

Abstract
Replicative hexameric helicases are thought to unwind duplex DNA by steric exclusion (SE) where one DNA strand is encircled by the hexamer and the other is excluded from the central channel. However, interactions with the excluded strand on the exterior surface of hexameric helicases have also been shown to be important for DNA unwinding, giving rise to the steric exclusion and wrapping (SEW) model. For example, the archaeal Sulfolobus solfataricus minichromosome maintenance (SsoMCM) helicase has been shown to unwind DNA via a SEW mode to enhance unwinding efficiency. Using single-molecule FRET, we now show that the analogous Escherichia coli (Ec) DnaB helicase also interacts specifically with the excluded DNA strand during unwinding. Mutation of several conserved and positively charged residues on the exterior surface of EcDnaB resulted in increased interaction dynamics and states compared with wild type. Surprisingly, these mutations also increased the DNA unwinding rate, suggesting that electrostatic contacts with the excluded strand act as a regulator for unwinding activity. In support of this, experiments neutralizing the charge of the excluded strand with a morpholino substrate instead of DNA also dramatically increased the unwinding rate. Of note, although the stability of the excluded strand was nearly identical for EcDnaB and SsoMCM, these enzymes are from different superfamilies and unwind DNA with opposite polarities. These results support the SEW model of unwinding for EcDnaB that expands on the existing SE model of hexameric helicase unwinding to include contributions from the excluded strand to regulate the DNA unwinding rate.
Keywords: DNA helicase, DNA replication, DNA-protein interaction, nucleic acid enzymology, single-molecule biophysics, DnaB, steric exclusion and wrapping, unwinding
Introduction
Hexameric helicases are structurally conserved toroidal enzyme complexes capable of translocating and separating double-stranded DNA (dsDNA) into two single strands (ssDNA),3 providing templates for DNA replication. They utilize the inherent energy from ATP hydrolysis to translocate along an encircled strand, physically displacing the opposing excluded strand. The translocation polarity of hexameric helicases differs among helicase superfamilies (SFs), defined by the organization and conservation of various folds (1). SF4 helicases from bacteria and associated phages (T4 and T7) include RecA-like folds and have 5′–3′ unwinding polarity, translocating on the lagging strand, whereas SF6 helicases from archaea and eukaryotes have AAA+ folds and 3′–5′ unwinding polarity, translocating on the leading strand (2). Although these two well-studied helicase families have globally conserved structural features, their amino acid sequences, structural folds, and unwinding polarities are not. Moreover, the precise contacts with each DNA strand to facilitate duplex unwinding are not known.
The bacterial replicative helicase, DnaB, has been shown to encircle the 5′-lagging strand in its central channel. Orientation of DnaB binding on ssDNA was shown to place the RecA motor C-terminal domain (CTD) adjacent to the duplex region and the N-terminal domain (NTD) toward the 5′-end (3). DnaB unwinds dsDNA in a steric exclusion (SE) mechanism and can even translocate over two or three strands of DNA, indicating plasticity within the central channel of the hexamer (4, 5). Currently, various X-ray and EM structures of hexameric DnaB (with and without DNA or accessory proteins) show the hexamer as either a closed ring (6–8) or a split lock washer (9, 10). ssDNA bound in the central channel adopts an A-form right-handed spiral conformation making contacts with multiple interior DNA-binding loops from different subunits to pass DNA along in a hand-over-hand mechanism (9).
In addition to specific DNA contacts that exist within the central channel, further exterior contact sites are proposed to exist for hexameric helicases to aid in DNA loading and unwinding (2). Previously, we have identified an interaction path on the external surface of the archaeal (AAA+) SsoMCM helicase with the excluded strand that both protects and stabilizes the complex in a forward unwinding mode (11, 12). This interaction expanded the widely accepted SE model of unwinding to include contributions of the excluded strand in the mechanism. This new unwinding model was termed steric exclusion and wrapping (SEW). Recently, interactions with the excluded strand have been uncovered from a variety of hexameric helicase complexes in addition to archaeal MCM including papillomavirus E1 (13), SV40 large T (14), T7gp4 (15, 16), EcDnaB (17, 18), TWINKLE (19), and the eukaryotic Cdc45/MCM2–7/GINS complex (20, 21). It is hypothesized that external interactions with the excluded strand will not only protect ssDNA but also stabilize the helicase/DNA complex and modulate the unwinding rate.
In this study, we examined whether EcDnaB has a similar specificity for exterior interactions with the excluded single-strand DNA. Single-molecule FRET (smFRET) experiments were used to directly detect EcDnaB binding to the excluded strand and to compare it with SsoMCM binding. The absolute FRET states, transition probabilities, and dwell times were strikingly similar between EcDnaB and SsoMCM even though they reside in different superfamilies and have opposite unwinding polarities. Mutation of several conserved external positively charged residues on EcDnaB differentially altered the observed FRET states and binding dynamics, consistent with disruption of the excluded strand binding path. Notably, those external SEW mutations on EcDnaB dramatically increased the dsDNA unwinding rate compared with wild type (WT). Neutralizing the negative charge on the excluded stand with a morpholino substrate increased the unwinding rate for wild-type EcDnaB, consistent with electrostatic interactions regulating activity. This is the first molecular explanation for regulation of the rate of DNA unwinding through specific external surface interactions on the helicase with the excluded strand.
Results
EcDnaB interacts with the excluded strand
Previously, we have shown that interactions with the excluded strand exist for the 3′–5′ hexameric SsoMCM helicase (11, 22). Using a similar smFRET approach, we sought to examine whether analogous contacts on the exterior of EcDnaB also interact with the excluded strand despite the opposing 5′–3′ translocation polarity. Three separate model fork substrates, 30/30 (DNA43/DNA44), 40/30 (DNA111/DNA44), and 50/30 (DNA116/DNA44) composed of an 18-bp duplex with (dT)30 on the 5′-lagging strand and (dT)30, (dT)40, and (dT)50 on the 3′-leading strand, respectively, were used. DNA forks alone result in low FRET signals as a result of Cy3 and Cy5 on the termini of the fork arms not being in close proximity (Fig. 1A). Additional nucleotides on the leading strand 3′-arm of the fork further decrease the FRET efficiency as expected. Addition of EcDnaB to each of these substrates shifts the signal to higher FRET states (Fig. 1B). EcDnaB preferentially encircles the 5′-strand (3), and a titration of EcDnaB onto 30/30 showed little to no variation in the resulting histogram profiles, suggesting that only one hexamer can be accommodated by the fork substrate over a large concentration range (Fig. 1C). Should a second hexamer encircle the opposing 3′-strand, the FRET values would decrease. The occurrence of a high-FRET state is consistent with an interaction of the excluded Cy3 3′-strand on the external surface of EcDnaB, analogous to SsoMCM binding to DNA and consistent with the proposed SEW model of helicase interaction and unwinding (11).
Figure 1.
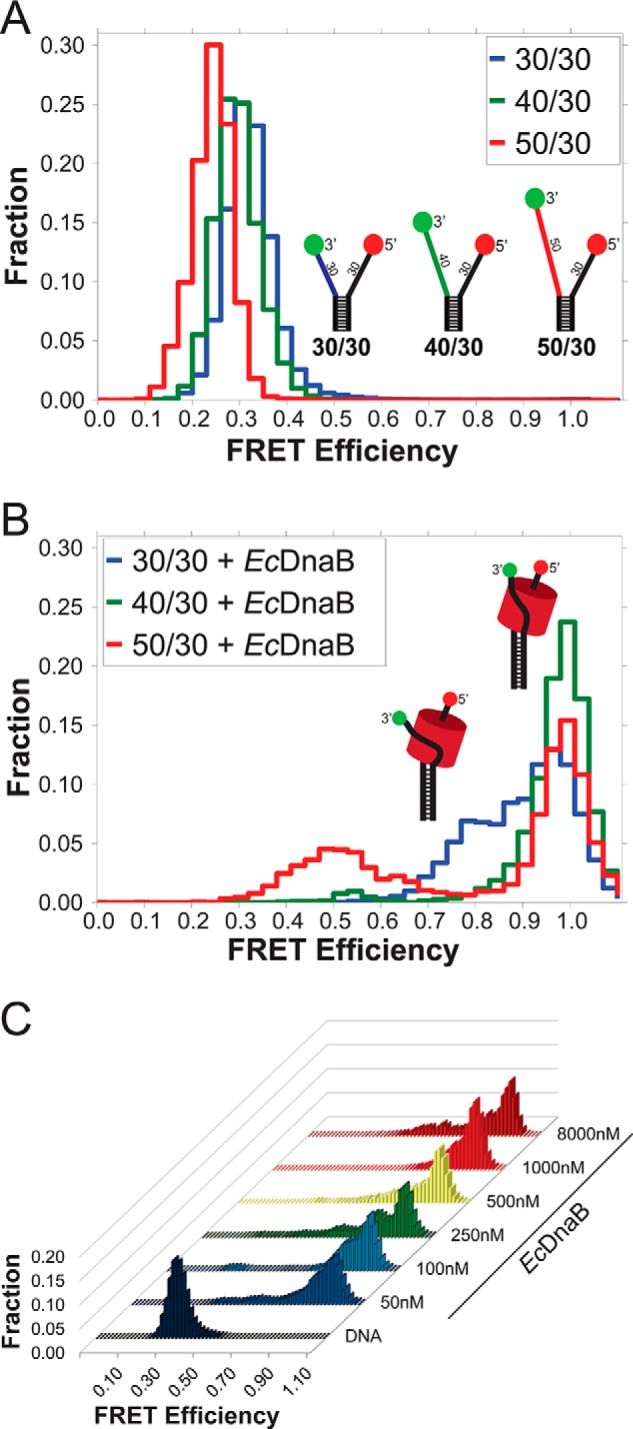
Single-molecule FRET monitoring of EcDnaB binding to DNA fork substrates. A, histograms of the smFRET signal from the DNA fork substrates alone, colored to match schematic models of the DNA forks with a static 30-base encircled 5′-strand and variable excluded strand 3′-arm lengths (30, 40, and 50 nucleotides) shown in blue, green, and red, respectively. B, histograms of the three DNA substrates in the presence of 250 nm WT EcDnaB. C, histogram profiles from a titration of WT EcDnaB onto the 30/30 fork substrate from 50 nm to 8 μm are shown. 30/30 alone exhibits low FRET (shown in dark blue). Adding WT EcDnaB shifts the FRET signal to higher FRET values in all cases without significantly altering the histogram profile.
EcDnaB loaded onto the 40/30 substrate produced an almost exclusively high-FRET state (>0.9), the 50/30 fork produced a bimodal distribution of high- (>0.9) and medium (∼0.5)-FRET states, and the 30/30 fork yielded a bimodal distribution of two high-FRET (0.8 and >0.9) states in the presence of EcDnaB. The interaction with the excluded strand likely differs from varying excluded strand lengths sampling slightly different external binding paths. However, in all cases, the shifts to high-FRET states correspond to a stably wrapped excluded strand that places the Cy3 dye near the Cy5 dye on the encircled strand.
Explicit probability and rate transition (ExPRT) analysis of excluded strand dynamics
To better visualize the FRET states, explicit transition probabilities, and dwell times on a single plot to compare different conditions, mutants, and helicases, we developed ExPRT plots (Fig. 2A). The positions of the circular markers correspond to transitions between specific FRET states with the initial FRET state on the x axis and the final FRET state on the y axis. The initial and final FRET states for a particular transition refer to the observed FRET states immediately preceding and immediately following the transition of interest, respectively. The size and color of each marker correspond to the probability of that transition occurring within a measured trace and the average dwell time of the state preceding the transition, respectively.
Figure 2.
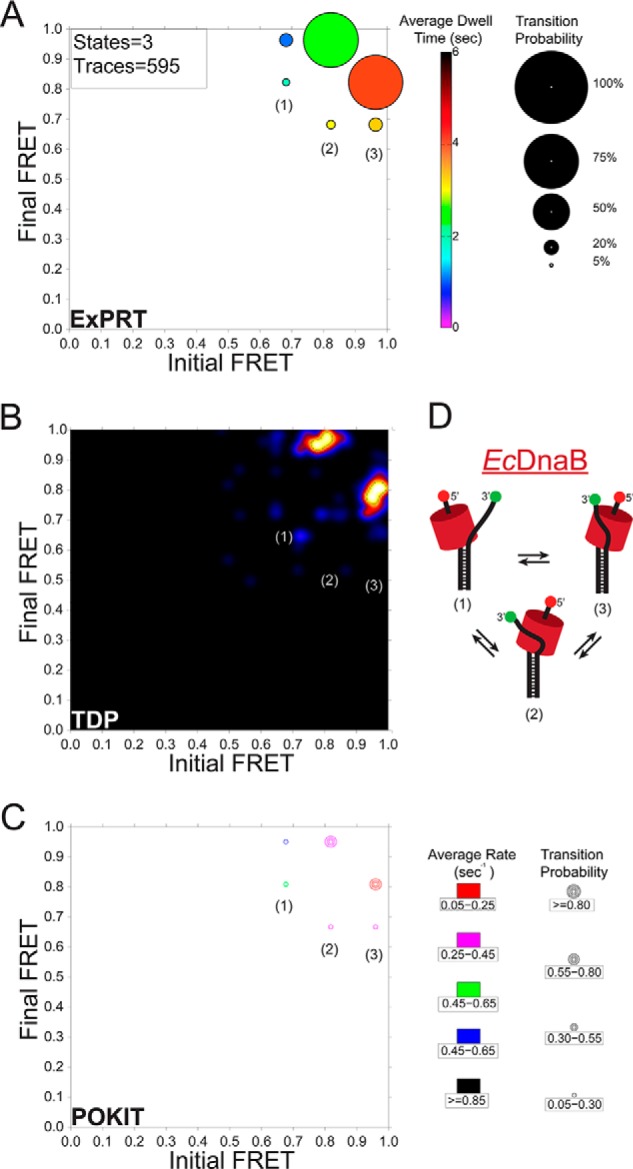
Comparison of smFRET dynamics for excluded strand interactions on EcDnaB. A, ExPRT plot showing the probability (size) and dwell time (color) of transitions for EcDnaB (250 nm) on 30/30 fork DNA. The numbers of states and traces fit by the data are in the upper left-hand corner for each plot. B, the smFRET data set from WT EcDnaB on 30/30 was also analyzed and fit using HaMMY and subsequently analyzed and visualized by the TDP. C, separately the same data were stitched together and fit using vbFRET and visualized using the POKIT analysis program. D, schematic representation of the hypothesized three states (1–3) of bound helicase to DNA fork identified in the ExPRT plots and indicated on each plot.
A comparison of analyses between established programs, the HaMMY and transition density plot (TDP) programs (Fig. 2B) (23) as well as the population-weighted and kinetically indexed transition (POKIT) density program (Fig. 2C) (24), and the ExPRT program is shown. Each program analyzed and visualized identical data corresponding to EcDnaB bound to the 30/30 substrate and can distinguish states (Fig. 2D). Each plot illustrates that the transitions between FRET states of ∼0.8 and ∼0.95 are the most frequent. The TDP program analysis works on a trace-by-trace basis and is able to reveal heterogeneities in the transition data that can be missed by programs that work on stitched data sets such as the POKIT and ExPRT programs. However, despite the TDP program's ability to gather probability and rate values, these values are not directly visualized by the resulting plot. The POKIT program bins the probabilities and rates of each transition into user-defined ranges and produces plots that allow for some level of quantitative comparison between experimental conditions. However, these plots fail to display explicit probabilities and rate values. Determining explicit transitions, probabilities, and rates without binning is an inherent advantage of single-molecule methods that allows for extensive insights into the dynamics and kinetics of molecular interactions and enzymatic activities. The ExPRT analysis program extracts these explicit values from the smFRET data and visualizes them directly in a single plot. This allows users and readers to easily make comparisons between data sets on the most detailed level. Therefore, the ExPRT plots provide a useful advance in the investigation and comparison of the probability and kinetics of smFRET dynamics.
EcDnaB and SsoMCM interact with the excluded strand similarly
The bimodal distribution observed for EcDnaB on 30/30 fork substrate was very similar to the distribution produced by the archaeal MCM helicase on the same substrate (Fig. 3A) (11). In fact, the single-molecule traces for EcDnaB and SsoMCM exhibited strikingly similar dynamics between two high-FRET states (∼0.95 and ∼0.8) (Fig. 3, B and C). This is highlighted by the ExPRT plots for EcDnaB compared with SsoMCM on the 30/30 fork substrate (Fig. 3, D–F). In both cases, the excluded strand has a reversible transition between two high-FRET states (∼0.8 and ∼0.95) that is exhibited by ∼70% of the molecules analyzed. For both EcDnaB and SsoMCM, there is a preference for the ∼0.95 FRET state, indicated by the longer dwell times measured for that state (shades of orange versus green/yellow). In addition, both data sets exhibit a reversible lower probability transition between each of the high-FRET states and a medium-FRET state of ∼0.55. Altogether, it is noteworthy that similarities in the FRET states, transitions, and dwell times exist for EcDnaB and SsoMCM on the 30/30 fork even though these two helicases belong to different superfamilies with low sequence homology, exist in different domains of life (Bacteria versus Archaea), and have opposite unwinding polarities. The similarities are suggestive of a common SEW unwinding mechanism across diverse replicative helicases.
Figure 3.

Comparison of the excluded strand interactions of SsoMCM and EcDnaB by smFRET. A shows the overlaid histograms of both SsoMCM and EcDnaB on the 30/30 fork. B and C, representative single-molecule traces for EcDnaB (B) and SsoMCM (C) on the 30/30 DNA template. The top panels show the Cy3 (green) and Cy5 (red) signals. The bottom panels show the corresponding FRET signal (blue) with overlaid ideal states (red) for each trace as fit by vbFRET (see “Experimental procedures”). D–F, ExPRT plots (D) showing the probability (size) and dwell time (color) of transitions for EcDnaB (250 nm) (E) and SsoMCM (1.3 μm) (F), respectively, on 30/30 fork DNA. The numbers of states and traces fit by the data is in the upper left-hand corner for each plot. The insets are schematic representations of the hypothesized three states (1–3) of bound helicase to DNA fork identified in the ExPRT plots.
Exterior surface mutations of EcDnaB alter excluded strand wrapping
As positively charged residues on the surface of SsoMCM had previously been shown to support an external interaction with the excluded strand (11), similar surface-exposed and conserved residues were identified based on a homology model for EcDnaB (Fig. 4). Four EcDnaB surface positions (Arg-74, Arg-164, Lys-180, and Arg-328/Arg-329) that exist in positively charged electrostatic patches were mutated to alanine, overexpressed, and purified. All mutant proteins were consistent with a hexamer as the major peak after gel filtration (data not shown). smFRET DNA fork binding assays were performed for the wild type and each mutant on each of the three fork templates (30/30, 40/30, and 50/30). The results were analyzed and compared using traditional histograms and ExPRT plots. The mutations gave rise to several important differences in the hexamer-excluded strand interactions and dynamics. Across all the excluded strand lengths tested, EcDnaB (R74A) does not sample the highest observed FRET state (Eapp = ∼0.95) observed for the WT and the other mutants. This can be seen clearly in the histograms (Fig. 5, B versus A) and the ExPRT plots (Fig. 5, G, L, and Q versus F, K, and P). R74A also produces fewer transitions between FRET states compared with the WT for the 30/30 and 40/30 forks because the most frequent transitions observed for the WT data are those between the highest FRET state (Eapp = ∼0.95) and lower FRET states. The absence of the 0.95 FRET state for R74A across all substrates tested suggests that Arg-74 is necessary to close the connection of the excluded strand to the NTD traversing the entire longitudinal length of EcDnaB (9).
Figure 4.
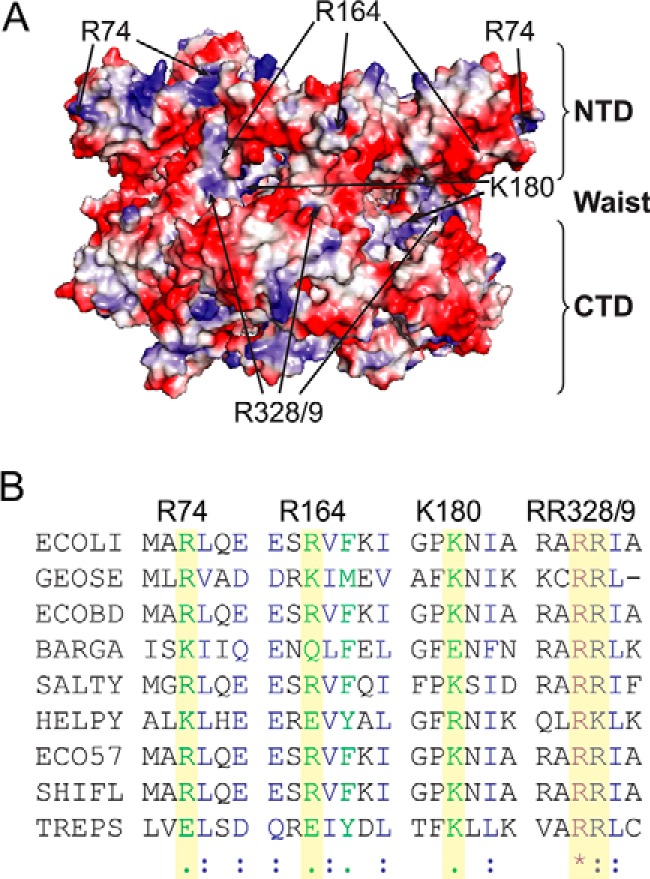
Identification of exterior electrostatic SEW sites for EcDnaB. A, position of the SEW mutations (from multiple subunits) mapped onto the homology model for EcDnaB colored with an electrostatic surface identifying the NTD, RecA CTD, and waist. B, multiple amino acid alignment of DnaB helicases using ClustalW2. Identical (*), similar (:), and somewhat similar (.) residues are indicated. ECOLI, Escherichia coli strain (K-12); GEOSE, G. stearothermophilus; ECOBD, E. coli strain (BL21-DE3); BARGA, Bartonella grahamii (strain as4aup); SALTY, Salmonella enterica serovar Typhimurium; HELPY, Helicobacter pylori strain (26695); ECO57, E. coli O157:H7; SHIFL, Shigella flexneri; TREPS, Treponema pallidum SS14.
Figure 5.

Histograms and ExPRT plots of WT EcDnaB and mutants bound to DNA forks. Histograms (A–E) report the population of molecules as a function of FRET states on DNA forks with a 30-base 5′-strand and a 30-base (blue), 40-base (green), or 50-base (red) 3′-strand for WT, R74A, R164A, K180A, and R328A/R329A, respectively. Yellow, blue, and red regions highlight low-, medium-, and high-FRET populations, respectively. Corresponding ExPRT plots are shown for 30/30 (F–J), 40/30 (K–O), and 50/30 (P–T) forks for each of the respective EcDnaB helicases.
EcDnaB (K180A) bound to the 30/30 fork produces similar FRET states and dynamics when compared with the WT EcDnaB; however, there are now five states compared with three (Fig. 5, I versus F). Examples traces for individual molecules for WT compared with K180A as well as other mutants are shown in Fig. 6. As an example, for molecule 51, there are 10 transitions between two states, and for molecule 177, there are 17 transitions between three states. A greater number of FRET states and transitions are indicative of a less stable and less precise interaction between the exterior surface of the helicase and the excluded strand, leading to alternative binding paths. Similarly, the R164A mutant also samples a greater number of states than WT on the 40/30 (five versus two states) and 50/30 substrates (four versus two states) (Fig. 5, M and R versus K and P). The ExPRT plots for both K180A (Fig. 5, I versus F) and R164A (Fig. 5, H and F) on the 30/30 substrate somewhat resemble the WT on the same substrate. However, the histograms of K180A and R164A (Fig. 5, C and D) show populations that are broader than those seen for the WT and R74A (Fig. 5, A and B), which is indicative of less stable or precise interactions between the helicase and the excluded strand as visualized in the ExPRT plots. For the R164A mutation on the 30/30 substrate (Fig. 5H), the most probable transitions occur at approximately the same rate reversibly. Similar to the WT, K180A on 40/30 and 50/30 substrates show fewer transitions to and from the high-FRET state and a significantly longer dwell time for the high-FRET state (Fig. 5, N and S). This may indicate greater stabilization of an interaction toward the NTD of the helicase. Therefore, Lys-180 and Arg-164 contribute to but do not solely mediate the helicase-excluded strand interactions that give rise to the FRET states we observe for the WT.
Figure 6.

Example smFRET kinetic traces. A–F, comparison of WT (A–C) and K180A (D), R74A (E), and R328A/R329A (F) EcDnaB FRET efficiencies as a function of time on the 30/30, 40/30, and 50/30 forks, respectively. The calculated FRET values (blue) are overlaid with the ideal state fits (red).
The EcDnaB (R328A/R329A) mutant shows similar FRET state transitions, probabilities of those transitions, and dwell times on the 30/30 fork when compared with the WT (Fig. 5, J versus F). However, there are extreme differences in the binding states and dynamics on the longer DNA strands compared with WT (Fig. 5, T versus P). WT EcDnaB bound to the 50/30 fork shows a small fraction of traces that transition between high- and medium-FRET states. In comparison, EcDnaB (R328A/R329A) produces almost entirely medium-FRET states that are very dynamic with many transitions and relatively short dwell times, indicative of severe destabilization of binding. In contrast to WT, very little high-FRET signal from the R328A/R329A mutant on the 30/50 substrate is observed. These results indicate that Arg-328 and Arg-329 may be required to stabilize longer excluded strands (40- and 50-mers) along the waist of the hexamer and mediate interactions between the excluded strand and other regions such as NTD where Arg-74 is located and responsible for the highest FRET state (Eapp = ∼0.95).
Although we primarily tested the effect of eliminating positive charge on the exterior surface of EcDnaB and its effect on excluded strand binding, there may also be additional noncovalent binding interactions defining a path. To directly test whether electrostatic interactions exclusively define the excluded strand binding path, we titrated NaCl into WTEcDnaB prebound to a 30/30 fork in our smFRET experiments. Increased salt concentrations resulted in increased dynamics (shorter dwell times), but the FRET states were not significantly affected as visualized using ExPRT plots (Fig. 7). Even though the FRET states and transition probabilities remained constant, the decreased dwell times suggest that EcDnaB utilizes electrostatic interactions to mediate wrapping but that other noncovalent interactions are also important.
Figure 7.
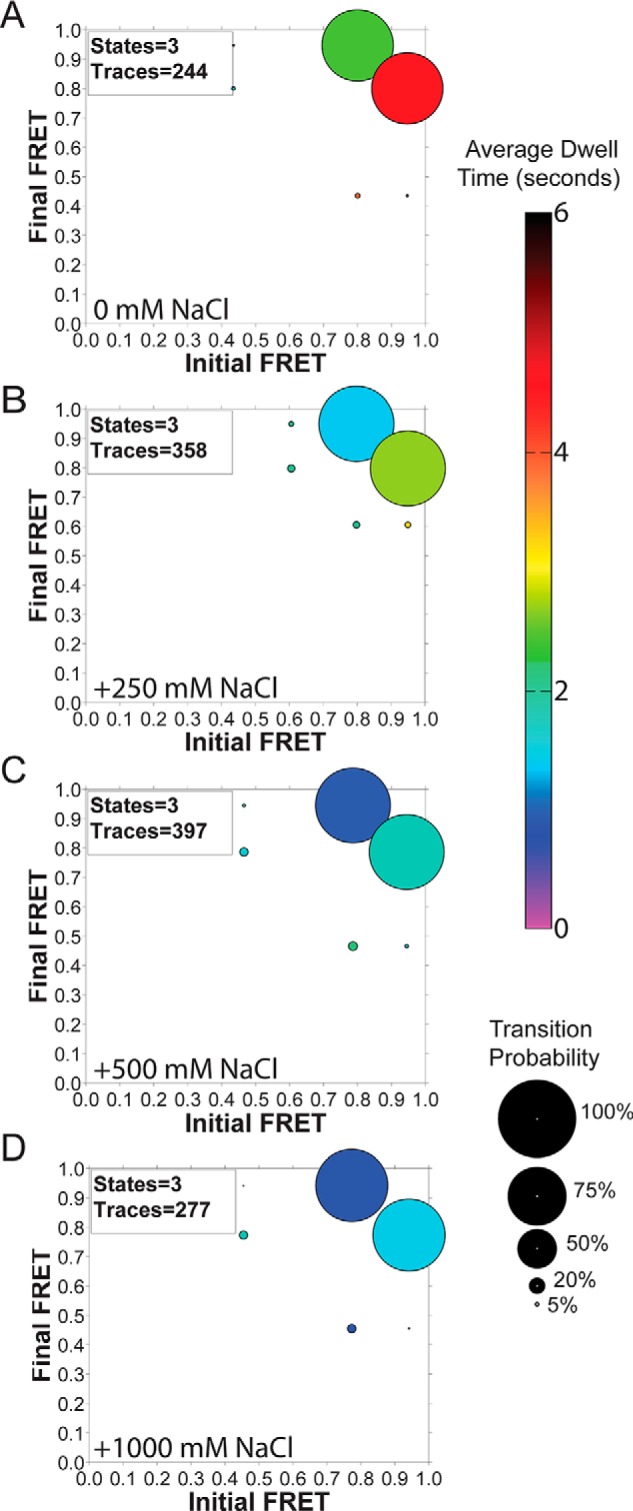
Titration of NaCl onto EcDnaB-bound 30/30 fork. A–D, ExPRT plots of [NaCl] titration onto EcDnaB (250 nm) prebound to 30/30 DNA fork.
SEW mutants of EcDnaB have enhanced DNA unwinding activity
Mutating positively charged residues involved in the excluded strand interaction inhibited SsoMCM's unwinding activity (11, 22). Gel-based fluorescent DNA unwinding assays were performed to determine whether these EcDnaB SEW mutants have any effect on activity. Fig. 8A shows a representative 6-min time point; however, quantification of the steady-state unwinding rates occurred over multiple time points for each mutant (Fig. 8B). All mutants had increased unwinding rates compared with the WT EcDnaB. Specifically, R74A (26 ± 1 nm s−1) and R164A (53 ± 13 nm s−1) have 3- and 6-fold increases, respectively, over WT (9 ± 1 nm s−1); whereas K180A (199 ± 16 nm s−1) and R328A/R329A (191 ± 13 nm s−1) have more than 20-fold increases in unwinding activity.
Figure 8.
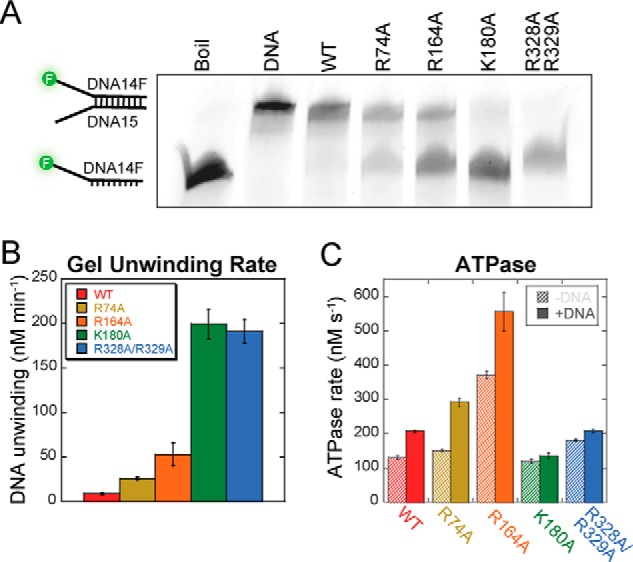
Biochemical properties of SEW mutants of EcDnaB. A and B, a representative gel for 6-min time point is shown for EcDnaB (WT and mutants, 3 μm) unwinding assays performed on a fluorescein-labeled fork DNA (DNA14F/DNA15) (15 nm) (A) and quantified over multiple time points (B) is shown. Throughout, data for EcDnaB constructs are consistently colored (WT, red; R74A, ochre; R164A, orange; K180A, green; R328A/R329A, blue). Error bars represent the standard error from at least three independent experiments. C, quantification of the ATP hydrolysis rate in the absence (diagonal hash) and presence (solid) of fork DNA (DNA14/DNA15).
ATPase assays were performed for WT and mutant EcDnaB proteins, and the rates were quantified in the absence and presence of DNA (Fig. 8C). R74A, K180A, and R328A/R329A have similar basal rates to WT; however, R164A had a 2.5-fold enhancement over WT. Similar to WT EcDnaB, both R74A and R164A were stimulated 1.5–2.0-fold in the presence of DNA, consistent with previous results (25). Interestingly, the fastest unwinding mutants, K180A and R328A/R329A, were only weakly stimulated further in the presence of DNA. Previously, the R328A/R329A mutant was investigated for its potential role in a leucine zipper motif (26), although it was later discounted (27) and also found not to have DNA-stimulated ATPase activity. No stimulation in ATPase rate with DNA is sometimes indicative of a perturbation in DNA binding; however, these mutants show stimulated unwinding abilities, and fluorescence anisotropy experiments showed no significant differences in Kd values measured for mutants binding to fork DNA compared with WT (data not shown).
To validate whether alteration of electrostatic interactions are responsible for the increased unwinding rates in the mutants compared with WT EcDnaB, we performed DNA unwinding reactions with a 3′-morpholino (Morph) strand. Morpholino nucleic acids have standard base pairing properties but have morpholine rings linked through phosphorodiamidate groups that lack negative charge and are as stable or more stable than an equivalent DNA duplex (28, 29). Previously, the homologous hexameric T7 gp4 DNA helicase was shown to unwind excluded strand morpholino substrates with a greater rate and efficiency than for DNA (16). They attributed this enhanced unwinding activity to the disruption of the helicase's interaction with the displaced strand that limits its activity.
Interestingly, WT EcDnaB also unwinds excluded strand morpholino substrates with a profoundly enhanced rate compared with a DNA/DNA duplex (Fig. 9). The rate of unwinding for the Morph/DNA is at least 1000-fold faster with a >0.7 amplitude after 1 min, whereas the DNA/DNA duplex is only ∼0.4 unwound after 45 min. No unwinding or strand separation is seen when ATP is excluded from the experiment. Accurate quantification of the unwinding rate would require rapid quench experiments, but it is apparent that EcDnaB unwinds excluded strand morpholinos rapidly. The unwinding rates for the Morph/DNA are also significantly faster (∼10-fold) than even those seen above for the fastest SEW mutant (K180A) on a DNA fork (Fig. 8). This may not be surprising as the SEW mutants only affect contact at one specific mutated site on the exterior surface, whereas the excluded strand Morph eliminates electrostatic contacts throughout the longitudinal length of EcDnaB.
Figure 9.
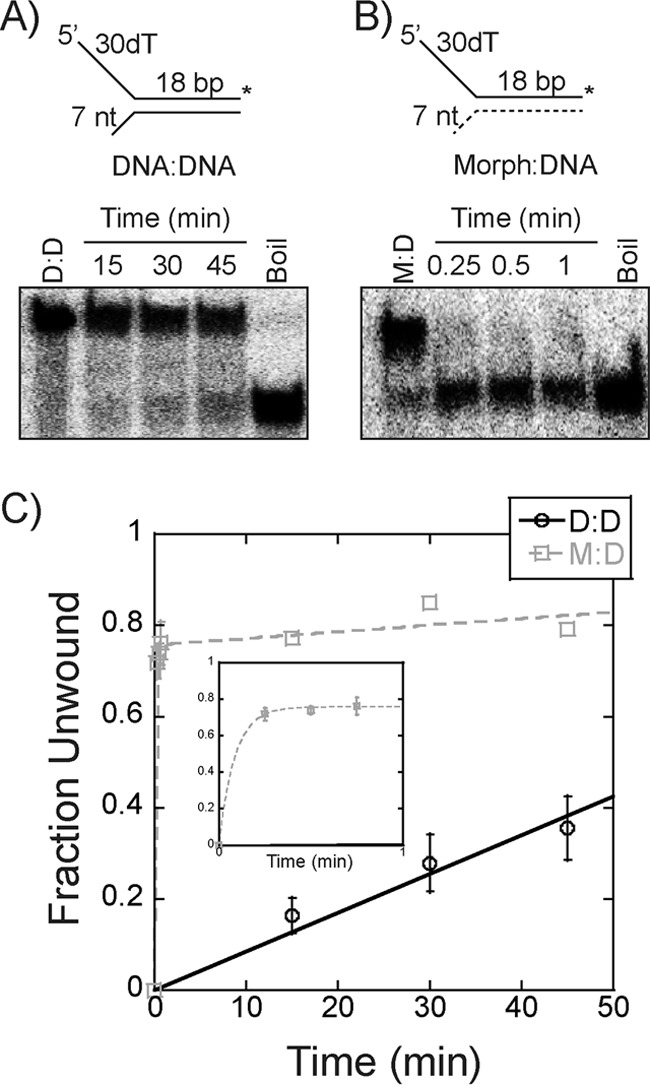
Unwinding of morpholino forked substrates. A and B, a representative unwinding time course for 250 nm EcDnaB on either an 18-bp DNA/DNA (D:D) (A) or Morph/DNA (M:D) (B) substrate (15 nm) with a 7-base 3′-excluded strand flap is shown. Single-turnover experiments were initiated with ATP and a single-strand trap identical to the radiolabeled strand as described under “Experimental procedures.” C, averaged unwinding data for DNA/DNA (black ○) were plotted and fit with a linear regression to give k = 0.009 min−1, and those for Morph/DNA (gray □) were plotted and fit using Equation 3 to give k1 = 11 min−1. The inset plot highlights data within the 1st min. Error bars are the standard error from three independent experiments.
Discussion
Although hexameric DNA replication helicases have global structural conservation, their amino acid sequences are not conserved, allowing for the classification of these helicases into different superfamilies. We have shown previously that the 5′-excluded strand makes important external surface interactions that aid in the mechanism of unwinding for the SF6 archaeal SsoMCM helicase (3′–5′) to develop the SEW of unwinding (11). In this report, we can now show that the bacterial replication helicase, EcDnaB, with opposite unwinding polarity (5′–3′) and of a different family (SF4) and organismal domain, has similar conformational states and dynamics of binding the excluded strand that also regulate DNA unwinding (Fig. 10). The combined results highlight the importance and conservation of the SEW model for hexameric helicase unwinding of DNA and reveal external surface residues required for regulating the activity of the EcDnaB helicase. Importantly, the SEW effects on the mechanism of unwinding are opposing for SsoMCM and EcDnaB.
Figure 10.
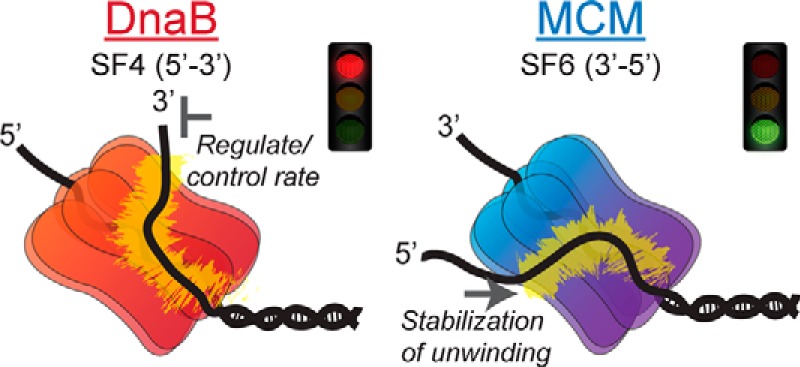
SEW models for hexameric helicase unwinding. Bacterial DnaB and archaeal MCM encircle the lagging or leading strand, respectively, and interact with the excluded strand on the exterior surface to either regulate the unwinding rate using this electrostatic brake or stabilize unwinding in a forward direction.
It is striking that the absolute FRET states, transition probabilities, and dwell times visualized by the ExPRT plots are extremely similar between SsoMCM and EcDnaB hexameric helicases bound to fork DNA. In both cases, the large increase in FRET observed is consistent with encircling of the translocating strand and exclusion of the other along the exterior surface. For EcDnaB, there have been reports of hexamers loading on opposing strands in opposite orientations (3, 30–32). The consequence of loading two hexamers would ultimately separate the strands further, resulting in a decrease in the FRET signal, which is opposite to what we observe in the smFRET experiments even at high concentrations of EcDnaB (Fig. 1B). Both helicases are of similar size and oligomeric state and are thought to engage their respective translocating strands in a similar way, so the DNA-bound states of each helicase may be structurally equivalent even with opposing translocation polarities. Therefore, EcDnaB binding of DNA includes both the encircling of the 5′-strand and the exclusion and external interaction of the 3′-strand in a similar manner to SsoMCM and the SEW model for unwinding.
Without an appropriate DNA-bound crystal structure of EcDnaB, we had to infer binding positions for the excluded 3′-strand based on amino acid homology and electrostatics from crystal structures that represented a closed ring (6, 7) or a split lock washer structure (9). Because ssDNA was contained in the central channel of the split lock washer structure, we used this conformation as a primary model to interpret interactions with the excluded strand. This restricted the definition of any precise or specific exterior binding path, and rather we can only conclude general binding to the CTD, waist, and NTD. That along with the specific residues that were mutated and the smFRET data informed our interpretation of excluded strand binding.
To test the specificity of this external interaction, mutations of conserved residues were found to both disrupt and alter the binding states. In particular, Arg-74 was found to be necessary for stable interactions of the excluded strand at the NTD, giving rise to the highest FRET state (∼0.95). R164A and K180A exhibited somewhat different dynamics than WT, generally fitting to more FRET states, which may reflect alternate binding paths on the helicase exterior. It is likely that these two residues partially contribute to the excluded strand interaction. Arg-328 and Arg-329 both seem to be important for wrapping longer ssDNA substrates at the waist whereupon mutation the values shift from a medium- (∼0.8) to a lower (∼0.5) FRET state, consistent with decreased wrapping. Altogether, these mutants individually alter the interaction between the excluded strand and the exterior of the helicase to varying degrees, and the amount of destabilization or altered external DNA binding paths can depend on excluded strand length. The data provide information on the contacts all along the longitudinal length of the hexamer, defining a minimal binding path.
Previously, mutation of external positively charged residues on SsoMCM reduced DNA unwinding, presumably through a slippage mechanism where the mutant helicase was unable to stabilize forward unwinding steps (11). However, for EcDnaB, mutation of external positively charged residues generally increased unwinding rates. The enzymatic effects of the mutations largely correlate with two classes of results. Two of the mutations (R74A and R164A) show slightly enhanced unwinding activity, and their ATPase rates are stimulated with DNA similarly to WT. For R164A, the increased ATPase activity of this mutant could account for the DNA unwinding enhancement, but R74A has similar ATPase rates to WT. In both cases, the mutation to alanine has disrupted or altered the external interaction as measured by smFRET. We propose that releasing some of the electrostatic wrapping interaction frees the helicase to unwind faster.
The more intriguing class of mutations (K180A and R328A/R329A) exhibits more than 20-fold increases in DNA unwinding. Interestingly, they do not show as significant DNA-dependent stimulation of ATP hydrolysis rates. For these mutants, the most significant differences are an increase in the number of conformations for K180A with shorter fork arms and a global change in FRET states and an increase in dynamics for R328A/R329A with the longer excluded strand. Overall, a general trend of increased unwinding activity emerges as we neutralize positively charged residues found on the exterior of the hexamer. Therefore, exterior electrostatic interactions with the excluded strand restrict the unwinding activity of EcDnaB.
By using excluded strand morpholino substrates instead, we have the added benefit of testing the total effect of exterior electrostatics on unwinding instead of contributions at specific amino acid sties. Disruption of the electrostatic interaction of the excluded strand through this morpholino chemistry was strongly stimulatory to unwinding. A similar stimulation in unwinding of morpholino strands was also seen with the homologous T7 gp4 hexameric helicase (16). In both cases, interactions on the outer surface of the helicase with the excluded strand will act to regulate the unwinding rate. However, this external interaction is not entirely electrostatic as increasing ionic strength in the smFRET experiments resulted in decreased dwell times but did not significantly affect the FRET states.
Previous single-molecule work has detailed the single-molecule force contributions of each DNA strand to unwinding by EcDnaB using either a hairpin or fork substrate (33). In that study, it was concluded that the unwinding rate was controlled by both force-induced destabilization of the duplex and interactions of the excluded strand with the exterior surface. The main apparent discrepancy between our work and theirs is that when the excluded strand is sequestered because of constraints in the hairpin assay the rate is slower than when it is allowed to interact with the exterior surface in the fork assay. This would imply that contacts of the excluded strand with the external surface of EcDnaB increase unwinding, whereas we show that specific external contacts restrain unwinding. However, it is probable that the force applied to the excluded strand in the fork assay artificially alters the interaction with the exterior surface in a way analogous to the altered DNA binding paths and kinetics for the R74A and R164A mutants. Therefore, measured increases in unwinding in both studies can be explained by altered DNA binding paths on the exterior surface. It is notable that a variety of recent biophysical techniques monitoring EcDnaB activity and binding have detected an elusive external interaction with the excluded strand (18, 33). We can now conclude that this SEW interaction and the precise binding path regulate the speed of unwinding for EcDnaB.
Clearly, interactions with the excluded strand are acting as a regulator to control the speed of unwinding. It remains to be seen whether this is because of a greater increased force applied by the motor domain for EcDnaB that is modulated by the excluded strand or whether discrete external binding paths or polarity dictates the rate of unwinding. Further experiments will be needed to more specifically define the exterior binding path. For EcDnaB, the excluded strand interaction may act as a “molecular brake” to control the amount of exposed ssDNA or provide a platform for accessory helicases, e.g. Rep, to assemble and rescue stalled forks (34, 35). Coupled DNA synthesis by the leading strand polymerase (polymerase III) could sequester the excluded strand from the exterior surface of EcDnaB and explain the increased rate of unwinding by the coupled replisome (36–38). In fact, a recent report has shown that when the bacterial helicase and polymerase become decoupled, the unwinding rate is reduced by 80% as a fail-safe “dead man's switch” (39). This can be explained at the molecular level by our data in which decoupling of the polymerase engages the excluded strand with the exterior surface of EcDnaB, slowing its unwinding progression. In addition to controlling the unwinding rate, the external DNA-binding sites on both helicases are likely to contribute during the loading mechanism for encircling of the translocating strand to maintain strand separation during the action and conformational changes induced by the initiation enzymes.
Experimental procedures
Materials
Oligonucleotides used (supplemental Table S1) were purchased from Integrated DNA Technologies, Inc. (Coralville, IA). Fluorescently labeled DNA was HPLC-purified (Integrated DNA Technologies, Inc.), and non-labeled oligos were gel-purified (40). Morpholinos were from GeneTools (Philomath, OR). SsoMCM was purified as described previously and reported as hexamer concentrations (11). All other chemicals were analytical grade or better.
Cloning and protein purification of EcDnaB
The R74A, R164A, K180A, and R328A/R329A mutations of EcDnaB were created by overlap extension from pET11b-EcDnaB. The DNA primers are listed in supplemental Table S1. Mutations were confirmed by the DNA sequencing facility at the University of Pittsburgh. Mutants and WT EcDnaB were expressed in Rosetta 2 cells (EMD Millipore, Billerica, MA) or C41 cells (Lucigen, Middleton, WI) using autoinduction (41) or by induction with 0.1 mm isopropyl β-d-1-thiogalactopyranoside. Cells were pelleted, resuspended in EcDnaB lysis buffer (10% sucrose, 50 mm Tris-HCl (pH 7.5), 50 mm NaCl, 5 mm dithiothreitol (DTT)), and lysed using lysozyme and sonication. Ammonium sulfate was added to the resulting supernatant at 0.2 g/ml, pelleted, and then resuspended in EcDnaB buffer A (10% glycerol, 0.1 mm EDTA, 50 mm Tris-HCl (pH 7.5), 50 mm NaCl, 5 mm DTT). The supernatant was purified using an ÄKTA Prime FPLC equipped with a HiTrap Mono Q column (GE Healthcare) and eluted with a stepwise gradient of EcDnaB buffer A with 500 mm NaCl followed by a similar procedure using a HiTrap heparin column (GE Healthcare). The purified fractions were combined and applied to a Superdex S-200 26/60 gel filtration column (GE Healthcare) with buffer C (50 mm Tris-HCl (pH 7.5), 50 mm NaCl, 5 mm DTT) to isolate the hexamer. An extinction coefficient (185,000 cm −1 m−1) was used to quantify the fractions containing purified hexameric EcDnaB (42). All concentrations for EcDnaB are indicated as hexamer throughout.
Single-molecule fluorescence resonance energy transfer
DNA substrates labeled with Cy3 and Cy5 fluorophores were immobilized on a PEGylated quartz slide utilizing biotin-streptavidin interactions (43). A prism-based total internal reflection microscope was used to collect all smFRET data (44, 45). A 532 nm diode laser was used to excite Cy3 fluorophore, and subsequent Cy3 and Cy5 emission signals were separated by a 610-nm dichroic long-pass mirror, a 580/40-nm bandpass filter, and a 660-nm long-pass filter. An electron-multiplying charge-coupled device iXon camera (Andor, Belfast, UK) was used to image the signals. Data were acquired at 10 fps for 10 or more regions with each region containing 50–250 molecules in the presence of an oxygen radical-scavenging solution (1 mg/ml glucose oxidase, 0.4% (w/v) d-glucose, 0.04 mg/ml catalase) and 2 mm trolox. EcDnaB (250 nm) was added and given a 5-min equilibration period. All single-molecule experiments were performed in reaction buffer as described previously (11).
Single-molecule FRET signals were identified by fitting individual regions of signal intensity to a 2D Gaussian function and measuring the goodness of fit. These peaks were corrected for thermal drift and local background intensity using ImageJ (46) and the image stabilizer plugin for ImageJ. The resulting signal was used to calculate the apparent FRET efficiency, Eapp, according to the following equation.
| (Eq. 1) |
in which IA and ID are the intensity of the acceptor and donor signals, respectively.
Single-molecule FRET data analysis and ExPRT plots
Data analysis and visualization were performed using manually selected single-molecule traces that displayed anticorrelation between the donor and acceptor fluorophores and single-step fluorophore photobleaching. Traces collected under identical experimental conditions were stitched together and fit to ideal states via hidden Markov modeling using the vbFRET software package (48). Stitched traces were fit to a given number of states based on those states being more than Eapp = 0.1 tk;4apart from one another and the variation of one state not overlapping with another. Traces were then unstitched and fed into the ExPRT analysis program. This MATLAB executable program produces transition plots where the markers are sized based on the probability of transition occurring within an observed single-molecule trace and colored based on the dwell time(s) of the state preceding the transition. The average of all dwell times for a given transition was used to determine the color of the marker. Only dwell times that were both preceded and followed by transitions were included. Stitched data as fit by vbFRET were also analyzed by the POKIT analysis program, and the resulting plot contains a legend for the ranges of rates and probabilities (24). The data were also fit using the HaMMY program, allowing the program to fit the data to up to five states. The output of the HaMMY program was subsequently analyzed by the transition density plot program (23).
EcDnaB structural homology model
Global sequence alignments were performed using ClustalW2 analysis (http://www.ncbi.nlm.nih.gov/blast/bl2seq/wblast2.cgi). The homology model of EcDnaB was created by threading the alignment onto the structure of Geobacillus stearothermophilus DnaB (Protein Data Bank code 4ESV) (9) using SWISS-MODEL (49).
ATPase assay
EcDnaB variants (350 nm) were incubated in the absence and presence of 4 μm forked DNA (DNA14/DNA15) as described previously (47). Briefly, 25-μl reactions were incubated at 37 °C for 5 min in unwinding buffer (50 mm HEPES (pH 8.0), 10 mm Mg(OAc)2, 5 mm DTT, 0.2 mg/ml BSA), and 1 mm ATP with trace amounts of [γ-32P]ATP was added to initiate the reaction. Samples were quenched at 2, 5, 10, and 15 min after initiation in equal volumes of 0.7 m formic acid. A total of 1 μl of quenched reaction was spotted on Millipore TLC PEI Cellulose F, allowed to dry, resolved in 0.6 m potassium phosphate (pH 3.5), phosphorimaged, and quantified for the linear ATPase rate (pmol/min).
Gel-based DNA unwinding assays
Helicase assays were assembled in unwinding buffer at 37 °C. 15 nm fluorescent forked DNA (DNA14/F/DNA15) was incubated with 500 nm EcDnaB at 37 °C for 5 min before initiation with 5 mm ATP. Reactions were quenched with an equal volume of quench solution (50% glycerol, 1% SDS, 100 mm EDTA (pH 8.0), 300 nm ssDNA trap (unlabeled strand with same sequence as radiolabeled strand)) at various time points from 1 to 15 min. Reactions were kept on ice until loading and were resolved on 20% (29:1 acrylamide:bisacrylamide) tris borate-EDTA gels electrophoresed in tris borate-EDTA buffer. The gels were imaged on a Typhoon 9400 phosphorimaging system (GE Healthcare), and the fraction unwound was calculated after background subtraction using ImageQuant software according to the following equation.
| (Eq. 2) |
where Is(t) and Id(t) are the intensities of the single- and double-strand bands, respectively, at time t; subscript 0 and b indicate equivalent counts at t = 0 and the boiled sample, respectively. The fraction unwound or unwinding rate (nm min−1) was calculated from a linear regression fit of the fraction unwound for each time point.
Single-turnover DNA unwinding assays
Single-turnover unwinding experiments were also performed by initiating the reaction simultaneously with 5 mm ATP and 150 nm ssDNA trap (unlabeled strand with the same sequence as the radiolabeled strand). Experiments were performed with 250 nm EcDnaB and 15 nm forked 3′-DNA (DNA163/DNA161) or 3′-morpholino (DNA163/DNA160m) substrates at 37 °C. The 5′-end of DNA163 was labeled with [γ-32P]ATP (PerkinElmer Life Sciences) with Optikinase (Affymetrix, Cleveland, OH) according to the manufacturers' directions. The reaction was quenched at various times using an equal volume of quench solution (20% Ficoll, 1.0% SDS, 200 mm EDTA (pH 8.0), 2 mg/ml Proteinase K) followed by incubation at 37 °C for 10 min. After electrophoresis as above, the gels were imaged on a Storm 820 (GE Healthcare), and the fraction unwound was calculated using ImageQuant software according to Equation 2. Single-turnover data were further quantified using the following equation.
| (Eq. 3) |
where A1 is the amplitude associated with the initial burst rate increase (k1), kss is a steady-state rate, and C is a constant.
Author contributions
S. M. C., S. H. L., and M. A. T. designed the experiments. S. M. C. performed the cloning, protein purification, biochemical assays, and single-molecule experiments. S. M. C. and S. G. performed DNA unwinding assays and protein purifications. M. A. T. directed the project and together with S. M. C. wrote the manuscript. All authors edited the manuscript.
Supplementary Material
Acknowledgments
pET11b-EcDnaB was a generous gift from Dan Kaplan. We thank the Walter laboratory for access to the POKIT analysis program. We thank Brian Graham, Heather McFarland, Grant Schauer, and Valerie O'Shea for helpful discussions.
This work was supported by Baylor University, The University of Pittsburgh, American Cancer Society Research Scholar Grant RSG-11-049-01-DMC (to M. A. T.), National Science Foundation Division of Molecular and Cellular Biosciences Grant NSF1613534 (to M. A. T.), and National Institutes of Health Grants AI081571 and GM068406 (to S. H. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Table S1.
- ssDNA
- single-stranded DNA
- AAA+
- ATPase associated with diverse cellular activities
- CTD
- C-terminal domain
- Cy
- cyanine dye
- Ec
- E. coli
- ExPRT
- explicit rate and probability transition
- GINS
- Go Ichi Nii San (5,1,2,3 in Japanese) complex
- MCM
- minichromosome maintenance protein
- NTD
- N-terminal domain
- SE
- steric exclusion
- SEW
- steric exclusion and wrapping
- SF
- super family
- smFRET
- single-molecule FRET
- Sso
- Sulfolobus solfataricus
- TDP
- transition density plot
- POKIT
- population-weighted and kinetically indexed transition density
- Morph
- morpholino.
References
- 1. Thomsen N. D., and Berger J. M. (2009) Running in reverse: the structural basis for translocation polarity in hexameric helicases. Cell 139, 523–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Trakselis M. A. (2016) Structural mechanisms of hexameric helicase loading, assembly, and unwinding. F1000Res. 5, F1000 Faculty Rev-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jezewska M. J., Rajendran S., Bujalowska D., and Bujalowski W. (1998) Does single-stranded DNA pass through the inner channel of the protein hexamer in the complex with the Escherichia coli DnaB Helicase? Fluorescence energy transfer studies. J. Biol. Chem. 273, 10515–10529 [DOI] [PubMed] [Google Scholar]
- 4. Kaplan D. L., and O'Donnell M. (2004) Twin DNA pumps of a hexameric helicase provide power to simultaneously melt two duplexes. Mol. Cell 15, 453–465 [DOI] [PubMed] [Google Scholar]
- 5. Kaplan D. L., and O'Donnell M. (2002) DnaB drives DNA branch migration and dislodges proteins while encircling two DNA strands. Mol. Cell 10, 647–657 [DOI] [PubMed] [Google Scholar]
- 6. Bailey S., Eliason W. K., and Steitz T. A. (2007) Structure of hexameric DnaB helicase and its complex with a domain of DnaG primase. Science 318, 459–463 [DOI] [PubMed] [Google Scholar]
- 7. Wang G., Klein M. G., Tokonzaba E., Zhang Y., Holden L. G., and Chen X. S. (2008) The structure of a DnaB-family replicative helicase and its interactions with primase. Nat. Struct. Mol. Biol. 15, 94–100 [DOI] [PubMed] [Google Scholar]
- 8. Liu B., Eliason W. K., and Steitz T. A. (2013) Structure of a helicase-helicase loader complex reveals insights into the mechanism of bacterial primosome assembly. Nat. Commun. 4, 2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Itsathitphaisarn O., Wing R. A., Eliason W. K., Wang J., and Steitz T. A. (2012) The hexameric helicase DnaB adopts a nonplanar conformation during translocation. Cell 151, 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arias-Palomo E., O'Shea V. L., Hood I. V., and Berger J. M. (2013) The bacterial DnaC helicase loader is a DnaB ring breaker. Cell 153, 438–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Graham B. W., Schauer G. D., Leuba S. H., and Trakselis M. A. (2011) Steric exclusion and wrapping of the excluded DNA strand occurs along discrete external binding paths during MCM helicase unwinding. Nucleic Acids Res. 39, 6585–6595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rothenberg E., Trakselis M. A., Bell S. D., and Ha T. (2007) MCM forked substrate specificity involves dynamic interaction with the 5′-tail. J. Biol. Chem. 282, 34229–34234 [DOI] [PubMed] [Google Scholar]
- 13. Lee S. J., Syed S., Enemark E. J., Schuck S., Stenlund A., Ha T., and Joshua-Tor L. (2014) Dynamic look at DNA unwinding by a replicative helicase. Proc. Natl. Acad. Sci. U.S.A. 111, E827–E835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang H., Zhao K., Arnett D. R., and Fanning E. (2010) A specific docking site for DNA polymerase α-primase on the SV40 helicase is required for viral primosome activity, but helicase activity is dispensable. J. Biol. Chem. 285, 33475–33484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Satapathy A. K., Kulczyk A. W., Ghosh S., van Oijen A. M., and Richardson C. C. (2011) Coupling dTTP hydrolysis with DNA unwinding by the DNA helicase of bacteriophage T7. J. Biol. Chem. 286, 34468–34478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jeong Y. J., Rajagopal V., and Patel S. S. (2013) Switching from single-stranded to double-stranded DNA limits the unwinding processivity of ring-shaped T7 DNA helicase. Nucleic Acids Res. 41, 4219–4229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Manhart C. M., and McHenry C. S. (2015) Identification of subunit binding positions on a model fork and displacements that occur during sequential assembly of the Escherichia coli primosome. J. Biol. Chem. 290, 10828–10839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Galletto R., Jezewska M. J., and Bujalowski W. (2004) Unzipping mechanism of the double-stranded DNA unwinding by a hexameric helicase: the effect of the 3′ arm and the stability of the dsDNA on the unwinding activity of the Escherichia coli DnaB helicase. J. Mol. Biol. 343, 101–114 [DOI] [PubMed] [Google Scholar]
- 19. Khan I., Crouch J. D., Bharti S. K., Sommers J. A., Carney S. M., Yakubovskaya E., Garcia-Diaz M., Trakselis M. A., and Brosh R. M. Jr. (2016) Biochemical characterization of the human mitochondrial replicative Twinkle helicase: substrate specificity, DNA branch migration, and ability to overcome blockades to DNA unwinding. J. Biol. Chem. 291, 14324–14339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bleichert F., Botchan M. R., and Berger J. M. (2015) Crystal structure of the eukaryotic origin recognition complex. Nature 519, 321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Costa A., Ilves I., Tamberg N., Petojevic T., Nogales E., Botchan M. R., and Berger J. M. (2011) The structural basis for MCM2–7 helicase activation by GINS and Cdc45. Nat. Struct. Mol. Biol. 18, 471–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Graham B. W., Tao Y., Dodge K. L., Thaxton C. T., Olaso D., Young N. L., Marshall A. G., and Trakselis M. A. (2016) DNA interactions probed by hydrogen-deuterium exchange (HDX) Fourier transform ion cyclotron resonance mass spectrometry confirm external binding sites on the minichromosomal maintenance (MCM) helicase. J. Biol. Chem. 291, 12467–12480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McKinney S. A., Joo C., and Ha T. (2006) Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys. J. 91, 1941–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abelson J., Blanco M., Ditzler M. A., Fuller F., Aravamudhan P., Wood M., Villa T., Ryan D. E., Pleiss J. A., Maeder C., Guthrie C., and Walter N. G. (2010) Conformational dynamics of single pre-mRNA molecules during in vitro splicing. Nat. Struct. Mol. Biol. 17, 504–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Biswas E. E., and Biswas S. B. (1999) Mechanism of DnaB helicase of Escherichia coli: structural domains involved in ATP hydrolysis, DNA binding, and oligomerization. Biochemistry 38, 10919–10928 [DOI] [PubMed] [Google Scholar]
- 26. Biswas E. E., and Biswas S. B. (1999) Mechanism of DNA binding by the DnaB helicase of Escherichia coli: analysis of the roles of domain γ in DNA binding. Biochemistry 38, 10929–10939 [DOI] [PubMed] [Google Scholar]
- 27. Bailey S., Eliason W. K., and Steitz T. A. (2007) The crystal structure of the Thermus aquaticus DnaB helicase monomer. Nucleic Acids Res. 35, 4728–4736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Summerton J., and Weller D. (1997) Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 7, 187–195 [DOI] [PubMed] [Google Scholar]
- 29. Tackett A. J., Wei L., Cameron C. E., and Raney K. D. (2001) Unwinding of nucleic acids by HCV NS3 helicase is sensitive to the structure of the duplex. Nucleic Acids Res. 29, 565–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jezewska M. J., Rajendran S., and Bujalowski W. (1998) Functional and structural heterogeneity of the DNA binding site of the Escherichia coli primary replicative helicase DnaB protein. J. Biol. Chem. 273, 9058–9069 [DOI] [PubMed] [Google Scholar]
- 31. Bujalowski W., and Jezewska M. J. (1995) Interactions of Escherichia coli primary replicative helicase DnaB protein with single-stranded DNA. The nucleic acid does not wrap around the protein hexamer. Biochemistry 34, 8513–8519 [DOI] [PubMed] [Google Scholar]
- 32. Jezewska M. J., Rajendran S., and Bujalowski W. (1997) Strand specificity in the interactions of Escherichia coli primary replicative helicase DnaB protein with a replication fork. Biochemistry 36, 10320–10326 [DOI] [PubMed] [Google Scholar]
- 33. Ribeck N., Kaplan D. L., Bruck I., and Saleh O. A. (2010) DnaB helicase activity is modulated by DNA geometry and force. Biophys. J. 99, 2170–2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Atkinson J., Gupta M. K., and McGlynn P. (2011) Interaction of Rep and DnaB on DNA. Nucleic Acids Res. 39, 1351–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guy C. P., Atkinson J., Gupta M. K., Mahdi A. A., Gwynn E. J., Rudolph C. J., Moon P. B., van Knippenberg I. C., Cadman C. J., Dillingham M. S., Lloyd R. G., and McGlynn P. (2009) Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol. Cell 36, 654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim S., Dallmann H. G., McHenry C. S., and Marians K. J. (1996) Coupling of a replicative polymerase and helicase: a tau-DnaB interaction mediates rapid replication fork movement. Cell 84, 643–650 [DOI] [PubMed] [Google Scholar]
- 37. Manosas M., Spiering M. M., Ding F., Croquette V., and Benkovic S. J. (2012) Collaborative coupling between polymerase and helicase for leading-strand synthesis. Nucleic Acids Res. 40, 6187–6198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Indiani C., Langston L. D., Yurieva O., Goodman M. F., and O'Donnell M. (2009) Translesion DNA polymerases remodel the replisome and alter the speed of the replicative helicase. Proc. Natl. Acad. Sci. U.S.A. 106, 6031–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Graham J. E., Marians K. J., and Kowalczykowski S. C. (2017) Independent and stochastic action of DNA polymerases in the replisome. Cell 169, 1201–1213.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sambrook J., and Russel D. W. (2001) Molecular Cloning: A Laboratory Manual, pp. 5.51–5.54, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 41. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 42. Bujalowski W., Klonowska M. M., and Jezewska M. J. (1994) Oligomeric structure of Escherichia coli primary replicative helicase DnaB protein. J. Biol. Chem. 269, 31350–31358 [PubMed] [Google Scholar]
- 43. Roy R., Hohng S., and Ha T. (2008) A practical guide to single-molecule FRET. Nat. Methods 5, 507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fagerburg M. V., and Leuba S. H. (2011) Optimal practices for surface-tethered single molecule total internal reflection fluorescence resonance energy transfer analysis, in DNA Nanotechnology, Methods and Protocols (Zuccheri G., and Samori B., eds) 1st Ed., pp. 273–289, Humana Press, New York, NY: [DOI] [PubMed] [Google Scholar]
- 45. Schauer G. D., Huber K. D., Leuba S. H., and Sluis-Cremer N. (2014) Mechanism of allosteric inhibition of HIV-1 reverse transcriptase revealed by single-molecule and ensemble fluorescence. Nucleic Acids Res. 42, 11687–11696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rasband W. S. (1997–2016) ImageJ, National Institutes of Health, Bethesda, MD [Google Scholar]
- 47. McGeoch A. T., Trakselis M. A., Laskey R. A., and Bell S. D. (2005) Organization of the archaeal MCM complex on DNA and implications for the helicase mechanism. Nat. Struct. Mol. Biol. 12, 756–762 [DOI] [PubMed] [Google Scholar]
- 48. Bronson J. E., Fei J., Hofman J. M., Gonzalez R. L. Jr, and Wiggins C. H. (2009) Learning rates and states from biophysical time series: a Bayesian approach to model selection and single-molecule FRET data. Biophys. J. 97, 3196–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Arnold K., Bordoli L., Kopp J., and Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.