

Abstract
Precise control of the cell cycle allows for timely repair of genetic material prior to replication. One factor intimately involved in this process is checkpoint kinase 1 (Chk1), a DNA damage repair inducing Ser/Thr protein kinase that contains an N-terminal kinase domain and a C-terminal regulatory region consisting of a ∼100-residue linker followed by a putative kinase-associated 1 (KA1) domain. We report the crystal structure of the human Chk1 KA1 domain, demonstrating striking structural homology with other sequentially diverse KA1 domains. Separately purified Chk1 kinase and KA1 domains are intimately associated in solution, which results in inhibition of Chk1 kinase activity. Using truncation mutants and site-directed mutagenesis, we define the inhibitory face of the KA1 domain as a series of basic residues residing on two conserved regions of the primary structure. These findings point to KA1-mediated intramolecular autoinhibition as a key regulatory mechanism of human Chk1, and provide new therapeutic possibilities with which to attack this validated oncology target with small molecules.
Keywords: cell cycle, checkpoint control, crystal structure, DNA damage response, enzyme inactivation, enzyme mechanism, protein domain, serine/threonine protein kinase, structural biology
Introduction
Checkpoint kinase 1 (Chk1),3 a Ser/Thr protein kinase first described in yeast (1), plays a crucial role in cell cycle regulation across eukaryotes. Chk1 is activated upon phosphorylation by the upstream kinase ataxia telangiectasia and Rad3-related protein (ATR) when the cell senses DNA damage from various agents (2, 3). Chk1 activity pauses the cell cycle at the G2/M checkpoint and induces DNA damage repair, most canonically through phosphorylation of the CDC25 family phosphatases, inhibiting them and preventing activation of cyclin-dependent kinases (CDKs) (4). Chk1 also phosphorylates and activates Wee1 kinase, whose activity in turn inhibits CDK1 and pauses the cell cycle (5). Because rapidly dividing cancer cells are more reliant on the ATR/Chk1 pathway for survival than surrounding cells, these kinases have long been potential therapeutic targets in cancer alongside traditional chemotherapies or radiotherapies (to prevent repair of DNA damage in these cells), or as a monotherapy to induce replicative stress through duplication of damaged DNA (6–8). Chk1 activation has also been proposed to be a mechanism by which select cancers can be specifically targeted, as cancer cells with constitutively active Chk1 cannot divide (4, 9).
Chk1 belongs to the Ca2+/calmodulin–regulated kinase–like (CAMKL) family of Ser/Thr kinases, other prominent members include AMPK and the MARKs (10). Conserved structural features within this family include an N-terminal protein kinase domain, a linker region of varying length, and a C-terminal kinase–associated 1 (KA1) domain (Fig. 1A, top). The KA1 domain itself was first described in yeast, and exhibits remarkable structural homology despite relatively low sequence identity (11–13). Conserved features among all KA1 domains appear to include a four-stranded β-sheet flanked by two α-helices, which come together to form a hydrophobic core and generally basic surface (Fig. 1A, bottom cyan). Given that crystal structures of the Chk1 kinase domain show an open “active” conformation (14) (Fig. 1A, bottom gray), and activation loop phosphorylation has not been described for Chk1, we wondered whether the Chk1 KA1 domain could autoregulate the kinase domain (15–17). C-terminal truncations of human Chk1 result in substantially more active kinase in vitro (14), and kinase and C-terminal domains from the Chk1 ortholog in Xenopus laevis can associate with one another (18), suggesting that the human Chk1 KA1 domain may act as an autoinhibitory domain as recently described for MARK1 (16). Recent in vivo FRET experiments indicate that the N and C termini of Chk1 separate following DNA damage or in response to particular KA1 domain mutants (19), and Chk1 is constitutively activated when predicted secondary structure elements of the KA1 domain are disrupted (20). In each case, Chk1 activation was concurrent with phosphorylation of the linker region (Fig. 1A, bottom). Among KA1 domains from Chk1 orthologs, variations of two sequence-conserved motifs denoted CM1 and CM2 have been noted that reside at the N and C termini of the KA1 domain, respectively (4, 21) (Fig. 1, A, bottom cyan, and B). CM1 and CM2 of human Chk1 have been assigned as a non-canonical nuclear export sequence and nuclear localization sequence, respectively, bestowing upon the KA1 domain a role of spatial regulator (22), which may be linked to kinase regulation (21, 23, 24).
Figure 1.

Domain architecture and structure of Chk1. A, domain architecture (top) and crystal structures (bottom) of Chk1. Chk1 consists of a protein kinase domain (gray) (PDB code 1NVR) and a KA1 (cyan) domain (this work) connected by a ∼100-residue linker. Phosphorylation of the linker region (yellow arrows) by ATR kinase activates Chk1. The crystal structure of the KA1 domain reported here highlights the location of conserved motifs CM1 and CM2 (blue). B, multiple sequence alignment of KA1 domains from Chk1 orthologs with secondary structure (arrows for β-strands, bars for α-helices) of human Chk1 indicated. Identical (red, asterisk) and similar (gray, strongly similar: colon, or weakly similar, dot) positions are noted. Locations of CM1 and CM2 are indicated in blue. C, structural alignment of the KA1 domains of Homo sapiens Chk1 (this work, residues 377–468), MARK1 (PDB code 3OSE, 696–795, 18.7% sequence identity), AMPK (PDB code 4CFE, 404–473, 532–551, 18.1%), Mus musculus SAD-A (PDB code 4YOM, 533–636, 15.7%), and A. thaliana SOS2 (PDB code 2EHB, 337–430, 20.4%).
Here, we report the preparation and X-ray crystal structure determination of recombinant Chk1 KA1 domain, revealing a strikingly similar fold to other structurally characterized KA1 domains. Kinetic and biophysical studies reveal a high-affinity intramolecular autoinhibitory interaction of the Chk1 kinase domain emanating from the KA1 domain. Extensive site-directed mutagenesis implicates CM1 and CM2 as playing a central role in autoinhibitory interactions, especially basic residues within these regions, pinpointing the likely interface of autoinhibition among all Chk1 orthologs and linking Chk1 autoinhibition to that previously described for MARK1 (16). Intimate knowledge of the mechanism of KA1-mediated Chk1 autoinhibition may lead to novel strategies to modulate activity of this validated oncology target.
Results
Crystal structure of the human Chk1 KA1 domain
A human Chk1 KA1 domain construct containing amino acids 377–476 was expressed in Escherichia coli, affinity-purified in 6 m guanidine HCl, and refolded. Optimized crystals originally obtained in 0.1 m sodium acetate, pH 4.5, and 2 m ammonium sulfate diffracted to 2.5 Å. After phasing by molecular replacement, the structure was refined to Rwork/Rfree values of 21.6/24.6%, respectively, with two molecules in the asymmetric unit (Table 1). The final model contained Met-377 to Ser-468 of chain A (Fig. 1A, bottom cyan), along with an additional N-terminal serine in chain B left over from TEV-cleavage of the polyhistidine tag. The model closely resembles other KA1 domain structures, with a four-stranded β-sheet flanked by two α-helices, along with a short N-terminal β-strand (Fig. 1, A, bottom cyan, and C). The CM1 and CM2 nuclear export and localization sequences reside at the N terminus of the domain (CM1) and the final turn-helix of the C terminus (CM2), respectively. Residues N-terminal to Met-377 may be unstructured, explaining our inability to crystallize KA1 constructs that included more of the N terminus. Overall, the crystal structure confirms that the Chk1 KA1 domain retains a similar fold to other structurally characterized KA1 domains despite sequence identity of 20.4% or less (Fig. 1C).
Table 1.
Summary of crystallographic statistics
| Data collection | |
|---|---|
| Space group | P 3 |
| Unit cell (a, b, c (Å)) | 76.8, 76.8, 31.8 |
| Wavelength (Å) | 1.0 |
| Resolution (Å) | 50.0–2.5 (2.54–2.50)a |
| Rmerge | 0.097 (0.376) |
| I/σ | 24.5 (3.6) |
| Completeness (%) | 99.9 (97.8) |
| Redundancy | 5.0 (4.7) |
| No. of unique reflections | 7,313 |
| Refinement | |
| Rwork/Rfree (%) | 21.6/24.6 |
| No. of atoms (average B factor (Å2)) | |
| Protein | 1449 (63.2) |
| Water | 30 (64.9) |
| Acetate, sulfate, glycerol | 30 (81.6) |
| Ramachandran plot (%) | |
| Favored | 95.7 |
| Allowed | 4.3 |
| Root mean square deviation | |
| Bond length (Å) | 0.01 |
| Bond angles (deg) | 0.74 |
a Values in parentheses indicate highest-resolution shell.
Truncation mutants implicate the KA1 domain in intramolecular Chk1 autoinhibition
Given evidence that the KA1 domains are involved in autoinhibition of Chk1 (18, 19) and other CAMKL kinases (16), we assessed the activity of C-terminal domain variants to ask whether previous reports of increased kinase activity upon removal of the C terminus (14) reflected KA1 domain deletion. The Chk1 variants were purified and assayed for kinase activity using a CDC25C-derived peptide substrate (Fig. 2A). Compared with the kinase domain alone, any construct that contained the KA1 domain, including linker region deletions (see domain architecture schematic, Fig. 1A), showed reduced activity by at least 40-fold. To confirm that this putative autoinhibitory interaction is intramolecular, sedimentation equilibrium-analytical ultracentrifugation (SE-AUC) was used to demonstrate that full-length, linker-deleted, or kinase domain Chk1 constructs behave as monomers in solution (Fig. 2B), with fitted molecular masses close to the expected values of 55.0, 46.8, and 32.6 kDa, respectively. Thus, as in previous studies of MARK1 (16), KA1-derived autoinhibition appears to occur within a single polypeptide chain.
Figure 2.

Evidence for a KA1-mediated intramolecular autoinhibitory mechanism for Chk1. A, activity of recombinant Chk1 kinase domain (KD, 1–277), full-length Chk1 (FL, 1–476), and linker region deletion mutants (FL Δ270–342, FL Δ290–364). All constructs were assayed at 1 μm except for KD, which was assayed at 0.05 μm. Values plotted are mean ± S.D. of three replicates. B, sedimentation equilibrium analytical ultracentrifugation of 4 μm FL, FL Δ290–364, or KD Chk1 at the indicated speeds. The black lines represent global fits of the three indicated speeds at two concentrations (4 and 8 μm for FL/KD, 2 and 4 μm for FL Δ290–364). Data are representative of two independent protein preparations. C, FL at 0.5 μm and KD at 0.05 μm were assayed in the presence of 30 to 1000 mm NaCl. Values plotted are mean ± S.D. of three replicates with lines connecting the data points for clarity.
Given the preponderance of basic side chains within the KA1 domain resulting in a high predicted isoelectric point (pI of 9.84 compared with 6.41 for the kinase domain), we asked whether charge–charge interactions might play a role in KA1-mediated autoinhibition and compared the effect of increasing NaCl concentration on activity of full-length Chk1 or the isolated kinase domain (Fig. 2C). Whereas isolated kinase domain showed a steady activity decline with increasing ionic strength, possibly due to effects on substrate affinity, full-length Chk1 was activated to ∼125% initial activity at 300 mm NaCl before declining with a similar trend. We infer that this small initial stimulation of full-length Chk1 activity reflects disruption of intramolecular autoinhibitory KA1/kinase domain interactions, which partially overcome the general inhibitory effect of increased salt below 400 mm NaCl (Fig. 2C). This finding supports our suggestion that electrostatic interactions play an important role in intramolecular KA1-mediated autoinhibition of Chk1.
Separately purified KA1 domain interacts with the Chk1 kinase domain
If the KA1 domain mediates Chk1 autoinhibition by interacting directly with the kinase domain, we reasoned that we should be able to detect interaction between kinase (residues 1–277) and KA1 domains (residues 366–476) in an intermolecular context, as was recently observed in the case of MARK1 (16). Consistent with this assertion, the kinase and KA1 domains were mixed and co-eluted off of a gel filtration column at a position consistent with formation of a stable KA1-kinase domain complex in solution (Fig. 3A). Furthermore, addition of the KA1 domain to the Chk1 kinase domain also enhanced its thermal stability by ∼12 °C as monitored by SYPRO Orange hydrophobic-interacting fluorescent dye, indicating binding and stabilization of the kinase domain by the KA1 domain (Fig. 3B). Moreover, SE-AUC sedimentation curves obtained when equimolar kinase and KA1 domain were mixed (at 4 and 8 μm) reported a single species sedimenting with a molecular mass of 45.0 kDa, which is close to the estimated heterodimeric molecular mass of 46.7 kDa (Fig. 3C). We confirmed that this increase in particle size was not due to KA1 domain oligomerization in SE-AUC studies of the KA1 domain alone, which sedimented as a 15.4-kDa monomer, comparable with the estimated molecular mass of 14.1 kDa (Fig. 3C). These results suggest the formation of a stoichiometric complex between the Chk1 kinase and KA1 domains. The fact that near complete heterodimerization of the isolated Chk1 kinase and KA1 domains is observed at 4 and 8 μm in solution indicates that the Kd for their association is likely to be submicromolar. This finding argues that the Chk1 KA1-kinase domain interaction is at least 100-fold stronger than that observed for MARK1 (16), and likely underlies the particular importance of KA1-kinase interactions for regulation of Chk1.
Figure 3.

Separately purified Chk1 kinase and KA1 domains interact in solution. A, size-exclusion chromatography profiles of recombinant Chk1 kinase domain (KD, 1–277) (gray), KA1 domain (366–476) (cyan), or a mixture of KD and KA1 domains (black) at a 1:2 molar ratio. Peaks were normalized to maximum absorbance at 280 nm. Peak positions and molecular weights of protein standards are indicated above the plot. An SDS-PAGE gel of fractions from the black trace is displayed below the chromatogram aligned with the curve. B, representative melting curves of KD alone (light blue and gray) or a mixture of KD and KA1 (blue and black) at 2.5 or 5 μm based on SYPRO Orange fluorescence. Mean ± S.D. melting temperature of six replicates were 36.6 ± 0.3 °C for kinase alone and 49.1 ± 0.4 °C for the mixture. Chk1 KA1 domain alone displayed a melting temperature of ∼50 °C (Fig. 5A). C, sedimentation equilibrium analytical ultracentrifugation of a 24 μm Chk1 KA1 domain or an equimolar mixture of 4 μm KD and KA1 at the indicated speeds. The black lines represent global fits of the three indicated speeds at two concentrations (8 and 24 μm for KA1, 4 and 8 μm for the mixture). Data are representative of two independent protein preparations.
The KA1 domain efficiently inhibits the Chk1 kinase domain in trans
Given the estimated submicromolar Kd of the Chk1 KA1-kinase domain interaction, we reasoned that trans inhibition of the kinase domain would be much more efficient than seen with MARK1, where a KA1 domain concentration of 100 μm was required to halve activity of the 5 μm kinase domain (16). Indeed, a KA1 domain construct including both CM1 and CM2 (366–476) was able to inhibit activity of the kinase domain when titrated into the Chk1 radioassay at concentrations below 1 μm (Fig. 4A, black data points). The inhibition profile was fit to calculate a Ki value of 0.29 ± 0.01 μm for a non-competitive model (15) with inclusion of a Hill coefficient. We propose that the fitted Hill coefficient of 3.08 ± 0.14 reflects nonspecific electrostatic interactions may partially contribute to KA1-mediated inhibition of the Chk1 kinase domain, as has been observed for small-molecule inhibitors (25). The analogous Ki calculated for the kinase–KA1 domain interaction of SAD-A was 20-fold higher than that of Chk1 (15), indicating that SAD-A, like many other CAMKL family members, may rely on additional regulatory elements aside from the KA1 domain. To assess specificity, we asked whether other KA1 domains could elicit the same effect and found that analogous KA1 domain constructs from Danio rerio Chk1 and Saccharomyces cerevisiae Chk1 also inhibited the human Chk1 kinase domain, although Ki values were 2- and 13-fold higher than human Chk1, respectively (Fig. 4A). By contrast, the human MARK1 KA1, which shares the same basic character as the Chk1 KA1 domain (pI of 9.1), failed to inhibit the Chk1 kinase domain significantly. The lack of significant inhibitory activity of the MARK1 KA1 domain (Fig. 4A, gray) indicates that sequence-specific requirements for autoinhibition exist that do not solely rely on charge. Indeed, previous studies have shown that a L449R mutation in the KA1 domain activates full-length Chk1 in vivo (9, 19), implying that highly specific interactions play key roles in autoinhibition.
Figure 4.
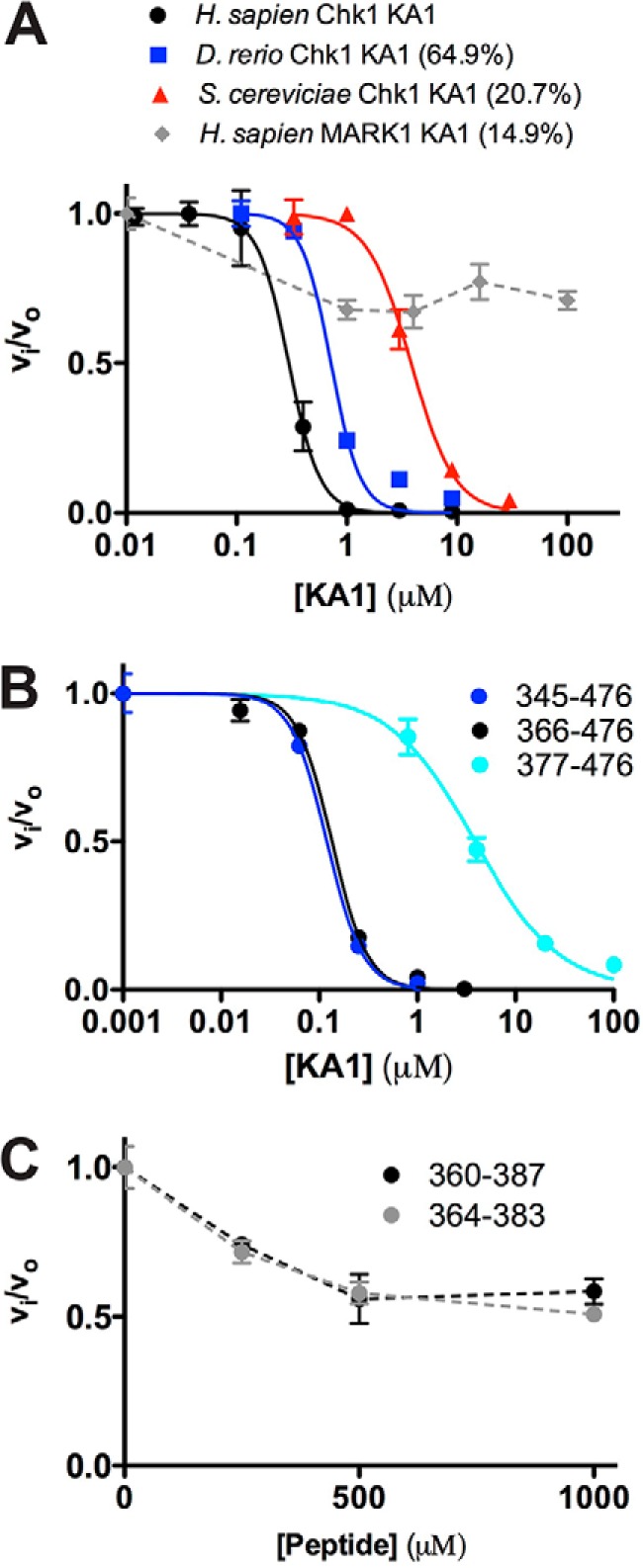
KA1 domains inhibit the Chk1 kinase domain in trans. A and B, human Chk1 kinase domain (1–277) at 0.05 μm was assayed in the presence of increasing concentration of KA1 domain constructs. Where applicable, plots of fractional velocity versus concentration (mean ± S.D. of three replicates) were fit (solid lines) to calculate Ki for non-competitive inhibition and a Hill coefficient (n, see “Experimental procedures”). A, fits of KA1 domains from H. sapiens (366–476, black), D. rerio (301–410, blue), and S. cerevisiae (412–527, red) resulted in Ki values of 0.29 ± 0.01, 0.72 ± 0.08, and 3.82 ± 0.24 μm with Hill coefficients of 3.08 ± 0.14, 3.27 ± 0.92, and 2.20 ± 0.28, respectively. The MARK1 KA1 domain (683–795, gray) did not display strong enough inhibition to fit, indicated by a dashed line. Sequence identities compared with the human Chk1 KA1 domain construct are indicated. B, fits of human Chk1 KA1 constructs consisting of 345–476 (blue), 366–476 (black), or 377–476 (cyan) resulted in Ki values of 0.12 ± 0.01, 0.13 ± 0.01, and 3.86 ± 0.39 μm with Hill coefficients of 2.38 ± 0.09, 2.43 ± 0.24, and 1.01 ± 0.10, respectively. C, two peptides made up of CM1 only plus flanking sequence consisting of 360–387 (black) or 364–383 (gray) did not display enough inhibitory activity to be fit. Mean ± S.D. of three replicates are plotted, with dashed lines connecting the data points for clarity.
Both CM1 and CM2 regions of the KA1 domain contribute to kinase inhibition
We next asked whether the presumed disordered region at the N terminus of the KA1 domain plays a role in Chk1 autoinhibition, as is the case for SAD-A and MARK1 (15, 16). Titrating three different KA1 domain constructs into the assay containing residues 345–476, 366–476, and 377–476 (the crystallized construct, lacking a portion of CM1) resulted in Ki values of 0.12 ± 0.01, 0.13 ± 0.01, and 3.86 ± 0.39 μm, respectively (Fig. 4B). Although, all KA1 constructs were able to inhibit kinase activity in a dose-response fashion, the constructs that contained the entire CM1 (345–376 or 366–476) showed a nearly ∼30-fold increase in inhibition capacity (lower Ki value), indicating that this region is required for maximal KA1-mediated autoinhibition. Based on this result, peptides containing the entire CM1 region were synthesized and when titrated into the assay showed only minimal inhibition of the Chk1 kinase domain at concentrations of up to 1 mm (Fig. 4C). In contrast to Chk1, the interaction between Arabidopsis thaliana SOS2 and calcium sensor SOS3 is mediated primarily by a sequence N-terminal to the KA1 domain (26). For murine SAD-A, the only structurally characterized kinase-KA1 domain interaction, an autoinhibitory sequence N-terminal to the KA1 domain was also shown to be the primary determinant of autoinhibition (15). Clearly, the mode of KA1-mediated autoinhibition of Chk1 differs from that of SOS2 and SAD-A despite close structural homology (Fig. 1C). Interestingly, the inhibition profile of the 377–476 KA1 construct (lacking part of CM1) was fitted with a lower Hill coefficient of 1 compared with the other constructs (Fig. 4B), indicating that the CM1 region may be contributing to nonspecific electrostatic interactions, whereas other regions of the molecule may bestow sequence specificity on the KA1-kinase domain interaction.
To determine whether specific conserved residues both outside and within CM1 and CM2 are involved in autoinhibition, we tested the trans inhibitory activity of a series of KA1 domain mutants. Point mutations were designed based on both sequence conservation and the crystal structure (Fig. 1) and assayed at various concentrations relative to the Chk1 kinase domain (Fig. 5C). Melting curves confirmed that each of these constructs were properly folded as they all displayed melting temperatures greater than 30 °C (Fig. 5A). Mutations at residues that contribute to the hydrophobic core (Phe-380, Phe-381, Ile-465, and Val-466) were the only variants that significantly reduced melting temperature (Fig. 5B). Although many of the KA1 variants exhibited an intermediate loss of inhibitory activity, four mutants covering eight positions of the KA1 domain (dashed lines), all within or near CM1 and CM2, clearly reduced KA1-mediated inhibition of Chk1 kinase domain activity (Fig. 5, C and D). Basic residues at Lys-375 and Arg-376 positions in CM1 are conserved among all Chk1 orthologs (Fig. 1B). The L449R variant has been shown to activate Chk1 in vivo and result in decreased FRET between the N and C terminus, likely due to loss of this physical autoinhibitory interaction (9, 19). Our in vitro results corroborate this finding, because the CM2 L449R mutation alone can reduce trans inhibitory activity of the KA1 domain (Fig. 5C). Mutation of conserved or partially conserved basic residues of CM2, or just outside this motif, positioned on the C-terminal helix (Lys-452, Arg-453, Lys-457, Lys-459, and Lys-461) also reduced inhibitory activity of the KA1 domain significantly (Fig. 5D). Despite lack of sequence identity (<20%), placement of these residues within the Chk1 KA1 domain structure closely mirrors that of the MARK1 KA1 domain (16), suggesting an analogous autoinhibitory mechanism involving basic residues of the N and C terminus of the KA1 domain (Fig. 5E). Interestingly, mutation of Ile-465/Val-466 to alanine, a variation analogous to an Schizosaccharomyces pombe Chk1 mutant that matches the phenotype of a KA1 domain deletion, retained inhibitory activity for human Chk1 (27). As appears to be the case in yeast, this C-terminal unstructured region may not be involved in autoinhibition per se, but may be required to interact with additional ligands once autoinhibition is broken. Multiple studies have noted the differences in regulation between Chk1 orthologs (4, 21), highlighting the need to understand the organism-specific details of KA1-mediated autoinhibition to best exploit this property in the human ortholog to target Chk1 with anticancer therapeutics.
Figure 5.

The autoinhibitory face of Chk1 KA1 domains includes CM1 and CM2. A, melting curves of Chk1 KA1 domain mutants at 25 to 50 μm based on SYPRO Orange fluorescence. All melting temperatures cluster from ∼45 to ∼55 °C except for F380A/F381A and I465A/V466V, which show reduced melting temperatures of ∼35 °C. The K365S/R376S, T378A/R379S, and R419S/R420S mutants likely show high fluorescence readings at 30 °C due to SYPRO Orange binding to exposed hydrophobic protein patches prior to temperature-induced protein unfolding. B, residues contributing to the hydrophobic core of the Chk1 KA1 domain (sticks) include Phe-380, Ile-465, and Val-466 (salmon sticks). C, Chk1 kinase domain (1–277) at 0.05 μm was assayed in the presence of up to 3 μm of various surface-mutated KA1 domain (366–476) constructs compared with WT (black line) with mean ± S.D. of three replicates plotted. Lines connecting the data points are included for clarity. Mutants that most significantly abrogated inhibitory activity (dashed lines) were located on or near CM1 (gray) or CM2 (black). D, residues most critical for KA1-mediated inhibition (magenta sticks) are mapped onto the Chk1 KA1 domain structure (cyan schematic) with the location of CM1 and CM2 indicated (blue). Because these were not part of the crystal construct, Lys-375 and Arg-376 were modeled in COOT (39) as was the side chain of Lys-457, which was disordered in the crystal structure. E, residues (magenta sticks) of the MARK1 KA1 domain (green schematic) previously implicated in autoinhibition (16).
Discussion
The crystal structure of the Chk1 KA1 domain confirms that this module adopts a similar fold as previously structurally characterized KA1 domains (Fig. 1C). This work represents the third case, to our knowledge, in which a direct autoinhibitory function has been linked to the KA1 domain of CAMKL kinases (15, 16), although the existence of this domain as part of larger regulatory complexes in SOS2 and AMPK (26, 28) indicates that the KA1-fold is implemented in numerous ways throughout biology. Based on our work, it is clear that the Chk1 KA1 domain operates differently from that of the structurally characterized KA1-kinase domain interaction of murine SAD-A, which relies on a sequence immediately N-terminal to the KA1 domain for autoinhibition (15). In human Chk1, both CM1 and CM2 appear to play a role in intramolecular autoinhibition, as mutations to one or the other do not fully abrogate inhibitory activity in vitro. Despite a lack of sequence homology, MARK1 employs basic residues in similar positions for its own KA1-mediated autoinhibition (Fig. 5E). In the case of MARK1, however, other regulatory inputs such as activation loop phosphorylation and the presence of a UBA domain (16, 29) might explain why the KA1-kinase domain interactions are substantially weaker than for Chk1.
Although KA1-kinase domain interactions in Chk1 appear to rely substantially on surface lysine and arginine residues in the KA1 domain (Fig. 5D), these clearly do not account for the entire story, as Leu-449 of CM2 remains a critical residue for inhibitory activity. The apparent reliance on charge–charge interactions for Chk1 autoinhibition suggested by partial activation of full-length Chk1 with increased salt (Fig. 2C) hints at the intriguing possibility that phosphorylation of the linker region between kinase and KA1 domains, a demonstrated activation mechanism of the kinase (4, 18), may disrupt these charge–charge interactions between the kinase and KA1 domains. Multiple phosphorylation of the linker region would effectively shield the kinase domain from inhibitory charge effects of the KA1 domain and promote the active “open” form as modeled in Fig. 6. The crystal structure of the KA1 domain makes readily apparent how recently intimated autophosphorylation events and Thr-378 and Thr-382 (20), two residues in CM1 (Fig. 1B), could disrupt the secondary structure of the KA1 domain and promote charge shielding, leading to loss of autoinhibition.
Figure 6.

A proposed model for the role of linker phosphorylation in Chk1 activation. It is not known how phosphorylation of the linker region between kinase (gray) and KA1 (cyan) domains of Chk1 by ATR results in release of autoinhibition and full activation of Chk1 (yellow lines), potentially assisted by additional binding partners (lower arrow, green and yellow proteins). Due to the reliance of KA1-mediated autoinhibition of Chk1 on basic residues of CM1 and CM2, we propose that phosphorylation of the linker region by ATR and subsequent autophosphorylation may interrupt the charge–charge interaction between KA1 and kinase domain by binding to CM1 and CM2 with negatively charged phosphates (middle panel). This conformation could activate Chk1 on its own, or act as an intermediate between the autoinhibited state and sustained activation through binding other partners at sites of Chk1 phosphorylation or the KA1 domain itself.
Phosphorylation at Ser-317 and Ser-345 by ATR may be the initial events that promote autophosphorylation, prolonged activation of Chk1, and retention in the nucleus (Fig. 6) (22, 30). Given that Chk1 is activated as part of a DNA damage response network, and the number of other proteins purported to bind Chk1 in the cellular context (4), it is possible that additional protein–protein interactions mediated by the KA1 domain further regulate Chk1 within these complexes (Fig. 6). Binding of 14-3-3 proteins to phosphorylated linker sites along with other protein–protein interactions may retain Chk1 in its active form (31). Mutations in the Chk1 CM1 that reduce proliferating cell nuclear antigen binding by this motif were also shown to impair phosphorylation of Ser-317 and Ser-345 by ATR in vivo (32). Additionally, interaction of the KA1 domain with F-box–only protein 6, an E3 ligase, promotes ubiquitination and degradation of Chk1 (33). Although various KA1 domains have been described as anionic phospholipid-dependent plasma membrane effector domains (13, 15, 16), it is difficult to imagine that such a regulatory mechanism exists for Chk1. The Chk1 DNA damage response role occurs in the nucleus surrounded by a neutral bilayer (34), and its primary substrates, the CDC25 phosphatases, are diffuse, soluble proteins. KA1 domains appear to have co-evolved with their conjugate catalytic domains to perform specific spatiotemporal regulatory functions of which autoinhibition is only a part. Targeting KA1 domain interactions may provide an avenue for generating novel small-molecule therapeutics that specifically act upon CAMKL kinases, reducing the off-target toxicity sometimes associated with more promiscuous ATP-competitive inhibitors.
Experimental procedures
Cloning, expression, and purification of Chk1 and KA1 constructs
DNA encoding the Chk1 kinase domain (amino acids 1–277, KD) and full-length Chk1 (amino acids 1–476, FL) was amplified from cDNA (Addgene) using PCR and ligated between the EcoRI and XhoI sites of pFastBac-HTA (ThermoFisher) to introduce a TEV-cleavable N-terminal polyhistidine tag. Truncations of the linker region in the full-length construct were generated using “round the horn” site-directed mutagenesis (35). All Chk1 kinase domain-containing constructs were produced in Sf9 cells using recombinant baculovirus, harvested after 3 days, and stored frozen. A temperature of 4 °C was maintained throughout the purification of Chk1 constructs. Pellets were resuspended in a lysis buffer consisting of 25 mm Tris, pH 8, 0.15 m NaCl, 5% glycerol (v/v), 5 mm β-mercaptoethanol (BME), 1 mm phenylmethylsulfonyl fluoride, and 10 mm imidazole. Clarified lysates following sonication were subjected to Ni-NTA affinity chromatography with wash and elution buffers containing 50 and 300 mm imidazole, pH 8, respectively, along with the lysis buffer components. Ni-NTA elution fractions were subjected to concurrent TEV cleavage of the polyhistidine tag and overnight dialysis at 4 °C in 0.15 m NaCl, 5% glycerol (v/v), 5 mm BME, and either 25 mm Tris, pH 8, or MES, pH 6, for kinase domain or KA1-containing constructs, respectively. Chk1 constructs were then subjected to ion exchange chromatography using HiTrap Q (kinase domain construct) or HiTrap SP (KA1-containing constructs) (GE Healthcare) columns with a gradient in these same buffers from 0.15 to 1 m NaCl. Eluted fractions were concentrated and subjected to size exclusion chromatography (SEC) on a Superdex 75 column (GE Healthcare) with a 20 mm HEPES, pH 7.5, 0.15 m NaCl, and 1 mm tris(2-carboxylethyl)phosphine as running buffer.
Human Chk1 KA1 domain constructs (residues 345–476, 366–476, or 377–476) were cloned from cDNA into a modified pET28 (EMD Millipore) vector containing a polyhistidine tag and TEV cleavage sequence immediately upstream of the BamHI restriction site, resulting in a minimal Gly-Ser scar after TEV cleavage in these constructs. Non-cleavable N-terminal His6-containing constructs were also generated in pET21 vectors encoding these peptides along with D. rerio Chk1 KA1(301–410). cDNA encoding D. rerio Chk1 was a generous gift of Dr. Michael Lampson (University of Pennsylvania, Philadelphia, PA). Previously implemented KA1 constructs of S. cerevisiae Chk1(412–527) and human MARK1(683–795) were also obtained (16). KA1 domain point mutants were generated for the pET21(366–476) human Chk1 KA1 domain construct using round the horn site-directed mutagenesis (35). KA1 constructs were transformed into the Rosetta 2(DE3) E. coli expression strain (EMD Millipore). 6-ml overnight constructs were used to inoculate each liter of LB shaking at 37 °C, followed by isopropyl β-d-1-thiogalactopyranoside induction at A600 of 0.6. Cells were harvested after 4–6 h and stored frozen. Purification of KA1 domains took place at 4 °C where possible. Frozen E. coli was resuspended in PBS, sonicated, and centrifuged to pellet inclusion bodies and other insoluble material. Pellets were homogenized, briefly sonicated in 6 m guanidine HCl, 25 mm Tris, pH 8, 0.25 m NaCl, 5 mm BME, and 10 mm imidazole, and subjected to Ni-NTA affinity chromatography with wash and elution buffers containing 25 and 300 mm imidazole, pH 8, respectively, along with the other homogenization buffer components. Elution fractions were dialyzed against 25 mm Tris, pH 8, 0.15 m NaCl, 10% glycerol (v/v), and 5 mm BME overnight to remove the denaturant. 0.5 m arginine could be included in the dialysis buffer to increase refolding yield, but was not necessary. An additional dialysis step against this same buffer (without arginine) was implemented for TEV site-containing constructs to facilitate cleavage of the affinity tag. Following dialysis, all KA1 constructs were subjected to SEC as with the kinase constructs. Aside from crystallization and comparison of inhibitory activity of KA1 truncation mutants, all KA1 constructs used included the non-cleavable N-terminal polyhistidine tag.
Crystallization and structure determination of the Chk1 KA1 domain
The purified human Chk1 KA1 domain (377–476) concentrated to 7 mg/ml was incubated in hanging drops in a 1:1 ratio with reservoir solution containing 0.1 m sodium acetate trihydrate, pH 4.5, and 2 m ammonium sulfate in which small triangular crystals appeared within 3 days at 20 °C. Optimization yielded 0.2 m sodium acetate, pH 4.8, and 2 m ammonium sulfate as a condition in which much larger crystals grew within 4 weeks. These were cryoprotected in 3 m ammonium sulfate, 5% glycerol (v/v) and flash frozen in liquid nitrogen. Diffraction data were collected at the GM/CA 23-ID-D beamline at the Advanced Photon Source, and reduced and scaled using HKL-2000 (36). Phases were obtained by molecular replacement through PHASER within the PHENIX software suite (37). Phyre2 (38) generated the reference model using the KA1 domain from A. thaliana SOS2 as the molecular replacement template (26) (PDB code 2EHB). The model was manually rebuilt in COOT (39) between rounds of iterative maximum-likelihood refinement using PHENIX. The final model was validated using MOLPROBITY (40).
Kinase assays
The Chk1 radioassay consisted of monitoring of 32P transfer from γ-labeled ATP (∼20 μCi per experiment) to a CDC25C-derived peptide substrate (Chktide, SignalChem). Enzyme in SEC buffer was diluted 5-fold to initiate the reaction at 25 °C, whose conditions also included 25 mm HEPES, pH 7.5, 1 mm DTT, 5 mm MgCl2, and 100 μm of both ATP and peptide substrate. Peptides were captured by spotting onto phosphocellulose paper followed immediately by quenching with a 0.5% phosphate solution. After three washes in phosphate solution and one more in acetone, phosphocellulose squares were subjected to scintillation counting where radioactivity incorporation was measured. Initial velocities were calculated as peptide phosphorylated per enzyme molecule per minute. For assays involving trans inhibition by KA1 domains, these modules were added to the mastermix and not preincubated with enzyme. Where applicable, inhibition data were fit to the following non-competitive model for Ki modified with a Hill coefficient (15, 25), where v0 is uninhibited velocity, vi is inhibited velocity, and n is the Hill coefficient, using GraphPad Prism (GraphPad Software, Inc., San Diego, CA).
| (Eq. 1) |
Analytical ultracentrifugation
Sedimentation equilibrium analytical ultracentrifugation experiments were performed at 4 °C on a Beckman Optima XL-I instrument, monitoring absorbance at 280 nm along a 1.2-cm path length using an An-Ti 60 four-hole rotor containing six-channel centerpieces and quartz windows. Datasets were fit globally to two concentrations at the three indicated speeds using Heteroanalysis (J. Cole and J. Lary, University of Connecticut, Mansfield, CT). Buffer and sample-dependent variables were calculated with SEDNTERP (41). All samples were measured suspended in SEC buffer.
Thermal shift assays
15 μl of protein sample in SEC buffer was mixed with 5 μl of 300-fold diluted SYPRO Orange dye (Invitrogen) in 384-well white microplates. A real-time PCR machine read dye fluorescence as the sample temperatures were increased from 20 to 95 °C over 90 min. Traces were normalized to minimum and maximum fluorescent signal. The first derivative of the melting curves was used to calculate melting temperature where applicable.
Author contributions
R. E. designed, performed, and analyzed all experiments, and wrote the manuscript. M. S. constructed expression vectors of the Chk1 KA1 domain mutants and performed pilot kinase assays under the supervision of R. E. K. F. and R. M. contributed to experimental design, analysis, and writing of the manuscript.
Acknowledgments
We thank members of the Marmorstein laboratory and Mark A. Lemmon for valuable discussion and reviewing the manuscript. We thank Elliot Dean for Sf9 cell culture. D. rerio Chk1 cDNA was provided by Michael Lampson.
This work was supported by National Institutes of Health Postdoctoral Fellowship F32-GM115098 (to R. P. E.) and National Institutes of Health Grant P01-CA114046 (to R. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 5WI2) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Chk1
- checkpoint kinase 1
- AMPK
- AMP-activated protein kinase
- ATR
- ataxia telangiectasia and Rad3-related protein
- BME
- β-mercaptoethanol
- CAMKL
- Ca2+/calmodulin-regulated kinase-like
- CDC25
- M-phase inducer phosphatase
- CDK
- cyclin-dependent kinase
- CM
- conserved motif
- FL
- full-length
- KD
- kinase domain
- LB
- lysogeny broth
- SAD
- synapses of amphids defective
- SE-AUC
- sedimentation equilibrium-analytical ultracentrifugation
- SEC
- size exclusion chromatography
- SOS
- salt overly sensitive
- TEV
- tobacco etch virus
- Ni-NTA
- nickel-nitrilotriacetic acid
- PDB
- Protein Data Bank
- KA1
- kinase-associated 1.
References
- 1. Walworth N., Davey S., and Beach D. (1993) Fission yeast Chk1 protein kinase links the rad checkpoint pathway to cdc2. Nature 363, 368–371 [DOI] [PubMed] [Google Scholar]
- 2. Sanchez Y., Wong C., Thoma R. S., Richman R., Wu Z., Piwnica-Worms H., and Elledge S. J. (1997) Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 277, 1497–1501 [DOI] [PubMed] [Google Scholar]
- 3. Smits V. A., and Gillespie D. A. (2015) DNA damage control: regulation and functions of checkpoint kinase 1. FEBS J. 282, 3681–3692 [DOI] [PubMed] [Google Scholar]
- 4. Zhang Y., and Hunter T. (2014) Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer 134, 1013–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee J., Kumagai A., and Dunphy W. G. (2001) Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol. Biol. Cell 12, 551–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garrett M. D., and Collins I. (2011) Anticancer therapy with checkpoint inhibitors: what, where and when? Trends Pharmacol. Sci. 32, 308–316 [DOI] [PubMed] [Google Scholar]
- 7. Zhang Y., Lai J., Du Z., Gao J., Yang S., Gorityala S., Xiong X., Deng O., Ma Z., Yan C., Susana G., Xu Y., and Zhang J. (2016) Targeting radioresistant breast cancer cells by single agent CHK1 inhibitor via enhancing replication stress. Oncotarget 7, 34688–34702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rundle S., Bradbury A., Drew Y., and Curtin N. J. (2017) Targeting the ATR-CHK1 axis in cancer therapy. Cancers 9, e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang J., Han X., and Zhang Y. (2012) Autoregulatory mechanisms of phosphorylation of checkpoint kinase 1. Cancer Res. 72, 3786–3794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marx A., Nugoor C., Panneerselvam S., and Mandelkow E. (2010) Structure and function of polarity-inducing kinase family MARK/Par-1 within the branch of AMPK/Snf1-related kinases. FASEB J. 24, 1637–1648 [DOI] [PubMed] [Google Scholar]
- 11. Tassan J. P., and Le Goff X. (2004) An overview of the KIN1/PAR-1/MARK kinase family. Biol. Cell 96, 193–199 [DOI] [PubMed] [Google Scholar]
- 12. Tochio N., Koshiba S., Kobayashi N., Inoue M., Yabuki T., Aoki M., Seki E., Matsuda T., Tomo Y., Motoda Y., Kobayashi A., Tanaka A., Hayashizaki Y., Terada T., Shirouzu M., Kigawa T., and Yokoyama S. (2006) Solution structure of the kinase-associated domain 1 of mouse microtubule-associated protein/microtubule affinity-regulating kinase 3. Protein Sci. 15, 2534–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moravcevic K., Mendrola J. M., Schmitz K. R., Wang Y. H., Slochower D., Janmey P. A., and Lemmon M. A. (2010) Kinase associated-1 domains drive MARK/PAR1 kinases to membrane targets by binding acidic phospholipids. Cell 143, 966–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen P., Luo C., Deng Y., Ryan K., Register J., Margosiak S., Tempczyk-Russell A., Nguyen B., Myers P., Lundgren K., Kan C. C., and O'Connor P. M. (2000) The 1.7 A crystal structure of human cell cycle checkpoint kinase Chk1: implications for Chk1 regulation. Cell 100, 681–692 [DOI] [PubMed] [Google Scholar]
- 15. Wu J. X., Cheng Y. S., Wang J., Chen L., Ding M., and Wu J. W. (2015) Structural insight into the mechanism of synergistic autoinhibition of SAD kinases. Nat. Commun. 6, 8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Emptage R. P., Lemmon M. A., and Ferguson K. M. (2017) Molecular determinants of KA1 domain-mediated autoinhibition and phospholipid activation of MARK1 kinase. Biochem. J. 474, 385–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang Z., Xue B., Umitsu M., Ikura M., Muthuswamy S. K., and Neel B. G. (2012) The signaling adaptor GAB1 regulates cell polarity by acting as a PAR protein scaffold. Mol. Cell 47, 469–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Katsuragi Y., and Sagata N. (2004) Regulation of Chk1 kinase by autoinhibition and ATR-mediated phosphorylation. Mol. Biol. Cell 15, 1680–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Han X., Tang J., Wang J., Ren F., Zheng J., Gragg M., Kiser P., Park P. S., Palczewski K., Yao X., and Zhang Y. (2016) Conformational change of human checkpoint kinase 1 (Chk1) induced by DNA damage. J. Biol. Chem. 291, 12951–12959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gong E. Y., Smits V. A., Fumagallo F., Piscitello D., Morrice N., Freire R., and Gillespie D. A. (2015) KA1-targeted regulatory domain mutations activate Chk1 in the absence of DNA damage. Sci. Rep. 5, 10856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tapia-Alveal C., Calonge T. M., and O'Connell M. J. (2009) Regulation of Chk1. Cell Div. 4, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang J., Han X., Feng X., Wang Z., and Zhang Y. (2012) Coupling cellular localization and function of checkpoint kinase 1 (Chk1) in checkpoints and cell viability. J. Biol. Chem. 287, 25501–25509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kosoy A., and O'Connell M. J. (2008) Regulation of Chk1 by its C-terminal domain. Mol. Biol. Cell 19, 4546–4553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Palermo C., Hope J. C., Freyer G. A., Rao H., and Walworth N. C. (2008) Importance of a C-terminal conserved region of Chk1 for checkpoint function. PloS One 3, e1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Copeland R. A. (2005) Evaluation of enzyme inhibitors in drug discovery: a guide for medicinal chemists and pharmacologists. Methods Biochem. Anal. 46, 1–265 [PubMed] [Google Scholar]
- 26. Sánchez-Barrena M. J., Fujii H., Angulo I., Martínez-Ripoll M., Zhu J. K., and Albert A. (2007) The structure of the C-terminal domain of the protein kinase AtSOS2 bound to the calcium sensor AtSOS3. Mol. Cell 26, 427–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Caparelli M. L., and O'Connell M. J. (2013) Regulatory motifs in Chk1. Cell Cycle 12, 916–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xiao B., Sanders M. J., Carmena D., Bright N. J., Haire L. F., Underwood E., Patel B. R., Heath R. B., Walker P. A., Hallen S., Giordanetto F., Martin S. R., Carling D., and Gamblin S. J. (2013) Structural basis of AMPK regulation by small molecule activators. Nat. Commun. 4, 3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marx A., Nugoor C., Müller J., Panneerselvam S., Timm T., Bilang M., Mylonas E., Svergun D. I., Mandelkow E. M., and Mandelkow E. (2006) Structural variations in the catalytic and ubiquitin-associated domains of microtubule-associated protein/microtubule affinity regulating kinase (MARK) 1 and MARK2. J. Biol. Chem. 281, 27586–27599 [DOI] [PubMed] [Google Scholar]
- 30. Okita N., Minato S., Ohmi E., Tanuma S., and Higami Y. (2012) DNA damage-induced CHK1 autophosphorylation at Ser296 is regulated by an intramolecular mechanism. FEBS Lett. 586, 3974–3979 [DOI] [PubMed] [Google Scholar]
- 31. Jiang K., Pereira E., Maxfield M., Russell B., Goudelock D. M., and Sanchez Y. (2003) Regulation of Chk1 includes chromatin association and 14-3-3 binding following phosphorylation on Ser-345. J. Biol. Chem. 278, 25207–25217 [DOI] [PubMed] [Google Scholar]
- 32. Scorah J., Dong M. Q., Yates J. R. 3rd, Scott M., Gillespie D., and McGowan C. H. (2008) A conserved proliferating cell nuclear antigen-interacting protein sequence in Chk1 is required for checkpoint function. J. Biol. Chem. 283, 17250–17259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Y. W., Brognard J., Coughlin C., You Z., Dolled-Filhart M., Aslanian A., Manning G., Abraham R. T., and Hunter T. (2009) The F box protein Fbx6 regulates Chk1 stability and cellular sensitivity to replication stress. Mol. Cell 35, 442–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van Meer G., Voelker D. R., and Feigenson G. W. (2008) Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell. Biol. 9, 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hemsley A., Arnheim N., Toney M. D., Cortopassi G., and Galas D. J. (1989) A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res. 17, 6545–6551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 37. Adams P. D., Afonine P. V., Bunkóóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Laue T. M., Shah B. D., Ridgeway T. M., and Belletier S. L. (1992) Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding S., and Rowe A., eds) pp. 90–125, Royal Society of Chemistry, London [Google Scholar]