

Abstract
Pleiotrophin (PTN) is a multifunctional, cationic, glycosaminoglycan-binding cytokine and growth factor involved in numerous physiological and pathological processes, including tissue repair and inflammation-related diseases. PTN has been shown to promote leukocyte responses by inducing their migration and expression of inflammatory cytokines. However, the mechanisms through which PTN mediates these responses remain unclear. Here, we identified the integrin Mac-1 (αMβ2, CD11b/CD18) as the receptor mediating macrophage adhesion and migration to PTN. We also found that expression of Mac-1 on the surface of human embryonic kidney (HEK) 293 cells induced their adhesion and migration to PTN. Accordingly, PTN promoted Mac-1–dependent cell spreading and initiated intracellular signaling manifested in phosphorylation of Erk1/2. While binding to PTN, Mac-1 on Mac-1–expressing HEK293 cells appears to cooperate with cell-surface proteoglycans because both anti-Mac-1 function-blocking mAb and heparin were required to block adhesion. Moreover, biolayer interferometry and NMR indicated a direct interaction between the αMI domain, the major ligand-binding region of Mac-1, and PTN. Using peptide libraries, we found that in PTN the αMI domain bound sequences enriched in basic and hydrophobic residues, indicating that PTN conforms to the general principle of ligand-recognition specificity of the αMI domain toward cationic proteins/peptides. Finally, using recombinant PTN-derived fragments, we show that PTN contains two distinct Mac-1–binding sites in each of its constitutive domains. Collectively, these results identify PTN as a ligand for the integrin Mac-1 on the surface of leukocytes and suggest that this interaction may play a role in inflammatory responses.
Keywords: adhesion, cell migration, integrin, nuclear magnetic resonance (NMR), pleiotrophin, CD11b/CD18, Mac-1, αMβ2
Introduction
Pleiotrophin (PTN)3 is a glycosaminoglycan-binding cytokine and growth factor with potent mitogenic and angiogenic activities. Together with the related protein midkine, it forms a unique family of cytokines that are normally expressed during embryogenesis and neonatal development but are also produced by injured tissues during repair and regeneration (1, 2). PTN has also been identified as crucial in the maintenance of hematopoietic stem cells (3, 4). Moreover, PTN levels have been shown to be highly elevated in a large number of cancer cell lines as well as being correlated with their metastatic abilities (5–13). Several studies have also associated PTN with inflammation and leukocyte recruitment (13, 14). There are now efforts both to develop PTN inhibitors to treat cancer and to use PTN itself for prevention of tissue injury during ischemia (15–18). Several proteins have been proposed as receptors of PTN. The most studied among these include the heparan sulfate proteoglycan (HSPG) N-syndecan and the chondroitin sulfate proteoglycan receptor-type protein tyrosine phosphatase ζ (19–21). Several non-proteoglycan receptors are also known to bind PTN, including nucleolin (22, 23) and the integrin αVβ3 (24).
Structurally, PTN is made up of two thrombospondin type-1 repeat (TSR) domains flanked by unstructured termini, both of which are highly basic (25, 26). The TSR domains also contain a number of basic amino acid clusters with two clusters in the C-terminal TSR domain having the highest affinity for glycosaminoglycans (25, 27). It is thought that the basic nature of PTN is crucial to its ability to interact with receptors and initiate signaling.
Although the role of PTN in mitogenesis and angiogenesis has been extensively researched, its function in inflammation has not been widely explored. Given the fact that injured tissues express PTN, it is likely that leukocytes are exposed to it at sites of inflammation. Indeed, some studies have suggested that PTN plays a role in recruitment of leukocytes during inflammation (13, 14). However, the identity of the PTN receptor(s) on leukocytes that mediate this response and the role of PTN signaling in these cells remain unknown. We hypothesized that integrin αMβ2 (Mac-1, CD11b/CD18, CR3) can serve as a receptor for PTN. This hypothesis was based on our recent finding that Mac-1 binds cationic proteins, many of which, similar to PTN, interact with heparin (28–30).
Mac-1 is an adhesion receptor expressed on the surface of myeloid cells such as monocyte/macrophages and neutrophils. As an adhesion receptor, Mac-1 is crucial to numerous leukocyte responses, including migration, phagocytosis, degranulation, adherence to microorganisms, and others (31, 32). Among integrins, which are in general notorious for their capacity to recognize multiple ligands, Mac-1 is its most promiscuous member. In contrast to many other integrins that recognize the RGD motif, Mac-1 does not bind this sequence. Instead, as we have recently revealed, Mac-1 has preferences for sequences enriched in basic and hydrophobic amino acid residues (29, 30, 33, 34). The identification of Mac-1 recognition motifs has led to the prediction that other cationic proteins and peptides released from leukocytes and other cells during tissue injury can serve as Mac-1 ligands (29). Most of Mac-1's ligand binding and thus its broad ligand binding specificity can be attributed to the αMI domain, a region of ∼200 amino acid residues inserted into the αM subunit, inasmuch as its removal in Mac-1 eliminates ∼85% of ligand binding (35).
In this study, we present biochemical and cell biology evidence that PTN is a ligand for Mac-1. Using various Mac-1–expressing cells, we demonstrate that Mac-1 supports adhesion to PTN, promotes spreading, and induces activation of the MAP kinase Erk1/2. Furthermore, PTN induces a potent migratory response in Mac-1–expressing human embryonic kidney 293 (Mac-1 HEK293) cells and in isolated murine macrophages. Direct interaction between PTN and the αMI domain, the major ligand-binding region of Mac-1, has been shown using biolayer interferometry analyses and confirmed by solution NMR spectroscopy. Finally, using peptide array analysis, we identified the putative αMI domain-binding sites in both TSR domains of PTN and validated their importance in cell adhesion and signaling assays with individual recombinant PTN fragments.
Results
Mac-1 is involved in adhesion of Mac-1–expressing cells to PTN
To assess whether PTN can bind Mac-1, we initially examined the ability of Mac-1 HEK293 to adhere to immobilized PTN. As shown in Fig. 1A, Mac-1 HEK293 cells adhered to immobilized PTN in a concentration-dependent manner with saturable adhesion achieved with a PTN coating concentration of ∼100 nm. Wild-type HEK293 cells also strongly adhered to PTN. However, the presence of Mac-1 significantly augmented adhesion (Fig. 1A). Because PTN has high affinity for glycosaminoglycans and HSPG N-syndecan binds PTN, we examined the possibility that HSPGs on wild-type HEK293 cells mediate adhesion to PTN. Preincubation of cells with heparin (1 μg/ml) almost completely eliminated adhesion (Fig. 1B), indicating that on the surface of these cells HSPGs are entirely responsible for PTN binding. However, adhesion of Mac-1 HEK293 cells was reduced by only ∼60%, suggesting that Mac-1 on these cells may be involved in binding PTN. To investigate this possibility, Mac-1 HEK293 cells were treated with the function-blocking mAb 44a (directed against the αMI domain of Mac-1). Although adhesion tended to decrease, it was not significantly altered with the maximal blocking effect (19 ± 10%) observed at 10 μg/ml of mAb. Likewise, although heparin inhibited adhesion in a dose-dependent manner, the maximal blocking effect attained did not exceed 59 ± 6%. However, when Mac-1 HEK293 cells were treated with both mAb 44a and heparin, cell adhesion was inhibited by >95%. These results suggest both HSPG and Mac-1 can act as receptors for PTN.
Figure 1.

PTN supports adhesion and spreading of Mac-1–expressing HEK293 cells and IC-21 macrophages. A, aliquots (100 μl; 5 × 104/ml) of Mac-1 HEK293 and HEK293 cells labeled with calcein were added to microtiter wells coated with different concentrations of PTN and postcoated with 1% PVP. After 30 min at 37 °C, nonadherent cells were removed by washing, and fluorescence of adherent cells was measured in a fluorescence plate reader. Data shown are means ± S.E. from three separate experiments with triplicate measurements. *, p ≤ 0.05. Statistically insignificant differences are not labeled. B, Mac-1 HEK293 cells and HEK293 cells were preincubated with anti-αM mAb 44a (5 μg/ml), heparin (1 μg/ml), or a mixture of both for 20 min and added to wells coated with 100 nm PTN. Adhesion in the absence of Mac-1 inhibitors and heparin was assigned a value of 100%. Data shown are means ± S.E. Error bars represent S. E. from three separate experiments with triplicate measurements. ***, p ≤ 0.001 compared with control adhesion in the absence of inhibitors; n/s, not significant. C, Mac-1 HEK293 (upper panel) and HEK293 cells (lower panel) were plated on glass slides coated with 100 nm PTN and allowed to adhere for different durations at 37 °C. Nonadherent cells were removed, and adherent cells were fixed with 2% paraformaldehyde and permeabilized with 0.1% Triton X-100 in PBS followed by staining with Alexa Fluor 546 phalloidin (actin) and DAPI (DNA). The cells were imaged with a Leica SP5 laser-scanning confocal microscope with a 63×/1.4 objective. D, the diameter of 20–50 Mac-1 HEK293 and HEK293 cells was calculated for each time point from images captured with a 20×/0.50 objective. Data shown are means ± S.E. from five random images for each time point from one to four experiments. Error bars represent S. E. *, p ≤ 0.05; ***, p ≤ 0.001. E, IC-21 macrophages were preincubated with anti-mouse αM mAb M1/70 (10 μg/ml), heparin (1 μg/ml), or a mixture of both and added to wells coated with 100 nm PTN. Data shown are means ± S.E. from three separate experiments with six measurements. Error bars represent S. E. ***, p ≤ 0.001.
Consistent with the role of Mac-1 in adhesion to PTN, Mac-1 HEK293 cells spread with the formation of actin filaments as detected by staining with Alexa Fluor 546-conjugated phalloidin (Fig. 1C, upper panels). By contrast, spreading of wild-type HEK293 cells was visibly less even after 24 h (Fig. 1C, bottom panels). Quantification of cell spreading confirmed this observation (Fig. 1D).
To investigate the relevance of PTN–Mac-1 interaction in immune cells, we performed adhesion assays using IC-21 cells, a murine macrophage cell line naturally expressing Mac-1. As shown in Fig. 1E, IC-21 cells adhered to immobilized PTN, and ∼65% of adhesion was inhibited by mAb M1/70 against the murine αM integrin subunit. This is in contrast to Mac-1 HEK293 cells whose adhesion to PTN was reduced modestly by mAb 44a (Fig. 1B). Moreover, heparin, which greatly decreased adhesion of Mac-1 HEK293 cells to PTN, had little effect on adhesion of IC-21, suggesting that Mac-1 on the surface of IC-21 cells largely contributes to the interaction with PTN. Together, these results identify PTN as an adhesive ligand for Mac-1 and suggest that on the surface of different cells both Mac-1 and HSPGs are involved in PTN adhesion, albeit to a different extent.
PTN bound to extracellular matrix (ECM) proteoglycans supports adhesion of Mac-1 HEK293 cells
Because PTN is secreted from cells and deposited into the ECM, we explored whether PTN bound to the ECM components is capable of supporting adhesion. Because Mac-1 is known to interact with a number of ECM proteins, including fibronectin, vitronectin, collagens, Cyr61, and others (36), a heterogeneous mixture of these molecules such as those found in Matrigel was not suitable as a substrate for investigations of PTN–Mac-1 interactions. PTN is only known to bind proteoglycans; therefore, the most relevant form of PTN encountered by immune cells in vivo is likely to be PTN anchored to ECM proteoglycans. To simulate such an environment, we studied cell adhesion to PTN prebound to aggrecan, a common proteoglycan found in the ECM. Fig. 2 shows that at two concentrations of PTN tested both Mac-1 HEK293 and wild-type HEK293 (HEK293) cells adhered to aggrecan-bound PTN with Mac-1 HEK293 cells adhering at a significantly higher level than HEK293 cells (p < 0.001). Neither type of cells had affinity for aggrecan itself. Notably, binding of PTN to aggrecan did not reduce cell adhesion, indicating that PTN in its aggrecan-bound form remains an efficient Mac-1 ligand.
Figure 2.
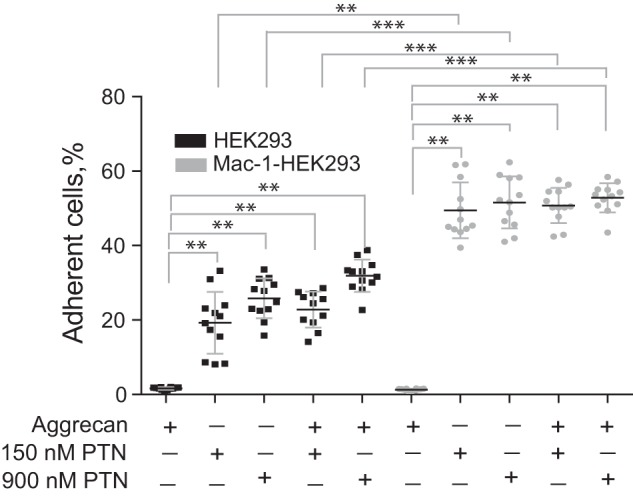
Effect of aggrecan on PTN-mediated adhesion of Mac-1–expressing HEK293 cells. Aggrecan (10 μg/ml) was used to coat the microtiter wells overnight before addition of PTN (150 and 900 nm). Aliquots (100 μl; 5 × 104/ml) of calcein-labeled Mac-1 HEK293 and wild-type HEK293 cells were added to microtiter wells. After 30 min at 37 °C, nonadherent cells were removed by washing, and fluorescence of adherent cells was measured in a fluorescence plate reader. Data shown are means ± S.E. from two separate experiments with six measurements. Error bars represent S. E. **, p ≤ 0.01; ***, p ≤ 0.001.
PTN induces migration of Mac-1–expressing cells
PTN is known to induce cell migration, and in some instances, this effect has been shown to be integrin-dependent (24, 37). Therefore, we investigated whether Mac-1 can support PTN-induced migration. In particular, using a Transwell system, we compared the ability of wild-type and Mac-1 HEK293 cells to migrate toward PTN. Previous studies reported that these cell lines are a useful system for assessing the role of Mac-1 in migration (38). PTN induced a potent migratory response (Fig. 3, A and B). In contrast, wild-type HEK293 migrated only slightly. Direct evidence that migration of Mac-1 HEK293 cells was dependent on Mac-1 was obtained in the experiments in which anti-Mac-1 mAb 44a was tested. Although mAb 44a eliminated cell migration, an IgG1 isotype control for this mAb had no significant effect (Fig. 3B). Heparin (1 μg/ml) also inhibited migration to a modest but significant degree.
Figure 3.

Migration of Mac-1–expressing cells to PTN in a Transwell system. A, Transwell inserts were coated with 67 nm PTN for 3 h at 37 °C. Mac-1 HEK293 cells (100 μl at 3 × 106/ml) were added to the upper wells of the Transwell chamber, and their ability to migrate was analyzed. After 16 h at 37 °C, the cells were labeled with calcein-AM for 30 min at 37 °C, and the cells from the upper chamber were removed by wiping with a cotton-tipped applicator. Images of the cells on the underside of the Transwell filter were taken with a 10×/0.25 objective. The figure is representative of three experiments. In separate experiments, cells were preincubated with 20 μg/ml 44a mAb or IgG1 isotype control mAb or 1 μg/ml heparin for 20 min before loading to the upper wells. B, quantification of cell migration. The number of migrated cells was determined using NIH ImageJ. Migration was expressed as percentage of pixel intensity of images from at least three random fields per well. The data shown are means and S.E. from three individual experiments. Error bars represent S. E. Migration of cells in the absence of inhibitors was assigned a value of 100%. ***, p ≤ 0.001; n/s, not significant. C, migration of thioglycolate-elicited macrophages isolated from the peritoneum of wild-type and Mac-1–deficient mice and purified as described under “Experimental procedures.” 100 μl of macrophages (3 × 106/ml) was placed in the upper chamber and allowed to migrate through the 5-μm membranes coated with PTN (100 nm) or buffer (control) for 90 min at 37 °C. Images of the cells on the underside of the Transwell filter were taken with a 40×/0.75 objective. D, quantification of macrophage migration. Data are presented as fluorescence of migrated cells per field ±S.E. for four random fields per well from three individual experiments. Error bars represent S. E. ***, p ≤ 0.001.
In a separate set of experiments, we tested whether PTN can induce migration of mouse macrophages isolated from the peritoneum of wild-type and Mac-1–deficient mice. Macrophages were purified from a total population of peritoneal cells, and their migration was examined in a Transwell system. As shown in Fig. 3, C and D, PTN induced a strong migratory response in wild-type macrophages. PTN also induced migration of Mac-1–deficient macrophages. However, compared with wild-type cells, migration of Mac-1–deficient macrophages was significantly impaired (by 76 ± 21%). These results indicate that Mac-1 is a crucial mediator of PTN-induced cell migration. Furthermore, because macrophages lacking Mac-1 can migrate, albeit at a significantly reduced rate, other integrins (most likely αVβ3) may contribute to migration.
PTN induces activation of MAP kinase (MAPK) Erk1/2
The interaction of integrins with ligands is known to induce activation of MAP kinases. To determine whether PTN activates MAPKs via binding to Mac-1, adherent Mac-1 HEK293 cells were treated with different concentrations of PTN, and phosphorylation of Erk1/2 was examined. A PTN concentration of 0.7 μm induced a significant increase in phosphorylation of Erk1/2, and this response was blocked by mAb 44a and heparin (Fig. 4, A and B). The simultaneous use of two reagents increased the inhibitory effect; however, the cumulative effect did not reach a statistical significance. Treatment of control HEK293 cells with PTN did not induce phosphorylation of Erk1/2 (Fig. 4A). These data suggest that in this system initiation of intracellular signaling by PTN requires both Mac-1 and HSPGs.
Figure 4.
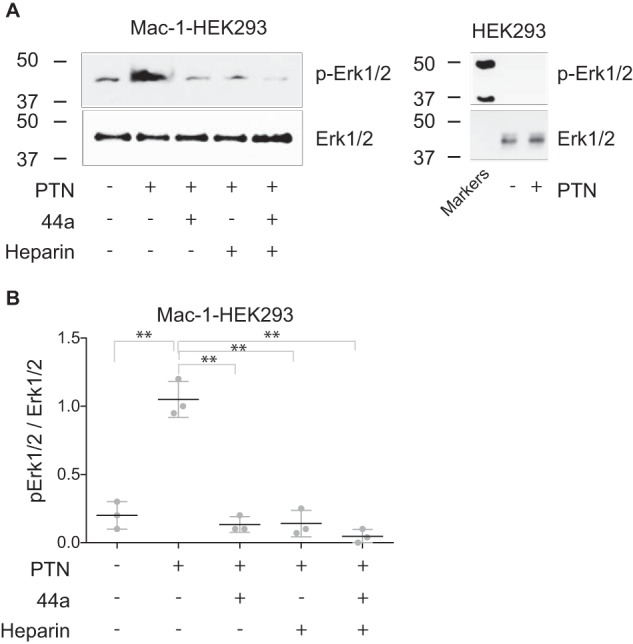
PTN-induced phosphorylation of Erk1/2 in wild-type HEK293 and Mac-1 HEK293 cells. A, adherent HEK293 or Mac-1 HEK293 cells were treated with 0.7 μm PTN in the absence and presence of mAb 44a (10 μg/ml), heparin (10 μg/ml), or a combination of both. After 30 min at 37 °C, cells were washed and lysed. The lysates were separated by 12.5% SDS-PAGE, and Western blotting was performed using mAb specific for the phosphorylated form of Erk1/2. The mAb against total Erk1/2 was used to equalize protein loading. A representative of three experiments is shown. The molecular weight markers are shown on the left of each set of blots. B, densitometry analysis of Erk1/2 phosphorylation for Mac-1 HEK293 cells. Erk1/2 activation data are shown as a ratio of the intensity of the phosphorylated Erk1/2 (p-Erk1/2) band to the total (Erk1/2) band. The phosphorylated Erk1/2-to-Erk1/2 ratio of the PTN-activated lane was set to 1.0. Data are calculated from three experiments. Error bars represent S. E. **, p ≤ 0.01.
Biochemical analyses of the interaction between PTN and αMI domain
To further characterize the Mac-1–PTN interactions and determine domains of Mac-1 responsible for PTN binding, we analyzed the binding parameters of the interaction between the αMI domain and PTN. We focused on the αMI domain because this domain is the major ligand-binding region in Mac-1, and previous studies have shown that several basic proteins and peptides interact with it (28–30, 34, 39). To measure the affinity of the αMI domain–PTN interaction, we used biolayer interferometry (BLI) in which PTN was coupled to the matrix coating the biosensor via lysines. The interaction between PTN with both active and nonactive forms of the αMI domain was measured in 20 mm HEPES buffer containing 150 mm NaCl, 1 mm MgCl2, and 0.05% Tween 20, pH 7.5. When PTN was allowed to interact with various concentrations of soluble active αMI domain, a dose-dependent binding was observed (Fig. 5, A and B). The dissociation rate constant (Kd) value determined from the maximal responses achieved in the equilibrium portion of the BLI sensorgrams was 1.2 ± 0.2 μm. The interaction of PTN with nonactive αMI domain was also detected (Fig. 5C); however, binding was lower than that of the active form, which precluded an accurate assessment of the Kd value. In contrast to the αMI domains, the active form of the I domain of integrin αLβ2, which has narrow ligand specificity and binds only intercellular adhesion molecule and JAM-1 molecules, did not bind PTN (Fig. 5C). To assess how divalent cations might affect the interaction of PTN with αMI domain, the binding of active αMI domain was measured in the presence of 5 mm EDTA. As shown in Fig. 5C, EDTA only modestly decreased binding of active αMI domain. The latter result indicates that the αMI domain binding to PTN is not dependent on the presence of Mg2+.
Figure 5.

Analyses of binding of the αMI domains to PTN by BLI. A, representative sensorgrams of titration of PTN immobilized on the ForteBio sensor with active αMI domain (0, 0.24, 0.48, 0.95, 1.9, 3.8, and 5.7 μm). B, saturable binding curve for the interaction of the αMI domain with PTN. Representative data from three independent experiments are shown. C, comparison of the ability of different I domains (5.7 μm) to bind PTN. The interaction between the active αMI domain and PTN in the presence of 5 mm EDTA is also shown. Representative data of three separate experiments are shown. D, representative binding curves of titration of biotinylated PTN immobilized on the ForteBio streptavidin sensor with active αMI domain (0, 0.15, 0.3, 0.75 1.5, and 3.0 μm).
Because recognition by the αMI domain of many of its ligands depends on basic amino acids, we sought to determine whether immobilization of PTN onto the BLI sensor using lysine side chains may potentially reduce PTN's binding to αMI domain. Therefore, we repeated the experiments by randomly biotinylating glutamate and aspartate side chains of PTN and attaching the biotinylated PTN to streptavidin-coated sensors. As determined from the sensorgrams shown in Fig. 5D, the alternative mode of immobilization did not change considerably the binding affinity of the two proteins (Kd = 3.9 ± 0.1 μm).
We also examined the interaction of PTN with the active αMI domain using solution NMR. Fig. 6A shows the 1H-15N HSQC spectrum of PTN in the presence and absence of 1 molar eq of active αMI domain. Addition of the αMI domain decreased the signal intensities of residues in the TSR domains of PTN by ∼40%, whereas PTN residues in the unstructured N and C termini were not affected by αMI domain (Fig. 6B). A signal intensity decrease often takes place when a small protein binds a larger protein or if the interactions between the two is dynamic and the kinetics of the interaction is on the intermediate NMR time scale (40). The observation of a signal intensity decrease therefore indicates that the TSR domains of PTN are involved in binding αMI domain. Together, these data indicate that PTN has a significant affinity for the active form of the αMI domain.
Figure 6.
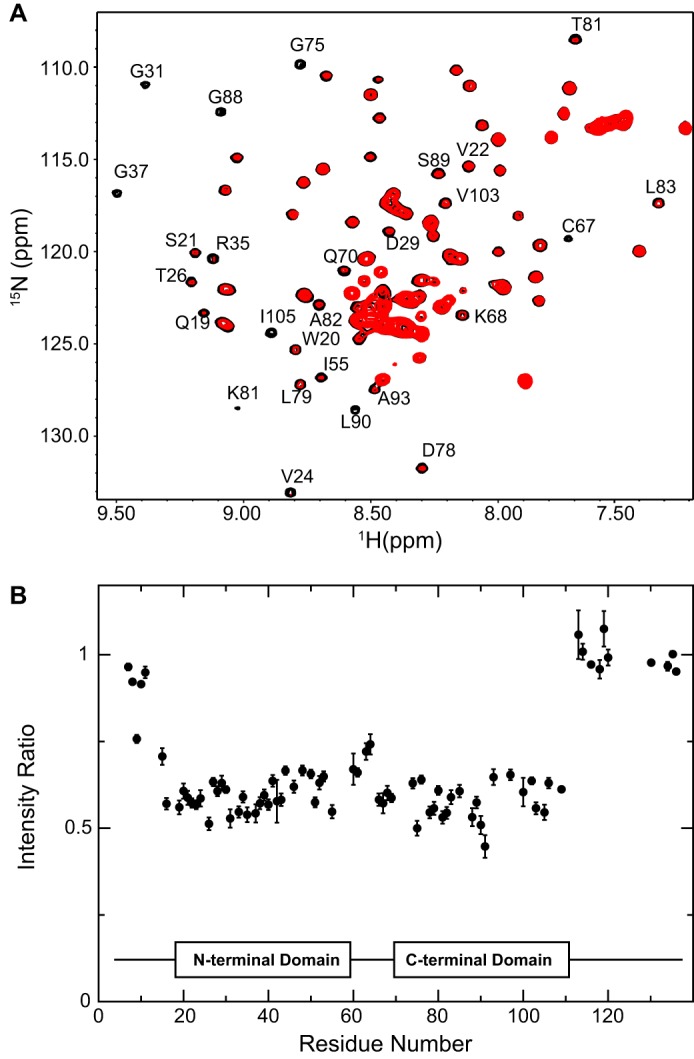
PTN binding to the αMI domain determined from the 1H-15N HSQC spectra of 15N-labeled PTN. A, superimposed 1H-15N HSQC spectra of 40 μm 15N-labeled PTN in the presence (red contour) and absence (black contour) of 40 μm active αMI domain. The presence of αMI domain led to significant decreases in signal intensities of many residues. Residues whose signals were most significantly altered have been labeled with their residue numbers. B, ratio of signal intensities for each PTN residue in the presence and absence of active αMI domain. Error bars represent sum of the noise intensities in the two spectra.
Screening of the PTN-derived peptide libraries for αMI domain binding
To localize the putative αMI domain-binding sites in PTN, we screened the cellulose-bound peptide library representing the complete sequence of PTN. The library, consisting of 9-mer peptides with a 3-residue offset (Fig. 7A), was probed with 125I-labeled recombinant αMI domain. The screening identified four peptide clusters that strongly bound αMI domain (Fig. 7B). These clusters correspond to the following PTN sequences: 1–15 (cluster 1; spots 1–3), 46–69 (cluster 2; spots 16–21), 82–99 (cluster 3; spots 28–31), and 106–132 (cluster 4; spots 36–42). Control spots containing only β-Ala spacer (spots 49 and 50) were negative as were many other peptides. The αMI domain-binding peptides were analyzed by a computer program designed to determine the capacity of peptides to interact with the αMI domain (29). The program assigns each peptide an energy value, which serves as a measure of probability that the αMI domain binds this sequence: the lower the energy, the higher the likelihood that the sequence binds the αMI domain. As previously determined, strong αMI domain-binding peptides derived from various Mac-1 ligands have energy values in the range of −20 to 2 kJ/mol. The analyses showed a good relationship between the energy scores and the αMI-binding activity of PTN-derived peptides revealed in peptide scans (Fig. 7A). In agreement with previous findings (29), peptides enriched in positively charged and hydrophobic residues had the highest affinities for the αMI domain (spots 2, 16–19, 29, 38, and 42). For example, the stretch of overlapping peptides KQTMKTQRCKIPCNWKKQ (spots 16–19) and peptide KTRTGSLKR (spot 29) contain several short motifs, HyB and BHy where Hy represents any hydrophobic residue and B (basic) is either arginine or lysine, that have been shown to be abundant in the αMI domain binders (29). When mapped onto the three-dimensional structure, the αMI domain-binding sequences were found in both TSR domains as well as the N- and C-terminal tails (Fig. 7C). These results indicate that, in the case of PTN, the αMI domain has affinity for its positively charged regions, and thus recognition specificity toward PTN conforms to the general principles of ligand recognition exhibited by Mac-1 toward cationic proteins (29).
Figure 7.

Screening the peptide library spanning the sequence of PTN for αMI domain binding. The assembled peptide library spanning the sequence of PTN consisting of 9-mer peptides with 3-residue offset (A) was incubated with 125I-labeled active αMI domain (residues Glu123–Lys315) and subjected to autoradiography (B). The αMI domain binding was observed as dark spots. Spots 49 and 50 contain only the β-Ala spacer. The numbers in the right columns in A show the relative binding of the αMI domain to peptides determined by densitometry and expressed as a percentage of the intensity of the most active peptide, KTRTGSLKR (spot 29). Peptide energies (middle columns in A) that serve as a measure of the probability each peptide can bind the αMI domain were calculated as described (29). C, the ribbon model of PTN based on Protein Data Bank code 2N6F. Side chains of Arg and Lys are shown in stick representations. The four clusters with peptides identified as positive for the αMI domain binding are shown in purple.
Individual TSR domains of PTN support Mac-1–dependent cell adhesion and induce activation of Erk1/2
The above data from peptide library screening suggest that PTN may contain several αMI domain-binding sites. These sites have been mapped to the N- and C-terminal (NTD and CTD) TSR domains of PTN as well as the C-terminal tail. To investigate the contribution of each domain to Mac-1 binding, we prepared truncated PTN fragments corresponding to the NTD (residues 1–57), CTD (residues 58–114), and PTN-short (residues 1–114) with the C-terminal tail deleted (Fig. 8A) and examined their ability to support cell adhesion and induce MAPK activation (Fig. 8, B–F). The data showed that removing the C-terminal tail of PTN did not reduce significantly the ability of PTN-short to support adhesion of Mac-1 HEK293 cells compared with intact PTN (Figs. 8B and 1A). Furthermore, the pattern of adhesion of HEK293 cells to PTN-short (Fig. 8B) was similar to that of wild-type PTN (Fig. 1A). Both NTD and CTD were also able to support adhesion (Fig. 8, C and D). However, removing either one of the structural domains of PTN resulted in the reduction of their adhesion-promoting activity (Fig. 8, C and D). In particular, although a 100 nm coating concentration of wild-type PTN was sufficient to achieve a saturable level of adhesion, ∼5- and ∼10-fold higher concentrations of CTD and NTD, respectively, were required to reach saturation. Furthermore, the maximal level of adhesion mediated by these fragments was lower than that mediated by intact PTN. The effect of the removal of either structural domain of PTN on proteoglycan binding was even more severe. Specifically, wild-type HEK293 cells adhered poorly to NTD and CTD even at a concentration of 1 μm (Fig. 8, C and D). To rule out the possibility that differences in the adhesion-promoting activity of PTN-derived fragments were due to their different coating efficiencies, we examined the ability of soluble fragments to inhibit adhesion. Wild-type PTN and all fragments blocked adhesion in a dose-dependent manner (supplemental Fig. S1). At 5 μm, wild-type PTN was the most potent inhibitor (78 ± 13% inhibition of adhesion) followed by PTN-short (68 ± 15%), CTD (57 ± 10%), and NTD (38 ± 12%) (Fig. 8E), thus corroborating the results of adhesion assays. Consistent with the presence of the Mac-1–binding sites in both NTD and CTD domains, treatment of Mac-1 HEK293 cells with each fragment resulted in phosphorylation of Erk1/2, whereas only basal phosphorylation was detected in untreated cells (Fig. 8F and supplemental Fig. S2).
Figure 8.

PTN fragments support adhesion, inhibit adhesion, and induce Erk1/2 activation in Mac-1–expressing HEK293 cells. A, a schematic representation of the domain structure of PTN and recombinant fragments used in the adhesion and Erk1/2 activation assays. B, aliquots (100 μl; 5 × 104/ml) of Mac-1 HEK293 and wild-type HEK293 cells labeled with calcein were added to microtiter wells coated with different concentrations of truncated PTN without the C-terminal tail (PTN-short) and postcoated with 1% PVP. After 30 min at 37 °C, nonadherent cells were removed by washing, and fluorescence of adherent cells was measured. Data shown are means ± S.E. from three separate experiments with triplicate measurements. Error bars represent S. E. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. C and D, adhesion assays with immobilized NTD (C) and CTD (D) were performed as described in B. E, Mac-1 HEK293 cells were preincubated with 5 μm PTN or PTN fragments for 20 min before loading into wells coated with 100 nm PTN and postcoated with 1% PVP. Adhesion in the absence of soluble PTN or PTN fragments was assigned a value of 100%. Data shown are means ± S.E. from three separate experiments with sextuplicate measurements. Error bars represent S. E. *, p ≤ 0.05; ***, p ≤ 0.001 compared with control adhesion in the absence of inhibitors. F, Mac-1 HEK293 cells were treated with 0.7 μm PTN or PTN fragments. After 30 min at 37 °C, cells were lysed, and lysates were separated by 12.5% SDS-PAGE. Western blotting was performed using the mAb specific for the phosphorylated form of Erk1/2 (p-Erk1/2). The mAb against total Erk1/2 was used to equalize protein loading. A representative of three experiments is shown.
Discussion
PTN is a 15-kDa basic heparin-binding protein that induces proliferation, cell growth, and angiogenesis in a wide variety of cells by interacting with specific receptors, including receptor-type protein-tyrosine phosphatase (RPTP) β/ζ, anaplastic lymphoma kinase, N-syndecan, and αVβ3 (3–5, 9, 24). PTN has also been implicated in mediating inflammation based on its ability to trigger in vivo migration of neutrophils and monocyte/macrophages (13, 14). In this study, we demonstrated that PTN induces a potent migratory response in macrophages in vitro and identified integrin Mac-1 as the major receptor mediating cell migration. In support of this finding, we showed that PTN induces migration of Mac-1–expressing HEK293 cells and wild-type mouse macrophages but not wild-type HEK293 cells or Mac-1–deficient macrophages. Mac-1 expressed on HEK293 cells also augmented adhesion to PTN compared with wild-type HEK293 cells and specifically promoted cell spreading. Similarly, PTN also supported adhesion of IC-21 mouse macrophages in a Mac-1–dependent manner. In addition, Mac-1 was essential for PTN-induced activation of Erk1/2. Finally, we showed direct interaction between αMI domain, the ligand-binding region of Mac-1, and PTN using BLI and NMR. These observations establish PTN as a novel ligand for integrin Mac-1 and suggest a role for this protein in the pathophysiologic functions of myeloid leukocytes, which specifically express this receptor.
Integrin Mac-1 is a member of the β2 subfamily of integrin adhesion receptors and is the major receptor on the surface of neutrophils and monocytes/macrophages. This receptor contributes to leukocyte adhesion to and diapedesis through the inflamed endothelium and controls leukocyte migration to sites of inflammation. Moreover, ligand engagement by Mac-1 initiates a variety of cellular responses, including phagocytosis, neutrophil degranulation and aggregation, and expression of cytokines/chemokines and many other pro- and anti-inflammatory molecules (31, 32, 41). Innumerable roles played by Mac-1 in leukocyte biology arise from its multiligand binding properties. Indeed, this receptor exhibits broad recognition specificity and is capable of binding an extremely diverse group of protein and nonprotein ligands. We recently showed that, within its many ligands, Mac-1 binds not to a specific amino acid sequence but rather has a preference for the sequence patterns consisting of a core of positively charged residues flanked by hydrophobic residues (29). In particular, the binding motifs for Mac-1 can be coded as HyBHy, HyHyBHy, HyBHyHy, and HyHyBHyHy. Other amino acids can also be found, but in general, their proportion within the Mac-1–binding motifs is very small, and negatively charged (acidic) residues are largely omitted.
Analyses of the PTN sequence by a previously developed program (29) that predicts the Mac-1–binding sites in its ligands showed that PTN contains several potential αMI domain-recognition sequences, and this prediction was confirmed experimentally (Fig. 7). Screening of the peptide library spanning the sequence of PTN demonstrated the presence of four clusters composed of overlapping αMI domain-binding peptides (Fig. 7, A and B). The αMI domain-binding peptides in clusters 2 and 3 correspond to the segments that form the structural NTD and CTD of PTN. Sequences in the C-terminal part of cluster 2 correspond to the segment in the hinge region (residues 58–66) connecting the TSR domains (Fig. 7, A and C). In addition, the αMI domain-binding sequences identified within clusters 1 and 4 are present in the N- and C-terminal tails, respectively (Fig. 7C). The PTN structure shows that each PTN domain possesses large basic surfaces formed by segments enriched in lysine and arginine that are brought into close proximity by PTN folding (Fig. 9) (25). Within CTD, Lys68, Lys84, Arg86, Lys91, Arg92, and Lys107 contribute to the formation of an extended basic surface (Fig. 9B). Within NTD, Arg35, Arg39, and Lys49 contribute to the basic surface on one side of the β-sheet forming the domain (Fig. 9A). Side chains of Tyr69, Leu90, Val103, and Ile105 in CTD form a small hydrophobic cluster in the middle of CTD (25). Furthermore, Phe63 in the hinge segment that connects NTD and CTD has significant contacts with side chains of residues in the hydrophobic cluster. Because of these interactions, the bend formed in the hinge places Lys60 and Lys61 close to the basic surface in CTD (Fig. 7C). Interestingly, all of the residues forming the basic surfaces in CTD and NTD as well as those found in the hydrophobic cluster and the hinge were found in the αMI domain-binding clusters 2 and 3, and two residues (Val103 and Lys105) are in the beginning of cluster 4. Because basic and hydrophobic amino acid residues were shown to be the main contributors to the interaction between the αMI domain and peptides in the previously screened libraries of Mac-1 ligands (29, 30, 34), it is possible that the basic surfaces in CTD and NTD form the binding sites for the αMI domain. The identification of the majority of the αMI domain-binding residues in NTD and CTD is in agreement with the NMR data showing that many residues in these domains were perturbed in the presence of αMI domain (Fig. 6).
Figure 9.
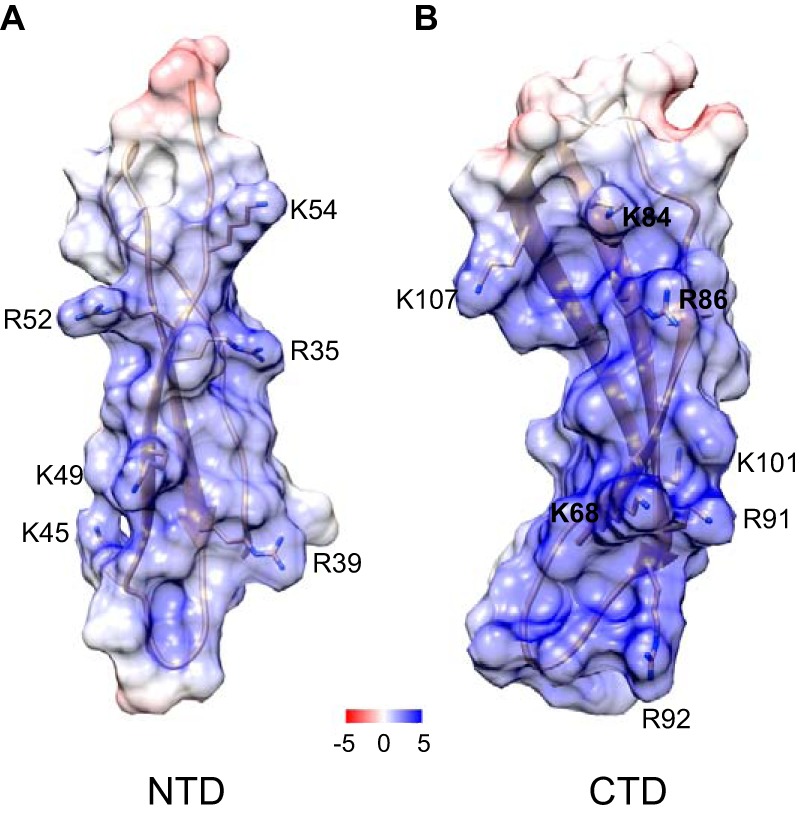
Location of the αMI domain-binding residues in NTD and CTD of PTN. The electrostatic potential mapped is onto the ribbon diagram of NTD (A) and CTD (B). The unit of potential is in kT·e−1. Positively charged residues identified in the αMI domain binders using peptide libraries are shown. The PTN models are based on Protein Data Bank code 2N6F.
To validate the relevance of the proposed Mac-1–binding sites, we investigated adhesion of Mac-1–expressing HEK293 cells to truncated PTN fragments. The results showed that immobilized NTD and CTD can independently support Mac-1–dependent adhesion, and soluble NTD and CTD can inhibit cell adhesion to intact PTN, albeit to a lower extent than soluble intact PTN (Fig. 8, B–E). Interestingly, removal of each NTD or CTD reduced PTN's affinity for cell-surface proteoglycans to a greater extent than its affinity for Mac-1 (Fig. 8, C and D), suggesting that proteoglycans are involved but not absolutely required for adhesion. Based on these data, it is reasonable to propose that each of the two structural domains of PTN can bind Mac-1 independently. Furthermore, it appears that both domains of PTN are required for the optimal proteoglycan binding, and thus the enhanced adhesion of Mac-1–expressing cells to intact PTN may result from the cooperative binding of Mac-1 and proteoglycans.
Consistent with the role of proteoglycans in Mac-1–mediated cell adhesion, previous studies demonstrated that soluble heparin partially blocks adhesion of Mac-1–expressing cells, including human monocytes to various ligands (28, 30, 34). Moreover, the inhibitory effect of anti-Mac-1 function-blocking reagents has been potentiated by heparin. We observed similar effects of heparin and anti-Mac-1 function-blocking mAb 44a on adhesion of Mac-1 HEK293 cells to PTN (Fig. 1). Heparin also partially inhibited migration of Mac-1 HEK293 cells. However, in contrast to adhesion, mAb 44a completely inhibited migration even in the absence of heparin, indicating that Mac-1 was absolutely required for migration (Fig. 3, C and D). These findings suggest that, although HSPGs may influence cell migration in this system, PTN engagement by Mac-1 is indispensable for triggering intracellular signaling that initiates migration. Heparin may exert its inhibitory effect by binding to the heparin-binding sites in PTN or by interacting with Mac-1. Indeed, both NTD and CTD of PTN contain sites that bind heparin with high affinity (27, 42). Furthermore, heparin has also been shown to interact with the αMI domain of Mac-1 (43), although the binding site(s) remains uncertain. These findings suggest that, if present, HSPGs on leukocytes may participate in optimal adhesion and migration to PTN. Moreover, PTN has high affinity for chondroitin sulfate (CS) A and E with a Kd in the low nm range (27, 42). This suggests that CS, which (similar to heparin) is covalently attached to several core proteins, creating a variety of CSPGs on the cell surface, can act cooperatively with Mac-1. The complex relationship among PTN, Mac-1, and cell-surface HS/CS proteoglycans remains poorly understood. Because PTN contains two glycosaminoglycan-binding sites in NTD and CTD and can bind the αMI domain through the same basic surfaces, it may potentially bridge Mac-1 and proteoglycans, inducing their clusterization. The additional binding site for glycosaminoglycans in the C-terminal tail of PTN may also participate in the formation of complexes between Mac-1 and HS/CSPG, further increasing the complexity of multimolecular clusters. Given noticeable differences in the affinity of PTN domains for glycosaminoglycans, it is reasonable to propose that this variability may influence the interactions of PTN with Mac-1 on leukocytes and thus affect cell adhesion, migration, and other Mac-1–dependent leukocyte responses. Interestingly, although HSPGs on Mac-1–expressing HEK293 cells and human monocytes (28) cooperate with Mac-1 in PTN binding, their role in adhesion of IC-21 mouse macrophages appears to be less important because heparin alone does not inhibit adhesion, whereas anti-Mac-1 mAb produces a strong inhibitory effect (Fig. 1E). The reason for the differential involvement of proteoglycans in PTN recognition is unclear but may stem from differences in the composition of glycosaminoglycans expressed on various cells. In addition to cell-surface proteoglycans, ECM proteoglycans may also play a role in storage and presentation of PTN to migrating cells. In this regard, we showed that PTN bound to aggrecan, a well-known CSPG in the ECM, supports adhesion, suggesting that PTN deposited in the ECM remains an active Mac-1 ligand.
Binding of several ligands to Mac-1 is controlled by the activation state of the receptor. Previous studies demonstrated that the αMI domain exists in two different conformations, active and nonactive, with the position of the C-terminal α7 helix regulating its activation state (44). In contrast to such Mac-1 ligands as C3bi, recognition of which requires the active state of the αMI domain (45), the nonactive form of the αMI domain bound PTN, albeit to a lower extent than active αMI domain (Fig. 5C). This finding suggests that the αMI domain binding to PTN is not strictly activation-dependent. Furthermore, in contrast to other integrin ligands, binding of PTN to the active αMI domain still occurred in the absence of Mg2+ or in the presence of EDTA (Fig. 5C). Similar characteristics, i.e. the lack of dependence on the activation state of the αMI domain and divalent cations, have been noted with some Mac-1 ligands, including opioid peptide dynorphin A (30), fibrinogen peptide P2-C (39), and cationic proteins myeloperoxidase and elastase.4 Because all these molecules are highly positively charged and their binding to the αMI domain is mediated by sequences enriched in basic residues (29), it is tempting to speculate that cationic peptides/proteins represent a unique group of Mac-1 ligands that bind the αMI domain through activation- and cation-insensitive mechanisms. Further structural studies of the αMI domain-PTN complex may help to define the mechanism of this interaction.
Although the biological significance of leukocyte interaction with PTN remains to be established, the expression of PTN in different cells during regeneration after injury (2, 46) suggests that it may play a role in inflammatory responses. In this regard, PTN has been shown to promote neutrophil and mononocyte/macrophage recruitment during liver regeneration and peritoneal fibrosis (13, 14). Our in vitro studies showing that Mac-1 can mediate migration of macrophages and Mac-1–expressing HEK293 cells to PTN suggest that leukocyte migration in vivo may also be driven by this receptor. Furthermore, the only known PTN homolog, midkine (MK), which shares 50% sequence identity and a similar tertiary structure with PTN, is expressed in damaged tissues and has a comparable proinflammatory profile (47). It has been reported that MK enhances migration of inflammatory leukocytes in a number of pathological conditions induced in experimental animals (14, 48–52). MK receptors responsible for triggering these responses are not known, but MK has been shown to activate both β1 and β2 integrins (53, 54). Furthermore, adhesion to immobilized MK was shown to be mediated by β2 integrins, although the nature of α subunit pairing with the β2 subunit has not been determined. Both PTN and MK have also been shown to induce expression of inflammatory cytokines in peripheral blood monocytes (47, 55). Because ligand engagement by Mac-1 is known to initiate intracellular signaling that regulates numerous leukocyte responses, including expression of cytokines, a receptor that mediates this effect upon PTN and MK binding is most likely to be Mac-1. The potential role of PTN in inducing other reactions of monocyte/macrophages that are known to be mediated by Mac-1 merits further investigation.
Experimental procedures
Reagents
The mouse mAb 44a directed against the human αM integrin subunit was purified from conditioned media of hybridoma cells obtained from the American Type Culture Collection (Manassas, VA) using protein A-agarose. Alexa Fluor 546-conjugated phalloidin was purchased from Life Technologies. The mouse mAb G3A1, an IgG1 isotype control for mAb 44a, was obtained from Cell Signaling Technology (Beverly, MA). BSA, polyvinylpyrrolidone (PVP), EDTA, heparin, and aggrecan were purchased from Sigma. Calcein-AM was purchased from Molecular Probes (Eugene, OR). mAbs directed to total Erk1/2 and the phosphorylated form of Erk1/2 were from Cell Signaling Technology. Goat anti-rabbit IgG (heavy + light)-HRP conjugated antibody was from Bio-Rad. Protease and phosphatase inhibitor mixture was from Thermo Scientific (Rockford, IL).
Expression and purification of recombinant proteins
Expression and purification of PTN were performed as described previously (25). Briefly, human PTN ORF cloned into the pET-15b vector was transformed into Origami B (DE3) cells (Novagen, Madison, WI). The transformed cells were grown in M9 medium at 37 °C until induction with 0.25 mm isopropyl 1-thio-β-d-galactopyranoside when A600 reached 0.8. The culture was then incubated with shaking overnight at room temperature. The PTN was purified from supernatant using heparin-affinity chromatography with a 5-ml HiTrap heparin column (GE Healthcare). Truncated PTN fragments, PTN-short (residues Gly1–Lys114), NTD (residues Gly1–Cys57), and CTD (residues Asn58–Lys114) were cloned into the pET-15b vector and expressed as described above. Expression of recombinant active (residues αMGlu123–Lys315) and nonactive (residues αMGln119–Glu333) αMI domains as well as active αLI domain (residues αLGly127–Tyr307; K287C,K294C) were described previously (39). Alternatively, the αMI domain ORF was cloned into pHUE vector (56) as a fusion protein with ubiquitin and transformed into BL21 (DE3) cells (Bioline). The αMI domains were purified with 5-ml HisTrap HP nickel columns (GE Healthcare). To remove the His-tagged ubiquitin, the fusion protein was incubated with the ubiquitinase USP2 overnight at a protein-to-enzyme ratio of 30:1. The pure αMI domain was separated from other impurities by size-exclusion chromatography using a Superdex 75 column (GE Healthcare). The αMI domain was biotinylated with EZ-Link Sulfo-NHS-LC-Biotin (Thermo Fisher Scientific, Inc., San Jose, CA). The efficiency of biotinylation was tested using a biotin quantitation assay kit (Thermo Fisher Scientific, Inc.) with biotinylated horseradish peroxidase as a standard.
Screening of peptide libraries for αMI domain binding
A peptide library was prepared by parallel spot synthesis on cellulose membranes as described previously (57, 58). Purified recombinant αMI domain was labeled with 125I using Iodo-Gen (Pierce). The library spanning the sequence of PTN was synthesized as 9-mer overlapping peptides with a 3-amino acid offset. Peptides were C-terminally attached to the cellulose via a (β-Ala)2 spacer and were acetylated N-terminally. The membrane with attached peptides was blocked with 1% BSA and then incubated with 10 μg/ml 125I-labeled αMI domain in TBS containing 1 mm MgCl2, 0.05% Tween 20, and 1 mm dithiothreitol. After washing, the membrane was dried, and the αMI domain binding was visualized by autoradiography and analyzed by densitometry.
NMR
PTN 1H-15N HSQC NMR data were collected using a Bruker 600-MHz Avance III HD spectrometer (Bruker Corp., Billerica, MA) equipped with a Prodigy probe. The NMR samples contained 40 μm PTN in PBS buffer at pH 7.5 and either 0 or 40 μm unlabeled αMI domain. Data were processed using NMRPipe (59) and analyzed with NMRView (60).
Cells
HEK293 and Mac-1 HEK293 cells were described previously (61). The cells were maintained in DMEM (Mediatech Inc., Manassas, VA) with 10% fetal bovine serum and antibiotics. The IC-21 mouse macrophage cell line was obtained from ATCC. Mouse peritoneal macrophages were isolated from the peritoneal lavage of wild-type (C57BL/6J) and Mac-1−/− (B6.129S4-Itgamtm1Myd/J) mice 3 days after thioglycolate injection using the EasySep Mouse selection kit (StemCell Technologies) with mAb F4/80 conjugated to FITC.
Cell adhesion assays
Cell adhesion assays were performed according to methods described previously (36, 61). Briefly, 96-well microtiter plates (Immulon 4HBX, Thermo) were coated with different concentrations of PTN and PTN fragments (PTN, PTN-short, CTD, and NTD) for 3 h at 37 °C and blocked with 1% PVP in PBS for 1 h at 37 °C. The cells were labeled with 7.5 μm calcein-AM (Thermo) for 30 min at 37 °C. The labeled cells were washed with Hanks' balanced salt solution containing 0.1% BSA and resuspended in the same buffer at a concentration of 5 × 105/ml. Aliquots (100 μl) of labeled cells were added to each well and incubated for 30 min at 37 °C. Non-adherent cells were removed with two washes with PBS, and fluorescence was measured with a fluorescence plate reader (Perseptive Biosystems, Framingham, MA). For inhibition experiments, labeled cells were treated with 5 μg/ml anti-Mac-1 antibodies (anti-αM mAb 44a and M1/70 for Mac-1 HEK293 and IC-21 macrophages, respectively) or heparin (1 μg/ml) for 15 min at 22 °C before they were added to the wells. For PTN inhibition experiments, cells were treated with either 2 or 5 μm soluble PTN or PTN fragments for 20 min at 22 °C before they were added to the wells. To test the effect of aggrecan-bound PTN, 10 μg/ml aggrecan in PBS was used to coat 96-well microtiter plates overnight at 4 °C as described (62). Then PTN was added to aggrecan-coated wells for 3 h at 37 °C. The remaining steps were the same as described above. The coating efficiency of aggrecan was determined using biotinylated aggrecan.
Cell migration assays
Cell migration assays were performed under sterile conditions using Transwell inserts (Costar, Corning, NY) with a pore size of 8 (HEK293 cells) or 5 μm (macrophages) as described (30, 38). Briefly, after coating PTN on the Transwell membrane at 37 °C for 3 h, cells (100 μl) were loaded in the upper chamber of the Transwell system at a concentration of 3 × 106/ml. For inhibition experiments, cells were pretreated with 20 μg/ml anti-αM mAb 44a or 1 μg/ml heparin for 20 min at 22 °C before loading into the upper chamber of the Transwell system. The lower chamber contained 600 μl of DMEM. Migration took place at 37 °C in a 5% CO2 humidified atmosphere for 16 h. For detection, the HEK293 cells were labeled with calcein-AM for 30 min at 37 °C, and macrophages were labeled with FITC during isolation. Cells from the upper chamber of the Transwells were removed by wiping with a cotton-tipped applicator. Images of the cells on the underside of the Transwell filter were taken with a Leica DM 4000B microscope camera (Leica, Buffalo Grove, IL).
Cell spreading assays
For cell spreading assays, glass coverslips coated with 1.5 μg/ml PTN were placed in 6-well plates (Costar), and 4 × 105 cells were added to each well. After incubation at 37 °C for different times (30 min, 1, 2, 8, and 24 h), cells were washed with PBS, fixed with 2% paraformaldehyde for 10 min at room temperature, and then treated with 0.1% Triton X-100. For actin staining, the cells were treated with 200 μl of 165 mm phalloidin solution for 20 min at room temperature. DAPI was added for DNA staining. The specimens were imaged with a Leica SP5 laser-scanning confocal microscope using 63×/1.4 objective. The diameters of 20–50 cells for each time point were counted using ImageJ software from images taken with a 20×/0.5 objective. The diameter was defined as the largest distance between two opposite sides of the cell.
Biolayer interferometry
The experiments were performed using an Octet K2 instrument (ForteBio, Pall Corp.). Purified PTN was immobilized on the amine-reactive second-generation (AR2G) biosensor using the amine coupling kit according to the manufacturer's protocol. Different concentrations of the active and nonactive forms of αMI domain and active αLI domain were applied in the mobile phase, and the association between the immobilized and flowing proteins was detected. Experiments were performed in 20 mm HEPES, 150 mm NaCl, 1 mm MgCl2, and 0.05% (v/v) Tween 20, pH 7.5, or in 20 mm HEPES, 150 mm NaCl, 5 mm EDTA, and 0.05% (v/v) Tween 20, pH 7.5. The PTN-coated surface was regenerated with 25 mm NaOH. Analyses of the binding kinetics were performed using ForteBio Data Analysis 9.0 software. The Kd was obtained by curve fitting of the association and dissociation phases of sensorgrams using a heterogeneous ligand model. BLI experiments were also carried out using PTN biotinylated through glutamate or aspartate side chains with amine-PEG2-biotin (Thermo Fisher Scientific). Biotinylated PTN at a concentration 50 μg/ml was immobilized on the streptavidin biosensor in 20 mm HEPES buffer supplemented with 150 mm NaCl, 0.1% BSA, 0.02% Tween 20, 1 mm MgCl2, and 1 mm CaCl2 for 15 min, which generated a saturated level of PTN on the biosensor. After immobilization, the biosensor was washed with HEPES buffer, and different concentrations of αMI domain were added to the immobilized PTN. After each association–dissociation cycle, the biosensor was regenerated using 20 mm NaOH.
Western blotting
HEK293 cells and Mac-1 HEK293 cells were added to the wells of 6-well culture plates at 2 × 106/well and cultured for 2 h in DMEM/F-12 + 10% FBS. Adherent cells were treated with PTN and PTN fragments (0.7 μm). After 30 min at 37 °C in a humidified 5% CO2 atmosphere, cells were washed with ice-cold PBS containing a protease and phosphatase inhibitor mixture. Next, 200 μl of the lysis buffer (50 mm Tris-HCl, pH 7.4, 1% Triton X-100, 150 mm NaCl, 1 mm EDTA, and protease and phosphatase inhibitor mixture) was added to each well, and cells were incubated on ice for 1 h. Protein concentration in the lysates was quantified by a BCA assay (Thermo Scientific), and equal amounts of protein (20 μg) were loaded onto 12.5% SDS-polyacrylamide gels. After the transfer of proteins onto nitrocellulose membranes, the membranes were blocked with skim milk and incubated with anti-Erk1/2 and anti-phospho-Erk1/2 (Tyr202 and Tyr204) rabbit mAbs. The mAb binding was detected using HRP-conjugated goat anti-rabbit antibodies and chemiluminescence (Thermo Scientific).
Statistical analysis
All data are presented as the mean ± S.E. The statistical differences were determined using one-way analysis of variance using SigmaPlot 11.0 software (Systat Software, San Jose, CA). For multiple comparisons, the Bonferroni correction method was used. Differences were considered significant if the p value was less than 0.05.
Author contributions
N. P. P., T. P. U., and X. W. designed the research study. D. S., N. P. P., A. B., C. L. A., and V. P. Y. performed experiments. D. S., N. P. P., A. B., V. P. Y., T. P. U., and X. W. analyzed data. T. P. U., N. P. P., and X. W. wrote the manuscript.
Supplementary Material
This work was supported by National Institutes of Health Grants GM118339 (to X. W.), GM118518 (to X. W.), HL63199 (to T. P. U.), and DK102020 (to V. P. Y.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1 and S2.
D. Shen, N. P. Podolnikova, V. P. Yakubenko, C. L. Ardell, A. Balabiyev, T. P. Ugarova, and X. Wang, unpublished data.
- PTN
- pleiotrophin
- HSPG
- heparan sulfate proteoglycan
- TSR
- thrombospondin type-1 repeat
- MAP
- mitogen-activated protein
- ECM
- extracellular matrix
- BLI
- biolayer interferometry
- HSQC
- heteronuclear single quantum coherence
- NTD
- N-terminal TSR domain
- CTD
- C-terminal TSR domain
- CS
- chondroitin sulfate
- CSPG
- chondroitin sulfate proteoglycan
- MK
- midkine
- PVP
- polyvinylpyrrolidone.
References
- 1. González-Castillo C., Ortuño-Sahagún D., Guzmán-Brambila C., Pallàs M., and Rojas-Mayorquín A. E. (2014) Pleiotrophin as a central nervous system neuromodulator, evidences from the hippocampus. Front. Cell. Neurosci. 8, 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perez-Pinera P., Berenson J. R., and Deuel T. F. (2008) Pleiotrophin, a multifunctional angiogenic factor: mechanisms and pathways in normal and pathological angiogenesis. Curr. Opin. Hematol. 15, 210–214 [DOI] [PubMed] [Google Scholar]
- 3. Himburg H. A., Harris J. R., Ito T., Daher P., Russell J. L., Quarmyne M., Doan P. L., Helms K., Nakamura M., Fixsen E., Herradon G., Reya T., Chao N. J., Harroch S., and Chute J. P. (2012) Pleiotrophin regulates the retention and self-renewal of hematopoietic stem cells in the bone marrow vascular niche. Cell Rep. 2, 964–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Himburg H. A., Yan X., Doan P. L., Quarmyne M., Micewicz E., McBride W., Chao N. J., Slamon D. J., and Chute J. P. (2014) Pleiotrophin mediates hematopoietic regeneration via activation of RAS. J. Clin. Investig. 124, 4753–4758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hatziapostolou M., Delbe J., Katsoris P., Polytarchou C., Courty J., and Papadimitriou E. (2005) Heparin affin regulatory peptide is a key player in prostate cancer cell growth and angiogenicity. Prostate 65, 151–158 [DOI] [PubMed] [Google Scholar]
- 6. Papadimitriou E., Mikelis C., Lampropoulou E., Koutsioumpa M., Theochari K., Tsirmoula S., Theodoropoulou C., Lamprou M., Sfaelou E., Vourtsis D., and Boudouris P. (2009) Roles of pleiotrophin in tumor growth and angiogenesis. Eur. Cytokine Netw. 20, 180–190 [DOI] [PubMed] [Google Scholar]
- 7. Jäger R., Noll K., Havemann K., Pflüger K. H., Knabbe C., Rauvala H., and Zugmaier G. (1997) Differential expression and biological activity of the heparin-binding growth-associated molecule (HB-GAM) in lung cancer cell lines. Int. J. Cancer 73, 537–543 [DOI] [PubMed] [Google Scholar]
- 8. Mentlein R., and Held-Feindt J. (2002) Pleiotrophin, an angiogenic and mitogenic growth factor, is expressed in human gliomas. J. Neurochem. 83, 747–753 [DOI] [PubMed] [Google Scholar]
- 9. Zhang N., Zhong R., Wang Z. Y., and Deuel T. F. (1997) Human breast cancer growth inhibited in vivo by a dominant negative pleiotrophin mutant. J. Biol. Chem. 272, 16733–16736 [DOI] [PubMed] [Google Scholar]
- 10. Weber D., Klomp H. J., Czubayko F., Wellstein A., and Juhl H. (2000) Pleiotrophin can be rate-limiting for pancreatic cancer cell growth. Cancer Res. 60, 5284–5288 [PubMed] [Google Scholar]
- 11. Kong Y., Bai P. S., Nan K. J., Sun H., Chen N. Z., and Qi X. G. (2012) Pleiotrophin is a potential colorectal cancer prognostic factor that promotes VEGF expression and induces angiogenesis in colorectal cancer. Int. J. Colorectal Dis. 27, 287–298 [DOI] [PubMed] [Google Scholar]
- 12. Koutsioumpa M., Poimenidi E., Pantazaka E., Theodoropoulou C., Skoura A., Megalooikonomou V., Kieffer N., Courty J., Mizumoto S., Sugahara K., and Papadimitriou E. (2015) Receptor protein tyrosine phosphatase β/ζ is a functional binding partner for vascular endothelial growth factor. Mol. Cancer 14, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yokoi H., Kasahara M., Mori K., Ogawa Y., Kuwabara T., Imamaki H., Kawanishi T., Koga K., Ishii A., Kato Y., Mori K. P., Toda N., Ohno S., Muramatsu H., Muramatsu T., et al. (2012) Pleiotrophin triggers inflammation and increased peritoneal permeability leading to peritoneal fibrosis. Kidney Int. 81, 160–169 [DOI] [PubMed] [Google Scholar]
- 14. Ochiai K., Muramatsu H., Yamamoto S., Ando H., and Muramatsu T. (2004) The role of midkine and pleiotrophin in liver regeneration. Liver Int. 24, 484–491 [DOI] [PubMed] [Google Scholar]
- 15. Besse S., Comte R., Fréchault S., Courty J., Joël de L, Delbé J. (2013) Pleiotrophin promotes capillary-like sprouting from senescent aortic rings. Cytokine 62, 44–47 [DOI] [PubMed] [Google Scholar]
- 16. Fang Q., Mok P. Y., Thomas A. E., Haddad D. J., Saini S. A., Clifford B. T., Kapasi N. K., Danforth O. M., Usui M., Ye W., Luu E., Sharma R., Bartel M. J., Pathmanabhan J. A., Ang A. A., et al. (2013) Pleiotrophin gene therapy for peripheral ischemia: evaluation of full-length and truncated gene variants. PLoS One 8, e61413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Silver K., Desormaux A., Freeman L. C., and Lillich J. D. (2012) Expression of pleiotrophin, an important regulator of cell migration, is inhibited in intestinal epithelial cells by treatment with non-steroidal anti-inflammatory drugs. Growth Factors 30, 258–266 [DOI] [PubMed] [Google Scholar]
- 18. Yu Y., Shi M. H., Xu X., and Hu H. C. (2010) Construction of siRNA lentiviral expressing vector targeting pleiotrophin gene and its impact on growth and apoptosis in H446 cells of human small cell lung cancer. Zhonghua Jie He He Hu Xi Za Zhi 33, 289–294 [PubMed] [Google Scholar]
- 19. Raulo E., Chernousov M. A., Carey D. J., Nolo R., and Rauvala H. (1994) Isolation of a neuronal cell surface receptor of heparin binding growth-associated molecule (HB-GAM). Identification as N-syndecan (syndecan-3). J. Biol. Chem. 269, 12999–13004 [PubMed] [Google Scholar]
- 20. Meng K., Rodriguez-Peña A., Dimitrov T., Chen W., Yamin M., Noda M., and Deuel T. F. (2000) Pleiotrophin signals increased tyrosine phosphorylation of β β-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase β/ζ. Proc. Natl. Acad. Sci. U.S.A. 97, 2603–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maeda N., Nishiwaki T., Shintani T., Hamanaka H., and Noda M. (1996) 6B4 proteoglycan/phosphacan, an extracellular variant of receptor-like protein-tyrosine phosphatase ζ/RPTPβ, binds pleiotrophin/heparin-binding growth-associated molecule (HB-GAM). J. Biol. Chem. 271, 21446–21452 [DOI] [PubMed] [Google Scholar]
- 22. Koutsioumpa M., Drosou G., Mikelis C., Theochari K., Vourtsis D., Katsoris P., Giannopoulou E., Courty J., Petrou C., Magafa V., Cordopatis P., and Papadimitriou E. (2012) Pleiotrophin expression and role in physiological angiogenesis in vivo: potential involvement of nucleolin. Vasc. Cell 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Take M., Tsutsui J., Obama H., Ozawa M., Nakayama T., Maruyama I., Arima T., and Muramatsu T. (1994) Identification of nucleolin as a binding protein for midkine (MK) and heparin-binding growth associated molecule (HB-GAM). J. Biochem. 116, 1063–1068 [DOI] [PubMed] [Google Scholar]
- 24. Mikelis C., Sfaelou E., Koutsioumpa M., Kieffer N., and Papadimitriou E. (2009) Integrin αvβ3 is a pleiotrophin receptor required for pleiotrophin-induced endothelial cell migration through receptor protein tyrosine phosphatase β/ζ. FASEB J. 23, 1459–1469 [DOI] [PubMed] [Google Scholar]
- 25. Ryan E., Shen D., and Wang X. (2016) Structural studies reveal an important role for the pleiotrophin C-terminus in mediating interactions with chondroitin sulfate. FEBS J. 283, 1488–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kilpelainen I., Kaksonen M., Kinnunen T., Avikainen H., Fath M., Linhardt R. J., Raulo E., and Rauvala H. (2000) Heparin-binding growth-associated molecule contains two heparin-binding β-sheet domains that are homologous to the thrombospondin type I repeat. J. Biol. Chem. 275, 13564–13570 [DOI] [PubMed] [Google Scholar]
- 27. Raulo E., Tumova S., Pavlov I., Pekkanen M., Hienola A., Klankki E., Kalkkinen N., Taira T., Kilpelaïnen I., and Rauvala H. (2005) The two thrombospondin type I repeat domains of the heparin-binding growth-associated molecule bind to heparin/heparan sulfate and regulate neurite extension and plasticity in hippocampal neurons. J. Biol. Chem. 280, 41576–41583 [DOI] [PubMed] [Google Scholar]
- 28. Schober J. M., Chen N., Grzeszkiewicz T. M., Jovanovic I., Emeson E. E., Ugarova T. P., Ye R. D., Lau L. F., and Lam S. C. (2002) Identification of integrin αMβ2 as an adhesion receptor on peripheral blood monocytes for Cyr61 (CCN1) and connective tissue growth factor (CCN2): immediate-early gene products expressed in atherosclerotic lesions. Blood 99, 4457–4465 [DOI] [PubMed] [Google Scholar]
- 29. Podolnikova N. P., Podolnikov A. V., Haas T. A., Lishko V. K., and Ugarova T. P. (2015) Ligand recognition specificity of leukocyte integrin αMβ2 (Mac-1, CD11b/CD18) and its functional consequences. Biochemistry 54, 1408–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Podolnikova N. P., Brothwell J. A., and Ugarova T. P. (2015) The opioid peptide dynorphin A induces leukocyte responses via integrin Mac-1 (αMβ2, CD11b/CD18). Mol. Pain 11, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Coxon A., Rieu P., Barkalow F. J., Askari S., Sharpe A. H., von Andrian U. H., Arnaout M. A., and Mayadas T. N. (1996) A novel role for the β2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity 5, 653–666 [DOI] [PubMed] [Google Scholar]
- 32. Lu H., Smith C. W., Perrard J., Bullard D., Tang L., Shappell S. B., Entman M. L., Beaudet A. L., and Ballantyne C. M. (1997) LFA-1 is sufficient in mediating neutrophil emigration in Mac-1 deficient mice. J. Clin. Investig. 99, 1340–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lishko V. K., Podolnikova N. P., Yakubenko V. P., Yakovlev S., Medved L., Yadav S. P., and Ugarova T. P. (2004) Multiple binding sites in fibrinogen for integrin αMβ2 (Mac-1). J. Biol. Chem. 279, 44897–44906 [DOI] [PubMed] [Google Scholar]
- 34. Lishko V. K., Moreno B., Podolnikova N. P., and Ugarova T. P. (2016) Identification of human cathelicidin peptide LL-37 as a ligand for macrophage integrin αMβ2 (Mac-1, CD11b/CD18) that promotes phagocytosis by opsonizing bacteria. Res. Rep. Biochem. 2016, 39–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Luo B. H., Carman C. V., and Springer T. A. (2007) Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25, 619–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lishko V. K., Yakubenko V. P., and Ugarova T. P. (2003) The interplay between integrins αMβ2 and α5β1 during cell migration to fibronectin. Exp. Cell Res. 283, 116–126 [DOI] [PubMed] [Google Scholar]
- 37. Lu K. V., Jong K. A., Kim G. Y., Singh J., Dia E. Q., Yoshimoto K., Wang M. Y., Cloughesy T. F., Nelson S. F., and Mischel P. S. (2005) Differential induction of glioblastoma migration and growth by two forms of pleiotrophin. J. Biol. Chem. 280, 26953–26964 [DOI] [PubMed] [Google Scholar]
- 38. Forsyth C. B., Solovjov D. A., Ugarova T. P., and Plow E. F. (2001) Integrin αMβ2-mediated cell migration to fibrinogen and its recognition peptides. J. Exp. Med. 193, 1123–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yakubenko V. P., Solovjov D. A., Zhang L., Yee V. C., Plow E. F., and Ugarova T. P. (2001) Identification of the binding site for fibrinogen recognition peptide γ383–395 within the αM I-domain of integrin αMβ2. J. Biol. Chem. 276, 13995–14003 [DOI] [PubMed] [Google Scholar]
- 40. Cavanagh J., Fairbrother W. J., Palmer A. G. III, Rance M. and Skelton N. J. (2007) Protein NMR Spectroscopy: Principles and Practice, 2nd Ed., Elsevier, Amsterdam [Google Scholar]
- 41. Ding Z. M., Babensee J. E., Simon S. I., Lu H., Perrard J. L., Bullard D. C., Dai X. Y., Bromley S. K., Dustin M. L., Entman M. L., Smith C. W., and Ballantyne C. M. (1999) Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J. Immunol. 163, 5029–5038 [PubMed] [Google Scholar]
- 42. Ori A., Free P., Courty J., Wilkinson M. C., and Fernig D. G. (2009) Identification of heparin-binding sites in proteins by selective labeling. Mol. Cell. Proteomics 8, 2256–2265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Diamond M. S., Alon R., Parkos C. A., Quinn M. T., and Springer T. A. (1995) Heparin is an adhesive ligand for the leukocyte integrin Mac-1 (CD11b/CD18). J. Cell Biol. 130, 1473–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee J.-O., Bankston L. A., Arnaout M. A., and Liddington R. C. (1995) Two conformations of the integrin A-domain (I-domain): a pathway for activation? Structure 3, 1333–1340 [DOI] [PubMed] [Google Scholar]
- 45. Ajroud K., Sugimori T., Goldmann W. H., Fathallah D. M., Xiong J. P., and Arnaout M. A. (2004) Binding affinity of metal ions to the CD11b A-domain is regulated by integrin activation and ligands. J. Biol. Chem. 279, 25483–25488 [DOI] [PubMed] [Google Scholar]
- 46. Yeh H. J., He Y. Y., Xu J., Hsu C. Y., and Deuel T. F. (1998) Upregulation of pleiotrophin gene expression in developing microvasculature, macrophages, and astrocytes after acute ischemic brain injury. J. Neurosci. 18, 3699–3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Muramatsu T. (2014) Structure and function of midkine as the basis of its pharmacological effects. Br. J. Pharmacol. 171, 814–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Takada T., Toriyama K., Muramatsu H., Song X. J., Torii S., and Muramatsu T. (1997) Midkine, a retinoic acid-inducible heparin-binding cytokine in inflammatory responses: chemotactic activity to neutrophils and association with inflammatory synovitis. J. Biochem. 122, 453–458 [DOI] [PubMed] [Google Scholar]
- 49. Horiba M., Kadomatsu K., Nakamura E., Muramatsu H., Ikematsu S., Sakuma S., Hayashi K., Yuzawa Y., Matsuo S., Kuzuya M., Kaname T., Hirai M., Saito H., and Muramatsu T. (2000) Neointima formation in a restenosis model is suppressed in midkine-deficient mice. J. Clin. Investig. 105, 489–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sato W., Kadomatsu K., Yuzawa Y., Muramatsu H., Hotta N., Matsuo S., and Muramatsu T. (2001) Midkine is involved in neutrophil infiltration into the tubulointerstitium in ischemic renal injury. J. Immunol. 167, 3463–3469 [DOI] [PubMed] [Google Scholar]
- 51. Kadomatsu K. (2005) The midkine family in cancer, inflammation and neural development. Nagoya J. Med. Sci. 67, 71–82 [PubMed] [Google Scholar]
- 52. Muramatsu T. (2010) Midkine, a heparin-binding cytokine with multiple roles in development, repair and diseases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 86, 410–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Muramatsu H., Zou P., Suzuki H., Oda Y., Chen G. Y., Sakaguchi N., Sakuma S., Maeda N., Noda M., Takada Y., and Muramatsu T. (2004) α4β1- and α6β1-integrins are functional receptors for midkine, a heparin-binding growth factor. J. Cell Sci. 117, 5405–5415 [DOI] [PubMed] [Google Scholar]
- 54. Weckbach L. T., Gola A., Winkelmann M., Jakob S. M., Groesser L., Borgolte J., Pogoda F., Pick R., Pruenster M., Müller-Höcker J., Deindl E., Sperandio M., and Walzog B. (2014) The cytokine midkine supports neutrophil trafficking during acute inflammation by promoting adhesion via β2 integrins (CD11/CD18). Blood 123, 1887–1896 [DOI] [PubMed] [Google Scholar]
- 55. Achour A., M'bika J. P., Baudouin F., Caruelle D., and Courty J. (2008) Pleiotrophin induces expression of inflammatory cytokines in peripheral blood mononuclear cells. Biochimie 90, 1791–1795 [DOI] [PubMed] [Google Scholar]
- 56. Catanzariti A. M., Soboleva T. A., Jans D. A., Board P. G., and Baker R. T. (2004) An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Sci. 13, 1331–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Frank R. (1992) Spot-synthesis: an easy technique for the positionally addressable, parallel chemical synthesis on a membrane support. Tetrahedron 48, 9217–9232 [Google Scholar]
- 58. Kramer A., and Schneider-Mergener J. (1998) Synthesis and screening of peptide libraries on continuous cellulose membrane supports. Methods Mol. Biol. 87, 25–39 [DOI] [PubMed] [Google Scholar]
- 59. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 60. Johnson B. A. (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278, 313–352 [DOI] [PubMed] [Google Scholar]
- 61. Yakubenko V. P., Lishko V. K., Lam S. C., and Ugarova T. P. (2002) A molecular basis for integrin αMβ2 ligand binding promiscuity. J. Biol. Chem. 277, 48635–48642 [DOI] [PubMed] [Google Scholar]
- 62. Paveliev M., Fenrich K. K., Kislin M., Kuja-Panula J., Kulesskiy E., Varjosalo M., Kajander T., Mugantseva E., Ahonen-Bishopp A., Khiroug L., Kulesskaya N., Rougon G., and Rauvala H. (2016) HB-GAM (pleiotrophin) reverses inhibition of neural regeneration by the CNS extracellular matrix. Sci. Rep. 6, 33916. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.