

Abstract
The nuclear factor κB (NF-κB) transcription factor family regulates genes involved in cell proliferation and inflammation. The promoters of these genes often contain NF-κB-binding sites (κB sites) arranged in tandem. How NF-κB activates transcription through these multiple sites is incompletely understood. We report here an X-ray crystal structure of homodimers comprising the RelA DNA-binding domain containing the Rel homology region (RHR) in NF-κB bound to an E-selectin promoter fragment with tandem κB sites. This structure revealed that two dimers bind asymmetrically to the symmetrically arranged κB sites at which multiple cognate contacts between one dimer to the corresponding DNA are broken. Because simultaneous RelA-RHR dimer binding to tandem sites in solution was anti-cooperative, we inferred that asymmetric RelA-RHR binding with fewer contacts likely indicates a dissociative binding mode. We found that both κB sites are essential for reporter gene activation by full-length RelA homodimer, suggesting that dimers facilitate DNA binding to each other even though their stable co-occupation is not promoted. Promoter variants with altered spacing and orientation of tandem κB sites displayed unexpected reporter activities that were not explained by the solution-binding pattern of RelA-RHR. Remarkably, full-length RelA bound all DNAs with a weaker affinity and specificity. Moreover, the transactivation domain played a negative role in DNA binding. These observations suggest that other nuclear factors influence full-length RelA binding to DNA by neutralizing the transactivation domain negative effect. We propose that DNA binding by NF-κB dimers is highly complex and modulated by facilitated association–dissociation processes.
Keywords: cooperativity, crystal structure, dimerization, DNA transcription, NF-kB transcription factor, E-selectin, RelA, kB site
Introduction
The NF-κB family of dimeric transcription factors regulates the expression of a multitude of genes involved in cell proliferation, survival, and inflammation. NF-κB dimers are combinatorially formed from five subunits: p50, p52, RelA/p65, cRel, and RelB. The dimers regulate transcription by sequence-specific binding to DNA that is collectively known as the κB site. The 3D X-ray structures of several NF-κB–DNA complexes identified a common DNA recognition mode by the dimers. The G:C-rich flanking sequences of κB sites contact NF-κB with sequence specificity, whereas the central sequences are recognized with less sequence specificity (1, 2). General rules for the DNA-binding strategy adopted by these dimers are depicted in Fig. 1A. The broad conservation in DNA-binding chemistry by the dimers arises from high sequence homology in the N-terminal DNA-binding domain (DBD)4 of NF-κB proteins. This homologous DBD is commonly referred to as the Rel homology region (RHR). The RHR contains two folded immunoglobulin-like domains connected by a short linker, the N-terminal domain (NTD), and the dimerization domain (DD) (Fig. 1B). The flexible connection between of the two domains confers a unique DNA-binding property to the NF-κB dimers where one of the two NTDs in a dimer may not contact DNA with sequence specificity (2). This NTD still can make nonspecific DNA backbone contacts; thus most of the DNA-binding affinity is retained. This results in much broader DNA sequence spectrum for recognition with only half-site binding specificity than that would be possible if both half-sites were absolutely required for recognition (3–5).
Figure 1.

Two RelA homodimers bind tandem κB sites of E-selectin promoter asymmetrically. A, a cartoon depicting the general rules for the DNA binding by NF-κB dimers. The G:C flanking sequences of κB sites contact NF-κB with sequence specificity, whereas the central sequences are primarily recognized indirectly. B, a schematic representation of RelA. Residues of the full-length protein as well as different domains are indicated. PRO-rich, proline-rich domain; TA2, transactivation domain 2; TA1, transactivation domain 1. C, top panel, NF-κB consensus sequence showing the specific and variable nucleotides. Nucleotide positions are indicated below the DNA sequence. Middle panel, natural tandem E-selectin κB sites are shown. Bottom panel, HIV, VCAM-1, and germline 1γ tandem κB sites are shown. D, top panel, ribbon representation of two RelA homodimers bound to a 26-mer DNA sequence containing two κB sites in tandem in two orientations. Subunits A (in green) and B (in cyan) represent RelA homodimer 1, and subunits C (in magenta) and D (in yellow) represent RelA homodimer 2. Middle panel, DNA sequence used for structure determination. κB sites are underlined. The blue dot represents 2-fold symmetry. Bottom panel, corresponding DNA sequence in the native E-selectin promoter. E, top panels, a close-up view of dimer-dimer interaction in the complex where residues of subunit B (cyan) in RelA homodimer 1 and subunit C (magenta) in RelA homodimer 2 interact (left panel). Specific residues involved in this contact are shown. The right panel shows the corresponding region in subunit A (green) in RelA homodimer 1 and subunit D (yellow) in RelA homodimer 2. Bottom panel, the holocomplex is shown in an orientation to highlight the dimer–dimer interaction (boxed). F, top panel, nonspecific DNA contacts across all four subunits highlighting only two residues (Lys218 and Lys221) are shown (right panel). Orientations of the same two residues (Lys218 and Lys221) are shown in a RelA–DNA complex structure where one RelA homodimer is bound to a single κB site (left panel). Bottom panel, same orientation of the complex as top right but showing only the side chains of Lys218, Gln220, and Lys221 projected to DNA.
Sequence variations in the central region often provide differential transcriptional specificity by the NF-κB dimers. Even one nucleotide change can switch transcriptional program from gene activation to repression without altering DNA binding specificity by a dimer (6, 7). The precise mechanism of how one nucleotide change confers such a dramatic change in transcriptional output is still unclear. Taken together, these observations suggest that κB sites play at least two critical roles in transcriptional regulation: to select one or a subgroup of NF-κB dimers over other dimers at the level of binding specificity and to dictate different transcriptional outcome without changing binding specificity by a dimer.
Most of the NF-κB target genes contain multiple κB sites in their promoters (8, 9). These κB sites are arranged randomly with respect to orientation and spacing between them (Fig. 1C). Differences in their sequences at few positions guide them to recruit specific but overlapping set of dimers at a specific site (5, 10). Three well-characterized promoters containing multiple κB sites are E-selectin, HIV, and IP-10. E-selectin promoter contains three κB sites; two of them are inverted repeats with no spacing between these two sites, and the third κB site is located 12 bp downstream of the tandem sites (11). The HIV promoter contains two κB sites direct repeat sequences separated by 4 bp (12), whereas the IP-10 promoter contains two different κB sites separated by 43 bp (6). It is unclear, however, whether and how the two sites cooperate. The X-ray crystal structure of p50–RelA heterodimer bound to the tandem HIV κB sites is known (13). Both dimers interact with each κB site nearly identically. Although the structure predicts probable cooperation between the dimers to stably interact with DNA, biochemical experiments suggest that the p50–RelA heterodimers binds HIV κB sites anti-cooperatively. Thus the relationship between transcriptional activation by NF-κB dimers and its DNA binding to multiple sites on a promoter remains unresolved. It is thought that these sites act independently (14). This is in contrast with stable binding assembly of multiple transcription factors on promoters containing multiple binding sites. The current study investigates how RelA homodimer recognizes tandem κB sequences and how its binding to these DNA sequences affect transcription.
Results
Asymmetric binding of two RelA homodimers to symmetrically arranged tandem κB sites
To understand how two RelA homodimers bind closely spaced tandemly arranged κB sites, we chose to study the E-selectin promoter using X-ray crystallography. E-selectin is a cell adhesion molecule expressed in endothelial cells that plays an important role in inflammation (15). Initial crystallization experiments were performed using native DNA fragments containing the non-consensus invertedly oriented tandem κB sites and the RelA-RHR. However, these protein–DNA complexes did not produce suitable crystals for diffraction studies. A self-complementary 26-bp-long DNA containing tandem optimized κB sites at the center and a 5′ thymine overhang generated diffracting crystals as complex with RelA (Fig. 1D) (16). In this DNA, the native sequence and spacing between the two proximal half-sites was maintained to preserve the putative interactions between the two RelA monomers bound to the proximal half-sites. Crystals diffracted to 2.5 Å resolution, and the structure was solved using molecular replacement (Table 1). RelA homodimer 1 is indicated in green (subunit A) and cyan (subunit B), whereas RelA homodimer 2 is indicated in magenta (subunit C) and yellow (subunit D) throughout this paper where the subunits A and C interact with two proximal 5′-bp half-sites at the center, whereas subunits B and D recognize the flanking 4-bp half-sites (Fig. 1D). The binding of two dimers did not cause significant change in geometries of the DNA duplex. The 26-mer DNA is essentially B form, with an average helical rise of 3.35 Å, a helical twist of 34.0°, and a total bend of 7.6°. A large twist of 42.4° was observed in the center C/G that connects two 5-bp sites (CURVES+ version 1.31) (17). In contrast, the DNA conformation of the tandem HIV-κB experienced significant bending at the spacer sequence between the two κB sites as revealed by its structure bound to the p50–RelA heterodimer (supplemental Fig. S1, A–C) (13). As anticipated, two RelA homodimers directly interact, burying ∼1000 A2 of accessible surface. The structure revealed that the NTD of subunit A of RelA homodimer 1 contacts the DD of subunit D of RelA homodimer 2. Reciprocal contacts are mediated by subunits B and C. The residues involved in these interactions are Asp53, Thr54, Thr55, and Lys56 from NTD and Gln225, Arg236, Phe239, Ser240, Gln241, and Pro275 from DD (Fig. 1E). In addition, there is a hydrogen bond connecting two adjacent N-terminals in each side. Lys93 of subunit A forms a hydrogen bond with Asp151 of subunit D. Lys93 of subunit C contacts Asp80 of subunit B. Interestingly, the chemistry at this protein–protein interface does not appear to be complementary exemplified by the presence of non-polar groups in the polar environment and similar charged groups in close proximity. This suggests that interactions between the two dimers might not provide cooperativity for protein binding to DNA.
Table 1.
Data collection and refinement statistics
| Data collection | |
|---|---|
| X-ray source | APS 19ID |
| Wavelength (Å) | 0.97935 |
| Space group | P21 |
| Unit cell (Å) | |
| a | 88.91 |
| b | 117.8 |
| c | 70.81 |
| β | 91.2 |
| Resolution range (Å)a | 30.0–2.50 (2.54–2.50) |
| Rsym (%) | 9.1 (74.5) |
| Observations | 98,837 |
| Unique reflections | 50,721 |
| Completeness (%) | 99.6 (99.7) |
| <I/σ> | 13.1 (2.0) |
| Refinement | |
| Refinement program | CNS 1.3 |
| Number of reflections used in refinementb | 43,343 |
| Data completeness (%) (in resolution range) | ∼85.4 (29.63–2.50) |
| Rcryst (%) (in resolution range 2.54–2.50) | 21.0 (38.1) |
| Rfree (%) (in resolution range 2.54–2.50)c | 27.2 (46.7) |
| Total atoms | 10,183 |
| Protein atoms | 8710 |
| DNA atoms | 1102 |
| Average B, all atoms (Å2) | 81 |
| Root mean square deviation | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.12 |
| Ramachandran plot (%) | |
| Favored | 80 |
| Allowed | 20 |
| Disallowed | 0 |
a The data in parentheses are for highest resolution shell.
b Reflections with |Fo|σ < 1.0 rejected.
c Calculated against a cross-validation set of 5.0% of data selected at random prior to refinement.
The most striking feature of the complex is that the two RelA homodimers interact with DNA in an asymmetrical fashion despite the fact that both κB sites are identical and are related by a local 2-fold axis of symmetry (Fig. 1F). Subunit C of RelA homodimer 2 (magenta) positions away from the DNA interface, resulting in the disruption of most nonspecific protein-DNA contacts. Nine residues from each of the three subunits A, B, and D make similar phosphate backbone contacts. These residues are Lys122, Lys123, and Arg124 from NTD, Pro189 from linker, and Lys218, Gln220, Lys221, Arg246, and Gln247 from DD. These very same residues were also involved in the nonspecific contacts in the X-ray structures of most NF-κB–DNA complexes reported to date (1). In subunit C, these residues shift away from the DNA surface to distances ranging from 4.4 to 7.2 Å. The structure of the current complex does not explain why the contacts between subunit C and DNA are broken. It is possible that all four subunits may not be able to make all cognate contacts with DNA if the two proximal subunits are engaged in protein–protein interactions. It is also possible that when one RelA homodimer binds to a κB site, the conformation or the dynamics of the neighboring DNA is altered, affecting how the second RelA homodimer binds to DNA that cannot be captured by static X-ray structure. The suboptimal protein–protein contacts between the two dimers and broken contacts between DNA and subunit C suggest that two RelA homodimers may not form a thermodynamically stable complex with DNA.
Variations in “DNA sequence–specific” contacts by RelA
Different RelA–DNA structures previously solved by our group and others have identified seven specific residues on RelA that mediate its interaction with DNA (2). The four invariant residues, Arg33, Arg35, Tyr36, and Glu39 of RelA NTD, form identical base contacts in all four subunits of the RelA–DNA complex observed in the current structure that are identical to the previously determined structures (Fig. 2A). The other three invariant residues, Arg41 and Ser42 of loop L1 and Arg187 of the linker, often display DNA sequence-sensitive variations in binding (supplemental Fig. S2). In our structure, only Arg41 of subunits A and C directly contact the inner 5-bp κB half-sites. This residue donates two hydrogen bonds to O6 and N7 of the guanine at position 5. Arg187 on these subunits does not make direct contact with DNA. In the case of Arg187, it forms a hydrogen bond with main chain oxygen of Phe34. The other two subunits, B and D, bind to the outer 4-bp κB half-sites displaying different binding fashion. In subunit B, Arg41 donates hydrogen bonds to O6 and N7 of a non-consensus guanine at position 5 in the opposite DNA strand. Arg41 of subunit D contacts backbone phosphate of C+4. Arg187 forms a hydrogen bond with O4 of T+2 of both half-sites (Fig. 2A). Ser42 on any of the four RelA subunits, however, does not interact with the DNA sequence. A schematic representation of the interactions described above is shown in Fig. 2B.
Figure 2.
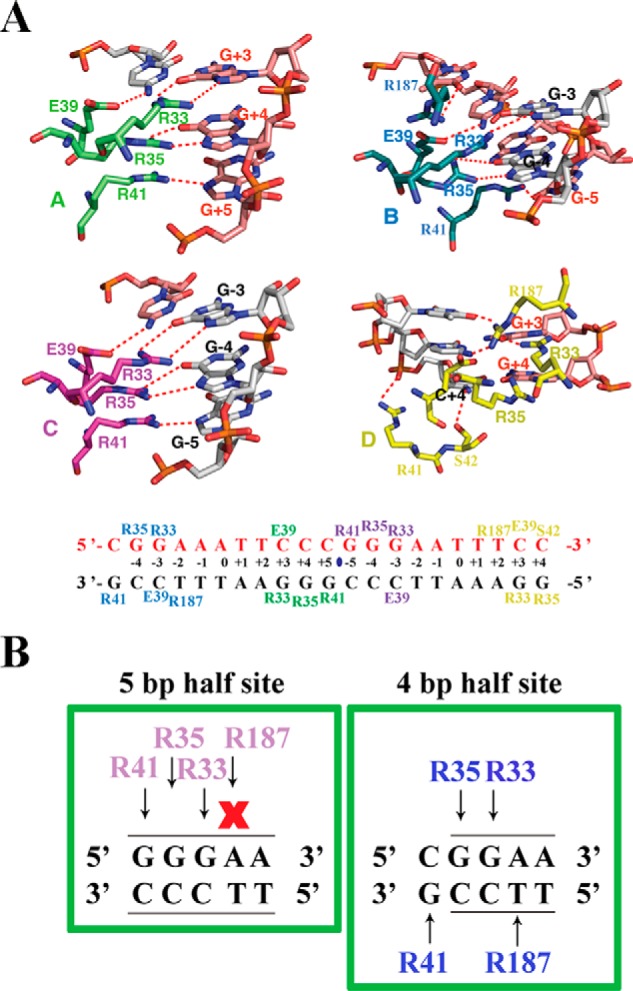
Arg187 of RelA binds 4- and 5-bp half-sites differently. A, top panel, close-up view of the amino acid residues of each subunit of the two RelA homodimers in contact with the 21-mer DNA sequence containing tandem κB sites. The dotted lines denote hydrogen bonds. Amino acid residues are shown in different colors according to the RelA subunit coloring in Fig. 1. DNA nucleotides participating in RelA–DNA contacts are also highlighted. Bottom panel, base-contacting residues are mapped on the DNA sequence in the complex. B, a cartoon highlighting how RelA interacts with half κB sites spanning 5 bp (left panel) or 4 bp (right panel), respectively. Amino acids involved in these contacts are colored in magenta and cyan, because these contacts correspond to subunit C and subunit B, respectively. Arrows indicate interaction, whereas X indicates no contact.
In general, these specific contacts confirm, as predicted previously, that RelA can bind both 5- and 4-bp κB half-sites. A single base pair switch at the first position from a G:C to non-G:C is responsible for this. In the case of the 5-bp GGGAN κB half-site, three Gs are contacted by Arg41, Arg35, and Arg33, and in the case of the 4-bp GGAN κB half-site, GGA is contacted by Arg35, Arg33, and Arg187 (T from the reverse strand). Our results reveal that if a G:C bp is present at the ±5 position, that will be specifically recognized by Arg41, and this contact alters the structure of the DNA in such a way that Arg187 cannot contact A:T bp at ±2 position. The result of their differential binding might explain the alteration of the overall conformation of the two half-complexes. The consequence of these conformational variations in gene expression has yet to be determined.
Tandemly arranged κB sites cooperate in activating transcription
To test whether the tandem arrangement of κB sites and protein–protein interaction between the two dimers observed in the current structure have any consequence in gene expression, we used luciferase reporter assay where the luciferase gene expression is driven by E-selectin promoter. As we mentioned above, the natural E-selectin promoter has three κB sites (site I, site II, and site III), where sites I and II are tandemly arranged. Initially, we tested the natural E-selectin promoter and a mutant version where the site III was deleted leaving behind the tandem sites (E-Sel I+II). Luciferase activity was measured 48 h after co-transfecting RelA and luciferase reporter into HeLa cells. Although the natural E-Sel I+II+III promoter showed the greatest activity, the E-Sel I+II promoter was transcriptionally active (Fig. 3A, see black bar corresponding to E-Sel I+II promoter). We next tested whether sites I and II synergize in activating RelA-mediated transcription. Mutation of either site, which are both weak-binding κB sites, reduced the reporter activity to near background levels, suggesting their functional cooperativity in cells (Fig. 3A, see gray and striped bars, respectively). These results also suggest that functional cooperativity might arise from direct interactions between the two dimers bound to two closely spaced κB sites.
Figure 3.

Tandem κB sites act synergistically to activate transcription by RelA. A, WT and variants of E-selectin promoter sequences used in luciferase reporter assay are shown. κB sites are boxed. Mutant κB sites are denoted by a large X, and specific base changes are shown in red. RelA homodimers effect was tested. Black, gray, and white boxes denote relative strengths of the sites with strong, intermediate, and weak binding affinities, respectively. Graphical representation of relative reporter activity is shown at the bottom of each panel. B, WT and variants of HIV-κB sequences used in luciferase reporter assay are shown at the top, and the relative luciferase is shown at the bottom. C, WT and IP-10-κB promoter variants used in luciferase reporter assay (top), and the relative luciferase is shown at the bottom. D, luciferase reporter assays performed using natural HIV sequence or mutants to test the importance of the spacing between κB sites. E, luciferase reporter assays performed using tandem κB sites on E-selectin or mutants to test the importance of the spacing between the κB sites and their relative orientation. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus control as indicated.
Two other NF-κB classical target promoters with tandemly arranged κB sites, HIV and IP-10, were analyzed in a similar manner. The HIV promoter has two near consensus identical κB sites for RelA homodimer separated by 4 bp. Mutation of one κB site decreased reporter activity more than 2-fold, suggesting that both κB sites functionally cooperate to activate transcription (Fig. 3B). Similar results were obtained when the IP-10 promoter was tested. This promoter contains two κB sites separated by 46 bp; one of them is a strong site for RelA homodimer binding, whereas the other is a weaker binding site. When both sites were individually mutated, reporter activity was greatly affected, and the effect was greater when the strong binding site was mutated. The non-additive nature of reporter activity suggests functional cooperation between the two sites physically separated by nearly 150 Å (Fig. 3C). This result suggests that two κB sites placed in tandem or separated by tens of bp can synergistically activate transcription but that synergy does not necessarily require physical interactions between the dimers bound to their respective sites.
We next evaluated whether changing the spacing between κB sites arranged in tandem could affect transcriptional outcome. We tested both HIV and E-selectin promoters. In the case of the HIV promoter, when the spacing was reduced by 1 bp from native 4 to 0 bp, we observed a significant decrease on RelA-dependent transcriptional activity in 1- and 0-bp spacing (Fig. 3D). These observations suggest that modification of the spacing between tandemly arranged κB sites prevents RelA homodimer binding, probably because of steric reasons. Reporter activity was also reduced when spacing between the two κB sites of E-selectin promoter was increased by 2 or 5 bp (Fig. 3E, see E-Sel I+II (2bp) or E-Sel I+II (5bp), respectively). We also analyzed the effect of swapping both κB sites on the E-selectin promoter, as well as increasing the spacing simultaneously of these already swapped κB sites (Fig. 3E, see E-Sel II+I SWAP or E-Sel II+I SWAP (2bp) or E-Sel II+I SWAP (5bp), respectively). Interestingly, whereas the E-Sel II+I SWAP promoter showed less transcriptional activity both the E-Sel II+I SWAP (2 bp) and E-Sel II+I SWAP (5 bp) promoters showed significantly enhanced transcriptional activity (Fig. 3E). According to our structural results shown in Fig. 1E, no direct protein–protein interaction between the two RelA homodimers is expected when the sites are separated by 5 bp both in E-Sel I+II (5bp) and E-Sel II+I SWAP (5bp). Thus direct dimer-dimer interactions cannot be responsible for this synergistic transcriptional activation observed in E-Sel II+I SWAP (5bp). These observations led us to propose that other proteins present endogenously might play a role that should be further addressed. Overall, our structural data, together with reporter assays, suggest that tandemly arranged κB sites in different RelA-dependent promoters cooperate to activate transcription. However, the cooperation is not unconditional but depends on their specific sequence, orientation, and spacing.
In vitro DNA-binding affinities of recombinant full-length or RelA-RHR fail to explain differential transcriptional outcomes
We wanted to test whether differential transcriptional outcomes of WT and mutant E-selectin promoters could be explained by different binding strengths of RelA homodimers for these promoters. We performed EMSA using RelA-RHR to determine their relative binding efficiency (Fig. 4A). Our results show that RelA-RHR binds the tandemly arranged κB sites (E-Sel I+II, E-Sel I+II (2bp), E-Sel II+I SWAP, and E-Sel II+I SWAP (2bp)) sequentially where binding of the first RelA dimer was detected at a concentration range between 0.12 and 3 nm to all probes. The second RelA-RHR was detected to bind DNA at concentrations between 15 and 60 nm to all the probes except for E-Sel II+I SWAP probe, in which the second κB site occupancy was detected at concentration around 500 nm. This observation suggests that the second RelA dimer binds the second site anti-cooperatively, while binding of the first dimer is inhibitory to the binding of the second dimer. More profound inhibition in the case of E-Sel II+I SWAP probe is perhaps due to strong steric repulsion (Fig. 4A). We also tested E-Sel I+II (5bp), E-Sel II+I SWAP (5bp), E-Sel I+II (mut), E-Sel I (mut)+II, and E-Sel I(mut)+II(mut) (supplemental Fig. S3A). No binding difference was observed between E-Sel I+II (5bp) or E-Sel II+I SWAP (5bp) probes. The last three probes tested showed the expected results: one site occupied when one κB single site was mutated and no binding when both κB sites were simultaneously mutated. These results suggest no correlation between in vitro DNA-binding affinity of RelA-RHR for κB sites and the cell-based reporter activity that used full-length RelA (Fig. 3). To examine whether full-length RelA differentially binds to the E-Sel promoters under study, we prepared full-length recombinant RelA from baculovirus-infected Sf9 cells (supplemental Fig. S3B). We tested the binding efficiency of full-length RelA for all probes discussed above using EMSA (Fig. 4B and supplemental Fig. S3C). Results of EMSA experiments led us to make two conclusions: first, full-length RelA binds poorly to all probes tested compared with the RelA-RHR (supplemental Fig. S3D), and second, in contrast to the sequential occupancy of both κB sites observed with RelA-RHR, two shifted complexes appeared simultaneously for full-length RelA (Fig. 4B and supplemental Fig. S3C).
Figure 4.

Activation domain of RelA inhibits DNA binding. A, binding of RelA-RHR (DBD) to different probes containing WT and mutant E-selectin κB sites. DNA sequences of the probes are shown in supplemental Table S2. Single arrowheads denote one RelA homodimer bound to the probe, whereas double arrowheads denote complex with two RelA homodimers. E-selectin I+II and E-selectin I+II (2bp) samples were run in one gel whereas E-selectin II+I SWAP and E-selectin II+I SWAP (2bp) were run in another gel. Panels were cropped and arranged according to the nomenclature used throughout the paper. B, binding of increasing concentrations of His-RelA full-length to different E-selectin probes. Specific DNA sequences tested are shown in supplemental Table S2. Single arrowheads indicate different RelA full-length–κB complexes formed. Asterisks indicate supershifted complexes detected by anti-His and anti-RelA specific antibodies. C, binding of RelA-RHR to the WT E-selectin DNA probe in presence of increasing concentrations of RelA AD.
To determine the specificity of the complexes, we performed antibody supershifts using anti-RelA and anti-His antibodies. The faster mobility complex was shifted in lesser degree than the slower mobility complex. It is possible that AD of one of the RelA subunits is degraded in the faster mobility complex (because the anti-RelA antibody used recognizes the RelA AD). However, this is unlikely because the protein appeared minimally degraded (supplemental Fig. S3B). Alternatively, the epitope recognized by the antibodies might be partly masked in the faster mobility complex. Strikingly, both shifted complexes appeared in probes where one site was mutated (supplemental Fig. S3C, see probes E-Sel I(mut)+II and E-Sel I+II(mut), respectively). It is likely that somehow recruitment of the homodimer at one site facilitates the recruitment of a second homodimer even when the second site is not a κB site. Mutation of both sites mostly abolished dimer binding (supplemental Fig. S3C, see probe E-Sel I(mut)+II(mut)), suggesting that the binding observed in probes containing only one consensus κB site is specific. In summary, our results suggest that recombinant full-length RelA poorly and indiscriminately binds tandem κB sites. We speculated whether that poor binding by full-length RelA might be due to the inhibitory effect of the AD (Fig. 1B). To test this, we assayed RelA-RHR binding to tandem E-selectin κB sites in the presence of increasing concentrations of RelA AD-TA (Fig. 4C). Our results showed that RelA-RHR was progressively released from the probe as the concentration of AD increased. Although it is not clear whether RelA AD competes with the DNA by directly binding to RelA-RHR or indirectly modulate DNA binding by RHR, the inhibitory effect of the AD in RelA DNA binding is clear in the purified in vitro system.
Discussion
The X-ray crystal structures of the NF-κB–DNA complexes known to date are mostly DNA sequences containing a single κB site bound to one dimer (2). Although these studies provided fundamental information on how different NF-κB dimers recognize κB sites, they failed to provide information on the relationship between the two dimers bound to tandem sites. This is an important question because a large number of NF-κB targets contain multiple κB sites (8, 9). Only the structure of tandem HIV-κB site bound to two p50–RelA heterodimers provide insights into the cooperativity between two dimers bound to nearby sites (13). Because differences in spacing, orientation, and sequence can result in numerous arrangements of dimers even with two κB sites, structural information from other multidimer–DNA complexes are required for better understanding of different assemblies that occur and their mechanism of formation. We chose E-selectin tandem κB sites as the model DNA for further study. The work presented here sheds new light on binding by two RelA homodimers to a specifically arranged tandem κB site and its effect on transcription. We show that two weak κB sites, which independently fail to induce transcription because they each bind RelA homodimer inefficiently, can drive transcription synergistically if placed in proper context. However, a random arrangement of the tandem sites is not optimal for transcription; their specific arrangement is important. Our study provides the first demonstration that not only the presence of multiple κB sites but also their arrangement could impact transcription, and these positional factors need to be considered while interpreting NF-κB–dependent transcriptional outcome.
Our effort to thoroughly explain differential RelA-driven transcription from differently arranged tandem sites using in vitro binding data and structural insights remained incomplete. Nonetheless, we made some intriguing observations; first, despite protein–protein interactions between the two dimers visualized in the structure, in vitro experiments indicate that the dimers do not cooperate for simultaneous stable interactions with DNA in solution. Second, the dimers could bind asymmetrically to symmetrically arranged sites in which one subunit of one dimer does not satisfy all DNA contacts. These broken DNA contacts suggest unstable DNA binding. This mode of binding possibly represents dissociating state of one dimer. Consistently, in vitro data for binding by the RelA-RHR (DBD) also shows the anti-cooperative mode of DNA binding by the two dimers. Finally, the full-length recombinant RelA binds DNA with poor specificity and weaker affinity as compared with RelA-RHR, suggesting an unexpected role for RelA AD. The AD domain being highly negatively charged might be involved in repulsive interactions with the negatively charged phosphates in the flanking exposed DNA. These observations are tallies with observations of earlier reports suggesting that RelA does not bind κB sites and functions as a non-DNA binding partner of p50 to activate transcription (18).
An important question now is how full-length RelA recognizes κB sites in vivo to activate transcription overcoming the negative effect of the AD. Multiple factors have been shown to influence transcriptional activity of RelA, including RPS3, OGG1, E2F1, and p53 (19–22). These later factors are DNA binding transcription factors, but their DNA binding activity is not essential to activate the ability of RelA to activate transcription (21, 22). We suggest that these factors, as shown in the cases of RPS3, OGG1, and p53, augment the ability of RelA to bind DNA at least in part by neutralizing the negative effect of the AD. However differential transcriptional activity observed with wild-type and various mutants of E-selectin promoters cannot be explained simply by neutralization of a negative effect of AD on RelA-RHR by these regulatory factors. We observe that multiple κB sites present in native promoters functionally cooperate among themselves to activate transcription by RelA, although artificially altering the arrangement of these κB sites resulted in transcriptional repression. At this stage, we are not clear about how RelA differentially selects these promoters. Perhaps NF-κB dimers in association with other regulatory factors facilitate each other in binding DNA through long-range electrostatics. Only when two sites are in close proximity does this facilitation rely on specific arrangement of the two sites. In contrast, when the sites are far apart as in IP-10, orientation of the sites may not be critical. A schematic representation of these multiple options is depicted in Fig. 5.
Figure 5.
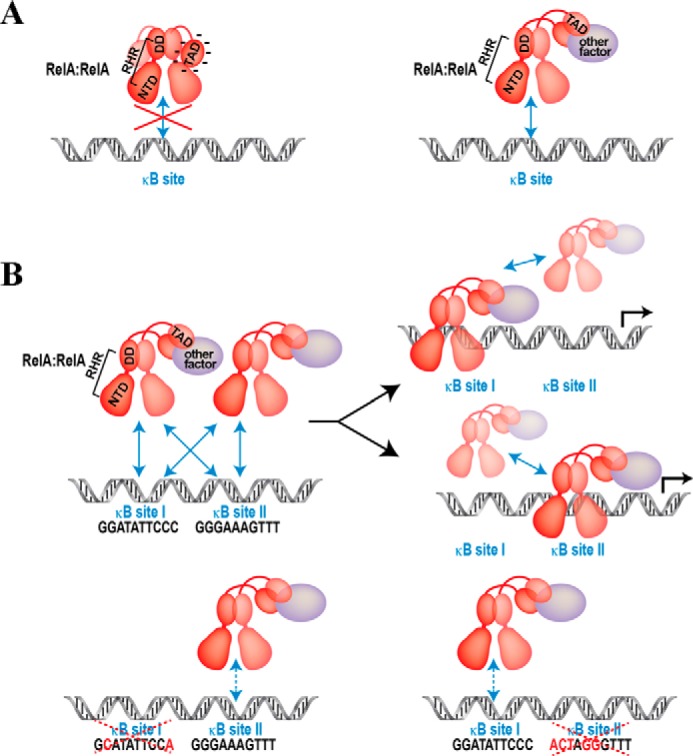
A schematic representation of transcription activation domain (TAD)–mediated inhibition of RHR binding to κB sites. A, the TAD engages in a highly dynamic interaction to inhibit DNA binding rather than forming a stable interaction with RHR. Other factor(s) must neutralize the negative effect of TAD to allow RHR to bind DNA. B, RelA homodimers facilitate each other for stable interaction of one homodimer to one κB site. However, under physiological conditions, both dimers cannot co-occupy the two κB sites. The model depicts cooperation between the two dimers at a long range but anti-cooperation on the DNA. The resultant effect is more profound when κB sites of tandem arrangements are weak. Therefore, when both sites are weak and one site is abolished, the other site cannot efficiently engage a dimer because of the absence of a long-range support from the other dimer.
Binding data reflecting anti-cooperative nature suggest that at the concentrations of RelA observed in the nucleus, both dimers could occupy the sites simultaneously. These results are consistent with hyperdynamicity of DNA binding by NF-κB as shown previously and opposed to a stable enhanceosome formation by multiple transcription factors bound to tandem sites (23, 24). Thus the dynamic engagement of RelA rather than a stable complex in equilibrium state is a critical determinant of transcriptional output. Combined effects of specificity of κB sequence, positional arrangement of multiple κB sites, and effect of other associated nuclear factors together determine the binding kinetics of RelA and other NF-κB factors. Future studies will address these intriguing possibilities.
Experimental procedures
Plasmids and antibodies
Untagged mouse RelA(19–304), pRC-HA-hRelA(1–551), and GST-RelA AD(429–551) have been previously described (3). E-selectin, HIV, and IP-10 luciferase reporters containing specific κB DNA promoter were cloned in CMXTK-Luciferase reporter (a kindly gift from Dr. D. Chakravarti, Northwestern University Feinberg School of Medicine) using SalI and BamHI restriction enzymes (New England Biolabs). Sequences of oligonucleotides containing the N-terminal SalI restriction site, the C-terminal BamHI restriction site, and the specific κB site used for reporter cloning are listed in supplemental Table S1. The antibody recognizing RelA (sc-372) was purchased from Santa Cruz. Anti-His antibody was a gift from Biobharati Life Science, Kolkata, India.
Protein expression
Recombinant untagged mouse RelA(19–304) was expressed and purified with modifications of a previously published protocol (25). Briefly, cells were lysed with lysis buffer (25 mm MES, pH 6.5, 50 mm NaCl, 0.5 mm EDTA, pH 8.0, 0.5 mm PMSF, and 10 mm 2-mercaptoethanol (BME)) and sonicated. Lysate was cleared by centrifugation at 13,000 rpm for 30 min at 4 °C. Supernatant was decanted into an ice-cold beaker, and 1/27 of streptomycin (10%) was added to a 0.3% final concentration while gently stirring. The sample was left stirring 20 min more at 4 °C and cleared by centrifugation at 13,000 rpm for 30 min at 4 °C. Supernatant was loaded onto an SP-Sepharose Fast flow column pre-equilibrated (Amersham Biosciences) with lysis buffer. Column was washed with 20 column volumes of Lysis buffer and then eluted at 4 °C with elution buffer (25 mm MES, pH 6.5, 300 mm NaCl, 0.5 mm EDTA pH 8.0, 0.5 mm PMSF, and 10 mm BME). Peak fractions were pooled and concentrated in an Amicon concentrator and further purified by size exclusion chromatography on a Superdex 75 column in 25 mm Tris-HCl, pH 7.5, 50 mm NaCl, and 1 mm DTT. GST-tagged RelA-AD was expressed in Escherichia coli Rosetta cells by growing cells harboring the expression plasmid (pGEX-4T containing RelA 429–551) to A600 0.4 followed by induction with 0.1 mm isopropyl β-d-thiogalactopyranoside overnight at room temperature. The fusion protein was purified in a single step using a glutathione-Sepharose column (gift from Biobharati Life Science, Kolkata, India) from the crude cell lysate (150 mm NaCl, 25 mm Tris-HCl, pH 7.5, glycerol 10% (v/v)) followed by elution with 10 mm glutathione.
Recombinant His-hRelA full-length baculovirus was kindly provided by Dr. James Kadonaga. Sf9 suspension cultures were infected with His-hRelA full-length baculovirus at a cell density of 1 × 106/ml and allowed to grow for 60 h postinfection. The cells were harvested and lysed in lysis buffer (10 mm Tris-HCl, pH 7.5, 500 mm NaCl, Nonidet P-40 0.1% (v/v), glycerol 10% (v/v), 15 mm imidazol, 10 mm BME, 2 mm PMSF, and protease inhibitor mixture (Sigma)) by sonication. The lysate was clarified by filtering with 0.22 μm and mixed with slurry of nickel-nitrilotriacetic acid resin (Qiagen) in batch in the cold room for 3 h. The resin was thoroughly washed with lysis buffer containing 30 mm imidazole and 300 mm NaCl prior to elution using the same buffer with 400 mm imidazole and 200 mm NaCl. Elution was done twice in the cold room for 30 and 10 min, respectively. Both elutions were pooled and dialyzed three times 1 h each. First and second dialysis were done against dialysis buffer 1 (10 mm Tris-HCl, pH 7.5, 200 mm NaCl, 10 mm BME, 10% glycerol (v/v)), and the third dialysis was against dialysis buffer 2 (10 mm Tris-HCl, pH 7.5, 200 mm NaCl, 10 mm BME, 5% glycerol (v/v)). Protein was concentrated by centrifugation using Centriprep 30-kDa cutoff membrane concentrator unit (Millipore) and loaded onto preparative Superdex 200 size exclusion column connected to an AKTA purifier (GE Healthcare) equilibrated with buffer containing 10 mm Tris-HCl, pH 7.5, 200 mm NaCl, 10 mm DTT, 5% glycerol (v/v) at room temperature. Peak fractions were concentrated again with Centriprep 30-kDa. Protein concentration was determined using Bradford reagent and was snap frozen in liquid N2 for long-term storage at −80 °C.
Crystallization, data collection, and structure solution
Protein–DNA complex was formed by mixing 2:1 (RelA dimer–DNA) to a final concentration of ∼10 mg/ml. The crystals were grown using hanging drop vapor diffusion method by mixing 1:1 ratio of the complex solution to reservoir solution (100 mm sodium citrate, pH 5.5, 10 mm CaCl2, 1 mm spermine, 10 mm DTT, 15% PEG3350 (v/v), 0.1% n-octyl-β-d-glucopyranoside (BOG) (v/v)). Before data collection, the crystals were soaked for about 1 min in a cryoprotectant buffer containing the original reservoir solution plus 25% PEG400 and flash-cooled under liquid nitrogen. X-ray diffraction data were collected at APS 19ID synchrotron source. The diffraction patterns revealed that the crystals belong to the monoclinic space group P21 with unit cell: a = 88.91, b = 117.80, c = 70.81 Å, and β = 91.20°. X-ray diffraction data were integrated and scaled to 2.50 Å by using HKL2000. The data processing statistics are included in Table 1.
The structure was determined by molecular replacement using MolRep, with the structure of RelA homodimer and IL-8 DNA complex as the search model. Two copies of the search model were located in the asymmetrical unit with DNA end-to-end stacking at the junction. The orientation and position of this initial model were refined by rigid body refinement in CNS (cns_solve_1.3). The structure was further refined using minimization and simulated annealing with a maximum likelihood target function and a flat bulk-solvent correction using the CNS system. The model rebuilding was performed based on 2Fo − Fc maps using Xtalview. After the individual temperature factors were included in the refinement, the R factor was 21.0%, and the free R factor was 27.2% for the final model. The detailed results of the refinement are included in Table 1. The coordinates have been deposited into the Protein Data Bank with entry code 5U01.
Luciferase reporter assays
HeLa cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin–glutamine. The cells were transiently transfected with pRC-HA-hRelA(1–551) or empty HA vector and the luciferase reporter DNA with specific κB DNA promoters. The total amount of plasmid DNA was kept constant for all assays. Transient transfections were carried out using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. Renilla luciferase expression plasmid was co-transfected as an internal control. The cells were collected 48 h after transfection. Luciferase activity assays were performed using a Dual-Luciferase reporter assay system (Promega) following the manufacturer's protocol. The results from promoter fold activation of the reporter plasmids are given as the ratio of relative luciferase activity (luciferase units/Renilla units) values from each sample relative to the corresponding control empty vector. The values represent the averages of three independent wells. The experiments were performed a minimum of three times with identical results. The data are represented as means ± S.D. κB sequence promoters are listed in supplemental Table S1.
Electrophoretic mobility shift assay
EMSA assays were performed as previously described (26). Briefly, E-selectin probes were radiolabeled and incubated with His-hRelA full-length or untagged mRelA-RHR for 20 min at room temperature. The amount of protein was quantified by Bradford assay (Bio-Rad). Protein complexes were analyzed by native electrophoresis on a 4% (w/v) non-denatured polyacrylamide gel. The probe sequences are listed in supplemental Table S2.
Author contributions
G. G. conceived the idea with help from M. C. M. and D.-B. H. determined the crystal structure. D.-B. H., G. G., and T. B. interpreted the structural results. M. C. M., H. T. N., V. Y. F. W., and Y. L. conducted biochemical experiments. G. G. and M. C. M. wrote the manuscript with help from T. B.
Supplementary Material
Acknowledgments
We thank Dr. Simpson Joseph for providing the fluorescence plate reader. We also thank Monique Pena and Sulakshana Mukherjee for helping with crystallization and Yunshu Song and Shandy Shahabi for protein purification.
This work was supported by National Institutes of Health Grant GM085490 (to G. G.) and Start-up Research Grant SRG2016–00084-FHS from the University of Macau (to V. Y. F. W). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Tables S1 and S2 and Figs. S1–S3.
The atomic coordinates and structure factors (code 5U01) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- DBD
- DNA-binding domain
- AD
- transactivation domain
- RHR
- Rel homology region
- NTD
- N-terminal domain
- DD
- dimerization domain
- BME
- 2-mercaptoethanol.
References
- 1. Chen F. E., and Ghosh G. (1999) Regulation of DNA binding by Rel/NF-κB transcription factors: structural views. Oncogene 18, 6845–6852 [DOI] [PubMed] [Google Scholar]
- 2. Ghosh G., Wang V. Y., Huang D. B., and Fusco A. (2012) NF-κB regulation: lessons from structures. Immunol. Rev. 246, 36–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen Y. Q., Ghosh S., and Ghosh G. (1998) A novel DNA recognition mode by the NF-κB p65 homodimer. Nat. Struct. Biol. 5, 67–73 [DOI] [PubMed] [Google Scholar]
- 4. Cheng C. S., Feldman K. E., Lee J., Verma S., Huang D. B., Huynh K., Chang M., Ponomarenko J. V., Sun S. C., Benedict C. A., Ghosh G., and Hoffmann A. (2011) The specificity of innate immune responses is enforced by repression of interferon response elements by NF-κB p50. Sci. Signal. 4, ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Siggers T., Chang A. B., Teixeira A., Wong D., Williams K. J., Ahmed B., Ragoussis J., Udalova I. A., Smale S. T., and Bulyk M. L. (2011) Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-κB family DNA binding. Nat. Immunol. 13, 95–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leung T. H., Hoffmann A., and Baltimore D. (2004) One nucleotide in a κB site can determine cofactor specificity for NF-κB dimers. Cell 118, 453–464 [DOI] [PubMed] [Google Scholar]
- 7. Wang V. Y., Huang W., Asagiri M., Spann N., Hoffmann A., Glass C., and Ghosh G. (2012) The transcriptional specificity of NF-κB dimers is coded within the κB DNA response elements. Cell Rep. 2, 824–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martone R., Euskirchen G., Bertone P., Hartman S., Royce T. E., Luscombe N. M., Rinn J. L., Nelson F. K., Miller P., Gerstein M., Weissman S., and Snyder M. (2003) Distribution of NF-κB-binding sites across human chromosome 22. Proc. Natl. Acad. Sci. U.S.A. 100, 12247–12252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xing Y., Yang Y., Zhou F., and Wang J. (2013) Characterization of genome-wide binding of NF-κB in TNFα-stimulated HeLa cells. Gene 526, 142–149 [DOI] [PubMed] [Google Scholar]
- 10. Kunsch C., Ruben S. M., and Rosen C. A. (1992) Selection of optimal κB/Rel DNA-binding motifs: interaction of both subunits of NF-κB with DNA is required for transcriptional activation. Mol. Cell. Biol. 12, 4412–4421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Whitley M. Z., Thanos D., Read M. A., Maniatis T., and Collins T. (1994) A striking similarity in the organization of the E-selectin and β interferon gene promoters. Mol. Cell. Biol. 14, 6464–6475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kawakami K., Scheidereit C., and Roeder R. G. (1988) Identification and purification of a human immunoglobulin-enhancer-binding protein (NF-κB) that activates transcription from a human immunodeficiency virus type 1 promoter in vitro. Proc. Natl. Acad. Sci. U.S.A. 85, 4700–4704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stroud J. C., Oltman A., Han A., Bates D. L., and Chen L. (2009) Structural basis of HIV-1 activation by NF-κB: a higher-order complex of p50:RelA bound to the HIV-1 LTR. J. Mol. Biol. 393, 98–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Giorgetti L., Siggers T., Tiana G., Caprara G., Notarbartolo S., Corona T., Pasparakis M., Milani P., Bulyk M. L., and Natoli G. (2010) Noncooperative interactions between transcription factors and clustered DNA binding sites enable graded transcriptional responses to environmental inputs. Mol. Cell 37, 418–428 [DOI] [PubMed] [Google Scholar]
- 15. Pober J. S. (1999) Immunobiology of human vascular endothelium. Immunol. Res. 19, 225–232 [DOI] [PubMed] [Google Scholar]
- 16. Joachimiak A., and Sigler P. B. (1991) Crystallization of protein-DNA complexes. Methods Enzymol. 208, 82–99 [DOI] [PubMed] [Google Scholar]
- 17. Lavery R., and Sklenar H. (1989) Defining the structure of irregular nucleic acids: conventions and principles. J. Biomol Struct. Dyn. 6, 655–667 [DOI] [PubMed] [Google Scholar]
- 18. Nolan G. P., Ghosh S., Liou H. C., Tempst P., and Baltimore D. (1991) DNA binding and IκB inhibition of the cloned p65 subunit of NF-κB, a Rel-related polypeptide. Cell 64, 961–969 [DOI] [PubMed] [Google Scholar]
- 19. Wan F., Anderson D. E., Barnitz R. A., Snow A., Bidere N., Zheng L., Hegde V., Lam L. T., Staudt L. M., Levens D., Deutsch W. A., and Lenardo M. J. (2007) Ribosomal protein S3: a KH domain subunit in NF-κB complexes that mediates selective gene regulation. Cell 131, 927–939 [DOI] [PubMed] [Google Scholar]
- 20. Pan L., Zhu B., Hao W., Zeng X., Vlahopoulos S. A., Hazra T. K., Hegde M. L., Radak Z., Bacsi A., Brasier A. R., Ba X., and Boldogh I. (2016) Oxidized guanine base lesions function in 8-Oxoguanine DNA glycosylase-1-mediated epigenetic regulation of nuclear factor κB-driven gene expression. J. Biol. Chem. 291, 25553–25566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lim C. A., Yao F., Wong J. J., George J., Xu H., Chiu K. P., Sung W. K., Lipovich L., Vega V. B., Chen J., Shahab A., Zhao X. D., Hibberd M., Wei C. L., Lim B., et al. (2007) Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-κB upon TLR4 activation. Mol. Cell 27, 622–635 [DOI] [PubMed] [Google Scholar]
- 22. Choy M. K., Movassagh M., Siggens L., Vujic A., Goddard M., Sánchez A., Perkins N., Figg N., Bennett M., Carroll J., and Foo R. (2010) High-throughput sequencing identifies STAT3 as the DNA-associated factor for p53-NF-κB-complex-dependent gene expression in human heart failure. Genome Med. 2, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bosisio D., Marazzi I., Agresti A., Shimizu N., Bianchi M. E., and Natoli G. (2006) A hyper-dynamic equilibrium between promoter-bound and nucleoplasmic dimers controls NF-κB-dependent gene activity. EMBO J. 25, 798–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thanos D., and Maniatis T. (1995) Virus induction of human IFNβ gene expression requires the assembly of an enhanceosome. Cell 83, 1091–1100 [DOI] [PubMed] [Google Scholar]
- 25. Huxford T., Malek S., and Ghosh G. (2000) Preparation and crystallization of dynamic NF-κB·IκB complexes. J. Biol. Chem. 275, 32800–32806 [DOI] [PubMed] [Google Scholar]
- 26. Moorthy A. K., Huang D. B., Wang V. Y., Vu D., and Ghosh G. (2007) X-ray structure of a NF-κB p50/RelB/DNA complex reveals assembly of multiple dimers on tandem κB sites. J. Mol. Biol. 373, 723–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.