

Abstract
Thioredoxin 1 (Trx1) is a 12-kDa oxidoreductase that catalyzes thiol-disulfide exchange reactions to reduce proteins with disulfide bonds. As such, Trx1 helps protect the heart against stresses, such as ischemia and pressure overload. Mechanistic target of rapamycin (mTOR) is a serine/threonine kinase that regulates cell growth, metabolism, and survival. We have shown previously that mTOR activity is increased in response to myocardial ischemia–reperfusion injury. However, whether Trx1 interacts with mTOR to preserve heart function remains unknown. Using a substrate-trapping mutant of Trx1 (Trx1C35S), we show here that mTOR is a direct interacting partner of Trx1 in the heart. In response to H2O2 treatment in cardiomyocytes, mTOR exhibited a high molecular weight shift in non-reducing SDS-PAGE in a 2-mercaptoethanol-sensitive manner, suggesting that mTOR is oxidized and forms disulfide bonds with itself or other proteins. The mTOR oxidation was accompanied by reduced phosphorylation of endogenous substrates, such as S6 kinase (S6K) and 4E-binding protein 1 (4E-BP1) in cardiomyocytes. Immune complex kinase assays disclosed that H2O2 treatment diminished mTOR kinase activity, indicating that mTOR is inhibited by oxidation. Of note, Trx1 overexpression attenuated both H2O2-mediated mTOR oxidation and inhibition, whereas Trx1 knockdown increased mTOR oxidation and inhibition. Moreover, Trx1 normalized H2O2-induced down-regulation of metabolic genes and stimulation of cell death, and an mTOR inhibitor abolished Trx1-mediated rescue of gene expression. H2O2-induced oxidation and inhibition of mTOR were attenuated when Cys-1483 of mTOR was mutated to phenylalanine. These results suggest that Trx1 protects cardiomyocytes against stress by reducing mTOR at Cys-1483, thereby preserving the activity of mTOR and inhibiting cell death.
Keywords: heart, mechanistic target of rapamycin (mTOR), oxidative stress, redox regulation, thioredoxin
Introduction
Reactive oxygen species (ROS),3 such as hydrogen peroxide (H2O2), are generated by various biological reactions, including mitochondrial respiration and activation of ROS-producing enzymes such as NADPH oxidases. Increased ROS are frequently observed under various pathological and stressed conditions and are generally considered to promote the pathology because of oxidative damage of proteins, lipids, and DNAs. To protect against ROS, cells express antioxidant systems, including thioredoxins and glutathione, and reducing enzymes, such as superoxide dismutase, and catalase.
Thioredoxin 1 (Trx1) is a reducing enzyme evolutionarily conserved from prokaryotes to mammals that regulates a wide range of biological reactions, including DNA synthesis, immune reaction, transcription, and stress resistance (1). Trx1 possesses two cysteine residues in its catalytic center comprising a CXXC motif, which reduces the disulfides of oxidized proteins through disulfide formation with the catalytic center. Oxidized Trx1 is then reduced by Trx reductase. Trx1 protects against oxidative stress (2). The cytoprotective effect of Trx1 renders resistance to ischemic injury in cardiomyocytes (3, 4) and drug resistance in cancer cells (5). Although Trx1 itself does not react effectively with H2O2, Trx1 reduces H2O2 through peroxiredoxins, Trx1-dependent peroxidases (6). In addition, Trx1 directly reduces specific substrates, including NFκB, class II histone deacetylases, caspase, and AMP-activated protein kinase (AMPK), through thiol-disulfide exchange reactions (7). Trx1 is a unique redox regulator that is deeply involved in signal transduction. Identification and characterization of Trx1 substrates may allow discovery of novel biological reactions that serve to counteract oxidative stress.
The mechanistic target of rapamycin (mTOR) is an evolutionarily conserved serine/threonine kinase that regulates cell growth, survival, metabolism, and protein synthesis (8). There are two distinct mTOR complexes: mTOR complex 1 (mTORC1) and mTORC2. Although both complexes contain mTOR as a common kinase subunit, mTORC1 and mTORC2 phosphorylate distinct substrates, thereby regulating distinct biological reactions. mTOR has been shown to be essential in cardiac function, with cardiac-specific knockout resulting in fatal dilated cardiomyopathy (9). On the other hand, either genetically induced or pharmacological mTOR inhibition protects the heart in several pathological contexts (10). Several studies have demonstrated that mTOR is regulated by cellular redox status. There are conflicting reports showing that mTOR is alternatively activated or inhibited by oxidative stress, which could be cell type- and oxidant-dependent (11–13). We demonstrated previously that mTOR activity is increased in response to myocardial ischemia–reperfusion injury (14). However, whether mTOR directly senses cellular redox status and the potential impact of mTOR redox regulation in cardiac pathophysiology remain unknown.
Using cardiac-specific Trx1 substrate-trapping mutant (Trx1C35S) mice, we here identified that mTOR is a Trx1 substrate in the heart (4). In this study, we show that mTOR was subjected to redox modification. mTOR formed intermolecular disulfides in response to oxidation that were reduced by Trx1. mTOR oxidation in cardiomyocytes was linked to mTOR inhibition. We propose that the cardioprotective effects of Trx1 are partly mediated through mTOR.
Results
Identification of mTOR as a Trx1 substrate in cardiomyocytes
Trx1 possesses two cysteines (Cys-32 and Cys-35) in its catalytic center. During the catalytic reaction, Cys-32 forms a disulfide bond with a substrate, and then Cys-35 cleaves it. Thus, a Trx1C35S mutant stably forms a disulfide with substrates (Fig. 1A). To gain insight into how Trx1 facilitates cardioprotective effects under ischemic conditions, we screened potential Trx1 substrates using cardiac-specific transgenic mice with N-terminal FLAG-tagged and C-terminal HA-tagged Trx1C35S (FLAG-Trx1C35S-HA) subjected to ischemia. Trx1C35S was immunoprecipitated with anti-FLAG antibody, and the substrates were screened with Western blot analyses. As shown in Fig. 1B, mTOR was immunoprecipitated with Trx1C35S under both basal and ischemic conditions. Interaction between mTOR and FLAG-TrxC35S-HA was also observed in primary cultured cardiomyocytes, and the interaction was abolished with a lysis buffer containing DTT, a reducing agent (Fig. 1C), suggesting that the interaction between Trx1 and mTOR is mediated through an intermolecular disulfide bond. The interaction between mTOR and FLAG-TrxC35S-HA was not significantly enhanced under stress conditions such as ischemia or H2O2 treatment, suggesting that Trx1 constantly reduces mTOR, with or without oxidative stress (Fig. 1, B and C). Interaction between Trx1 without C35S mutation and mTOR was not detected even in the presence of H2O2 (Fig. 1D), suggesting that the interaction between Trx1 and mTOR is transient.
Figure 1.
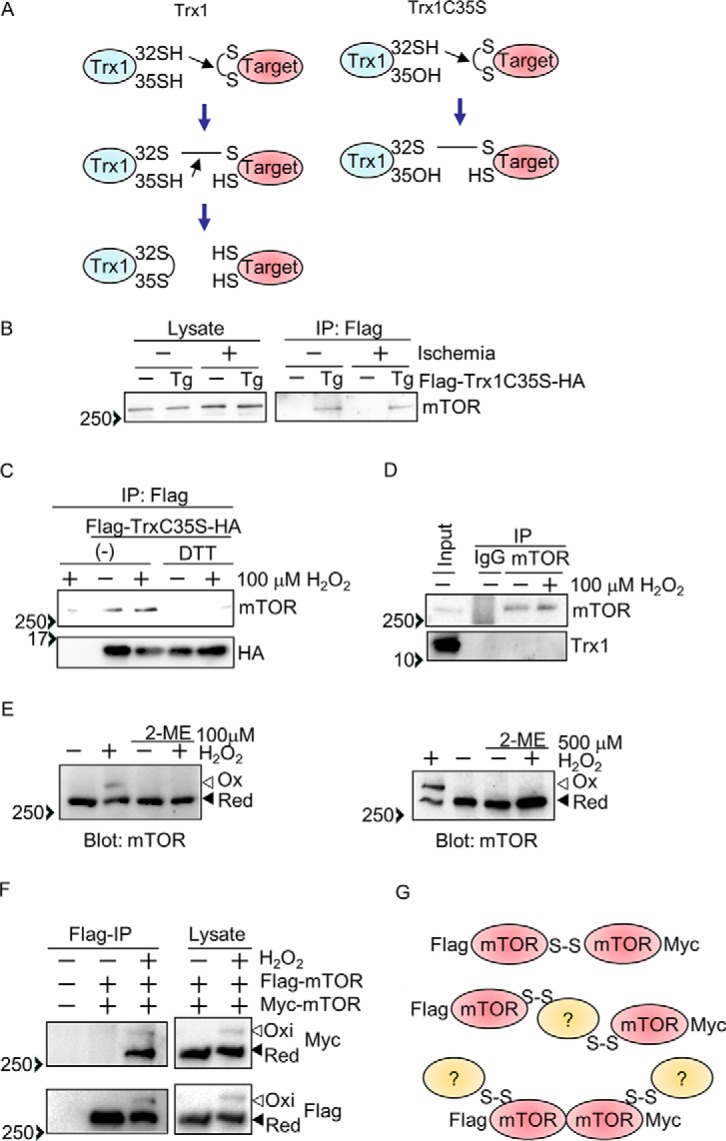
The mTOR kinase is a Trx1 substrate. A, schematic of how the Trx1 substrate-trapping mutant (Trx1C35S) works. B, Trx1C35S traps mTOR in the heart. After 3 h of ischemia, FLAG-Trx1C35S was immunoprecipitated (IP) with anti-FLAG antibody. C, Trx1C35S traps mTOR in cultured cardiomyocytes. FLAG-Trx1C35S-HA was expressed via an adenovirus vector. Cardiomyocytes were incubated with 100 μm H2O2 for 30 min, and then FLAG-Trx1C35S-HA was immunoprecipitated with anti-FLAG antibody with or without 1 mm DTT. D, stable interaction between endogenous mTOR and Trx1 is not observed in cultured cardiomyocytes. Cardiomyocytes were treated with 100 μm H2O2 for 30 min, and endogenous mTOR was immunoprecipitated. E, mTOR intermolecular disulfide formation in primary cultured cardiomyocytes. Cardiomyocytes were incubated with 100 μm H2O2 for 30 min (left panel) or 500 μm H2O2 for 10 min (right panel). F, mTOR forms disulfide bonds with either mTOR itself or other proteins. FLAG-mTOR and Myc-mTOR were expressed in HEK293 cells. After H2O2 treatment (3 mm, 30 min), FLAG-mTOR was immunoprecipitated with anti-FLAG antibody. G, data interpretation of the results shown in F. E and F, Western blot analyses were performed after SDS-PAGE under non-reducing (without 2ME) conditions. Ox, oxidized form; Red, reduced form. Western blot results were verified by two (B–D and F) and three (E) additional independent experiments.
mTOR forms intermolecular disulfide bonds
To test whether mTOR forms an intermolecular disulfide bond in response to oxidation, Western blot analyses were performed with SDS-PAGE under non-reducing conditions. In response to H2O2 treatment, some mTOR in cardiomyocytes exhibited a band shift to a high molecular weight, which was abolished in the presence of a loading buffer containing 2-mercaptoethanol, a reducing agent, suggesting that mTOR forms intermolecular disulfide bonds (Fig. 1E). To test the possibility that one mTOR molecule forms a disulfide bond with another mTOR molecule, FLAG- and Myc-tagged mTOR were co-expressed in HEK293 cells. We used a lysis buffer with a high salt content (1 m NaCl) to dissociate any non-covalently bound mTOR binding proteins. After H2O2 treatment, FLAG-mTOR was immunoprecipitated with anti-FLAG antibody, and the immune complex was probed with anti-Myc antibody. As shown in Fig. 1F, Myc-mTOR was not immunoprecipitated with FLAG-mTOR from cells without H2O2 treatment, suggesting that the high-salt lysis buffer dissociates Myc-mTOR from FLAG-mTOR. However, with H2O2 treatment, Myc-mTOR was detected as both shifted and non-shifted bands, indicating that mTOR oligomerization through disulfide bonding (as reflected by the shifted band) and non-covalent binding (as reflected by the non-shifted band) is enhanced by H2O2. These results suggest three possibilities. First, FLAG-mTOR forms an intermolecular disulfide bond with Myc-mTOR (Fig. 1G, top model). Second, two molecules of mTOR form disulfide bonds with the same other molecule without binding to one another directly (Fig. 1G, center model). Third, FLAG-mTOR binds to Myc-mTOR but not through a disulfide bond, and Myc-mTOR and FLAG-mTOR form an intermolecular disulfide bond with another molecule (Fig. 1G, bottom model).
H2O2 inhibits phosphorylation of mTOR substrates
To examine how mTOR is regulated by oxidation, the effect of H2O2 upon phosphorylation of mTOR at Ser-2481 (known to be autophosphorylated (15)) and Ser-2448 (known to correlate with mTORC1 activity (16)) and upon known mTOR substrates was examined in cardiomyocytes. As shown in Fig. 2, A and B, H2O2 dose-dependently increased oxidation of mTOR. There was a trend that phosphorylation of mTOR and mTORC1 substrates, such as S6K and 4EBP1, was increased in the presence of H2O2 at concentrations ranging from 10 to 100 μm (mTOR and S6K) and from 10 to 300 μm (4EBP1) at 10 min. However, their phosphorylation was significantly inhibited in the presence of H2O2 at concentrations ranging from 100 to 500 μm (mTOR) and from 300 to 500 μm (S6K and 4EBP1) at the later time point of 30 min. In contrast, phosphorylation of Akt, a substrate of mTORC2, was slightly increased in the presence of H2O2 at concentrations ranging from 30 to 500 μm at 10 min and from 10 to 300 μm at 30 min without reaching statistical significance. H2O2 (100 μm) time-dependently induced oxidation of mTOR, peaking at 60 min. Phosphorylation of mTOR at Ser-2448 and Ser-2481 and of S6K was inhibited by H2O2 in a time-dependent manner. H2O2 time-dependently inhibited phosphorylation of 4EBP1, peaking at 60 min, with a partial recovery at 120 and/or 240 min (Fig. 2, C and D). Phosphorylation of the mTORC2 substrate Akt was increased at 10 min, normalized at 30 and 60 min, and increased at 120 to 240 min, although none of the time points showed statistical significance relative to the basal level. Thus, mTOR oxidation is associated with reduced phosphorylation of mTORC1 substrates. We also tested whether the phosphorylation of mTOR substrates induced by activation of mTOR by upstream stimuli is inhibited by H2O2. To this end, we stimulated mTOR with insulin. As shown in Fig. 2, E and F, insulin-induced phosphorylation of S6K, 4EBP1, and Akt was significantly inhibited in the presence of H2O2. Taken together, these results suggest that H2O2 inhibits insulin-induced phosphorylation of mTORC1 and mTORC2 substrates.
Figure 2.

H2O2 inhibits phosphorylation of mTOR substrates. A, dosage effect of H2O2 on phosphorylation of mTOR substrates. C, time-dependent effect of 100 μm H2O2 on phosphorylation of mTOR substrates. E, H2O2 inhibits insulin-induced mTOR activation. Western blot analyses with the indicated antibodies were performed. B, D, and F, relative signal intensities of the phosphorylated/total proteins shown in A (B), C (D), and E (F). n = 3–6 (B), 3–8 (D), and 3–4 (F). ★p < 0.05 versus basal levels (B and D). NR, Western blot analyses with non-reducing SDS-PAGE; Ox, oxidized form; Red, reduced form. Western blot results were verified by two to three (A), two (C), and three to four (E) additional independent experiments.
Oxidative stress inhibits mTOR kinase activity
To test whether mTOR kinase activity is inhibited by H2O2, an in vitro kinase assay was performed. mTOR was immunoprecipitated from H2O2-treated cardiomyocytes and incubated with recombinant 4EBP1 and ATP. As shown in Fig. 3, mTOR-induced phosphorylation of 4EBP1 was inhibited by H2O2. These results suggest that oxidative stress inhibits mTOR kinase activity.
Figure 3.
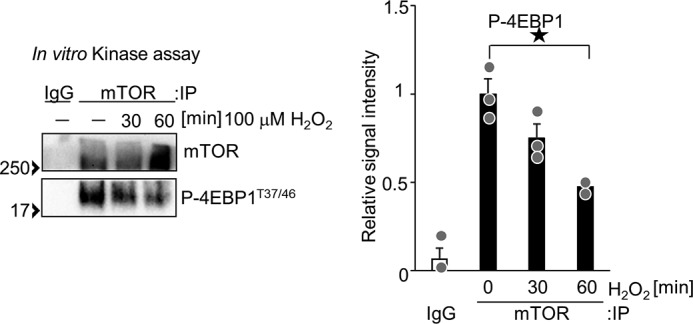
H2O2 inhibits mTOR kinase activity. Cardiomyocytes were treated with 100 μm H2O2 for 0.5 and 1 h. Immunoprecipitation (IP) of mTOR was performed with anti-mTOR antibody. An in vitro kinase assay was performed with immunoprecipitated mTOR and recombinant 4EBP1. n = 3. ★p < 0.05.
Common and specific effects of oxidants in mTOR regulation
In contrast to the H2O2-induced mTOR inhibition in cardiomyocytes, other oxidants, such as PAO, a thiol-cross-linking reagent that covalently binds to thiol groups, and diamide, a thiol-oxidizing reagent that induces disulfide bonds, are reported to activate mTOR signaling in HEK293 cells and mouse embryonic fibroblasts (11, 12). Whether and how mTOR is regulated by different oxidants in cardiomyocytes is an important consideration. To test whether PAO and diamide induce mTOR disulfide bond formation, mTOR oxidation (band shift) was examined. Unlike H2O2, PAO did not induce a band shift of mTOR, probably because PAO covalently binds to thiol groups to prevent disulfide bond formation. In contrast, diamide did induce a band shift, similar to the effect of H2O2 (Fig. 4C). PAO and diamide inhibited phosphorylation of mTOR and S6K in a time- and dosage-dependent manner. However, the lower doses of the oxidants, such as 0.5 μm PAO and 30 mm diamide, significantly promoted the phosphorylation of S6K at 30 min (Fig. 4, A–D). Phosphorylation of mTOR tended to increase under these conditions. Phosphorylation of mTOR and S6K were significantly inhibited by H2O2 in a time- and dosage-dependent manner, but the phosphorylation tended to increase with a lower dosage of H2O2 at 10 min (Fig. 2, A–C). Thus, the oxidants, including H2O2, PAO, and diamide, induce similar effects on phosphorylation of mTOR and S6K. On the other hand, PAO and diamide up-regulated both phosphorylated and total 4EBP1 (Fig. 4, A and C), which differs from H2O2. The ratio of phosphorylated/total 4EBP1 was not significantly inhibited at 30 min by any of the dosages we tested, but it was significantly inhibited by 5 μm PAO and 100 μm diamide in a time-dependent manner. The decreased ratio of phosphorylated/total 4EBP1 may indicate mTOR inhibition. Taken together, these results suggest that oxidants, including H2O2, PAO, and diamide, are able to inhibit mTOR signaling but that there are several oxidant-specific effects, such as mTOR disulfide bond formation and 4EBP1 regulation, in cardiomyocytes.
Figure 4.

Phenylarsine oxide (PAO) and diamide inhibit phosphorylation of mTOR and S6K. A, dosage effect of PAO (left panel) and time-dependent effect of 5 μm PAO (right panel) on phosphorylation of mTOR substrates. C, dosage effect of diamide (left panel) and time-dependent effect of 100 μm diamide (right panel) on phosphorylation of mTOR substrates. B and D, relative signal intensities of the phosphorylated/total proteins shown in A (B) and C (D). n = 3–8. ★p < 0.05 versus basal levels (B and D). NR, Western blot analyses with non-reducing SDS-PAGE. Open and closed arrows indicate oxidized and reduced mTOR, respectively. Western blot results were verified by two to three additional independent experiments.
Trx1 potentiates mTORC1 activity under oxidative stress conditions
To test whether Trx1 has a regulatory role in mTOR oxidation and activity, the effects of Trx1 overexpression and knockdown upon phosphorylation of mTORC1 substrates were examined. Although the oxidation status of mTOR as indicated by the band shift was not affected by overexpression or down-regulation of Trx1 alone, H2O2-induced mTOR oxidation was inhibited by Trx1 overexpression and enhanced by Trx1 knockdown, suggesting that Trx1 reduces disulfide bonds in mTOR (Fig. 5A). As shown in Fig. 5, A and B, Trx1 overexpression alone did not significantly affect phosphorylation of mTOR and its substrates, whereas Trx1 knockdown alone inhibited phosphorylation. Trx1 overexpression normalized the decrease in phosphorylation of mTOR and its substrates in the presence of H2O2, whereas the decrease in phosphorylation was enhanced by Trx1 knockdown. These results suggest that Trx1 potentiates mTORC1 activity in cardiomyocytes in the presence of H2O2.
Figure 5.
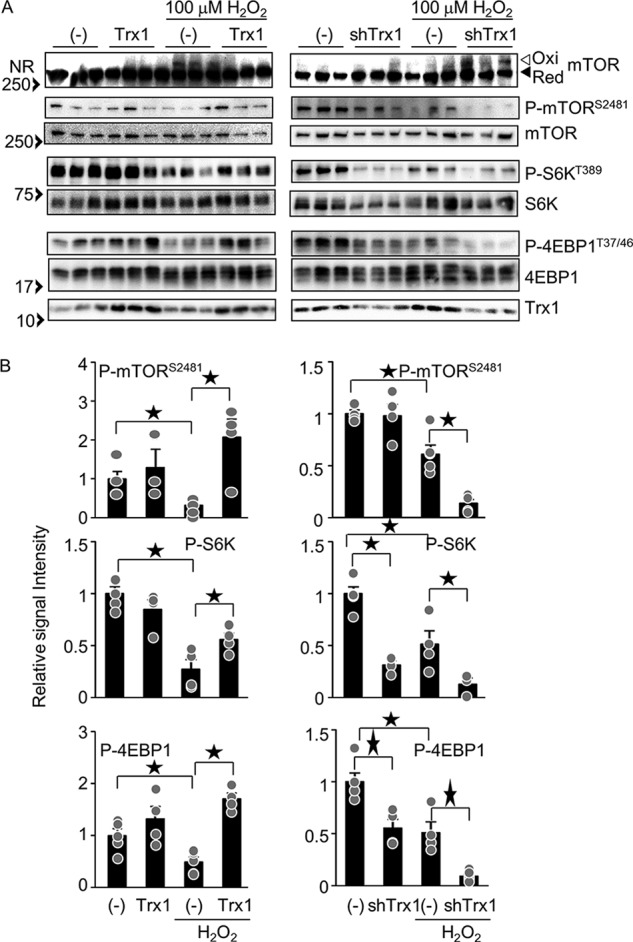
Trx1 normalizes H2O2-induced mTOR oxidation and inhibition. A, left panel, overexpression of Trx1 normalizes H2O2-induced mTOR oxidation and inhibition. After 2 days of Trx1 adenovirus vector transduction, cardiomyocytes were incubated with 100 μm H2O2 for 30 min. Right panel, knockdown of Trx1 enhances H2O2-induced mTOR oxidation and inhibition. After 3 days of shTrx1 transduction, cardiomyocytes were incubated with 100 μm H2O2 for 30 min. Each lane corresponds to an independent sample (n = 3). NR, non-reducing SDS-PAGE; Ox, oxidized form; Red, reduced form. B, relative signal intensities of the phosphorylated/total proteins shown in A. n = 4–11. ★p < 0.05.
The role of Trx1-mediated mTOR regulation
To test whether Trx1 potentiates mTOR activity in the heart, phosphorylation of mTOR substrates was examined in Trx1 heterozygous knock-out (Trx1+/−) mice under ischemic conditions. The band shift of mTOR because of oxidation was detected in Trx1+/− but not in wild-type mice under ischemic conditions, suggesting that endogenous Trx1 reduces disulfide bonds in mTOR in the heart in vivo (Fig. 6A). Phosphorylation of mTOR and its substrates was inhibited by ischemia, an effect that was further enhanced in Trx1+/− mice (Fig. 6, B and C). These results suggest that endogenous Trx1 plays an important role in preserving mTORC1 activity in the heart during ischemia in vivo. Simulated ischemia–reperfusion (IR) induces oxidative stress in cultured cardiomyocytes (17). As shown in Fig. 6D, IR-induced mTOR oxidation was enhanced by Trx1 knockdown. In addition, phosphorylation of mTOR substrates under IR conditions was inhibited by Trx1 knockdown (Fig. 6, D and E). As expected, IR-induced cell death was enhanced by Trx1 knockdown (Fig. 6F). Taken together, these results suggest that endogenous Trx1 preserves mTOR activity under oxidative stress conditions, which may, in turn, promote cell survival during IR.
Figure 6.

Trx1 has a regulatory role in mTOR signaling under ischemia and ischemia-reperfusion conditions. A, ischemia-induced mTOR oxidation in Trx1+/− mice. B, haploinsufficiency of Trx1 promotes ischemia-induced mTOR inhibition. Heart lysates were prepared after 3 h of ischemia. Each lane corresponds to an individual mouse. C, relative signal intensities of the Western blot results shown in B. n = 4–6. D, knockdown of Trx1 enhances IR-induced mTOR oxidation. Cardiomyocytes were transduced with shTrx1 for 5 days. The myocytes were incubated with 95% nitrogen 5% CO2 for 4 h and then incubated under normal cell culture conditions (5% CO2) for 30 min. C and E, relative signal intensities of the phosphorylated/total proteins shown in B (C) and D (E). n = 4–6 (C) and 4–8 (E). F, knockdown of Trx1 enhances IR-induced cell death. After 16 h of reoxygenation, cardiomyocyte viability was examined by trypan blue dye exclusion. n = 4–6. NR, non-reducing SDS-PAGE; Ox, oxidized form; Red, reduced form. Western blot results were verified by two (A and B), and two to three (D) additional independent experiments. ★p < 0.05.
Trx1 potentiates mTOR-mediated mitochondrial function
To test whether Trx1-induced cytoprotection is mediated by mTOR, cardiomyocyte viability was examined. H2O2-induced decreases in cardiomyocyte viability were normalized by Trx1 overexpression, but this protective effect was abolished in the presence of rapamycin, an mTORC1 inhibitor (Fig. 7A). These results suggest that Trx1-induced cytoprotection is mediated by mTOR. Expression of nucleus-encoded mitochondrial genes is positively regulated by mTOR at both the transcriptional and translational levels (18, 19). To test whether Trx1 potentiates mitochondrial function in an mTOR-dependent manner, ATP content and mitochondrial ATP production were examined. Both ATP content and mitochondrial ATP production were significantly inhibited by H2O2, whereas Trx1 conferred partial normalization, which was abolished in the presence of rapamycin (Fig. 7, B and C). Mitochondrial damage promotes mitochondrial ROS production (20). To investigate whether Trx1 prevents mitochondrial ROS production in an mTOR-dependent manner, we used HyPer-mito, a fluorescence-based H2O2 indicator that localizes to mitochondria. Induction of mitochondrial H2O2 production by exogenous H2O2 treatment was inhibited by Trx1, whereas the inhibitory effect of Trx1 was abolished in the presence of rapamycin (Fig. 7D). This result suggests that Trx1 prevents mitochondrial H2O2 production in an mTOR-dependent manner. Consistently, Trx1 knockdown promoted H2O2-induced cell death and exacerbated H2O2-induced decreases in ATP content (Fig. 7, E and F). H2O2-induced down-regulation of mitochondrial genes such as Atp5a, Uqcrc2, Mtco1, Sdhb, and Ndufb8 was normalized by Trx1, an effect that was abolished in the presence of rapamycin (Fig. 7G). Trx1 knockdown promoted H2O2-induced down-regulation of mitochondrial genes (Fig. 7H). Taken together, these results suggest that Trx1-induced cytoprotection against oxidative stress in cardiomyocytes is partly mediated through normalization of mTOR activation.
Figure 7.

The Trx1-induced cytoprotective effect is mediated by mTOR. A, Trx1 protects against H2O2-induced cell death through mTOR activation. After 2 days of Trx1 adenovirus vector transduction, cardiomyocytes were treated with 30 μm H2O2 and 10 nm rapamycin for 6 h. Cardiomyocyte viability was examined by trypan blue dye exclusion. n = 4–10. B–D, Trx1 protects against H2O2-induced mitochondrial damage through mTOR activation. After 2 days of Trx1 adenovirus vector transduction, cardiomyocytes were treated with 100 μm H2O2 and 10 nm rapamycin for 6 h. ATP content (B), ATP production in isolated mitochondria (C), and mitochondrial H2O2 production (D) were inhibited by H2O2 but were normalized by Trx1 overexpression. The Trx1-mediated normalization was abolished by rapamycin. n = 4–10 (B and C) and 4 (D). E and F, Trx1 protects against H2O2-induced cell death and reduction of ATP content. After 4–5 days of shTrx1 adenovirus vector transduction, cardiomyocytes were treated with 30 μm (E) or 100 μm (F) H2O2 for 6 h. Cardiomyocyte viability (E) and ATP content (F) were examined. n = 4 (E) and 3–5 (F). G, left panel, H2O2 inhibited mitochondrial protein expression, which was normalized by Trx1 overexpression. The Trx1-mediated normalization of mitochondrial protein expression was abolished by rapamycin. Right panel, relative signal intensities of the Western blot. n = 3–4. H, left panel, H2O2 inhibited mitochondrial protein expression, an effect that was enhanced by Trx1 knockdown. Right panel, relative signal intensities of the Western blot. n = 3–4. ★p < 0.05.
The Cys-1483 residue is responsible for mTOR oxidation and inhibition
To identify a cysteine residue responsible for mTOR oxidation and inhibition, we first investigated which part of mTOR forms a disulfide bond using Myc-tagged mTOR-truncated mutants (Fig. 8A). The molecular weight shift caused by intermolecular disulfide bond formation was observed most prominently in mTOR(1271–2008), suggesting that the region flanking the FRAP-ATM-TTRAP (FAT) domain contains a redox-sensitive cysteine (Fig. 8B). It has been reported that oxidation-resistant mutations at Cys-1483, including C1483F, C1483P, and C1483Y, are highly recurrent in cancer patients and enhance mTOR activity (21). We hypothesized that Cys-1483 is a residue responsible for mTOR redox regulation. Cys-1483 is evolutionally conserved in multiple cellular organisms (Fig. 8C). To test whether Cys-1483 mediates disulfide bond formation in mTOR, Myc-mTOR-C1483F was expressed in HEK293 cells. H2O2-induced oxidation of mTOR was attenuated in Myc-mTOR-C1483F, suggesting that Cys-1483 is involved in disulfide bond formation (Fig. 8D). To test whether Cys-1483 is reduced by Trx1, the interaction between mTOR-C1483F and Trx1C35S was examined. The interaction between mTOR and Trx1C35S was attenuated in the presence of mTOR-C1483F compared with intact mTOR under both basal and H2O2-treated conditions, suggesting that Trx1C35S forms an intermolecular disulfide bond with mTOR at Cys-1483 (Fig. 8E). To test the extent to which Cys-1483 mediates H2O2-induced mTOR inhibition, phosphorylation of mTOR substrates was examined. As shown in Fig. 8F, mTOR-C1483F conferred resistance against H2O2-induced dephosphorylation of S6K and 4EBP1, suggesting that Cys-1483 mediates H2O2-induced mTOR inhibition (Fig. 8F). Similarly, mTOR-C1483F conferred resistance against H2O2-induced cell death (Fig. 8G) and down-regulation of mitochondrial proteins (Fig. 8H). Taken together, these results suggest that mTOR Cys-1483 is subjected to oxidation and can be reduced by Trx1.
Figure 8.

The Cys-1483 residue is responsible for mTOR redox regulation. A, schematic of mTOR-truncated mutants. B, the indicated mTOR mutants were expressed in HEK293 cells. After 3 mm H2O2 treatment for 30 min, Western blot analyses were performed after SDS-PAGE under non-reducing (without 2ME) and reducing (with 2ME) conditions. C, Cys-1483 is evolutionarily conserved among multi-cellular organisms. D, Cys-1483 is responsible for H2O2-induced mTOR oxidation. Myc-mTOR and Myc-mTOR-C1483F were expressed in HEK293 cells. After 1 and 3 mm H2O2 treatment for 30 min, Western blot analyses were performed after SDS-PAGE under non-reducing conditions. LE, long exposure; SE, short exposure; Ox, oxidized form; Red, reduced form. E, Cys-1483 is a Trx1 reduction site. The indicated Myc-mTOR mutants and FLAG-Trx1C35S-HA were expressed in HEK293 cells. After 3 mm H2O2 treatment for 30 min, co-immunoprecipitation assays were performed with anti-FLAG antibody. Western blot analyses were performed with anti-Myc and anti-HA antibodies. F, left panel, Cys-1483 is responsible for H2O2-induced mTOR inhibition. Myc-mTOR-C1483F and HA-GST-S6K were expressed in HEK293 cells. After H2O2 treatment (1 and 3 mm, 30 min), Western blot analyses were performed with the indicated antibodies. Right panel, relative signal intensities of phosphorylated/total protein. n = 4–7. G, the C1483F mutation confers resistance against H2O2-induced cell death. HEK293 cells expressing Myc-mTOR or Myc-mTOR-C1483F were treated with 1 mm H2O2 for 6 h. Cell viability was examined. n = 4. H, left panel, the C1483F mutation confers resistance against H2O2-induced down-regulation of mitochondrial proteins. Right panel, relative signal intensities of the Western blot. n = 3–6. Western blot results were verified by two (B), three (D and E), and two to three (F) additional independent experiments. ★p < 0.05.
Discussion
In this study, we show that mTOR undergoes intermolecular disulfide formation in response to oxidative stress and that intermolecular disulfide bond formation at Cys-1483 is responsible for H2O2-induced mTOR inhibition. Trx1 serves as an electron donor to reduce the disulfide in mTOR, thereby positively regulating phosphorylation of mTOR substrates (Fig. 9).
Figure 9.
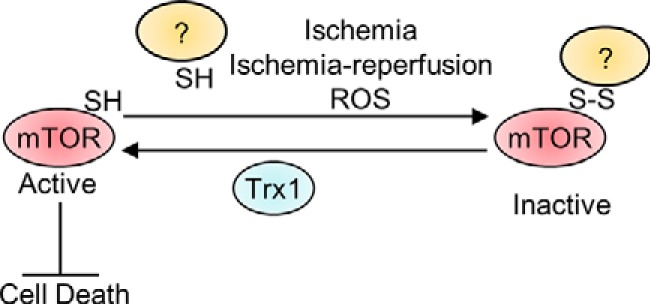
Schematic model. The mTOR kinase is inhibited by oxidative stress, possibly through intermolecular disulfide formation. Trx1 protects against oxidative stress, partly through mTOR.
The mechanism by which Cys-1483 oxidation inhibits mTOR is currently unknown. It is likely that mTOR forms intermolecular disulfide bonds either with another mTOR molecule or with a distinct molecule (Fig. 1G). The identity of the molecule with which mTOR forms an intermolecular disulfide bond remains to be elucidated. Because oxidation of mTOR negatively affects mTOR phosphorylation at Ser-2448 and Ser-2481, it is likely that the disulfide bond formation negatively regulates phosphorylation of mTOR by upstream kinases or by mTOR itself (autophosphorylation) through changes in the structure of mTOR because of either the oxidation itself or protein–protein interaction. The Cys-1483 mutation abolishes binding to Deptor, an inhibitory protein for mTOR (21). It will be interesting to test whether oxidation of Cys-1483 enhances the binding of Deptor to mTOR.
Missense mutations at Cys-1483 such as the C1483F mutation are highly recurrent in cancer patients (21). Resistance against oxidative stress may be an underlying mechanism by which the mutations at Cys-1483 contribute to cancer development. It has been proposed that Trx1 confers stress resistance, thereby increasing the malignancy of cancer (5). Trx1-mediated maintenance of mTOR function may confer stress resistance in cancer cells.
We showed that H2O2 inhibits mTORC1 activity, as evidenced by reduced phosphorylation of endogenous substrates and by impaired mTOR kinase activity, determined by immune complex kinase assays. On the other hand, H2O2 alone did not significantly inhibit phosphorylation of Akt, an mTORC2 substrate. However, H2O2 did inhibit insulin-induced Akt phosphorylation. How H2O2 affects the overall function of mTORC2 remains to be clarified.
Although mTOR activity was consistently suppressed after 30 min of H2O2 treatment at multiple concentrations, phosphorylation of mTOR targets tended to be increased transiently around 10 min in response to low concentrations of H2O2 despite oxidation of mTOR (Fig. 2, A–D). We speculate that another H2O2-sensitive mechanism may independently stimulate mTOR. There are conflicting reports showing that oxidative stress either activates or inhibits mTOR. H2O2 inhibits mTOR in primary cultured neurons and A549, PC12, and RAW264.7 cells (13, 22, 23). On the other hand, several oxidants, such as PAO and diamide, activate mTOR in HEK293 cells and mouse embryonic fibroblasts (11, 12). In this study, we showed that mTOR signaling is inhibited by multiple dosages of PAO and diamide and at multiple time points but that mTOR signaling tended to be activated transiently at low concentrations (Fig. 4). Although previous studies have shown that both PAO and diamide activate mTOR, the experiments were conducted with only one dosage at one time point (11, 12). Thus, we speculate that mTOR signaling may be activated transiently by a low level of oxidative stress, whereas it may be inhibited by higher levels of oxidative stress, with the extent of oxidative stress and time course of activation or inhibition of mTOR varying in each cell type.
Our results show that Trx1 interacts with mTOR through an intermolecular disulfide bond. Because this interaction is hardly observed between endogenous Trx1 and mTOR (Fig. 1D) and becomes obvious only when a Trx1-trapping mutant is used (Fig. 1, B and C), it is likely that Trx1 interacts with mTOR transiently to reduce mTOR with disulfide bonds at Cys-1483. Because down-regulation of endogenous Trx1 exacerbates oxidation of mTOR and suppresses phosphorylation of mTOR targets, we propose that Trx1 acts as an endogenous co-factor of mTOR, thereby protecting mTOR from oxidation and inactivation during oxidative stress. Although this investigation strongly suggests that Trx1 regulates the oxidation status of mTOR through direct protein–protein interaction, it is formally possible that Trx1 also indirectly prevents mTOR oxidation by scavenging H2O2 through Trx1-dependent peroxidases (6).
Thioredoxin-binding protein-2 (TBP-2)/thioredoxin-interacting protein (TXNIP) was originally identified as a Trx1 inhibitor (24). Consistent with our results, there are several reports showing that TBP-2 inhibits mTOR (25, 26). However, it has also been reported that TBP-2 is required for diabetes-induced mTOR activation (27). We have reported previously that TBP-2 serves as a scaffolding protein and is required for Trx1-mediated thiol exchange reactions for specific Trx1 substrates, including class II histone deacetylases (28). Thus, TBP-2 may serve as a scaffolding protein for mTOR to undergo thiol exchange reactions in a context-dependent manner. Whether TBP-2 promotes or inhibits Trx1-mediated thiol exchange reactions of mTOR remains to be elucidated.
Cardiac-specific overexpression of Trx1 ameliorates pressure overload–induced cardiac hypertrophy (29) and high-fat diet–induced exacerbation of ischemic injury (4). Cardiac-specific overexpression of Trx1 also attenuates mitochondrial dysfunction in septic mice (30). Trx1 acts as an antioxidant to reduce H2O2 through activation of peroxiredoxin (6). In addition, Trx1 reduces the oxidized form of histone deacetylase 4 (HDAC4) (28) and AMPK (4), thereby protecting the heart against cardiac hypertrophy and ischemic injury. We here propose that mTOR is another target of Trx1 that mediates the protective effect of Trx1 in cardiomyocytes. Although Trx1 negatively regulates cardiac hypertrophy (29, 31), mTOR can positively regulate cardiac hypertrophy by promoting protein translation (9). Thus, how Trx1-induced positive regulation of mTOR affects the overall level of cardiac hypertrophy may be determined by the balance between the direct effect upon cardiac hypertrophy and the improvement of cardiac performance. It is possible that Trx1-induced decreases in cell death and improvement of mitochondrial function secondarily inhibit pathological hypertrophy.
We show here that Trx1 positively affects mitochondrial function through mTOR in cardiomyocytes. Oxidative stress down-regulates expression of mitochondrial genes, including Atp5a, Uqcrc2, Mtco1, Sdhb, and Ndufb8, inhibits ATP production, and induces cell death in cardiomyocytes. These detrimental effects of oxidative stress are accompanied by suppression of mTOR, whereas they are alleviated in the presence of Trx1 through a rapamycin-sensitive mechanism, suggesting that the cardioprotective effect of the Trx1–mTOR pathway may be in part mediated through protection of mitochondria against oxidative stress. Consistently, Trx1 knockdown promoted H2O2-induced down-regulation of mitochondrial genes (Fig. 7H). It is possible that Trx1 knockdown and H2O2 (100 μm) have additive effects, and, thus, that their effects are mediated through independent mechanisms. However, because oxidation of mTOR by 100 μm H2O2 is further enhanced by Trx1 knockdown (Fig. 5A), Trx1 knockdown and H2O2 (100 μm) presumably act through the same targets. To test whether Trx1 knockdown and H2O2 act through independent mechanisms, we would have to test whether Trx1 knockdown can still enhance H2O2-induced down-regulation of mitochondrial genes at the maximum dose of H2O2. Unfortunately, higher dosages of H2O2 induced prominent cell death in the presence of Trx1 knockdown so that we were not able to address this issue in a reliable manner. Thus, the possibility that Trx1 knockdown and H2O2 independently down-regulate mitochondrial genes cannot be excluded.
mTOR up-regulates mitochondrial genes through PGC-1α–dependent transcriptional regulation and 4EBP1-dependent translational regulation (18, 19). We have reported that cardiac-specific overexpression of Trx1 up-regulates nucleus-encoded mitochondrial genes possessing a nuclear respiration factor-1 (NRF1)–binding element in their promoter regions (32). PGC-1α co-activates NRF1 (33). Thus, the Trx1–mTOR pathway may positively regulate mitochondrial gene expression through PGC-1α–NRF1 signaling. Moreover, the H2O2-induced decrease in 4EBP1 phosphorylation is attenuated by Trx1 in an mTOR-dependent manner (Fig. 7G). Taken together, these findings indicate that the Trx1–mTOR pathway maintains mitochondrial function in the presence of oxidative stress through multiple mechanisms.
Whether mTOR is protective or detrimental in the failing heart remains controversial (10). Although mTOR protects the heart by enhancing mitochondrial function and cell survival, it may negatively affect the heart through inhibition of autophagy. Increased protein translation is an energy-consuming process and can negatively affect survival of cardiomyocytes in the presence of myocardial ischemia (34). Thus, a novel strategy promoting only the protective aspects of mTOR function, such as the enhancement of mitochondrial function, while preventing mTOR-mediated inhibition of autophagy would potentially be beneficial for treatment of patients with cardiac stress. We have shown previously that Trx1 activates AMPK, an inducer of autophagy (4). Thus, enhancing the function of Trx1 may be an attractive strategy that enhances the salutary actions of mTOR while minimizing its detrimental aspects through stimulation of AMPK.
In summary, this study shows that mTOR is an important downstream target of Trx1. Trx1 acts as an endogenous co-factor of mTOR to inhibit disulfide bond formation and maintain the activity of mTOR. mTOR plays a critical role in mediating the protective effect of Trx1 in the heart, possibly by preserving mitochondrial function during oxidative stress.
Experimental procedures
Transgenic mice
Cardiac-specific Trx1C35S transgenic mice on the Friend virus B (FVB) background have been described previously (4). Trx1+/− mice were described previously (35). Trx1+/− mice were backcrossed to C57BL/6 mice more than 12 times. Ischemia was achieved by ligation of the anterior descending branch of the left coronary artery, with silicon tubing (1 mm outer diameter) placed on top of the artery, 2 mm below the border between the left atrium and left ventricle. All procedures involving animals were performed in accordance with protocols approved by Rutgers Biomedical and Health Sciences.
Immunoblot analysis
Heart homogenates or cell lysates were prepared using a lysis buffer (50 mm Tris-HCl (pH 7.6), 1% Triton X-100, 10 mm EDTA, 150 mm NaCl, 50 mm NaF, and protease inhibitor mixture (Sigma)). Total protein lysates (10–30 μg) were incubated with SDS sample buffer (final concentration: 100 mm Tris (pH 6.8), 2% SDS, 5% glycerol, 2.5% 2-mercaptoethanol, and 0.05% bromphenol blue) at 95 °C for 5–20 min. For mTOR redox status analyses, the lysates were prepared with lysis buffer containing 20 mm N-ethylmaleimide and SDS sample buffer without 2-mercaptoethanol. For immunoprecipitation, the lysates were incubated with FLAG-agarose or anti-mTOR antibody with protein A-agarose. Immunocomplexes were washed with lysis buffer three times and eluted with 2× SDS sample buffer. Antibodies used for this study were Trx1 (Cell Signaling Technology, 2429), mTOR (Cell Signaling Technology, 2797), phospho-mTOR(Ser-2448) (Cell Signaling Technology, 2796), phospho-mTOR(Ser-2481) (Cell Signaling Technology, 2774), p70 S6 kinase (Cell Signaling Technology, 9202), phospho-p70 S6 kinase (Cell Signaling Technology, 9205), 4EBP1 (Cell Signaling Technology, 9644), phospho-4EBP1 (Cell Signaling Technology, 2855), Akt (Cell Signaling Technology, 9272), phospho-Akt (Cell Signaling Technology, 9271), tubulin (Sigma, T6199), Myc (Cell Signaling Technology, 2272), FLAG (Cell Signaling Technology, 2368), and MitoProfile® containing antibodies against Atp4a, Uqcrc2, Mtco1, Sdhb, and Ndfub8 (Mitoscience, MS604). The signal intensity of Western blot signals was quantified using the ImageJ program. The signal intensity of non-phosphorylated proteins was normalized by a loading control, tubulin. The signal intensity of phosphorylated proteins was normalized by that of total protein. Differences in signal intensities derived from different Western blot membranes were reported as relative changes and combined to generate line or bar graphs.
In vitro kinase assay
Cardiomyocytes were treated with 100 μm H2O2 for 0.5 and 1 h. mTOR was immunoprecipitated with anti-mTOR antibody and protein A/G-agarose. The immunocomplex was washed with 1 ml of lysis buffer twice and with 1 ml of kinase buffer (50 mm HEPES (pH 7.4), 15 mm MgCl2, and 200 μm sodium vanadate) twice. In vitro kinase reactions were performed with mTOR in the kinase buffer, 2 μg of recombinant 4EBP1 (Novus) and 100 μm ATP at room temperature for 30 min in the kinase buffer. The kinase reaction was terminated with 2× SDS sample buffer. Phosphorylation levels of 4EBP1 were examined by Western blot analyses with anti-phospho-4EBP1 antibody.
Primary cultures of neonatal rat ventricular myocytes
Primary cultures of cardiomyocytes were prepared form 1-day-old Crl:(WI) BR-Wistar rats (Harlan). A cardiomyocyte-rich fraction was obtained by centrifugation through a discontinuous Percoll gradient. Cells were cultured in complete medium containing Dulbecco's modified Eagle's medium/F-12 supplemented with 5% horse serum, 4 μg/ml transferrin, 0.7 ng/ml sodium selenite, 2 g/liter bovine serum albumin (fraction V), 3 mm pyruvate, 15 mm HEPES (pH 7.1), 100 μm ascorbate, 100 mg/liter ampicillin, 5 mg/liter linoleic acid, and 100 μm 5-bromo-2′-deoxyuridine. Culture dishes were coated with 0.3% gelatin.
Simulated ischemia reperfusion
Cardiomyocytes were placed in a hypoxia chamber with 95% N2 and 5% CO2 for 4 h and then transferred to a standard CO2 incubator.
ATP content
Cardiomyocytes were lysed with somatic cell ATP releasing reagent (Sigma, FLSAR). The ATP content in the lysates was measured with an ATP bioluminescent assay kit (Sigma, FLAA).
ATP production in isolated mitochondria
Mitochondria were freshly isolated from cardiomyocytes (6-cm culture dish) using a mitochondrion isolation kit (Sigma, MITOISO1). The isolated mitochondria were suspended in 50 μl of storage buffer (10 mm HEPES (pH 7.4), 250 mm sucrose, and 2 mm K2HPO4). ATP production was measured by luminometric assay using an ATP bioluminescent assay kit (Sigma, FLAA). The ATP assay mixture was diluted with dilution buffer (1000-fold dilution). The diluted ATP assay mixture (25 μl) was combined with 25 μl of substrate buffer (5 mm HEPES (pH 7.9), 210 mm mannitol, 70 mm sucrose, 10 mm pyruvate, 10 mm malate, and 4 mm K2HPO4), and then 5 μl of mitochondrial solution was added. After 5 min of incubation at room temperature, the reaction was started by addition of 1 μl of 12.5 mm ADP. The luminescence was measured by luminometer for 10 s.
Mitochondrial H2O2 production
Adenovirus vector expressing HyPer-mito was transduced into cardiomyocytes. After 2–3 days of transduction, fluorescence (excitation, 500 nm; emission, 520 nm) was detected by fluorescence microcopy.
Adenovirus vectors
Adenoviruses harboring Trx1, shTrx1, FLAG-Trx1C35S-HA, and HyPer-mito were generated using the AdMax system (Microbix) as described previously (4, 36).
Plasmid vectors
The mammalian expression vectors for mTOR, including pRK5-Myc-mTOR, pRK5-Myc-mTOR(1–1482), pRK5-Myc-mTOR(1271–2008), pRK5-Myc-mTOR(1750–2549), and pRK5-HA-GST-p85S6K, were gifts from Dr. David Sabatini (Addgene; 1861, 21745, 21746, 21747, and 8466). The mTOR cDNA from pRK5-Myc-mTOR was inserted into pDC316 with an N-terminal FLAG tag to generate pDC316-FLAG-mTOR. pRK5-Myc-mTOR-C1483F was generated by site-directed mutagenesis.
Statistical methods and error bars
Statistical comparisons were made using Student's t test. p < 0.05 was defined as statistically significant and is indicated by a star. p > 0.05 is indicated by NS (not significant). All error bars represent S.E.
Author contributions
S. O., T. H., W. S., D. N., Y. C., A. C., H. Y., T. S., N. N., S. B., and K. S. carried out the experiments. P. Z. performed ischemia surgery. D. S. identified mTOR as a Trx1 substrate. J. Y. oversaw the generation of Trx1+/− mice. Y. H. backcrossed Trx1+/− mice to the C57BL6 genetic background. S. O. and J. S. developed the original concept and oversaw the study. S. O., S. S., and J. S. wrote the manuscript.
Acknowledgments
We thank Daniela Zablocki for critical reading of the manuscript.
This work was supported in part by Public Health Service Grants HL67724, HL91469, HL102738, HL112330, and AG23039 (to J. S.) and by the Leducq Foundation Transatlantic Network of Excellence (to J. S.). The authors declare that they have no conflicts of interest with the contents of this article.
- ROS
- reactive oxygen species
- Trx
- thioredoxin
- AMPK
- AMP-activated protein kinase
- mTOR
- mechanistic target of rapamycin
- PAO
- phenylarsine oxide
- IR
- ischemia–reperfusion
- 2ME
- 2-mercaptoethanol.
References
- 1. Holmgren A. (1985) Thioredoxin. Annu. Rev. Biochem. 54, 237–271 [DOI] [PubMed] [Google Scholar]
- 2. Masutani H., Ueda S., and Yodoi J. (2005) The thioredoxin system in retroviral infection and apoptosis. Cell Death Differ. 12, 991–998 [DOI] [PubMed] [Google Scholar]
- 3. Nagarajan N., Oka S., and Sadoshima J. (2017) Modulation of signaling mechanisms in the heart by thioredoxin 1. Free Radic. Biol. Med. 109, 125–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shao D., Oka S., Liu T., Zhai P., Ago T., Sciarretta S., Li H., and Sadoshima J. (2014) A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell metabolism 19, 232–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Powis G., and Kirkpatrick D. L. (2007) Thioredoxin signaling as a target for cancer therapy. Curr. Opin. Pharmacol. 7, 392–397 [DOI] [PubMed] [Google Scholar]
- 6. Rhee S. G. (2016) Overview on peroxiredoxin. Mol. Cells 39, 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu C., Parrott A. M., Fu C., Liu T., Marino S. M., Gladyshev V. N., Jain M. R., Baykal A. T., Li Q., Oka S., Sadoshima J., Beuve A., Simmons W. J., and Li H. (2011) Thioredoxin 1-mediated post-translational modifications: reduction, transnitrosylation, denitrosylation, and related proteomics methodologies. Antioxid. Redox Signal. 15, 2565–2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Laplante M., and Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang D., Contu R., Latronico M. V., Zhang J. L., Rizzi R., Catalucci D., Miyamoto S., Huang K., Ceci M., Gu Y., Dalton N. D., Peterson K. L., Guan K. L., Brown J. H., Chen J., et al. (2010) MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J. Clin. Invest. 120, 2805–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sciarretta S., Volpe M., and Sadoshima J. (2014) Mammalian target of rapamycin signaling in cardiac physiology and disease. Circulation research 114, 549–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sarbassov D. D., and Sabatini D. M. (2005) Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. The Journal of biological chemistry 280, 39505–39509 [DOI] [PubMed] [Google Scholar]
- 12. Yoshida S., Hong S., Suzuki T., Nada S., Mannan A. M., Wang J., Okada M., Guan K. L., and Inoki K. (2011) Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2-Rheb GTPase pathway. The Journal of biological chemistry 286, 32651–32660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang L., Kimball S. R., Jefferson L. S., and Shenberger J. S. (2009) Hydrogen peroxide impairs insulin-stimulated assembly of mTORC1. Free radical biology & medicine 46, 1500–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhai P., Sciarretta S., Galeotti J., Volpe M., and Sadoshima J. (2011) Differential roles of GSK-3β during myocardial ischemia and ischemia/reperfusion. Circ. Res. 109, 502–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peterson R. T., Beal P. A., Comb M. J., and Schreiber S. L. (2000) FKBP12-rapamycin-associated protein (FRAP) autophosphorylates at serine 2481 under translationally repressive conditions. J. Biol. Chem. 275, 7416–7423 [DOI] [PubMed] [Google Scholar]
- 16. Rosner M., Siegel N., Valli A., Fuchs C., and Hengstschläger M. (2010) mTOR phosphorylated at S2448 binds to raptor and rictor. Amino Acids 38, 223–228 [DOI] [PubMed] [Google Scholar]
- 17. Vanden Hoek T. L., Shao Z., Li C., Zak R., Schumacker P. T., and Becker L. B. (1996) Reperfusion injury on cardiac myocytes after simulated ischemia. Am. J. Physiol. 270, H1334–H1341 [DOI] [PubMed] [Google Scholar]
- 18. Cunningham J. T., Rodgers J. T., Arlow D. H., Vazquez F., Mootha V. K., and Puigserver P. (2007) mTOR controls mitochondrial oxidative function through a YY1-PGC-1α transcriptional complex. Nature 450, 736–740 [DOI] [PubMed] [Google Scholar]
- 19. Morita M., Gravel S. P., Chénard V., Sikström K., Zheng L., Alain T., Gandin V., Avizonis D., Arguello M., Zakaria C., McLaughlan S., Nouet Y., Pause A., Pollak M., Gottlieb E., et al. (2013) mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 18, 698–711 [DOI] [PubMed] [Google Scholar]
- 20. Chen Q., Vazquez E. J., Moghaddas S., Hoppel C. L., and Lesnefsky E. J. (2003) Production of reactive oxygen species by mitochondria: central role of complex III. J. Biol. Chem. 278, 36027–36031 [DOI] [PubMed] [Google Scholar]
- 21. Grabiner B. C., Nardi V., Birsoy K., Possemato R., Shen K., Sinha S., Jordan A., Beck A. H., and Sabatini D. M. (2014) A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 4, 554–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen L., Xu B., Liu L., Luo Y., Yin J., Zhou H., Chen W., Shen T., Han X., and Huang S. (2010) Hydrogen peroxide inhibits mTOR signaling by activation of AMPKα leading to apoptosis of neuronal cells. Lab. Invest. 90, 762–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seo G., Kim S. K., Byun Y. J., Oh E., Jeong S. W., Chae G. T., and Lee S. B. (2011) Hydrogen peroxide induces Beclin 1-independent autophagic cell death by suppressing the mTOR pathway via promoting the ubiquitination and degradation of Rheb in GSH-depleted RAW 264.7 cells. Free Radic. Res. 45, 389–399 [DOI] [PubMed] [Google Scholar]
- 24. Nishiyama A., Matsui M., Iwata S., Hirota K., Masutani H., Nakamura H., Takagi Y., Sono H., Gon Y., and Yodoi J. (1999) Identification of thioredoxin-binding protein-2/vitamin D3 up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J. Biol. Chem. 274, 21645–21650 [DOI] [PubMed] [Google Scholar]
- 25. Huang C., Zhang Y., Kelly D. J., Tan C. Y., Gill A., Cheng D., Braet F., Park J. S., Sue C. M., Pollock C. A., and Chen X. M. (2016) Thioredoxin interacting protein (TXNIP) regulates tubular autophagy and mitophagy in diabetic nephropathy through the mTOR signaling pathway. Sci. Rep. 6, 29196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jin H. O., Seo S. K., Kim Y. S., Woo S. H., Lee K. H., Yi J. Y., Lee S. J., Choe T. B., Lee J. H., An S., Hong S. I., and Park I. C. (2011) TXNIP potentiates Redd1-induced mTOR suppression through stabilization of Redd1. Oncogene 30, 3792–3801 [DOI] [PubMed] [Google Scholar]
- 27. Du C., Wu M., Liu H., Ren Y., Du Y., Wu H., Wei J., Liu C., Yao F., Wang H., Zhu Y., Duan H., and Shi Y. (2016) Thioredoxin-interacting protein regulates lipid metabolism via Akt/mTOR pathway in diabetic kidney disease. Int. J. Biochem. Cell Biol. 79, 1–13 [DOI] [PubMed] [Google Scholar]
- 28. Ago T., Liu T., Zhai P., Chen W., Li H., Molkentin J. D., Vatner S. F., and Sadoshima J. (2008) A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell 133, 978–993 [DOI] [PubMed] [Google Scholar]
- 29. Yamamoto M., Yang G., Hong C., Liu J., Holle E., Yu X., Wagner T., Vatner S. F., and Sadoshima J. (2003) Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J. Clin. Invest. 112, 1395–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sánchez-Villamil J. P., D'Annunzio V., Finocchietto P., Holod S., Rebagliati I., Pérez H., Peralta J. G., Gelpi R. J., Poderoso J. J., and Carreras M. C. (2016) Cardiac-specific overexpression of thioredoxin 1 attenuates mitochondrial and myocardial dysfunction in septic mice. Int. J. Biochem. Cell Biol. 81, 323–334 [DOI] [PubMed] [Google Scholar]
- 31. Yang Y., Del Re D. P., Nakano N., Sciarretta S., Zhai P., Park J., Sayed D., Shirakabe A., Matsushima S., Park Y., Tian B., Abdellatif M., and Sadoshima J. (2015) miR-206 mediates YAP-induced cardiac hypertrophy and survival. Circ. Res. 117, 891–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ago T., Yeh I., Yamamoto M., Schinke-Braun M., Brown J. A., Tian B., and Sadoshima J. (2006) Thioredoxin1 upregulates mitochondrial proteins related to oxidative phosphorylation and TCA cycle in the heart. Antioxid. Redox Signal. 8, 1635–1650 [DOI] [PubMed] [Google Scholar]
- 33. Scarpulla R. C. (2011) Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 1813, 1269–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Simon M. C., Liu L., Barnhart B. C., and Young R. M. (2008) Hypoxia-induced signaling in the cardiovascular system. Annu. Rev. Physiol. 70, 51–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Matsui M., Oshima M., Oshima H., Takaku K., Maruyama T., Yodoi J., and Taketo M. M. (1996) Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev. Biol. 178, 179–185 [DOI] [PubMed] [Google Scholar]
- 36. Matsushima S., Kuroda J., Zhai P., Liu T., Ikeda S., Nagarajan N., Oka S., Yokota T., Kinugawa S., Hsu C. P., Li H., Tsutsui H., and Sadoshima J. (2016) Tyrosine kinase FYN negatively regulates NOX4 in cardiac remodeling. J. Clin. Invest. 126, 3403–3416 [DOI] [PMC free article] [PubMed] [Google Scholar]