

Abstract
For decades, the enzymes of the fungus Hypocrea jecorina have served as a model system for the breakdown of cellulose. Three-dimensional structures for almost all H. jecorina cellulose-degrading enzymes are available, except for HjLPMO9A, belonging to the AA9 family of lytic polysaccharide monooxygenases (LPMOs). These enzymes enhance the hydrolytic activity of cellulases and are essential for cost-efficient conversion of lignocellulosic biomass. Here, using structural and spectroscopic analyses, we found that native HjLPMO9A contains a catalytic domain and a family-1 carbohydrate-binding module (CBM1) connected via a linker sequence. A C terminally truncated variant of HjLPMO9A containing 21 residues of the predicted linker was expressed at levels sufficient for analysis. Here, using structural, spectroscopic, and biochemical analyses, we found that this truncated variant exhibited reduced binding to and activity on cellulose compared with the full-length enzyme. Importantly, a 0.95-Å resolution X-ray structure of truncated HjLPMO9A revealed that the linker forms an integral part of the catalytic domain structure, covering a hydrophobic patch on the catalytic AA9 module. We noted that the oxidized catalytic center contains a Cu(II) coordinated by two His ligands, one of which has a His-brace in which the His-1 terminal amine group also coordinates to a copper. The final equatorial position of the Cu(II) is occupied by a water-derived ligand. The spectroscopic characteristics of the truncated variant were not measurably different from those of full-length HjLPMO9A, indicating that the presence of the CBM1 module increases the affinity of HjLPMO9A for cellulose binding, but does not affect the active site.
Keywords: electron paramagnetic resonance (EPR), enzyme mechanism, enzyme structure, Hypocrea jecorina, X-ray crystallography, auxiliary activity family 9 (AA9), lytic polysaccharide monooxygenase (LPMO)
Introduction
Lignocellulosic biomass represents an alternative source for fuels and materials and has the potential to replace fossil fuels as we strive to become a sustainable and carbon neutral society. Cellulose is the major structural polysaccharide of plant cell walls and is the most abundant and readily accessible source of renewable organic carbon on the planet. The enzymatic conversion of cellulose to glucose provides a carbon source for the biomanufacturing of fuels and chemicals. For decades, the secreted enzymes of the fungus Hypocrea jecorina (anamorph Trichoderma reesei) have served as a model system for the breakdown of cellulose. Major cellulases of H. jecorina, such as cellobiohydrolases Cel6A and Cel7A, endoglucanases Cel5A and Cel7B, and β-glucosidase Cel3A have been studied in detail, including determination of their 3D structures (1–5).
Lytic polysaccharide monooxygenases (LPMOs)3 boost the hydrolytic activity of cellulases (6–10). It is now well established that LPMOs are copper monooxygenases that introduce chain breaks through the oxidation of C1, C4, or both carbons of a sugar ring, using electrons provided by an external donor, which can be an enzyme (e.g. cellobiose dehydrogenase) or a chemical compound with antioxidant activity such as ascorbic acid (11–13). The boosting effect of LPMOs on cellulases has been attributed to the LPMO-mediated creation of free chain breaks, which are subsequently targeted by hydrolytic cellulases (14). LPMOs are currently categorized in carbohydrate-active enzyme (CAZy) families auxiliary activity family 9 (AA9), AA10, AA11, and AA13 (15). The AA9 family comprises fungal LPMOs that have been shown to be active on cellulose, soluble oligosaccharides (16), and hemicellulosic substrates such as xyloglucan and xylan (17). The H. jecorina genome has 3 AA9 encoding genes; HjLPMO9A (protein ID 73643), Cel61B/HjLPMO9B (protein ID 120961), and an AA9-like protein (protein ID 27554). HjLPMO9A and -B were shown to be significantly up-regulated along with other cellulases, when T. reesei QM6a and its hypercellulolytic mutant (Rut C30) were grown on corn stover and sawdust (18), indicating its importance in plant biomass decomposition. HjLPMO9B was the first fungal LPMO/AA9 enzyme structure to be published (19). HjLPMO9A was initially characterized as an endoglucanase EG4, due to a weak endoglucanase activity detected from the culture filtrate of an HjLPMO9A-expressing Saccharomyces cerevisiae strain (20). HjLPMO9A expression and endoglucanase activity were also reported in a subsequent study, where it was homologously overexpressed in T. reesei (21). HjLPMO9A expressed in Pichia pastoris was shown to oxidatively cleave phosphoric acid–swollen cellulose (PASC) and soluble oligosaccharides, yielding both C4- and C1-oxidized fragments (22). HjLPMO9A is a major component of enzyme mixtures that have been optimized for the degradation of pretreated biomass (23).
HjLPMO9A is a secreted modular protein of 323 amino acids, containing an AA9 domain (residues 1 to 220), a predicted linker region (residues 221 to 288), and a family 1 carbohydrate-binding module (CBM1). About 20% of AA9 enzymes occur as a catalytic domain C terminally coupled to a flexible linker followed by a CBM1 (PFAM database). CBM1 domains are compact cysteine knots, about 40 amino acid residues large, and have a flat surface with 2–3 aromatic residues that bind tightly to crystalline cellulose. They are associated with fungal enzymes and proteins involved in degradation of lignocellulose and appear most often with AA9 catalytic domains (PFAM database). Deletion of CBM1 modules from fungal (hydrolytic) cellulases reduce the ability of the enzyme to bind to crystalline cellulose although did not affect catalytic activity (24, 25).
The linker sequences connecting the CBM1 and the catalytic module display low sequence complexity with an overabundance of serine and threonine residues (26). These serine and threonine residues are often O-glycosylated, where glycosylation has been shown to protect the linker against proteolytic degradation and to increase the binding affinity (27). Linker sequences show little conservation and by all accounts are intrinsically disordered proteins that do not contain structural elements (27). Removal of linker sequences and CBM1 modules has been necessary to obtain crystals of cellulases for structure determination by X-ray crystallography (1, 4, 5, 28).
The role of CBM1 modules on the properties of AA9 enzymes has received little attention and most 3D structures are of AA9 LPMOs that natively do not contain a CBM1 or linker sequence (9, 19, 29–33). Neurospora crassa LPMO9C contains both a linker and a CBM1, and crystallized when produced without the predicted linker and CBM1 (29). Although NcLPMO9C displays broad specificity for polymeric and oligomeric β(1–4)-glycans, the mutant enzyme lacking a CBM1 domain showed reduced binding affinity for phosphoric acid–swollen cellulose and xyloglucan (29). LPMOs from Podospora anserina that occur natively with a CBM1 have been shown to release more oxidized products from cellulose than LPMOs without CBM1 (34). Interestingly, a 2016 study by Crouch et al. (35) showed that CBM2a and CBM3a modules had a measureable effect on activity and substrate specificity of AA10 cellulose-active LPMOs and were even found to modulate the mode of action of AA10 LPMOs.
Here we report the crystal structure of HjLPMO9A after removal of the CBM1 and part of the linker sequence. The structure was determined to 0.95-Å resolution and revealed a surprisingly ordered linker structure. The effects of the CBM1 module on substrate binding, activity on cellulose, and spectroscopic properties of the copper atom in the active site were determined in detail. This work expands our knowledge of the carbohydrate-active enzymatic toolkit of the industrially relevant fungus H. jecorina and its model system for the breakdown of cellulose.
Results and discussion
Removal of CBM and expression of HjLMPO9A-ΔCBM
A significant fraction, ∼17%, of the currently available AA9 sequences (PF03443 in Pfam, ∼409 of 2459) have a CBM1 attached to the C terminus of the catalytic AA9 domain via a variable linker region (pfamseq database, EBI Pfam version 30.0, http://pfam.xfam.org/family/PF03443).4 For other enzymes, such as cellobiohydrolase HjCel7A, the linker and CBM have been successfully removed by proteolytic cleavage. Structure determination of HjCel7A showed that the linker had been cleaved directly after the annotated catalytic domain (1). Genetic truncations of HjLPMO9A were designed to remove all or part of its linker. HjLPMO9B is a homolog from H. jecorina that lacks a linker and CBM and has a known structure (19). Fig. 1 shows the sequence alignment of HjLPMO9A and HjLPMO9B. HjLPMO9A was truncated after residue 229 based on this alignment, however, no expression was observed. Truncation after residue 231 resulted in very low expression levels of truncated protein (data not shown). Additional truncations were tested and a wild-type level of protein production was observed with 21 extra amino acid residues, i.e. when the protein was truncated after residue 252, denoted HjLPMO9A-gmΔCBM. In parallel, truncated HjLPMO9A was produced enzymatically using papain, resulting in HjLPMO9A-ΔCBM. Both HjLPMO9A-gmΔCBM and HjLPMO9A-ΔCBM crystallized. The structure of both HjLPMO9A-ΔCBM and HjLPMO9A-gmΔCBM were determined by X-ray crystallography and the structure of these are described later in this paper.
Figure 1.

Partial amino acid alignment of mature amino acid sequences of HjLPMO9A (9A), HjLPMO9A-gmΔCBM_1 (9AwoCBM1), HjLPMO9A-gmΔCBM (9AwoCBM2), HjLPMO9AΔCBM as resolved in the determined structure (9ADeltaCBM), and HjLPMO9B (9B). Sequences were aligned in MUSCLE using default settings.
Cellulose binding and activity of HjLPMO9A and HjLPMO9A-gmΔCBM
Removal of the CBM1 module from HjLPMO9A reduced its affinity for cellulose. Fixed concentrations of HjLPMO9A or HjLPMO9A-gmΔCBM were incubated with increasing amounts of Avicel and the amounts of unbound protein in the supernatant were determined (Fig. 2A). No electron donor was present, which ensured the inactivity of the enzymes. For both enzymes, the data fit well to a Langmuir adsorption isotherm. HjLPMO9A binds to almost full saturation (0.97 ± 0.01), whereas HjLPMO9A-gmΔCBM does not (0.83 ± 0.06). The binding constant of HjLPMO9A is 0.26 ± 0.02% dry solids; for HjLPMO9A-gmΔCBM it is 6.9 ± 1.2% dry solids. Thus, the CBM1 module was found to contribute significantly to the affinity of HjLPMO9A for cellulose and its removal reduced the affinity for cellulose by more than 20-fold compared with the full-length enzyme. This is similar to what has been found for NcLPMO9C that was modified to lack its CBM (29).
Figure 2.
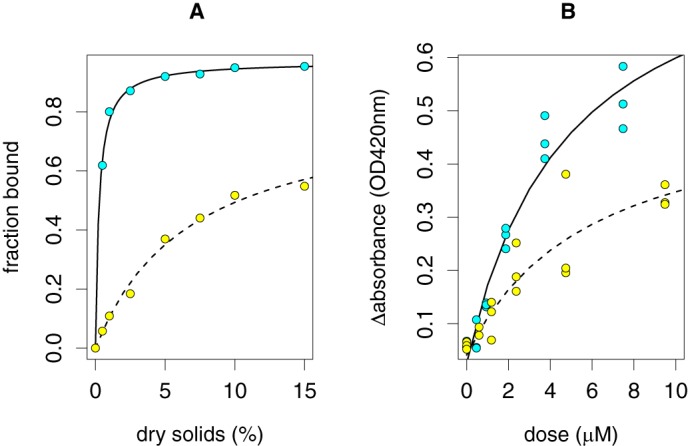
Cellulose binding and activity by HjLPMO9A (cyan, solid line) and HjLPMO9A-gmΔCBM (yellow, dashed line). A, truncated variants were incubated with various concentrations of Avicel and the unbound protein in the supernatant was determined by UPLC. The absence of an electron donor ensured inactivity of the truncated enzyme variants. Data were fit with a Langmuir adsorption function. B, PASC was incubated with various concentrations of HjLPMO9A and HjLPMO9A-gmΔCBM in the presence of ascorbic acid. Reduction in absorbance was used as a measure of LPMO activity. Data were fit with a Michaelis-Menten kinetic function.
Removal of the carbohydrate-binding module from HjLPMO9A also reduced activity on cellulose. Cellulose-degrading activity was determined using PASC at 0.25% dry solids. PASC is insoluble and forms an opaque suspension. The catalytic activity of HjLPMO9A solubilizes PASC, which was monitored by recording the OD420 (Fig. 2B). Only cellooligosaccharides up to a degree of polymerization of 8 are soluble in water (36). Enzymatic cuts that result in longer oligosaccharides are therefore not detected. It is likely they are formed since PASC is made from Avicel and the average degree of polymerization of Avicel is about 200 (37). For HjLPMO9A the maximum ΔOD420 is 0.85 ± 0.12, whereas it is 0.49 ± 0.13 for HjLPMO9A-gmΔCBM. The enzyme concentration with half of the maximum activity is not significantly different between HjLPMO9A and HjLPMO9A-gmΔCBM, i.e. 4.8 ± 1.6 and 5.9 ± 3.9 μm, respectively. Thus, deletion of the carbohydrate-binding domain reduced the PASC-solubilizing activity of the HjLPMO9A truncated variant by about 50%. A similar reduction in activity was observed when the CBM1 module was removed from NcLPMO9C (29).
Activity profiles analyzed by HPAEC-PAD
Comparison of the activity profiles on reduced PASC of HjLPMO9A and HjLPMO9A-ΔCBM (Fig. 3) shows that the removal of the CBM does not seem to alter the reaction specificity of HjLPMO9A on PASC. HjLPMO9A produces both C1 and C4 oxidized oligosaccharides, as was also reported by Tanghe et al. (22). Ratios of integrated peak areas of HjLPMO9A versus HjLPMO9A-ΔCBM for C1-oxidized products compared with C4-oxidized products were constant, which further indicates that the reaction specificity is not altered.
Figure 3.

HPAEC-PAD analyses of the glucan products after treating PASC with HjLPMO9A and without CBM. The overlaid chromatograms are show the release of oligosaccharides by full-length HjLPMO9A (black) and HjLPMO9A-ΔCBM (cyan). P. chrysosporium LPMO9D (gray) was used as a positive control with a C1-only oxidation pattern. PASC with no enzyme added, the negative control, is represented by a red chromatogram.
Overall structure of H. jecorina LPMO9A
The HjLPMO9A-ΔCBM (containing 17 of the linker residues) crystallized in space group P21 with unit cell parameters of a = 42.9 Å, b = 61.6 Å, and c = 47.8 with a β angle of 112.1°. The asymmetric unit of the crystal contains one enzyme molecule and the calculated Matthews coefficient was 2.2. The structure was solved by molecular replacement using the structure of H. jecorina LPMO9B as a template (PDB 2vtc) and was refined at 0.95-Å resolution. The refined and deposited structure model exhibits crystallographic R and R-free values of 11.5 and 12.7% and contains a total of 2,655 non-hydrogen atoms, including 248 amino acid residues, 1 copper atom, 2 N-acetyl-d-glucosamine residues, 15 mannose residues, 2 sulfate ions, and 372 water molecules. The amino acid residues are numbered according to the mature protein with the signal peptide removed, beginning at His-1. Statistics of the diffraction data and structure refinement are summarized in Table 1.
Table 1.
Data collection, processing, and refinement
| Data collection | ||
|---|---|---|
| PDB code | 5O2X | 5O2W |
| Data collection | ||
| Space group | P1211 | P1211 |
| Unit cell parameters | a = 42.9 Å | a = 42.9 Å |
| b = 61.6 Å | b = 62.1 Å | |
| c = 47.8 Å | c = 48.0 Å | |
| β = 112.1° | β = 111.8° | |
| X-ray source | ID23–1, ESRF | ID23, ESRF |
| Wavelength (Å) | 0.972425 | 0.87257 |
| Resolution range (Å) | 44.3 − 0.95 | 44.6 – 1.78 |
| Total No. of observations | ??? | 65,166 |
| Unique reflections | 118,793 | 21,394 |
| I/σ(I)a | 14.1 (1.9) | 4.6 (1.6) |
| Rmergeb | 0.058 (0.47) | 0.24 (0.60) |
| Multiplicity | 4.3 (1.5) | 4.3 (1.5) |
| Structure refinement | ||
| Resolution (Å) | 0.98 | 2.0 |
| Rwork/Rfree (%) | 11.5/12.7 | 19.7/23.1 |
| R.m.s. deviation for bond distances (Å) | 0.014 | 0.015 |
| R.m.s. deviation for bond angles (°) | 1.832 | 1.76 |
| No. of amino acid residues | 248 | 248 |
| No. of water molecules | 372 | 343 |
| No. of sugar residues | 17 | 17 |
| Ramachandran plotc | ||
| Most favored regions (%) | 98.2 | 99.6 |
| Outliers (%) | 1.8 | 0.4 |
| Disallowed regions (%) | 0 | 0 |
| Pyranose conformationsd (total/percentage) | ||
| Lowest energy conformation | 17/100 | 17/100 |
| Higher energy conformations | 0/0 | 0/0 |
a Values for the highest resolution shell are given in parentheses.
b Rmerge = ΣhklΣi|Ii(hkl) — 〈I(hkl)〉|/ΣhklΣi Ii(hkl), where Ii(hkl) is the intensity of the ith measurement of an equivalent reflection with indices hkl and hI(hkl)iis the mean intensity of Ii(hkl) for all i measurements.
c Calculated using a strict-boundary Ramachandran definition given by Kleywegt and Jones (3).
The overall fold of the HjLPMO9A catalytic domain is a β-sandwich fold consisting of two β-sheets formed by eight β-strands and common to all AA9 enzymes (19, 31). The extra residues near the N terminus compared with other AA9s, in the so-called L2 loop, place it as a Type-3 AA9 LPMO (32).
The copper atom at the catalytic center, modeled at 85% occupancy, is coordinated in one plane by three bonds to 2 histidine residues, His-1 and His-86. His-1 shows density of a methylation on the ϵ2 nitrogen atom (Fig. 4). There is also a water molecule 2.06 Å from the copper that could be modeled with only 40% occupancy. This indicates an ∼50–50 mixed state of Cu(II) and Cu(I) for the copper atom most likely due to photoreduction during X-ray data collection. Also near the active site, although not in the same plane and further from the copper, are the Oη-atom of the Tyr-174 side chain 2.8 Å from the copper, and one additional water molecule 2.4 Å from copper. A sulfate ion is also present at a distance of 3.9 Å from the copper. Two regions show increased temperature factors for the backbone, each ∼20 Å from the catalytic center and located on opposite sides of the potential cellulose-binding surface: residues 23–29 on one side and residues 120–126 and 211–217 on the other. The enzyme contain two cysteine bridges, both located on the same side of the β-sheets. The first, Cys-56 to Cys-177, is strictly conserved and located in the core of the enzyme and the second, between Cys-97 and Cys-101, is located in a loop near the enzyme surface.
Figure 4.
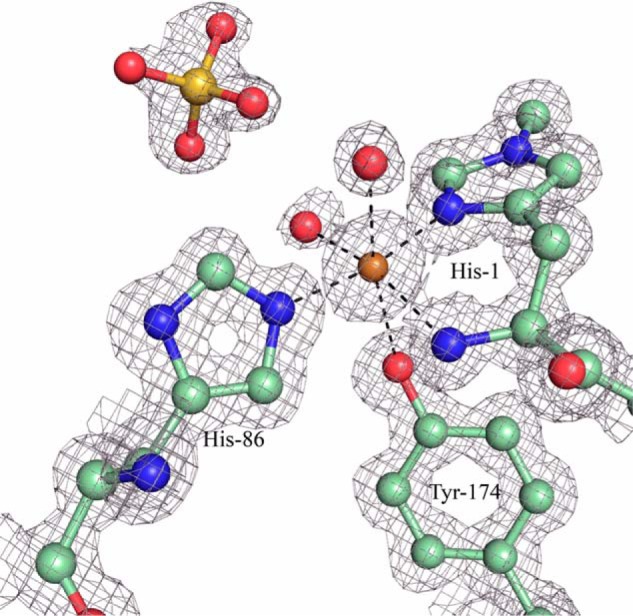
2Fo − Fc electron density map contoured at 0.63 e−/A3 showing the (non-hydrogen) atoms within the copper center of HjLPMO9A. Carbon atoms are shown in pale green, oxygen atoms are shown in red, nitrogen atoms in blue, sulfur atom in yellow, and copper atom in gold.
In HjLPMO9A, the last residue of the typical AA9-fold is a glycine (Gly-227). A glycine is also the last residue of HjLPMO9B and TaLPMO9A (9). Interestingly, all five N. crassa LPMOs with known structures as well as PcLPMO9D have a conserved cysteine as the last residue forming a disulfide bridge. The sequence corresponding to the AA9-fold suggests that HjLPMO9A belongs to Type-3 AA9 LPMOs (11, 32). However, the structured backbone of HjLPMO9A contains an additional 21 residues from the linker region in an extended loop conformation that wraps around the molecule and ends on the opposite side of the enzyme (Fig. 5, A and B). In Lentinus similis AA9A (LsAA9A; PDB 5acj), the C terminus is ∼15 residues longer than the typical LPMO fold, but this native enzyme does not contain any CBM (30). What is striking about the C-terminal extension in HjLPMO9A is the presence of eight O-glycosylations on the additional 21 residues. Altogether there are 17 O-glycosylation sites of which 15 could be modeled and refined resulting in reliable temperature factors. All of these sites are located within the last 58 residues when mapped to the sequence. In addition, there are two N-glycosylations at Asn-59 and Asn-137, each with one N-acetylglucosamine attached, and situated on opposite sides of the potential cellulose-binding surface. One of these, the glycosylation at Asn-137 (Fig. 6), has the same approximate location as the N-linked glycosylations of the other three Type-3 AA9 LPMOs (HjLPMO9B (19), TaLPMO9A (9), and NcLPMO9M (32)). Interestingly, these glycans have the same spatial location as the insert region II in Type-2 LPMOs, which is the signature sequence of these strict C4 oxidizing LPMOs (11, 32, 38). Notably, glycosylations are usually treated enzymatically prior to crystallization, thus what can be visualized in structure models are most likely remnants of much larger glycans. The C-terminal linker region showed only slightly higher temperature factors for the backbone atoms (average 7.3 Å2 for Cα's) compared with the catalytic domain itself (average 6.3 Å2 for the Cα's). Thirty-five residues on the catalytic domain interact with the linker over a surface spanning 928 Å2. The solvation-free energy, ΔiG, for a free peptide with the linker sequence, including the modeled O-glycosylations, bound to the catalytic domain is −13.3 kcal/mol (calculated using the Pisa module in CCP4i), with the negative value indicating a buried hydrophobic surface. Taken together, the glycosylated structured linker is likely an integral part of the HjLPMO9A catalytic module.
Figure 5.
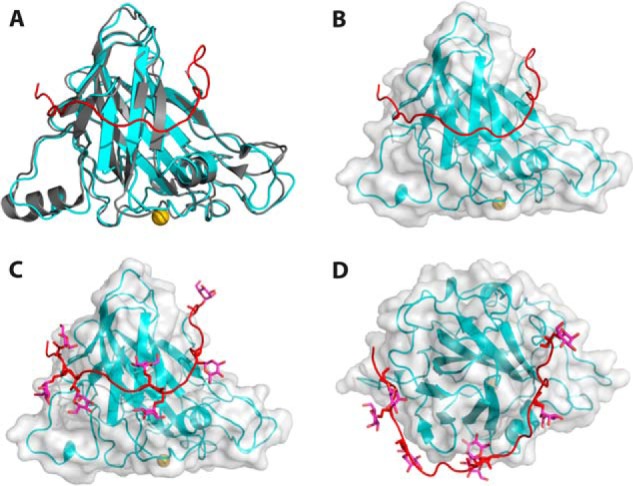
The C-terminal HjLPMO9A linker makes close contact with the catalytic core. A, the HjLPMO9B crystal structure (gray) and HjLPMO9A structure (cyan) with the HjLPMO9A C-terminal linker shown in red and copper shown in gold. B, surface rendering of the HjLPMO9A structure with the C terminus shown in red. C, surface rendering including glycosylated C terminus (red). D, HjLPMO9A as in C rotated 90 degrees.
Figure 6.
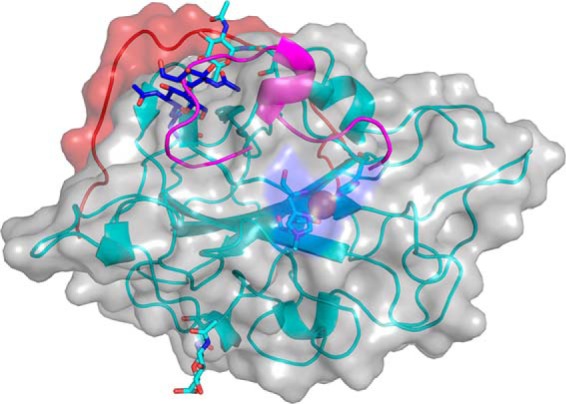
N-Linked glycosylations of the two Type-3 LPMOs of H. jecorina in comparison with Type-2 NcLPMO9D. HjLPMO9A (cyan), HjLPMO9B (blue, PDB code 2vtc) (19), and NcLPMO9D type-2 loop (magenta; PDB code 4eis) (32) are shown. The backbone schematic and the rendered surface is that of HjLPMO9A with the catalytic center highlighted in the middle and with the C-terminal linker in red.
This is to our knowledge the first observation of a linker sequence tightly bound to a fungal cellulosic enzyme. Similar O-glycosylated linkers wrapping around the catalytic module have previously been observed in both Aspergillus awamori and H. jecorina glucoamylases (39, 40). In both structures the GH18-fold leads into an extended loop that is highly O-glycosylated. In the H. jecorina glucoamylase, however, the structure of the complete linker and carbohydrate-binding module (CBM20) could be determined together with the catalytic domain revealing the relative orientation of the domains (40). The role of the structured part of the linker in HjLPMO9A may be similar in the sense that it could restrict the localization of the CBM to one side of the catalytic domain.
A superposition of HjLPMO9A-ΔCBM and HjLPMO9A-gmΔCBM structures using LSQ Superpose, the algorithm encoded in Coot, gave root mean square deviations of 0.136 and 0.164 Å for the main chain atoms 1–248 and all atoms 1–248, respectively. The main difference between the two structures is the loss of dual conformations of the side chains of some of the residues and sugar molecules in the structure of HjLPMO9A-gmΔCBM, which is most likely because of the difference in the resolution of the two structures.
Spectroscopy
Spectroscopic features of the copper active sites of HjLPMO9A and HjLPMO9A-ΔCBM were monitored by EPR, absorbance, CD, and MCD. EPR spin quantitation of the paramagnetic copper confirmed the loading of one Cu(II) per enzyme. These EPR spectra of copper-loaded samples of HjLPMO9A-ΔCBM show a single rhombically perturbed axial Cu(II) signal with spin Hamiltonian parameters similar to other AA9s including Ls(AA9)A (30), NcLPMO9C (29), and TaGH61A (41) (Fig. 7). Both X and Q band EPR spectra were simultaneously fit with gx = 2.047, gy = 2.067, gz = 2.280 and |Ax| = 13.3 × 10−4 cm−1, |Ay| = 0.5 × 10−4 cm−1, |Az| = 158.0 × 10−4 cm−1. These parameters (summarized in Table 2) are similar to those of the two Cu(II) sites in resting peptidylglycine α-hydroxylating monoxygenase, whereas the g‖ (2.280) and the g⊥,avg = 2.057 are slightly larger and the spectrum is slightly more rhombic (Δg⊥ = 0.03) compared with the EPR parameters of the Type-2 copper in the enzyme Fet3p (g‖ = 2.203, g⊥,avg = 2.048, Δg⊥ = 0.01, Fet3p Type-2 copper) (42, 43).
Figure 7.

A and B, X-band (9.65-GHz microwave frequency) (A) and Q-band (∼34-GHz microwave frequency) (B) EPR spectra of Cu(II)-HjLPMO9A-ΔCBM recorded at 77 K. The solid black curves are the experimental data and the solid red curves are simulations with parameters given in Tables 2 and 3.
Table 2.
EPR simulation parameters
| gx | gy | gz | Ax | Ay | Az |
|---|---|---|---|---|---|
| ×10−4 cm−1 | |||||
| 2.043 | 2.077 | 2.281 | 13.3 | 0.5 | 158.0 |
Room temperature Abs, CD, and low temperature (5 K) MCD spectra were obtained on copper-loaded HjLPMO9A-ΔCBM samples. These spectra were fit simultaneously with Gaussian functions at the energies given in Table 3. Energies were allowed to shift on the order of a few hundred wavenumbers in MCD relative to Abs and CD due to the temperature difference, and the bands sharpened at low temperature in the MCD spectrum. The peak fits (Table 3) are shown as the dashed colored lines in Fig. 8. The low temperature MCD spectrum of HjLPMO9A-ΔCBM shows features that are intense relative to the Abs spectrum and can therefore be assigned as ligand field transitions (44). The MCD spectrum can be fit with a minimum of 3 peaks centered at ∼12,900 cm−1, ∼13,900 cm−1, and ∼15,900 cm−1. The derivative shape of the low temperature MCD spectrum, referred to as a pseudo-A term, is comprised of two opposite signed C-terminal features assigned to dxz/yz → dx2-y2 ligand field transitions, which can spin orbit couple. The CD spectrum shown in Fig. 8B contains a negative feature at ∼13,900 cm−1 (blue dashed curve in Fig. 8B). The observation of this feature in the room temperature CD spectrum provides two important details. First, it indicates that there must be a corresponding peak at a comparable energy in the low temperature MCD spectrum of Fig. 8C. Second, as the dz2 → dx2-y2 transition is magnetic dipole forbidden it should not have significant CD intensity; therefore, this peak is assigned as the dxy → dx2-y2 transition.
Table 3.
Simultaneous Gaussian fit parameters for Abs, CD, and MCD energies
| Bands | Abs | CD | MCD | Assignment |
|---|---|---|---|---|
| cm−1 | ||||
| 1 | 12,995 | 13,081 | 12,896 | dxz/yz → dx2-y2 |
| 2 | 13,975 | 13,952 | 13,892 | dxy → dx2-y2 |
| 3 | 15,633 | 15,626 | 15,857 | dxz/yz → d-x2-y2 |
Figure 8.
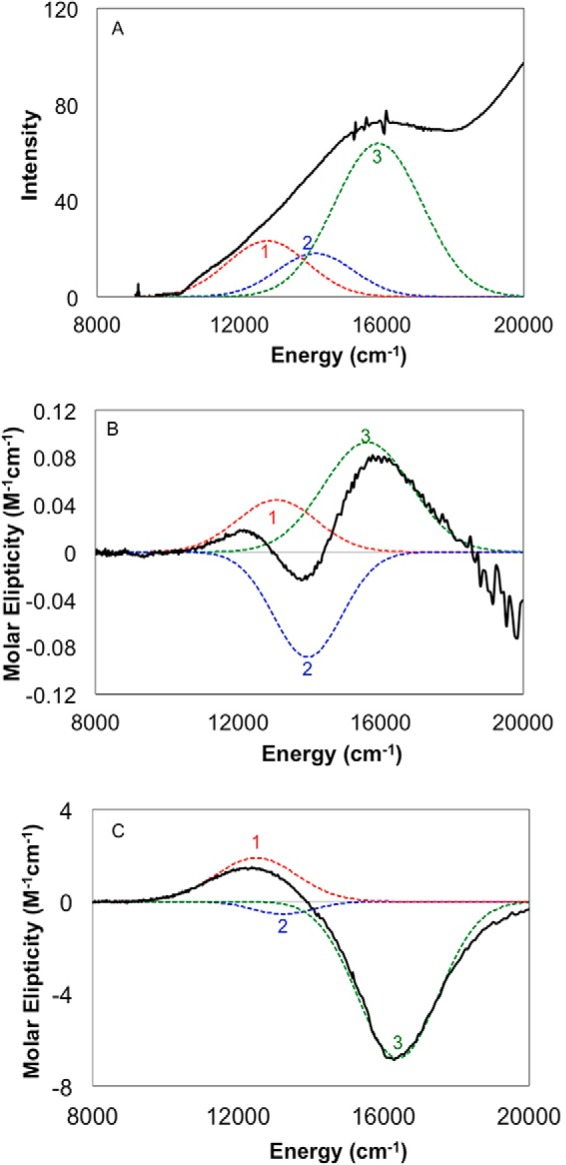
Absorption (A), room temperature CD (B), and low temperature, 5 K MCD (C) spectra of Cu(II)- HjLPMO9A-ΔCBM. The experimental data are shown as solid black lines. Peak fits are shown as dashed colored lines. The absorbance and CD spectra in panels A and B were obtained by subtracting the corresponding spectra of the apoenzyme from the copper-loaded spectra. The low temperature MCD spectrum of panel C was obtained by subtracting the 0 T spectrum from the 7 T data.
The spectra of Fig. 8 are similar to the ligand field spectra assigned in several tetragonal mononuclear copper sites including peptidylglycine α-hydroxylating monoxygenase and the [Cu(II)(1,2-dmIm)4](BF4)2 model complex (42). These spectroscopic data were then used to correlate the active site of the wild-type enzyme with that of HjLPMO9A-ΔCBM. The spectra of both are shown as overlays in Fig. 9. The X- and Q-band EPR spectra were simulated and the Abs, CD, and MCD spectra were fit with identical parameters to those of HjLPMO9A-ΔCBM. Importantly, the spectra of wild type do not exhibit measurable differences from HjLPMO9A-ΔCBM, showing that they have equivalent ligand field transition energies, spin Hamiltonian parameters, and ground-state copper covalency. This finding shows that the removal of the carbohydrate-binding module does not affect the copper active site.
Figure 9.

Overlay of the room temperature CD (A), low temperature, 5K MCD (B), 77K X-band EPR (C), and 77K Q-band EPR (D) spectra of wt-HjLPMO9A (blue) and Cu(II)-HjLPMO9A-ΔCBM (black). The CD spectra in panel A were obtained by subtracting the corresponding spectra of the apoenzymes from the copper-loaded spectra. The low temperature MCD spectra shown in panel B were obtained by subtracting the 0 T spectrum from the 7 T data.
Conclusions
Lytic polysaccharide monooxygenases enhance the efficiency of enzyme mixtures for the breakdown of cellulosic substrates and are important for the economic production of biofuels. The major enzymes of the cellulose-degrading H. jecorina have been studied in great detail and 3D structures have been determined for most of them. HjLPMO9A is the major LPMO from this well studied cellulase-producing fungus. We removed the carbohydrate-binding module both post-translationally with hydrolysis by papain and at the genetic level. Although the activity of HjLPMO9A-ΔCBM was found to decrease relative to the full-length enzyme, the spectroscopic comparison reveals that this difference is not reflected in the Cu(II) active site. The ligand field spectroscopy presented is highly sensitive to the ligand environment around the Cu(II) and is not perturbed by the removal of the CBM. Given the spectroscopic observations and the data indicating no change in C-1 and C-4 specificity, the decreased activity of HjLPMO9A-ΔCBM is likely a result of a decrease in affinity for cellulose. The structure of HjLPMO9A presented here not only adds to the set of existing structures, but also highlights a previously unknown structural importance of what was previously identified as a linker region. We have shown that ∼21 amino acid residues of the linker connecting the catalytic domain to the non-catalytic CBM should be retained for enzyme expression levels comparable with wild type, and that the crystal structures show an organized structure of an extensively glycosylated linker for a cellulose-active enzyme.
Experimental procedures
Generation of full-length and truncated proteins
H. jecorina LPMO9A (HjLPMO9A) full-length gene sequence (GenBankTM number CAA71999.1) was cloned, by PCR, from QM6a genomic DNA. Using Gateway cloning (Life Technologies), it was introduced into the pTTTpyr2 vector to produce the pTTTpyr2-HjLPMO9A plasmid (pTTTpyr2 is similar to the pTTTpyrG vector described before (PCT publication WO 2011/063308), except that the pyrG gene is replaced with the pyr2 gene). Using the pTTTpyr2-HjLPMO9A plasmid, HjLPMO9A-truncated genes were generated using primer combinations that truncated HjLPMO9A after residue 231 of the mature sequence, just after the catalytic domain and after residue 252, in the middle of the predicted linker (HjLPMO9A-gmΔCBM) (Fig. 1). The fragments with different linker lengths were cloned back into the pTTTpyr2 vector with Gateway® LR Clonase® (Life Technologies). The construct truncated at the end of the catalytic domain, completely lacking the linker sequence, did not result in expressed protein (data not shown). Protoplasts of H. jecorina strain Δ(cbhI, cbhII, egI, egII, egIII, egIV, egV, egVI, man1, bgl1) were transformed with the individual pTTTpyr2-HjLPMO9A or pTTTpyr2-HjLPMO9AΔCBM vectors and grown on selective agar containing acetamide at 28 °C for 7 days as previously described (PCT Publication WO 2009/048488). For HjLPMO9A production, a volume of 10 μl of spore suspension was added to 200 μl of a glycine minimal medium (PCT publication WO 2011/038019) supplemented with 2% glucose/Sepharose mixture (United States Patent 7,713,725). After sealing the plate with an oxygen-permeable membrane, the plates were incubated at 28 °C for 6 days, with shaking at 220 rpm (Infors incubator shaker). Protein samples were harvested by transferring the culture medium to a 96-well filter plate (Corning 3505) and collecting the filtrate under vacuum. Larger samples were prepared by fermentation in 1-liter DASGIP reactors (Eppendorf D76FB04MBPD) and centrifugation to generate cell-free supernatants.
Protein purification
Cell-free supernatants of H. jecorina cultures expressing HjLPMO9A were concentrated using a centrifugal concentrator with a 10-kDa cut-off (Vivascience, Littleton, MA). For crystallization experiments, 2 ml of the concentrated HjLPMO9A culture filtrate was loaded on an equilibrated (25 mm NaAc, pH 5.0) PD10 column (GE Healthcare) and eluted using the equilibration buffer following the instructions by the manufacturer. One ml of the eluted protein solution was directly applied to a Superdex 75 size exclusion purification column (GE Healthcare) equilibrated with 25 mm NaAc, pH 5.0.
In the subsequent step, the HjLPMO9A protein was treated with papain to remove the CBM. First, papain (Sigma) was activated in 0.1 m BisTris buffer (Sigma) at pH 6.5 containing 10 mm 2-mercaptoethanol (Merck Millipore) and 1 mm EDTA (Merck Millipore). Then, the activated papain was mixed with 3 mg/ml of HjLPMO9A in 0.1 m NaAc, pH 5.0, to a final papain concentration of 1:10 (papain:HjLPMO9A) and the mixture was incubated for 24 h at 37 °C. The papain-treated protein solution was then concentrated to 2 ml and applied to a Superdex 75 column equilibrated with 25 mm Tris-HCl, pH 7.5, and 150 mm NaCl. The fractions corresponding to HjLPMO9A-ΔCBM were pooled and the buffer of the protein solution was changed to 25 mm Tris-HCl, pH 7.5, and 25 mm NaCl. The sample was concentrated to a final protein concentration of 30 mg/ml as estimated by measuring the absorbance of the protein solution at 280 nm on a NanoDrop1000 (ThermoScientific, Thermo Fischer Scientific Inc.) and using the calculated extinction coefficient of 54,360 m−1 cm−1 for the full-length protein and 48,150 m−1 cm−1 for the HjLPMO9A-ΔCBM.
For biophysical characterization, Avicel binding assay, and PASC activity assay, protein purification was carried out in a different manner than described above for crystallization. Culture supernatants of H. jecorina expressing HjLPMO9A- and HjLPMO9A-truncated variants were starting materials for purification. Column purifications were performed using an Äkta Explorer FPLC system (GE Healthcare Biosciences). A 560-ml Sephadex G-25 desalting column (GE Healthcare BioSciences) was equilibrated with 20 mm MES, pH 6, containing 25 mm sodium chloride. Samples were loaded and eluted isocratically at a flow rate of 5 ml/min. Fractions containing protein were pooled for hydrophobic interaction chromatography. Samples were mixed 1:1 with a saturated ammonium sulfate solution (∼3 m) in 20 mm MES, pH 6, incubated at room temperature for 15 min, and centrifuged at 4000 rpm for 10 min. Supernatants were loaded onto a 30-ml phenyl-Sepharose column (GE Healthcare Biosciences) equilibrated with 20 mm MES, pH 6, containing 1.8 m ammonium sulfate and eluted at 0.5 ml/min with a linearly decreasing gradient of ammonium sulfate. Fractions containing the target protein, as determined by SDS-PAGE, were pooled. Samples were concentrated using Sartorius Vivaspin® concentrators with a 5-kDa membrane cutoff (Thermo Fisher Scientific, Waltham, MA). A Superdex 75 10/300 column (GE Healthcare) was equilibrated with 50 mm MES, pH 6, containing 100 mm sodium chloride. Samples with 1-ml volumes were filtered, loaded, and eluted isocratically at 0.6 ml/min. Fractions containing pure target protein as judged by SDS-PAGE were pooled for further study.
For the EPR experiment samples of HjLPMO9A and HjLPMO9A-ΔCBM were prepared with 10–20% EPR active Cu/Fe present. Apoenzyme was generated by dialysis in Slide-A-Lyzer dialysis cassettes (10,000 MWCO; purchased from Thermo Scientific) against a solution of 50 mm EDTA, 50 mm MES buffer, pH 6.0. After a 2-day dialysis, the cassettes were buffer exchanged in 50 mm MES buffer, pH 6.0, overnight. The apoenzyme was extracted from the dialysis cassettes and concentrated in Corning Spin-X ultracentrifuge filters. Copper reconstitution was performed by adding a 5-fold excess of Cu(II) to enzyme from a 3.48 mm Cu(NO3)2 in 50 mm MES stock, pH 4.0. Excess copper was removed by dialysis against MES buffer, pH 6.0, and samples were reconcentrated. Biquinoline copper assays and EPR spin integration confirmed binding of a single Cu(II) ion per molecule. Samples for magnetic circular dichroism (MCD) were prepared in MES buffer, pH 6.0, and mixed with 50–60% (v/v) glycerol to obtain high quality glasses. The samples were then injected into MCD cells, which consist of two quartz disks sealed with a 3-mm rubber spacer.
Protein crystallization, structure determination, and refinements
Initial protein crystallization screens were performed by sitting-drop vapor diffusion experiments of 0.3-μl drops in a 96-well plate, prepared using a Mosquito crystallization robot (TPP Labtech, Cambridge, United Kingdom). Small needle-like crystals started to appear after 1 week incubation at 28 °C, in an ammonium sulfate crystallization screen (Qiagen, Germany) by 1:1 mixing of 30 mg/ml of protein and crystal screen reservoir solution (1.6 m AmSO4 and 0.1 m citric acid, pH 4.0). Larger crystals were obtained by introducing nuclei from crushed crystals into new drops by streak seeding. Seeding resulted in crystals that grew to optimum size after 1 week. Prior to data collection, crystals were frozen in liquid N2 using the crystallization solution with 35% PEG 3350 and 30% m-PEG 2000 added as cryoprotectant. The crystals of the HjLPMO9A-ΔCBM enzyme belong to the space group P21 with unit cell parameters a = 42.9, b = 61.59, c = 47.83 Å, and β = 112.1°.
X-ray diffraction data on a HjLPMO9A-ΔCBM crystal were collected with a Pilatus detector at wavelength 0.972425 Å at 100 K on beam line ID23-1 at the European Synchrotron Radiation Facility, Grenoble, France. The structure of HjLPMO9A-ΔCBM was determined by molecular replacement using the program PHASER (45), and using the structure of H. jecorina LPMO9B (PDB code 2vtc) as the search model. The best solution was obtained with one molecule in the asymmetric unit.
Purification, crystallization, and structure determination of HjLPMO9A-gmΔCBM was carried out in a similar manner as for HjLPMO9A-ΔCBM. Two ml of the concentrated HjLPMO9A-gmΔCBM culture filtrate was loaded on a PD10 column. One ml of eluted protein was loaded onto a Superdex 75 size exclusion purification column (GE Healthcare) equilibrated with 25 mm NaAc, pH 5.0. Fractions corresponding to HjLPMO9A-gmΔCBM were pooled and concentrated to 27 mg/ml in 25 mm Tris-HCl, pH 7.5, and 10 mm NaCl. Crystals of HjLPMO9A-gmΔCBM were grown using hanging drop-vapor diffusion crystallization experiments incubated at 20 °C. The drops were prepared by mixing protein solution of 17 mg/ml with an equal amount of a well solution containing 1.6 m AmSO4 and 0.1 m citric acid, pH 4.0. Prior to data collection, crystals were frozen in liquid N2 using the crystallization solution with 35% PEG 3350 added as cryoprotectant. The crystals of the HjLPMO9A-gmΔCBM enzyme belong to the space group P21 with unit cell parameters a = 42.9, b = 62.1, c = 48.0 Å, and β = 111.8°. X-ray diffraction data from a HjLPMO9A-gmΔCBM crystal was collected on beam line ID23, ESRF with a Pilatus detector at a wavelength 0.87257 Å. The structure of HjLPMO9A-gmΔCBM was determined by molecular replacement using the structure of HjLPMO9A-ΔCBM as the search model.
X-ray diffraction data were processed using XDS (46, 47). The integrated data were then scaled using the scaling program SCALA (Collaborative Computational Project Number 4, 1994). For cross-validation and Rfree calculations (48), 5% of the reflections was set aside. The program REFMAC5 (49) was used for structure model refinements, and manual model rebuilding was performed with Coot (50, 51), with maximum likelihood σA-weighted 2Fobs − Fcalc electron density maps (50). Solvent molecules were automatically added using the automatic water picking function in the ARP/wARP package (52). Picked water molecules were selected or discarded manually by visual inspection of the 2Fobs − Fcalc electron density maps. The copper atoms bound in the active site of the enzyme in the two HjLPMO9A structures were introduced at a final stage of the structure refinement. Statistics from data processing and structure refinement are summarized in Table 1. The coordinates for the two final structure models, and the structure factors, have been deposited in the Protein Data Bank with accession codes 5O2X and 5O2W, respectively. PyMOL (version 1.5) was used for analysis of the structures and figure preparations.
Avicel-binding assay
Avicel (PH-101, FMC BioPolymer)-binding assays were conducted as described before (53). Various concentrations of Avicel solids were incubated at room temperature overnight with 0.125 mg/ml of LPMO (full-length or truncated) protein. The Avicel was removed by centrifugation and the supernatant concentration of the LPMO was determined by UHPLC. The fraction of bound LPMO was calculated from the starting and remaining concentrations.
Phosphoric acid–swollen cellulose–activity assay
Relative activity of LPMO on PASC was assayed essentially as previously described (54). Purified LPMO enzymes (full-length and truncated) were serially diluted in 96-well microtiter plate wells. PASC (0.25% solids) was combined with ascorbic acid (1 mm), copper sulfate (100 μm), and sodium acetate buffer (50 mm), pH 5, or acetate/BisTris buffer (120 mm), pH 7. Plates were sealed with foil and incubated at 50 °C with shaking for 6 h. LPMO-dependent PASC solubilization was measured by following the reduction of light scattering optical density at 420 nm. Activity is expressed as the (negative of the) change in absorbance relative to an initial, untreated PASC sample value.
HPAEC-PAD product characterizations
Potential differences in enzymatic behavior of HjLPMO9A and HjLPMO9A-ΔCBM on reduced PASC were analyzed using high performance anion exchange chromatography (HPAEC). PASC was prepared and reduced according to Westereng et al. (7). The LPMO enzymes were preincubated with CuSO4 corresponding to ×10 the enzyme concentration for 30 min at ambient temperature. HjLPMO9A and HjLPMO9A-ΔCBM were prepared as mentioned above. Phanerochaete chrysosporum LPMO9D was used as a C1-only oxidizer positive control and was produced and purified as described by Wu et al. (33).
0.2 ml of mixture containing (final concentrations) 0.1 mm ascorbic acid, 0.06–0.08 mm enzyme (no enzyme was added to the negative control), and 0.2% PASC was prepared in a 2-ml Eppendorf tube and incubated for 16 h at 50 °C and 1000 rpm in a Thermo mixer. After centrifugation, 19 μl of the supernatant was analyzed on a Dionex ICS3000 (Dionex, Sunnyvale, CA) equipped with a pulsed amperimetric detector using the Standard Quad waveform for detection. Separation of oligosaccharides was performed using PA1 guard and analytical columns at 30 °C as described by Westereng et al. (55) with the exception of an extra 3-min isocratic elution directly following the sample injection.
EPR, CD, and MCD characterizations
X-band EPR spectra were obtained with a Bruker EMX spectrometer, an ER 041 XG microwave bridge, and an ER4116DM cavity. A sample temperature of 77 K was maintained using a liquid nitrogen finger dewar. EPR settings were as follows: frequency ≈ 9.6 GHz, power ≈ 10 milliwatt, Modulation Amplitude = 3.00 G. All spectra were averaged over 3 scans. Q-band spectra were obtained using an ER 051 QR microwave bridge, an ER 5106QT resonator, and an Oxford Instruments continuous-flow CF935 cryostat that held the sample at 77 K. Q-band EPR settings were: frequency ≈ 34 GHz, power ≈ 0.37 milliwatt, Modulation Amplitude = 5.00 G. All spectra were averaged over 20 scans. EPR spin quantitation of the paramagnetic copper content was performed using a 0.945 mm AAS copper standard solution, Specpure (purchased from Alfa Aesar), in MES, pH 6.0, and 40% glycerol. EPR spectra were simulated using the SpinCount simulation software developed by Professor Michael Hendrich of Carnegie Mellon University (www.chem.cmu.edu/groups/hendrich/facilities/index.html).4
UV-visible absorption spectra were acquired on an Agilent 8453 diode array spectrophotometer, in the energy range 300–1100 nm. Circular dichroism (CD) spectra (4 °C) and magnetic CD (MCD) spectra (4 K) were measured on a Jasco 730 spectrapolarimeter with an S-20 photomultiplier tube or a liquid nitrogen-cooled InSb detector coupled via an 8-foot extended sample compartment to an Oxford Instruments SM4000–7T magnet. Zero-field baseline effects were eliminated in MCD by subtracting the 0 T scan from the +7 T scan. Simultaneous Gaussian fitting of the UV-visible, CD, and MCD data were done using PeakFit 4.0 (Jandel).
Author contributions
H. H., S. A. K., and N. M. planned and performed all structural experiments, interpreted data, and helped write the manuscript. N. D., S. T. K., A. L., and B. K. produced and purified the enzyme, planned and performed the activity studies, interpreted data, and helped write the manuscript. K. K. M. and S. M. J. planned and performed all EPR, absorption, CD, and MCD experiments, interpreted EPR, absorption, CD, and MCD data, and helped write the manuscript; T. K., M. S., and E. I. S. designed the study, planned the experiments, interpreted data, and wrote the manuscript. All authors have given approval to the final version of the manuscript.
This work was supported by the Swedish Energy Agency under award number 40144-1 (to M. S.), the NIDDK of the National Institutes of Health under Grant R01DK031450 (to E. I. S.), the Ruth L. Kirschstein National Research Service Award from the NIGMS of the National Institutes of Health under Grant F32GM116240 (to K. K. M.), and the Faculty for Natural Resources and Agriculture, Swedish University of Agricultural Sciences (to M. S., H. H., S. K., and N. M.) through the faculty research program MicroDrivE. N. D., S. T. K., A. L., and B. K. are employees of DuPont Industrial Biosciences, a producer of enzymes for industrial use. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5O2X and 5O2W) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- LPMO
- lytic polysaccharide monooxygenases
- AA9
- auxiliary activity family 9
- GH
- glycoside hydrolase
- MCD
- magnetic circular dichroism
- PASC
- phosphoric acid–swollen cellulose
- CBM1
- family 1 carbohydrate-binding module
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- HPAEC
- high performance anion exchange chromatography.
References
- 1. Divne C., Ståhlberg J., Reinikainen T., Ruohonen L., Pettersson G., Knowles J. K., Teeri T. T., and Jones T. A. (1994) The three-dimensional crystal structure of the catalytic core of cellobiohydrolase I from Trichoderma reesei. Science 265, 524–528 [DOI] [PubMed] [Google Scholar]
- 2. Karkehabadi S., Helmich K. E., Kaper T., Hansson H., Mikkelsen N. E., Gudmundsson M., Piens K., Fujdala M., Banerjee G., Scott-Craig J. S., Walton J. D., Phillips G. N. Jr., and Sandgren M. (2014) Biochemical characterization and crystal structures of a fungal family 3 β-glucosidase, Cel3A from Hypocrea jecorina. J. Biol. Chem. 289, 31624–31637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kleywegt G. J., and Jones T. A. (1996) Phi/psi-chology: Ramachandran revisited. Structure 4, 1395–1400 [DOI] [PubMed] [Google Scholar]
- 4. Lee T. M., Farrow M. F., Arnold F. H., and Mayo S. L. (2011) A structural study of Hypocrea jecorina Cel5A. Protein Sci. 20, 1935–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rouvinen J., Bergfors T., Teeri T., Knowles J. K., and Jones T. A. (1990) Three-dimensional structure of cellobiohydrolase II from Trichoderma reesei. Science 249, 380–386 [DOI] [PubMed] [Google Scholar]
- 6. Vaaje-Kolstad G., Westereng B., Horn S. J., Liu Z., Zhai H., Sørlie M., and Eijsink V. G. (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330, 219–222 [DOI] [PubMed] [Google Scholar]
- 7. Westereng B., Ishida T., Vaaje-Kolstad G., Wu M., Eijsink V. G., Igarashi K., Samejima M., Ståhlberg J., Horn S. J., and Sandgren M. (2011) The putative endoglucanase PcGH61D from Phanerochaete chrysosporium is a metal-dependent oxidative enzyme that cleaves cellulose. PLoS ONE 6, e27807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Phillips C. M., Beeson W. T., Cate J. H., and Marletta M. A. (2011) Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem. Biol. 6, 1399–1406 [DOI] [PubMed] [Google Scholar]
- 9. Quinlan R. J., Sweeney M. D., Lo Leggio L., Otten H., Poulsen J. C., Johansen K. S., Krogh K. B., Jørgensen C. I., Tovborg M., Anthonsen A., Tryfona T., Walter C. P., Dupree P., Xu F., Davies G. J., and Walton P. H. (2011) Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. U.S.A. 108, 15079–15084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim S., Ståhlberg J., Sandgren M., Paton R. S., and Beckham G. T. (2014) Quantum mechanical calculations suggest that lytic polysaccharide monooxygenases use a copper-oxyl, oxygen-rebound mechanism. Proc. Natl. Acad. Sci. U.S.A. 111, 149–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vu V. V., Beeson W. T., Phillips C. M., Cate J. H., and Marletta M. A. (2014) Determinants of regioselective hydroxylation in the fungal polysaccharide monooxygenases. J. Am. Chem. Soc. 136, 562–565 [DOI] [PubMed] [Google Scholar]
- 12. Hemsworth G. R., Johnston E. M., Davies G. J., and Walton P. H. (2015) Lytic polysaccharide monooxygenases in biomass conversion. Trends Biotechnol. 33, 747–761 [DOI] [PubMed] [Google Scholar]
- 13. Beeson W. T., Vu V. V., Span E. A., Phillips C. M., and Marletta M. A. (2015) Cellulose degradation by polysaccharide monooxygenases. Annu. Rev. Biochem. 84, 923–946 [DOI] [PubMed] [Google Scholar]
- 14. Eibinger M., Ganner T., Bubner P., Rošker S., Kracher D., Haltrich D., Ludwig R., Plank H., and Nidetzky B. (2014) Cellulose surface degradation by a lytic polysaccharide monooxygenase and its effect on cellulase hydrolytic efficiency. J. Biol. Chem. 289, 35929–35938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Levasseur A., Drula E., Lombard V., Coutinho P. M., and Henrissat B. (2013) Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 6, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Isaksen T., Westereng B., Aachmann F. L., Agger J. W., Kracher D., Kittl R., Ludwig R., Haltrich D., Eijsink V. G., and Horn S. J. (2014) A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J. Biol. Chem. 289, 2632–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frommhagen M., Sforza S., Westphal A. H., Visser J., Hinz S. W., Koetsier M. J., van Berkel W. J., Gruppen H., and Kabel M. A. (2015) Discovery of the combined oxidative cleavage of plant xylan and cellulose by a new fungal polysaccharide monooxygenase. Biotechnol. Biofuels 8, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adav S. S., Chao L. T., and Sze S. K. (2012) Quantitative secretomic analysis of Trichoderma reesei strains reveals enzymatic composition for lignocellulosic biomass degradation. Mol. Cell Proteomics 11, mcp.M111.012419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karkehabadi S., Hansson H., Kim S., Piens K., Mitchinson C., and Sandgren M. (2008) The first structure of a glycoside hydrolase family 61 member, Cel61B from Hypocrea jecorina, at 1.6-Å resolution. J. Mol. Biol. 383, 144–154 [DOI] [PubMed] [Google Scholar]
- 20. Saloheimo M., Nakari-Setälä T., Tenkanen M., and Penttilä M. (1997) cDNA cloning of a Trichoderma reesei cellulase and demonstration of endoglucanase activity by expression in yeast. Eur. J. Biochem. 249, 584–591 [DOI] [PubMed] [Google Scholar]
- 21. Karlsson J., Saloheimo M., Siika-Aho M., Tenkanen M., Penttilä M., and Tjerneld F. (2001) Homologous expression and characterization of Cel61A (EG IV) of Trichoderma reesei. Eur. J. Biochem. 268, 6498–6507 [DOI] [PubMed] [Google Scholar]
- 22. Tanghe M., Danneels B., Camattari A., Glieder A., Vandenberghe I., Devreese B., Stals I., and Desmet T. (2015) Recombinant expression of Trichoderma reesei Cel61A in Pichia pastoris: optimizing yield and N-terminal processing. Mol. Biotechnol. 57, 1010–1017 [DOI] [PubMed] [Google Scholar]
- 23. Banerjee G., Car S., Scott-Craig J. S., Borrusch M. S., and Walton J. D. (2010) Rapid optimization of enzyme mixtures for deconstruction of diverse pretreatment/biomass feedstock combinations. Biotechnol. Biofuels 3, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Srisodsuk M., Lehtiö J., Linder M., Margolles-Clark E., Reinikainen T., and Teeri T. T. (1997) Trichoderma reesei cellobiohydrolase I with an endoglucanase cellulose-binding domain: action on bacterial microcrystalline cellulose. J. Biotechnol. 57, 49–57 [DOI] [PubMed] [Google Scholar]
- 25. Ståhlberg J., Johansson G., and Pettersson G. (1993) Trichoderma reesei has no true exo-cellulase: all intact and truncated cellulases produce new reducing end groups on cellulose. Biochim. Biophys. Acta 1157, 107–113 [DOI] [PubMed] [Google Scholar]
- 26. Sammond D. W., Payne C. M., Brunecky R., Himmel M. E., Crowley M. F., and Beckham G. T. (2012) Cellulase linkers are optimized based on domain type and function: insights from sequence analysis, biophysical measurements, and molecular simulation. PLoS ONE 7, e48615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Payne C. M., Resch M. G., Chen L., Crowley M. F., Himmel M. E., Taylor L. E. 2nd, Sandgren M., Ståhlberg J., Stals I., Tan Z., and Beckham G. T. (2013) Glycosylated linkers in multimodular lignocellulose-degrading enzymes dynamically bind to cellulose. Proc. Natl. Acad. Sci. U.S.A. 110, 14646–14651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kleywegt G. J., Zou J. Y., Divne C., Davies G. J., Sinning I., Stâhlberg J., Reinikainen T., Srisodsuk M., Teeri T. T., and Jones T. A. (1997) The crystal structure of the catalytic core domain of endoglucanase I from Trichoderma reesei at 3.6-Å resolution, and a comparison with related enzymes. J. Mol. Biol. 272, 383–397 [DOI] [PubMed] [Google Scholar]
- 29. Borisova A. S., Isaksen T., Dimarogona M., Kognole A. A., Mathiesen G., Várnai A., Røhr Å. K., Payne C. M., Sørlie M., Sandgren M., and Eijsink V. G. (2015) Structural and functional characterization of a lytic polysaccharide monooxygenase with broad substrate specificity. J. Biol. Chem. 290, 22955–22969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frandsen K. E., Simmons T. J., Dupree P., Poulsen J. C., Hemsworth G. R., Ciano L., Johnston E. M., Tovborg M., Johansen K. S., von Freiesleben P., Marmuse L., Fort S., Cottaz S., Driguez H., Henrissat B., et al. (2016) The molecular basis of polysaccharide cleavage by lytic polysaccharide monooxygenases. Nat. Chem. Biol. 12, 298–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Harris P. V., Welner D., McFarland K. C., Re E., Navarro Poulsen J. C., Brown K., Salbo R., Ding H., Vlasenko E., Merino S., Xu F., Cherry J., Larsen S., and Lo Leggio L. (2010) Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: structure and function of a large, enigmatic family. Biochemistry 49, 3305–3316 [DOI] [PubMed] [Google Scholar]
- 32. Li X., Beeson W. T. 4th, Phillips C. M., Marletta M. A., and Cate J. H. (2012) Structural basis for substrate targeting and catalysis by fungal polysaccharide monooxygenases. Structure 20, 1051–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu M., Beckham G. T., Larsson A. M., Ishida T., Kim S., Payne C. M., Himmel M. E., Crowley M. F., Horn S. J., Westereng B., Igarashi K., Samejima M., Ståhlberg J., Eijsink V. G., and Sandgren M. (2013) Crystal structure and computational characterization of the lytic polysaccharide monooxygenase GH61D from the Basidiomycota fungus Phanerochaete chrysosporium. J. Biol. Chem. 288, 12828–12839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bennati-Granier C., Garajova S., Champion C., Grisel S., Haon M., Zhou S., Fanuel M., Ropartz D., Rogniaux H., Gimbert I., Record E., and Berrin J. G. (2015) Substrate specificity and regioselectivity of fungal AA9 lytic polysaccharide monooxygenases secreted by Podospora anserina. Biotechnol. Biofuels 8, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Crouch L. I., Labourel A., Walton P. H., Davies G. J., and Gilbert H. J. (2016) The contribution of non-catalytic carbohydrate binding modules to the activity of lytic polysaccharide monooxygenases. J. Biol. Chem. 291, 7439–7449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vejdovszky K., Warth B., Sulyok M., and Marko D. (2016) Non-synergistic cytotoxic effects of Fusarium and Alternaria toxin combinations in Caco-2 cells. Toxicol. Lett. 241, 1–8 [DOI] [PubMed] [Google Scholar]
- 37. Rojas J., Lopez A., Guisao S., and Ortiz C. (2011) Evaluation of several microcrystalline celluloses obtained from agricultural by-products. J. Adv. Pharm. Technol. Res. 2, 144–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moses V., Hatherley R., and Tastan Bishop Ö. (2016) Bioinformatic characterization of type-specific sequence and structural features in auxiliary activity family 9 proteins. Biotechnol. Biofuels 9, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aleshin A., Golubev A., Firsov L. M., and Honzatko R. B. (1992) Crystal structure of glucoamylase from Aspergillus awamori var. X100 to 2.2-Å resolution. J. Biol. Chem. 267, 19291–19298 [DOI] [PubMed] [Google Scholar]
- 40. Bott R., Saldajeno M., Cuevas W., Ward D., Scheffers M., Aehle W., Karkehabadi S., Sandgren M., and Hansson H. (2008) Three-dimensional structure of an intact glycoside hydrolase family 15 glucoamylase from Hypocrea jecorina. Biochemistry 47, 5746–5754 [DOI] [PubMed] [Google Scholar]
- 41. Kjaergaard C. H., Qayyum M. F., Wong S. D., Xu F., Hemsworth G. R., Walton D. J., Young N. A., Davies G. J., Walton P. H., Johansen K. S., Hodgson K. O., Hedman B., and Solomon E. I. (2014) Spectroscopic and computational insight into the activation of O2 by the mononuclear Cu center in polysaccharide monooxygenases. Proc. Natl. Acad. Sci. U.S.A. 111, 8797–8802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen P., Bell J., Eipper B. A., and Solomon E. I. (2004) Oxygen activation by the noncoupled binuclear copper site in peptidylglycine α-hydroxylating monooxygenase: spectroscopic definition of the resting sites and the putative CuIIM-OOH intermediate. Biochemistry 43, 5735–5747 [DOI] [PubMed] [Google Scholar]
- 43. Machonkin T. E., Quintanar L., Palmer A. E., Hassett R., Severance S., Kosman D. J., and Solomon E. I. (2001) Spectroscopy and reactivity of the type 1 copper site in Fet3p from Saccharomyces cerevisiae: correlation of structure with reactivity in the multicopper oxidases. J. Am. Chem. Soc. 123, 5507–5517 [DOI] [PubMed] [Google Scholar]
- 44. Gewirth A. A., and Solomon E. I. (1988) Electronic-structure of plastocyanin-excited-state spectral features. J. Am. Chem. Soc. 110, 3811–3819 [Google Scholar]
- 45. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brünger A. T. (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 [DOI] [PubMed] [Google Scholar]
- 49. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 51. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Langer G., Cohen S. X., Lamzin V. S., and Perrakis A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Georgelis N., Tabuchi A., Nikolaidis N., and Cosgrove D. J. (2011) Structure-function analysis of the bacterial expansin EXLX1. J. Biol. Chem. 286, 16814–16823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wood T. M., and Bhat K. M. (1988) Methods for measuring cellulase activities. Methods Enzymol. 160, 87–112 [Google Scholar]
- 55. Westereng B., Agger J. W., Horn S. J., Vaaje-Kolstad G., Aachmann F. L., Stenstrøm Y. H., and Eijsink V. G. (2013) Efficient separation of oxidized cello-oligosaccharides generated by cellulose degrading lytic polysaccharide monooxygenases. J. Chromatogr. A 1271, 144–152 [DOI] [PubMed] [Google Scholar]
- 56. Agirre J., Iglesias-Fernández J., Rovira C., Davies G. J., Wilson K. S., and Cowtan K. D. (2015) Privateer: software for the conformational validation of carbohydrate structures. Nat. Struct. Mol. Biol. 22, 833–834 [DOI] [PubMed] [Google Scholar]
- 57. Agirre J., Ariza A., Offen W. A., Turkenburg J. P., Roberts S. M., McNicholas S., Harris P. V., McBrayer B., Dohnalek J., Cowtan K. D., Davies G. J., and Wilson K. S. (2016) Three-dimensional structures of two heavily N-glycosylated Aspergillus sp. family GH3 β-d-glucosidases. Acta Crystallogr. D Struct. Biol. 72, 254–265 [DOI] [PMC free article] [PubMed] [Google Scholar]