phe-53 residue, which maps to the well-characterized negative regulatory region of MEK1. We found that these variants are phosphorylated by Raf faster than the wild-type enzyme, and this phosphorylation further increases their enzymatic activity. However, the maximal activities of fully phosphorylated wild-type and mutant enzymes are indistinguishable. On the basis of available structural information, we propose that the activating substitutions destabilize the inactive conformation of MEK1, resulting in its constitutive activity and making it more prone to Raf-mediated phosphorylation. Experiments in zebrafish revealed that the effects of activating variants on embryonic development reflect the joint control of the negative regulatory region and activating phosphorylation. Our results underscore the complexity of the effects of activating mutations on signaling systems, even at the level of a single protein.
Keywords: cancer biology, mitogen-activated protein kinase (MAPK), protein phosphorylation, Raf kinase, zebrafish, MEK1
Introduction
The Ras pathway is a highly conserved series of protein–protein interactions that is activated by extracellular signals binding to receptors at the cell surface, and it results in the activation of ERK (1, 2). Mutations in the components of the Ras pathway are found in both developmental abnormalities and cancers (3). The discovery of mutations proceeds at an ever increasing pace, motivating mechanistic studies of the emerging phenotypes at multiple levels of biological organization, from molecules to the organism (4). One of the challenges in achieving the understanding of how phenotypes emerge is that the affected components are involved in multiple interactions both upstream and downstream, each of which can be affected by the same mutation.
MEK1 is a core component of the pathway, playing a key role in transmitting signals from active Ras to ERK (5). Wild-type MEK1 (WT–MEK1) is activated when it is dually phosphorylated by an upstream kinase (e.g. Raf) on two specific serine residues within its activation sequence. Active MEK1 dually phosphorylates ERK2 on specific tyrosine and threonine residues within the activation loop. Both of these double-phosphorylation reactions are ordered, which is important for both the chemical aspects of catalysis and for systems-level properties of the signaling cascade (6–9). In addition to regulation by the activation loop, a region in the N terminus, helix A, has been demonstrated to negatively regulate MEK1 activity. Either deletion of the N terminus or phosphomimetic substitutions within the activation loop result in partial activation of MEK1 (10, 11). Activity is further enhanced when both of these modifications are combined, implying that the N terminus and the activation loop work synergistically to activate MEK1 (10, 11). Indeed, the crystal structure of MEK1 demonstrated that helix AL stabilizes the inactive configuration of the N-terminal domain and helix C of the kinase (Fig. 1, A and B) (12). The emerging picture is that the equilibrium between the low- and high-activity states of MEK1 can be shifted toward the active form by either phosphorylation of the activation loop or perturbations to the N terminus.
Figure 1.

Activating mutations affect thermal stability of MEK1 and its activity toward ERK2. A, location of position 53 in helix A from the structure of MEK1 in PDB code 5bx0 is indicated by a red dot. Shown are helix A (magenta), helix C (green), and helix AL (red), an inhibitory helix in the activation loop. B, top, close connection between helix A, helix C, and helix AL is shown in inactive MEK1. MEK1 is rotated 180° from A. Side chains involved in ionic interactions along helix C are shown. Phe-53 stabilizes these interactions through Lys-57. Glu-114, a catalytic residue, interacts with helix AL. Bottom, model of the three helices in the active configuration of MEK1 based on the structure of active PKA (PDB code 1ATP). Helix AL is refolded, building the active site. Glu-114 points back, adopting interactions in the active site (red dot, Glu-114). C, model for active MEK1, the whole protein based on PKA (PDB code 1ATP). Helix A adopts different interactions with helix C. D, thermal stability of wild-type MEK1 (black), F53L (red), and F53S (green) and ΔΝ60 (eliminating helix A) (gray) as visualized with SYPRO Orange dye. E and F, MEK1/F53L and MEK1/F53S are intrinsically active and, upon phosphorylation, become as active as phosphorylated wild-type MEK1. The normalized dpERK level is an average of four experimental replicates in E and F. Error bars, S.E.
Dozens of disease-related mutations in MEK1 have already been found in somatic tumors and developmental abnormalities, and many more are likely to be discovered by the sequencing of affected individuals and large-scale mutagenesis screens, but it is still unclear how these mutations affect regulation and function at the protein level (13, 14). Importantly, several of the disease-related mutations map to helix A or the adjacent parts of the structure, suggesting that they destabilize the inactive conformation and work in a manner similar to the N-terminal deletions (12). Here, we examine the effects of mutations of the commonly mutated Phe-53 residue of MEK1. Phe-53 forms part of helix A and as such might be predicted to contribute toward maintaining MEK1 in the inactive conformation. We focus on the MEK1/F53L and MEK1/F53S mutants, which are implicated in melanoma and the cardio-facio-cutaneous syndrome, respectively. Using mass spectrometry and in vitro assays, we examined the kinetics of Raf-mediated phosphorylation of MEK1 and MEK1 enzymatic activity toward ERK2. Our results reveal that activating mutations destabilize the inactive conformation of MEK1. They also make MEK1 more prone to Raf-mediated phosphorylation. Moreover, using zebrafish, we demonstrate an in vivo requirement for both the disease-associated activating mutation and the signal-dependent phosphorylation.
Results
Similar to the N-terminal deletion mutants, the MEK1/F53L and MEK1/F53S mutants are active in the absence of phosphorylation by Raf (15, 16). Importantly, their constitutive activity is not accompanied by phosphorylation of the activation loop of MEK1, as observed by mass spectrometry (supplemental Fig. S1). Fig. 1B (top) suggests the reason for constitutive activity caused by Phe-53 variants as follows: Phe-53 in helix A directly stabilizes helix C in the inactive structure of WT–MEK1, and the inactive configuration of helix C is further stabilized by helix AL, which houses the activating phosphorylation sites. A model for the active configuration is available from other protein kinases, and the expected rearrangement of the helices is shown in Fig. 1, B, bottom, and C. Apparently, the Phe-53 missense mutations in helix A destabilize inactive MEK1 and lead to its increased activity, even in the absence of phosphorylation within the activation loop. Consistent with the idea that the F53L and F53S mutations are destabilizing, we found that their thermal stability is reduced, to an extent that is even greater than that caused by the deletion of 60 N-terminal residues (Fig. 1D).
Based on the fact that phosphomimetic mutants act synergistically with the N-terminal deletion mutants to enhance activity (10), we anticipated that constitutive activity of point mutants should be further increased by their phosphorylation. Indeed, using in vitro assays, we found that phosphorylation of MEK1 by Raf leads to a significant increase of kinase activity toward ERK2. Notably, even though the activities of the unphosphorylated wild-type and mutant MEK1 proteins are clearly different, they become indistinguishable after phosphorylation by Raf, at least on the time scale of our experiments (Fig. 1, E and F). These results are consistent with the model in which the MEK1 enzyme exists in an equilibrium between active and inactive states. Apparently, the equilibrium is fully shifted toward the active form when MEK1 is phosphorylated by Raf, independently of mutations within the negative regulatory region. A tentative model of how MEK1 rearranges from the inactive to the active form can be obtained with an assumption that it resembles PKA in its active state. This model shows how the D-motif in the N terminus of active MEK1 can fit into the D-docking site of ERK2 (supplemental Fig. S2) (17, 18). Once in the active conformation, the activities of the wild-type and mutant forms of the MEK1 kinase become indistinguishable. Consistent with this model, we found using mass spectrometry that the mechanism of ERK2 phosphorylation by the MEK1 mutants is the same as that for the dually-phosphorylated WT–MEK1, i.e. ordered, with the tyrosine phosphorylated before threonine (data not shown).
The crystal structure of the MEK1–B-Raf complex reveals that the inactive conformation of helix AL is maintained even in the complex and must melt in order for the active site of B-Raf to gain access to the phosphorylation sites within the activation site of MEK1 (Fig. 2A) (19). Based on this, we reasoned that if helix AL is already destabilized in the MEK1 variants, they should be better substrates for active Raf. Accordingly, we found that the MEK1/F53S and MEK1/F53L variants are phosphorylated faster than the WT–MEK1 protein for two different MEK1 concentrations (Fig. 2, B and C, and supplemental Fig. S3). In all of these cases, the mechanism of MEK1 phosphorylation by Raf follows the strictly ordered mechanism established for the wild-type protein (supplemental Fig. S4). Thus, mutations in MEK1 result in its constitutive activity and at the same time affect the rate with which the MEK1 variants are phosphorylated by Raf.
Figure 2.
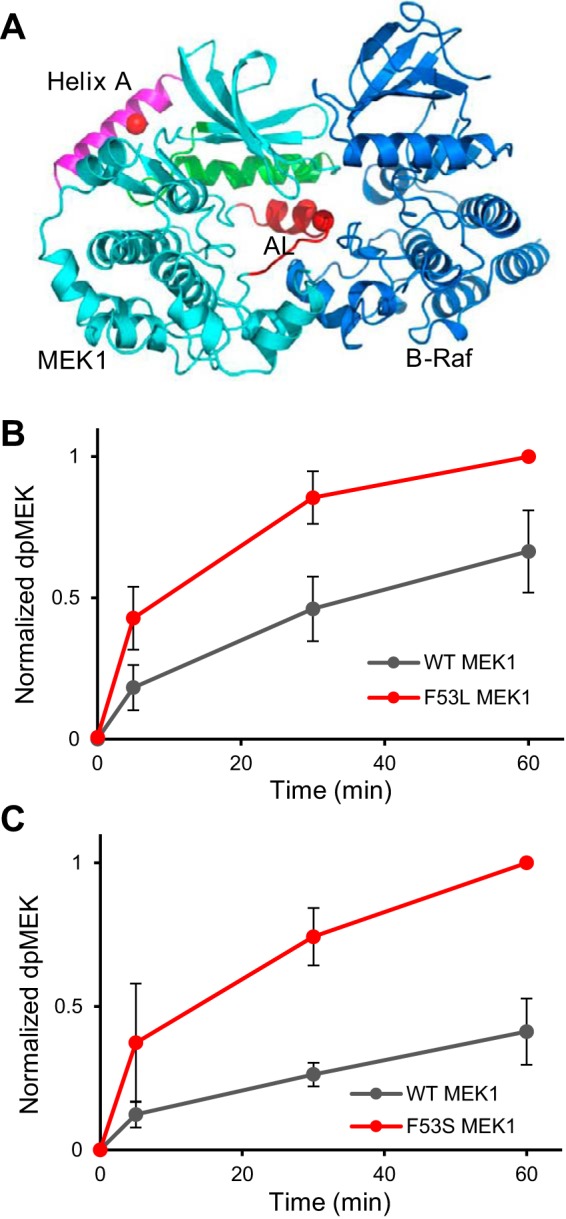
Activating mutations enhance the rate of MEK1 phosphorylation by Raf. A, complex between B-Raf and MEK1 is based on a structure (PDB code 4mne), with helix A modeled from PDB code 5bx0 (a more complete structure of MEK1). B and C, phosphorylation of MEK1/F53L and MEK1/F53S occurs faster than phosphorylation of wild-type MEK1. The normalized dpMEK is an average of three experimental replicates. The reaction was run at a Raf/MEK ratio of 1:∼217. Error bars, S.E.
What are the differential contributions of mutations within the negative regulatory region and phosphorylation of the active site for MEK1 activity in vivo? This question can be addressed in the early zebrafish embryo, which provides a convenient model for quantifying the effects of activating mutations in the Ras pathway components (20, 21). Earlier work established that injection of mRNA encoding MEK1 variants into the one-cell-stage embryos leads to an oval shape of the embryo at 11 h post-fertilization, resulting from defects in convergence and extension cell movements during gastrulation (21–24). The aspect ratio of the yolk can be used to compare the severities of different mutations (supplemental Fig. S6A) (21, 25).
To assess the relative contributions of upstream activation and mutations within helix A, we examined embryos injected with mRNA encoding either a Phe-53 variant or the variant that combines the Phe-53 mutation and the double-alanine substitution of the two serines within the activation loop. The unphosphorylatable variant should be capable of only constitutive activity, whereas the activity of the Phe-53 variant should be further increased by phosphorylation.
As expected, we found that the MEK1/F53L and MEK1/F53L/S218A/S222A variants are indistinguishable in their ability to phosphorylate ERK2 in vitro (Fig. 3A, and supplemental Fig. S5). However, their effects in vivo are remarkably different. The MEK1/F53L and MEK1/F53S variants lead to severe oval embryo phenotypes, whereas the aspect ratios of embryos injected with the MEK1/F53L/S218A/S222A and MEK1/F53S/S218A/S222A variants are indistinguishable from the uninjected embryos (Fig. 3, B and C, and supplemental Fig. S6). Apparently, constitutive activity of the F53L variant is insufficient for causing the oval embryo phenotype.
Figure 3.
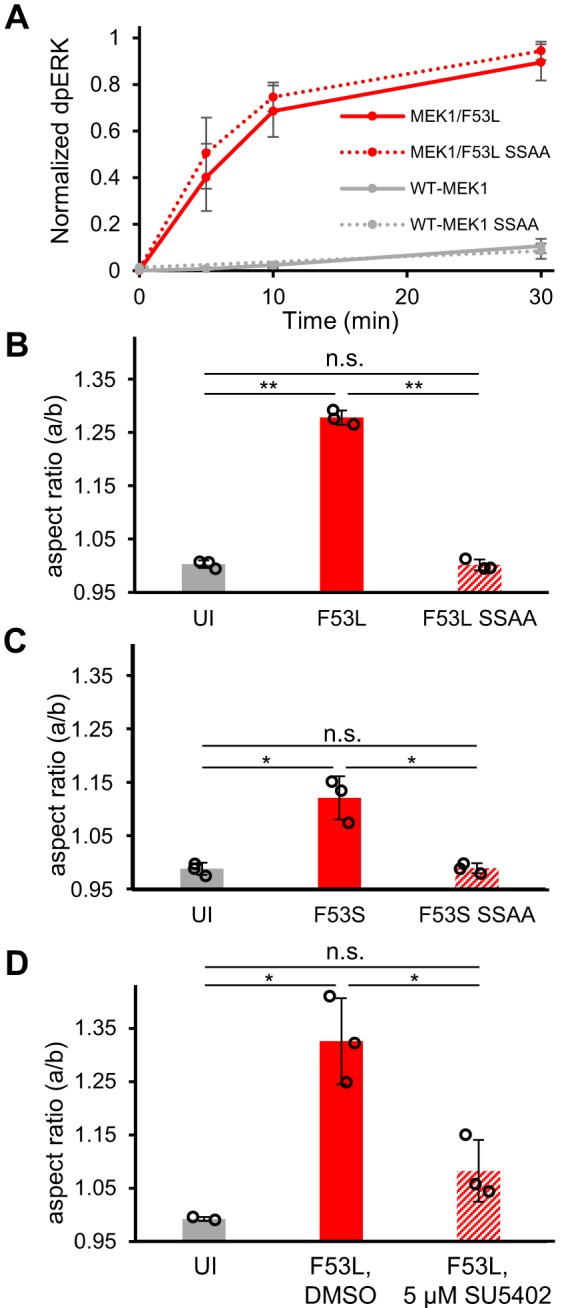
Joint control of MEK1 by mutations and phosphorylation. A, MEK1/F53L and MEK1/F53L/S218A/S222A are indistinguishable in their ability to phosphorylate ERK2 in vitro. The normalized dpERK is an average of three experimental replicates. Error bars, S.E. B, MEK1/F53L variant leads to a severe oval embryo phenotype, whereas the aspect ratio of embryos injected with the MEK1/F53L/S218A/S222A variant is indistinguishable from the uninjected embryos. NUI = 103, NF53L = 100, NF53L SSAA = 101 (three experimental replicates). C, MEK1/F53S variant leads to a milder oval embryo phenotype, whereas the aspect ratio of embryos injected with the MEK1/F53S/S218A/S222A variant is indistinguishable from the uninjected embryos. NUI = 61, NF53S = 83, NF53S SSAA = 80 (three experimental replicates). D, inhibition of FGFR signaling dramatically reduces elongation of the embryo. NUI = 37 (two experimental replicates), NF53L, DMSO = 56 (three experimental replicates), NF53L, 5 μm SU5402 = 56 (three experimental replicates). B–D, S218A/S222A is abbreviated as SSAA in the figure. B–D, pairwise Student's t tests (two-sided, homoscedastic) were performed to compare groups: **, p < 0.0005; *, p < 0.05, n.s., not significant. B–D, Error bars, S.D.
As a further test of the idea that the oval embryo phenotype induced by the F53L variant depends on its upstream phosphorylation, we tested whether this phenotype may be counteracted by inhibiting upstream signaling. This can be accomplished using an inhibitor of the kinase activity of the fibroblast growth factor receptor 1 (FGFR1), which acts upstream of MEK1 in the early embryo and other stages of zebrafish development (21, 26, 27). If the normalizing effect of the unphosphorylatable F53L variant reflects its inability to respond to upstream signals, it should be mimicked by inhibiting FGFR6 signaling. Consistent with this prediction, we found that pharmacological inhibition of FGFR signaling in embryos injected with the F53L variant results in a dramatic reduction of the oval embryo phenotype (Fig. 3D). This demonstrates that in vivo, the effects of activating mutations in MEK1 reflect its joint control by the negative regulatory region and phosphorylation within the activation loop.
Discussion
Taken together with previous structural and functional studies, our results can be summarized using the following analogy: the WT–MEK1 can be seen as a faucet with two settings: “off,” corresponding to the unphosphorylated state, and “on,” corresponding to the active dually-phosphorylated state. Activating mutations destabilize the off state, favoring the active state, making the faucet “drip.” At the same time, the faucet becomes easier to “open”, which corresponds to the increased rate within which these variants are phosphorylated by Raf. Once the faucet is on, the flow rate is the same as for the properly functioning unit, corresponding to the indistinguishable activities of the dually phosphorylated wild-type and mutant forms. The activating mutations simultaneously cause MEK1 to drip and make it “easier to open.” Both of these effects are consequences of the destabilization of helix AL.
Our model emphasizes the close linkage between helix A and helix AL through helix C. This model suggests that Lys-57 should also be activating, and indeed Lys-57 mutants have appeared in melanoma (28). Future tests of this model require structural studies of the various mutant MEK1–ERK2 complexes and a more resolved analysis of the effects of activating mutations on the molecular dynamics of the MEK1 protein.
Overall, we find that the mechanisms of MEK1 activation by Raf and ERK2 activation by MEK1 are not affected by the activating mutations. Both of these reactions proceed through the same steps, but with increased rates. This scenario is certainly not the only one possible; one can imagine that a mutant enzyme could be activated (or act on its substrates) following a completely different mechanism. For instance, the ordered nature of dual phosphorylation can be disrupted and can become random or even reversed. Interestingly enough, we found evidence of a change in the mechanism of MEK1 phosphorylation by MEKK1 using mass spectrometry; the order of phosphorylations was completely reversed between the wild-type and mutant proteins (supplemental Fig. S7). Although the functional significance of this change is unclear, it underscores the complexity of the effects of activating mutations on the dynamics of signaling networks, already at the level of a single protein.
Going forward, it will be important to investigate whether other activating mutations in MEK1 fall in the same category as the F53L and F53S mutants analyzed in this paper: whether they go through the same mechanism with altered rates or follow a completely different mechanism. These studies may reveal new mechanisms associated with activating mutations and explain the quantitative differences in the severities of the effects caused by different activating mutations in the organism (25). This information is critical for providing rational guidelines for the personalized treatments of diseases associated with the activating mutations in MEK1 and may provide a framework for the mechanistic analysis of mutations in other components of the Ras pathway.
Materials and methods
Plasmid construction, protein expression, and protein purification for in vitro reactions
Plasmids encoding N-terminally His6-tagged ERK and N-terminally His6-tagged MEK mutants were constructed as described previously (15). To express the MEK variants, pET28 plasmids encoding different mutations were transformed into BL21(DE3)-competent Escherichia coli. 5-ml overnight cultures were grown in LB medium supplemented with a 1:1000 dilution of 50 mg/ml kanamycin overnight. The culture was then split into three 1-liter portions of Terrific Broth medium with a 1:1000 dilution of 50 mg/ml kanamycin and grown at 37 °C with agitation at 220 rpm until an A600 of 0.9–1.1 was reached. Protein expression was induced by adding isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mm, and cultures were grown overnight at 25 °C with agitation at 180 rpm. Cells were collected by centrifugation, and the pellets were stored at −20 °C.
To express ERK2, the pQE80 plasmid with His6-tagged ERK2 was transformed into BL21(DE3)-competent E. coli. The overnight culture was sub-cultured into 1 liter of LB medium with 100 μg/ml ampicillin to a starting A600 of 0.02, and cultures were grown at 37 °C with agitation at 250 rpm until an A600 of 1.0 was reached. Then, protein expression was induced with 1 mm IPTG, and the cultures were grown at 22 °C with agitation at 250 rpm for 6 h. After centrifugation, the cell pellets were stored at −20 °C.
To purify the protein of interest, the cell pellets were resuspended in 40 ml of a solution of 10 mm imidazole, 300 mm NaCl, 50 mm NaH2PO4, pH 8.0, lysed with lysozyme, and sonicated while on ice. Cell debris was removed by centrifugation, and the supernatant was filtered through a 45-μm sterile filter. The lysate was then further purified with nickel-nitrilotriacetic acid-agarose resin (Qiagen) following the manufacturer's instructions, and the elution underwent buffer exchange into a solution made with 50 mm HEPES, 100 mm NaCl, 20 mm MgCl2, 10% glycerol, pH 7.4, using PD-10 desalting columns (Bio-Rad). The protein was concentrated using the Vivaspin 20, 30-kDa concentrator (Sartorius), until a final concentration of 0.5 mm for MEK variants and a concentration of 1.5 mm for ERK2. Aliquots of 25–50 μl were snap-frozen in liquid nitrogen and stored in −80 °C until use.
In vitro phosphorylation reactions
The Raf–MEK phosphorylation reactions were carried out in phosphorylation buffer composed of 50 mm HEPES, 100 mm NaCl, 20 mm MgCl2, 10% glycerol, pH 8.0, in a 30 °C water bath. For Fig. 2, B and C, the reaction is initiated by the addition of 3.25 μm MEK to a master mix containing the phosphorylation buffer, 1 mm ATP, and 0.015 μm Raf (c-Raf 14-325-D, Millipore) and run for 30 min. For supplemental Fig. S3, the reaction was initiated by the addition of 2.5 μm MEK to a master mix containing the phosphorylation buffer, 1 mm ATP, and 0.05 μm Raf (c-Raf 14-325-D, Millipore) and run for 90 min. The reaction was quenched by diluting 4-fold in 8 m urea. Similarly, in Fig. 1, E and F, MEK–ERK and ppMEK–ERK reactions were performed in the same conditions but contained 1 μm MEK/ppMEK and 10 μm ERK and were run for 60 min.
For Fig. 3 and supplemental S5, MEK–ERK phosphorylation reactions were performed in kinase buffer containing 25 mm HEPES, 100 mm NaCl, 20 mm MgCl2, pH 8.0, in a 30 °C water bath. The reaction was initiated by addition of a 1 mm final concentration of ATP to a master mix containing 2 μm MEK and 10 μm ERK. The reaction was quenched by diluting 4-fold in 8 m urea.
Immunoblotting
4× SDS loading buffer was added to the reaction samples and then immunoblotted to detect the phosphorylated forms and silver-stained to detect total proteins. Immunoblotting was performed using standard techniques onto LF PVDF membrane. Primary anti-dpMEK antibody (Cell Signaling Technology, 9121S) and anti-ERK T*Y* antibody (Cell Signaling Technology, 4370S) were used to track the formation of MEK S*S* and ERK T*Y*, respectively. Alexa Fluor conjugates (1:2000 dilution, Invitrogen) were used as the secondary antibodies. The presence of total MEK and total ERK was tracked using standard silver-staining techniques and developed to the desired exposure. Membranes and gels were imaged using the Bio-Rad ChemiDoc MP Imaging System.
Differential scanning fluorimetry analysis of thermal denaturation
MEK proteins (5 μm) were incubated with 10 mm Tris, pH 8.0, SYPRO® Orange (diluted 5000× from Thermo Fisher Scientific reagent) in 20-μl reaction volumes loaded onto a 96-well clear-bottom plate. The plate was centrifuged at 800 × g to remove bubbles. The temperature in each well was increased from 4 to 80 °C using 0.5 °C increments in a Bio-Rad CFX96 real-time PCR machine. The fluorescence intensity of SYPRO Orange was probed using the fluorescein amidite channel of the PCR instrument. The denaturation temperature (Tm) was taken at the point of greatest dRFU/dT during differential scanning fluorimetry, where RFU is relative fluorescence units (29).
Protein digestion and HPLC peptide separation
MEK1 proteins were derivatized with iodoacetamide (Sigma A3221) for 1 h at 30 °C and then quenched with excess dithiothreitol as described by the manufacturer. Trypsin protease reaction mix (50 mm NH4HCO3, 50 mm Tris-HCl, pH 8.0, and sequencing grade trypsin (Promega)) was used to dilute out the urea to 1 m and digest substrate proteins. Proteolysis reactions were conducted at 80:1 to 50:1 protein/protease molar ratio. Activation loop peptides were resolved by RP-C18 chromatography using an Agilent 1100 series LC system (Agilent Technologies, Palo Alto, CA) and RP-C18 microbore HPLC column (Phenomenex Aeris Widepore 150 × 2.1 mm, 3-μm particle size, 200-Å pore diameter). Peptides were eluted using a water/acetonitrile gradient with 0.1% formic acid. MEK1 peptides of interest were eluted at ∼24–26% acetonitrile.
LC-MS/MS analysis
HPLC-MS/MS analysis was performed on an LCQ DECA XP ion-trap mass spectrometer (Thermo Finnigan, San Jose, CA) in which the HPLC was coupled in line to an orthogonal electrospray ionization source. Integration under ion traces corresponding to mass ranges for activation loop peptides was used to acquire raw MS detector responses using Xcalibur software (Thermo Finnigan). Responses for activation loop peptides were summed and then scaled to indicate percent abundance for each peptide species. Time courses following peptide phosphorylation were conducted either singly or as independent assays in triplicate. MS/MS spectra were acquired in a data-dependent mode (Thermo Finnigan). MASCOT software (Matrix Science Ltd., London, UK) (30) or Mass Matrix (31) was used for identifying peptides from their MS spectrum.
Zebrafish experiments
Mutant MEK1–mCherry plasmids were synthesized using site-directed mutagenesis on the original MEK1–mCherry construct (25). mRNA (55 pg) encoding human MEK1 tagged with mCherry with either the Phe-53 mutation or the Phe-53 mutation in combination with the S218A and S222A mutations was injected into the yolk at the one-cell stage. PWT (32) and Tg(myl:EGFP) adults were bred to generate embryos for mRNA injection and were subsequently screened for mCherry fluorescence to confirm injection success. Embryos were imaged on their side at 11 hpf, and the aspect ratio was calculated (length/width) using ImageJ as described previously (25). For the drug treatments, embryos collected from PWT and Tg(myl:memGFP) adults were injected with mRNA as above. Injected embryos between 4.5 and 5.5 hpf were exposed to 5 μm SU5402 (an inhibitor of the FGF receptor) in DMSO, or DMSO as a vehicle control, in E3 buffer. The 4.5–5.5 hpf time window was previously demonstrated to be critical for rescuing the oval phenotype using MEK inhibitor (25). Drug was washed off, and the embryos were returned to blue water until they were imaged at 11 hpf. Three separate clutches were injected in three separate sessions. The averages and standard deviations were based on the mean aspect ratios for each clutch. Established zebrafish protocols were adhered to in accordance with the Princeton University Institutional Animal Care and Use Committee.
Author contributions
Y. G., J. M. H., G. A. J., R. D. B., S. Y. S., and E. J. G. conceived and designed the project. Y. G., K. T., and E. Y. analyzed constitutive activity of the mutants and their activation by Raf. E. Y. analyzed the activity of the MEK1 variant combining activating mutations and alanine substitutions. G. A. J. and V. L. P. analyzed the effects of MEK1 variants in zebrafish as well as the sensitivity of these effects to drug treatments. J. M. H. collected the MS kinetics data. H. H. and Y. G. purified proteins. G. A. J., Y. G., S. Y. S., and E. J. G. wrote the manuscript with input from all authors.
Supplementary Material
Acknowledgments
We thank Phillip Johnson, Heather McAllister, and the LAR staff for zebrafish care. We thank Rony Seger for helpful discussions and Alan Futran for contributions during the early stages of this work.
This work was supported in part by National Institutes of Health Grant R01 GM086537 (to J. M. H., H. H., E. J. G., Y. G., K. T., V. L. P., E. Y., R. D. B., and S. Y. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S7.
- FGFR
- fibroblast growth factor receptor
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- hpf
- hours post-fertilization
- PDB
- Protein Data Bank.
References
- 1. Kolch W. (2000) Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 351, 289–305 [PMC free article] [PubMed] [Google Scholar]
- 2. Johnson H. E., Goyal Y., Pannucci N. L., Schüpbach T., Shvartsman S. Y., and Toettcher J. E. (2017) The spatiotemporal limits of developmental Erk signaling. Dev. Cell 40, 185–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rauen K. A. (2013) The RASopathies. Annu. Rev. Genomics Hum. Genet. 14, 355–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jindal G. A., Goyal Y., Burdine R. D., Rauen K. A., and Shvartsman S. Y. (2015) RASopathies: unraveling mechanisms with animal models. Dis. Model. Mech. 8, 769–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seger R., Ahn N. G., Posada J., Munar E. S., Jensen A. M., Cooper J. A., Cobb M. H., and Krebs E. G. (1992) Purification and characterization of mitogen-activated protein kinase. Activator(s) from epidermal growth factor-stimulated A431 cells. J. Biol. Chem. 267, 14373–14381 [PubMed] [Google Scholar]
- 6. Ferrell J. E. Jr., and Bhatt R. R. (1997) Mechanistic studies of the dual phosphorylation of mitogen-activated protein kinase. J. Biol. Chem. 272, 19008–19016 [DOI] [PubMed] [Google Scholar]
- 7. Piala A. T., Humphreys J. M., and Goldsmith E. J. (2014) MAP kinase modules: the excursion model and the steps that count. Biophys. J. 107, 2006–2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burack W. R., and Sturgill T. W. (1997) The activating dual phosphorylation of MAPK by MEK is nonprocessive. Biochemistry 36, 5929–5933 [DOI] [PubMed] [Google Scholar]
- 9. Resing K. A., Mansour S. J., Hermann A. S., Johnson R. S., Candia J. M., Fukasawa K., Vande Woude G. F., and Ahn N. G. (1995) Determination of v-Mos-catalyzed phosphorylation sites and autophosphorylation sites on MAP kinase kinase by ESI/MS. Biochemistry 34, 2610–2620 [DOI] [PubMed] [Google Scholar]
- 10. Mansour S. J., Candia J. M., Matsuura J. E., Manning M. C., and Ahn N. G. (1996) Interdependent domains controlling the enzymatic activity of mitogen-activated protein kinase kinase 1. Biochemistry 35, 15529–15536 [DOI] [PubMed] [Google Scholar]
- 11. Mansour S. J., Matten W. T., Hermann A. S., Candia J. M., Rong S., Fukasawa K., Vande Woude G. F., and Ahn N. G. (1994) Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 265, 966–970 [DOI] [PubMed] [Google Scholar]
- 12. Fischmann T. O., Smith C. K., Mayhood T. W., Myers J. E., Reichert P., Mannarino A., Carr D., Zhu H., Wong J., Yang R.-S., Le H. V., and Madison V. S. (2009) Crystal structures of MEK1 binary and ternary complexes with nucleotides and inhibitors. Biochemistry 48, 2661–2674 [DOI] [PubMed] [Google Scholar]
- 13. Bromberg-White J. L., Andersen N. J., and Duesbery N. S. (2012) MEK genomics in development and disease. Brief. Funct. Genomics 11, 300–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Couto J. A., Huang A. Y., Konczyk D. J., Goss J. A., Fishman S. J., Mulliken J. B., Warman M. L., and Greene A. K. (2017) Somatic MAP2K1 mutations are associated with extracranial arteriovenous malformation. Am. J. Hum. Genet. 100, 546–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goyal Y., Jindal G. A., Pelliccia J. L., Yamaya K., Yeung E., Futran A. S., Burdine R. D., Schüpbach T., and Shvartsman S. Y. (2017) Divergent effects of intrinsically active MEK variants on developmental Ras signaling. Nat. Genet. 49, 465–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goyal Y., Levario T. J., Mattingly H. H., Holmes S., Shvartsman S. Y., and Lu H. (2017) Parallel imaging of Drosophila embryos for quantitative analysis of genetic perturbations of the Ras pathway. Dis. Model. Mech. 10, 923–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tanoue T., Adachi M., Moriguchi T., and Nishida E. (2000) A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell Biol. 2, 110–116 [DOI] [PubMed] [Google Scholar]
- 18. Goldsmith E. J., Akella R., Min X., Zhou T., and Humphreys J. M. (2007) Substrate and docking interactions in serine/threonine protein kinases. Chem. Rev. 107, 5065–5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Haling J. R., Sudhamsu J., Yen I., Sideris S., Sandoval W., Phung W., Bravo B. J., Giannetti A. M., Peck A., Masselot A., Morales T., Smith D., Brandhuber B. J., Hymowitz S. G., and Malek S. (2014) Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell 26, 402–413 [DOI] [PubMed] [Google Scholar]
- 20. Jopling C., van Geemen D., and den Hertog J. (2007) Shp2 knockdown and Noonan/LEOPARD mutant Shp2-induced gastrulation defects. PLoS Genet. 3, e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Runtuwene V., van Eekelen M., Overvoorde J., Rehmann H., Yntema H. G., Nillesen W. M., van Haeringen A., van der Burgt I., Burgering B., and den Hertog J. (2011) Noonan syndrome gain-of-function mutations in NRAS cause zebrafish gastrulation defects. Dis. Model. Mech. 4, 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anastasaki C., Estep A. L., Marais R., Rauen K. A., and Patton E. E. (2009) Kinase-activating and kinase-impaired cardio-facio-cutaneous syndrome alleles have activity during zebrafish development and are sensitive to small molecule inhibitors. Hum. Mol. Genet. 18, 2543–2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Anastasaki C., Rauen K. A., and Patton E. E. (2012) Continual low-level MEK inhibition ameliorates cardio-facio-cutaneous phenotypes in zebrafish. Dis. Model. Mech. 5, 546–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fürthauer M., Thisse C., and Thisse B. (1997) A role for FGF-8 in the dorsoventral patterning of the zebrafish gastrula. Development 124, 4253–4264 [DOI] [PubMed] [Google Scholar]
- 25. Jindal G. A., Goyal Y., Yamaya K., Futran A. S., Kountouridis I., Balgobin C. A., Schüpbach T., Burdine R. D., and Shvartsman S. Y. (2017) In vivo severity ranking of Ras pathway mutations associated with developmental disorders. Proc. Natl. Acad. Sci. 114, 510–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krens S. F., He S., Lamers G. E., Meijer A. H., Bakkers J., Schmidt T., Spaink H. P., and Snaar-Jagalska B. E. (2008) Distinct functions for ERK1 and ERK2 in cell migration processes during zebrafish gastrulation. Dev. Biol. 319, 370–383 [DOI] [PubMed] [Google Scholar]
- 27. Mohammadi M., McMahon G., Sun L., Tang C., Hirth P., Yeh B. K., Hubbard S. R., and Schlessinger J. (1997) Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science 276, 955–960 [DOI] [PubMed] [Google Scholar]
- 28. Forbes S. A., Beare D., Gunasekaran P., Leung K., Bindal N., Boutselakis H., Ding M., Bamford S., Cole C., Ward S., Kok C. Y., Jia M., De T., Teague J. W., Stratton M. R., McDermott U., and Campbell P. J. (2015) COSMIC: Exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 43, D805–D811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pantoliano M. W., Petrella E. C., Kwasnoski J. D., Lobanov V. S., Myslik J., Graf E., Carver T., Asel E., Springer B. A., Lane P., and Salemme F. R. (2001) High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen. 6, 429–440 [DOI] [PubMed] [Google Scholar]
- 30. Perkins D. N., Pappin D. J., Creasy D. M., and Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 31. Xu H., and Freitas M. A. (2010) A dynamic noise level algorithm for spectral screening of peptide MS/MS spectra. BMC Bioinformatics 11, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schottenfeld J., Sullivan-Brown J., and Burdine R. D. (2007) Zebrafish curly up encodes a Pkd2 ortholog that restricts left-side-specific expression of southpaw. Development 134, 1605–1615 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.