

Abstract
Background
Both defective DNA methylation and active DNA demethylation processes are emerging as important risk factors in chronic lymphocytic leukemia (CLL). However, associations between 5-cytosine epigenetic markers and the most frequent chromosomal abnormalities detected in CLL remain to be established.
Methods
CLL patients were retrospectively classified into a cytogenetic low-risk group (isolated 13q deletion), an intermediate-risk group (normal karyotype or trisomy 12), and a high-risk group (11q deletion, 17p deletion, or complex karyotype [≥ 3 breakpoints]). The two 5-cytosine derivatives, 5-methylcytosine (5-mCyt) and 5-hydroxymethylcytosine (5-hmCyt), were tested by ELISA (n = 60), while real-time quantitative PCR was used for determining transcriptional expression levels of DNMT and TET (n = 24).
Results
By using global DNA methylation/demethylation levels, in the low-risk disease group, two subgroups with significantly different clinical outcomes have been identified (median treatment-free survival [TFS] 45 versus > 120 months for 5-mCyt, p = 0.0008, and 63 versus > 120 months for 5-hmCyt, p = 0.04). A defective 5-mCyt status was further associated with a higher percentage of 13q deleted nuclei (> 80%), thus suggesting an acquired process. When considering the cytogenetic intermediate/high-risk disease groups, an association of 5-mCyt status with lymphocytosis (p = 0.0008) and the lymphocyte doubling time (p = 0.04) but not with TFS was observed, as well as a reduction of DNMT3A, TET1, and TET2 transcripts.
Conclusions
Combining cytogenetic studies with 5-mCyt assessment adds accuracy to CLL patients’ prognoses and particularly for those with 13q deletion as a sole cytogenetic abnormality.
Keywords: Chronic lymphocytic leukemia, Cytogenetics, DNA methylation, Active DNA demethylation, DNMT, TET
Introduction
Clinical heterogeneity is the hallmark of chronic lymphocytic leukemia (CLL), with some patients surviving for decades without needing treatment, while others present early progression and a short overall survival, if untreated [1]. Therefore, great efforts have been made in order to best predict CLL evolution at the time of diagnosis, and for this, the most widely used markers are cytogenetic parameters [2]. To this end, Dohner’s prognostic algorithm for clinical aggressiveness was proposed by combining conventional chromosome banding with the four main cytogenetic alterations detected by fluorescence in situ hybridization (FISH) since these abnormalities are present in more than 80% of CLL cases [3]. Indeed, patients with deletion (del)17p, del(11q), and a complex karyotype (≥ 3 chromosomal abnormalities) most frequently have an unfavorable outcome (high risk) followed by patients with trisomy 12 or a normal karyotype (intermediate risk). In contrast, over 30% of the cases are represented by patients with an isolated del(13q) and exhibit a favorable evolution (low risk). The main limitation of this cytogenetic approach is related to the fact that cytogenetic abnormalities can evolve during the course of the disease with new subclones which can substitute for those previously established, by presenting either new deletions (e.g., del(17p) or del(11q)) or driver mutations (e.g., TP53, SF3B1), and such an effect can be enhanced by chemotherapy [4].
In parallel, studies have investigated the outcome of CLL patients depending on epigenetic modifications as determined by epigenome-wide association studies (EWAS), genome-wide chromatin accessibility maps, and microRNA analysis showing that loss of epigenetic stability is associated with disease progression [5, 6]. The link between DNA methylation loss in promoters and a higher probability of harboring a subclonal driver mutation was highlighted, the consequence being an adverse clinical outcome [5]. The list of tumor regulator genes and markers with high prognostic significance, like TCL1, CD20, and LPL, which display altered DNA methylation regulation, is continuously growing [7–9]. DNA demethylation also occurs at repetitive elements, suggesting that global DNA demethylation reported in CLL largely accounts for repetitive elements that represent half of the chromatin [10, 11]. The mechanisms underlying aberrant DNA methylation in CLL are complex and do not rely only on the control of DNA methylation by DNA methyl transferases (DNMT). DNA demethylation, catalyzed by the ten-eleven translocation (TET) enzymes, has also been shown to be altered in CLL [12, 13].
The goal of the present work was to ascertain the associations between the main cytogenetic risk factors of the disease and the two cytosine derivatives: 5-methylcytosine (5-mCyt) and 5-hydroxymethylcytosine (5-hmCyt). DNMT and TET transcript levels were also studied by real-time quantitative (RTq)-PCR. The main observation highlighted by this study is that combining a conventional cytogenetic approach with global 5-mCyt assessment drastically improves CLL outcome prediction in patients with del(13q).
Results
Population characteristics according to cytogenetic risk groups
Conventional chromosome and FISH analyses were performed, prior to treatment initiation, on the 127 CLL patients retrospectively included in the study at Brest University Hospital. Among them, 110 (86.6%) exhibited del(13q) which was isolated in 64 (50%) patients, 8 (6.3%) presented a normal karyotype, 11 (8.7%) had trisomy 12, 13 (10.2%) had a del(11q), 12 (9.4%) had a del(17p), and 15 (11.8%) had a complex karyotype with 3 or more cytogenetic abnormalities. Patients were segregated into cytogenetic risk groups according to the following classification: 64 (50%) patients with isolated del(13q) were in the low-risk group, 19 (15%) with normal karyotype or trisomy 12 were in the intermediate-risk group, and 30 (23.6%) with del(11q), del(17p), or complex karyotype were in the high-risk group. To be consistent with the cytogenetic classification criteria, 14 patients (11%) presenting 1–2 cytogenetic abnormalities, other than those aforementioned, were excluded from further analysis [2, 14]. In order to validate the cohort, we next evaluated the CLL outcome from the time of diagnosis, and as anticipated, the low-risk group had the longest median time to progression (PFS) and treatment initiation (TFS) (both > 120 months), followed by the intermediate-risk group (72 and 77 months, respectively) and the high-risk group (40 and 72 months, p = 0.003 and p = 0.001, respectively) (Fig. 1a, b). We further evaluated the association between the cytogenetic classification and prognostic factors such as age at diagnosis, sex, Binet stage, lymphocytosis, lymphocyte doubling time (LDT), immunoglobulin heavy chain variable region (IGHV) mutational status, and CD38 positivity (> 30%) (Table 1). Among these factors, lymphocytosis significantly progressed from the low-risk group to the high-risk group, and the highest levels were observed in the intermediate-risk group (p = 0.006). Additionally, median LDT was progressively shorter from the low-risk group to the high-risk group (p < 0.001), confirming that LDT, rather than lymphocytosis, better predicted disease progression.
Fig. 1.
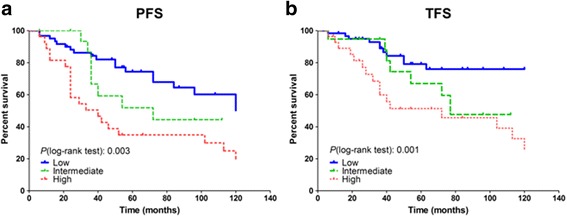
a, b Progression (PFS)- and treatment-free survival (TFS) according to cytogenetic risk groups. Kaplan–Meier survival curves from the date of diagnosis depicted for 127 chronic lymphocytic leukemia patients. Statistical differences between the curves were calculated using the log-rank test
Table 1.
Patients’ characteristics according to cytogenetic risk groups
| Cytogenetic risk groups | Low (n = 64) | Intermediate (n = 19) | High (n = 30) | Statistics (p) |
|---|---|---|---|---|
| Age at diagnosis, mean (±SD) | 64.4 (± 9.4) | 67.8 (± 8.2) | 64.9 (± 10.7) | NS |
| Age at study entry, mean (±SD) | 68.7 (± 9.3) | 72.2 (± 8.4) | 69.7 (± 9.8) | NS |
| Binet stage, No. of patients (%) | 0.05 | |||
| A | 50/64 (78.1%) | 11/19 (57.9%) | 16/30 (51.7%) | |
| B | 8/64 (12.5%) | 5/19 (26.3%) | 11/30 (37.9%) | |
| C | 6/64 (9.4%) | 3/19 (15.8%) | 3/30 (10.3%) | |
| Lymphocytosis (Giga/L), mean (±SD) | 30.7 (± 29.3) | 74.7 (± 71.1) | 49.6 (± 40.60) | 0.006 |
| IGHV mutational status, No. of patients (%) | NS | |||
| Unmutated (≥ 98% homology) | 1/22 (4.5%) | 2/12 (16.7%) | 3/10 (30%) | |
| Mutated (< 98% homology) | 21/22 (95.5%) | 10/12 (83.3%) | 7/10 (70%) | |
| CD38 > 30%, No. of patients (%) | 12/58 (20.7%) | 9/19 (47.4%) | 6/25 (24%) | NS |
| LDT from diagnosis, median (months)a | 48 | 24 | 17 | 0.0004 |
| PFS, median (months)a | > 120 | 72 | 40 | 0.003 |
| TFS, median (months)a | > 120 | 77 | 72 | 0.001 |
Abbreviations: NS not significant, No. number, SD standard deviation, IGHV immunoglobulin heavy-chain variable region, LDT lymphocyte doubling time, PFS progression-free survival, TFS treatment-free survival
aKaplan–Meier survival analysis
DNA methylation/hydroxymethylation and cytogenetic abnormalities
Next, for a comprehensive evaluation of DNA methylation/hydroxymethylation in CLL B cells and their interplay with the cytogenetic status, global 5-mCyt (Fig. 2a) and 5-hmCyt (Fig. 2b) were quantified by ELISA in purified peripheral blood B cells from 60 CLL patients randomly selected from the initial cohort and 15 healthy controls. The methylation status of the low-risk group (n = 30; 5-mCyt mean index 1.05 ± 0.02) and the high-risk group (n = 14; index 0.96 ± 0.05) were similar to healthy controls (index 1.09 ± 0.04, NS both), while a significant reduction was reported in the intermediate-risk group (n = 16, index 0.88 ± 0.05, p = 0.02). Regarding the hydroxymethylation status, levels of the low-risk group were similar to healthy controls (5-hmCyt mean index 0.22 ± 0.005 versus 0.24 ± 0.01, NS), while significant differences were observed for the intermediate-risk group (index 0.18 ± 0.04; p = 0.002) and the high-risk group (index 0.19 ± 0.03; p = 0.03).
Fig. 2.
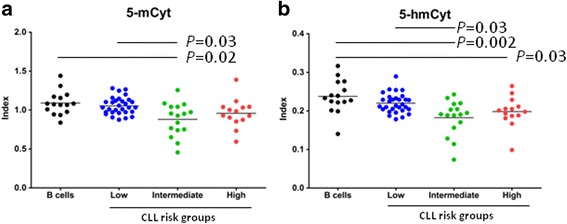
Global DNA methylation and hydroxymethylation according to cytogenetic risk groups a 5-methylcytosine (5-mCyt) and b 5-hydroxymethylcytosine (5-hmCyt) were determined by ELISA in purified B cells from 15 healthy controls and 60 untreated chronic lymphocytic leukemia (CLL) patients classified according to their cytogenetic risk group. ELISA results are expressed as indexes using a DNA reference sample for normalization (see “Materials and methods” section). The means and statistical differences were indicated when Dunn’s correction test was statistically significant (p < 0.05)
Bivariate analysis of cytogenetic and epigenetic characteristics
In order to further test the interplay between cytogenetic risk groups and epigenetic factors, a Cox regression analysis was performed on TFS for 5-mCyt and 5-hmCyt levels to segregate the 60 CLL patients tested by ELISA into subgroups with low and high levels. Next, two groups of patients were considered: first, the low-risk group with isolated del(13q) (n = 30) and, second, the cytogenetic intermediate/high-risk groups (n = 30). Concerning the latter, results were combined for statistical purposes, as the number of patients belonging to these two cytogenetic risk groups and classified as high for 5-mCyt or 5-hmCyt levels, was reduced (n = 5 and 7, respectively).
As depicted in Fig. 3a and presented in Table 2, the Kaplan–Meier survival curves reveal that the 5-mCyt low subgroup had markedly reduced PFS and TFS when considering the CLL patients presenting an isolated del(13q) (PFS 44.5 versus > 120 months, p = 0.01, and TFS 45 versus > 120 months, p = 0.0008). For 5-hmCyt (Fig. 3c and Table 2), differences were also significant when considering patients with del(13q) as a sole cytogenetic abnormality (PFS 56 versus > 120 months, p = 0.02; TFS 63 versus > 120 months, p = 0.04). Regarding the cytogenetic intermediate/high-risk group (Fig. 3b, d and Table 3), the same trend was observed for both 5-mCyt and 5-hmCyt but did not reach significance. We conclude from these experiments that combining cytogenetic and epigenetic parameters improves CLL patient outcome prediction and that 5-mCyt levels have a higher impact predominantly on those patients with isolated del(13q).
Fig. 3.
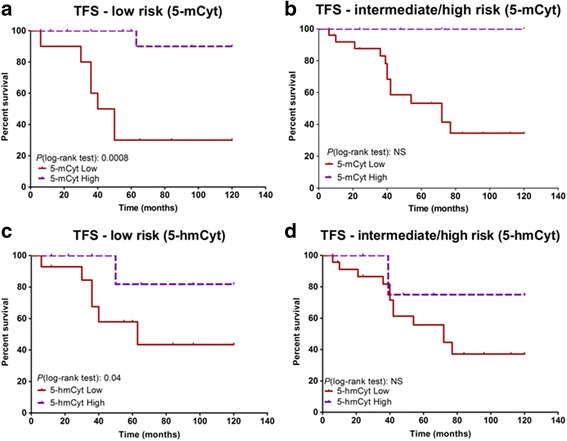
Treatment-free survival (TFS) in a bivariate analysis of cytogenetic and epigenetic characteristics. Influence of DNA methylation (5-methylcytosine, 5-mCyt) on TFS in a low-risk chronic lymphocytic leukemia (CLL) patients with isolated deletion (del)13q (n = 30) and in b intermediate/high-risk CLL patients (n = 30). Influence of active DNA demethylation (5-hydroxymethylcytosine, 5-hmCyt) on TFS in c low-risk CLL patients with isolated deletion (del)13q and in d intermediate/high-risk CLL patients. The Cox regression model of TFS for 5-mCyt and 5-hmCyt was used to identify the optimal cutoff level in order to dichotomize CLL patients into high versus low subgroups. Statistical differences between the Kaplan–Meier curves were calculated using the log-rank test. NS not significant
Table 2.
Characteristics of the cytogenetic low-risk group according to DNA methylation/hydroxymethylation levels
| CLL patients with del(13q) | ||||||
|---|---|---|---|---|---|---|
| 5-mCyt low (n = 12) | 5-mCyt high (n = 18) | Statistics (p) | 5-hmCyt low (n = 15) | 5-hmCyt high (n = 15) | Statistics (p) | |
| Age at diagnosis, mean (±SD) | 65.8 (± 8.5) | 61.4 (± 8.0) | NS | 66.2 (± 8.6) | 60.2 (± 7.2) | NS |
| Age at study entry, mean (±SD) | 69.8 (± 7.6) | 67.2 (± 8.4) | NS | 69.6 (± 8.4) | 66.9 (± 7.7) | NS |
| Binet stage, No. of patients (%) | 0.02 | NS | ||||
| A | 6/12 (50%) | 16/18 (88.9%) | 9/15 (60%) | 12/15 (80%) | ||
| B/C | 6/12 (50%) | 2/18 (11.1%) | 6/15 (40%) | 3/15 (20%) | ||
| Lymphocytosis (Giga/L), mean (±SD) | 53.53 (± 38.30) | 33.93 (± 19.9) | NS | 34.6 (± 28.6) | 48.9 (± 30.0) | NS |
| IGHV status, No. of patients (%) | NS | NS | ||||
| Unmutated (≥ 98% homology) | 1/7 (14.3%) | 0/12 (0%) | 1/9 (11.1%) | 0/10 (0%) | ||
| Mutated (< 98% homology) | 6/7 (85.7%) | 12/12 (100%) | 8/9 (88.9%) | 10/10 (100%) | ||
| CD38 > 30%, No. of patients (%) | 5/12 (41.7%) | 4/16 (25%) | NS | 6/15 (40%) | 3/13 (23%) | NS |
| del(13q), > 80% nuclei (%) | 7/12 (58.3%) | 3/18 (16.7%) | 0.02 | 4/15 (26.7%) | 6/15 (40%) | NS |
| del(13q), biallelic | 4/12 (33.3%) | 12/18 (66.7%) | NS | 6/15 (40%) | 10/15 (66.7%) | NS |
| LDT from diagnosis, median (months)a | 12 | 36 | NS | 24 | 18 | NS |
| PFS, median (months)a | 44.5 | 120 | 0.01 | 56 | > 120 | 0.02 |
| TFS, median (months)a | 45 | > 120 | 0.0008 | 63 | > 120 | 0.04 |
Abbreviations: 5-mCyt 5-methylcytosine, 5-hmCyt 5-hydroxymethylcytosine, NS not significant, No. number, SD standard deviation, IGHV immunoglobulin heavy-chain variable region, LDT lymphocyte doubling time, PFS progression-free survival, TFS treatment-free survival, del deletion
aKaplan–Meier survival analysis
Table 3.
Characteristics of the cytogenetic intermediate/high-risk group according to DNA methylation/hydroxymethylation levels
| CLL patients with cytogenetic intermediate/high risk | ||||||
|---|---|---|---|---|---|---|
| 5-mCyt low (n = 20) | 5-mCyt high (n = 10) | Statistics (p) | 5-hmCyt low (n = 23) | 5-hmCyt high (n = 7) | Statistics (p) | |
| Age at diagnosis, mean (±SD) | 65.8 (± 10.3) | 65.7 (± 8.0) | NS | 69.6 (± 10.5) | 71.1 (± 7.8) | NS |
| Age at study entry, mean (±SD) | 70.3 (± 10.9) | 69.1 (± 7.7) | NS | 69.6 (± 10.9) | 71.1 (± 7.8) | NS |
| Binet stage, No. of patients (%) | NS | NS | ||||
| A | 8/20 (40%) | 6/10 (60%) | 9/23 (39.1%) | 5/7 (71.4%) | ||
| B/C | 12/20 (60%) | 4/10 (40%) | 14/23 (60.9%) | 2/7 (28.6%) | ||
| Lymphocytosis (Giga/L), mean (±SD) | 93.5 (± 58.3) | 27.9 (± 22.8) | 0.0008 | 83.7 (± 60.0) | 32.1 (± 26.5) | 0.02 |
| IGHV status, No. of patients (%) | NS | NS | ||||
| Unmutated (≥ 98% homology) | 4/13 (30.8%) | 1/5 (20%) | 5/15 (33.3%) | 0/5 (0%) | ||
| Mutated (< 98% homology) | 9/13 (69.2%) | 4/5 (80%) | 10/15 (66.7%) | 5/5 (100%) | ||
| CD38 > 30%, No. of patients (%) | 7/18 (38.9%) | 4/10 (40%) | NS | 7/21 (33.3%) | 4/7 (57.1%) | NS |
| LDT from diagnosis, median (months)a | 12 | 24 | 0.04 | 16.5 | 30 | NS |
| PFS, median (months)a | 40 | 46 | NS | 40 | 83 | NS |
| TFS, median (months)a | 72 | > 120 | NS | 72 | > 120 | NS |
Abbreviations: 5-mCyt 5-methylcytosine, 5-hmCyt, 5-hydroxymethylcytosine, NS not significant, No. number, SD standard deviation, IGHV immunoglobulin heavy-chain variable region, LDT lymphocyte doubling time, PFS progression-free survival, TFS treatment-free survival, del deletion
aKaplan–Meier survival analysis
DNA methylation/hydroxymethylation low and high subgroups
We further evaluated, in the cytogenetic low-risk group (Table 2), whether a defective global DNA 5-mCyt status or 5-hmCyt status was associated with other established prognostic factors (age, Binet stage, lymphocytosis, LDT, IGHV mutational status, CD38 positivity) and with the subclonal heterogeneity of del(13q). Regarding prognostic factors, patients from the 5-mCyt low subgroup, but not those from the 5-hmCyt low subgroup, had a significantly higher proportion of Binet stage B and C disease (p = 0.02). Moreover, as previous studies have shown that CLL patients with isolated del(13q) have a more aggressive disease when presenting a high percentage of 13q deleted nuclei (> 80%) or biallelic deletions in the 13q14 locus, we tested whether these factors were associated with the global DNA 5-mCyt and 5-hmCyt status [15–17]. The 5-mCyt low subgroup presented an increased percentage of 13q deleted nuclei (p = 0.02). We failed to observe any difference between the two subgroups (5-mCyt or 5-hmCyt low and high) when biallelic deletions were taken into consideration. Altogether, these results suggest that a defective global DNA methylation status contributes to del(13q) clonal selection and in turn impacts disease outcome in CLL patients with isolated del(13q).
Second, the same analysis was conducted in CLL patients from the intermediate/high-risk groups showing an association between 5-mCyt status, lymphocytosis (p = 0.0008), and the LDT (p = 0.04) (Table 3). No differences were observed regarding the other prognostic factors (age, Binet stage, IGHV mutational status, and CD38 positivity) or when the intermediate/high-risk patients were segregated according to the 5-hmCyt status.
DNA methylation and demethylation regulators
Finally, transcript quantification of three members of the DNMT family (DNMT1, DNMT3A, and DNMT3B) responsible for the methylation of 5-Cyt to 5-mCyt and of the three members of the TET family (TET1, TET2, and TET3) responsible for the oxidation of 5-mCyt to 5-hmCyt was performed by RTq-PCR in 11 CLL patients from the low-risk group, 13 CLL patients from the intermediate/high-risk group, and 9 healthy controls (Fig. 4).
Fig. 4.
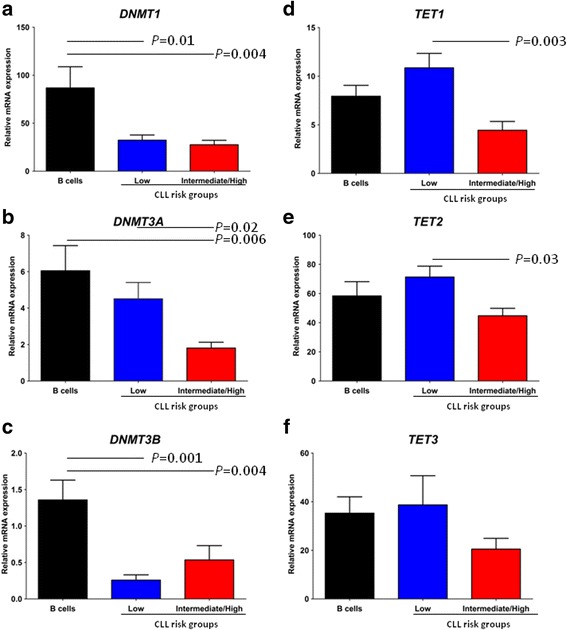
DNA methylation and demethylation regulators according to cytogenetic risk groups a DNMT1, b DNMT3A, c DNMT3B, d TET1, e TET2, and f TET3 were tested by real-time quantitative PCR and transcripts expressed relative to GAPDH. The mean ± standard error of the mean (SEM) values are indicated, and statistical differences were indicated when Dunn’s correction test was statistically significant (p < 0.05)
Compared to the low-risk group, CLL patients from the intermediate/high-risk group were characterized by a significant reduction of DNMT3A (p = 0.02), TET1 (p = 0.003), and TET2 (p = 0.03) transcripts. No significant differences were observed for DNMT1, DNMT3B, and TET3 transcripts when comparing the CLL intermediate/high-risk group with the CLL low-risk group. However, in opposition to the 5-mCyt results that did not reveal any differences when comparing healthy controls with the low-risk group, reductions were observed in the low-risk group for DNMT1 (p = 0.01) and DNMT3B (p = 0.001). Concerning the intermediate/high-risk group comparison to healthy controls, differences were observed for DNMT1 expression (p = 0.004), DNMT3A (p = 0.006), and DNMT3B (p = 0.04).
Discussion
Assessment of the cytogenetic status in CLL at diagnosis is routinely performed by using karyotype and FISH analysis for prognostic purposes. However, such an approach poorly integrates clonal evolution, thus indicating that the well-established Dohner’s classification can be improved. Accordingly, we have hypothesized that coupling cytogenetic and epigenetic factors would help predict disease outcome, and in addition, we have confirmed the importance of epigenetic modifications in CLL outcome.
Interplay exists between genetic and epigenetic mechanisms during CLL evolution, and two models were proposed for this co-evolution, one based on a stepwise acquisition model from early clonal mutations/cytogenetic abnormalities (e.g., del(13q)), while in the second model, a simultaneous acquisition was proposed (e.g., del(17p)) [18, 19]. The simultaneous acquisition model is supported by the observation that del(17p) involving the tumor suppressor TP53 is associated with global DNA demethylation in CLL B cells [10, 20] and by our observation that CLL patients from the cytogenetic intermediate- and/or high-risk groups present a reduction in 5-mCyt and 5-hmCyt levels and in DNMT3A and TET1/2 transcripts. In the second model, as observed in CLL patients with low-risk cytogenetic abnormalities such as del(13q), the global DNA methylation pattern is suspected to be relatively stable over time, and when disease progression occurs, selection of novel global DNA methylation patterns evolves independently of the genetic heterogeneity [18, 21]. Indeed, del(13q) is suspected to be an early event as confirmed by the detection of this cytogenetic abnormality in the CD34+ cell population in CLL patients [22]. Our study further supports the stepwise dynamic model of clonal populations in CLL with the del(13q) as an early event which can be associated with epigenetic dysregulations as this clonal population expands. This is highlighted by the observation that CLL patients with a higher percentage (above 80%) of 13q deleted nuclei also have a defective DNA methylation process and a shorter PFS and TFS. Further on, this is also in agreement with the report that CLL patients with a higher percentage of 13q deleted nuclei have a transcriptome pattern similar to del(11p) and del(17q) patients, abnormal miRNA expression, and dysregulated pathways implicated in apoptosis and proliferation (BCR and NF-kappaB signaling) [23]. Altogether, our data and previously published data support a chronology of molecular events involving first the del(13q) and afterwards, as the clone expends, important epigenetic modifications. However, one question remains to be answered which concerns the possible contribution of suppressor genes from the deleted 13q locus to the regulation of the methylation process. Moreover, in this locus, differentially methylated regions have been identified in CLL B cells compared to controls, and they are located within tumor suppressor genes including the deleted in lymphocytic leukemia (DLEU)1 and the DLEU2 variant ALT1 [20]. Further experiments are required to test whether these suppressor genes are in turn involved in the global DNA methylation process and whether DNA demethylation leads to an adverse clinical outcome per se or through subclonal driver mutations.
DNMT3A is thought to be critical in the control of DNA methylation in CLL, and such an assertion is supported by the observation that DNMT3A knockdown in rodent hematopoietic stem cells results in CLL development [24] and that DNMT3A is present in the top 1% of the under-expressed genes in human CLL B cells [25]. An interaction between the two CLL high-risk factors TCL1 and NOTCH1 with DNMT3A to inhibit its function is also reported in CLL B cells [26, 27]. Accordingly, we have investigated DNMTs in CLL B cells and observed that DNMT1 and DNMT3B are downregulated in the cytogenetic low- and intermediate/high-risk groups while DNMT3A reduction was restricted to CLLs carrying cytogenetic intermediate/high-risk factors. This result differs from previous publications, which have compared CLL B cells with peripheral blood normal B cells to show, in one case, normal DNMT1 expression, an upregulation of DNMT3A, and a reduction of DNMT3B in CLL B cells [28], and in the other case, researchers have reported a reduction in DNMT1 [10]. These discrepancies could be most likely due to the selection of the control cells as important variations in DNMTs are reported during B cell ontogeny and to the reference gene used for correction [29–31].
Despite its stability, 5-mCyt can be reversed firstly during DNA replication in a passive way or, secondly, by TET proteins which can mediate 5-mCyt oxidation to 5-hmCyt, and in a less efficient fashion to 5-formylcytosine (5-fCyt), and 5-carboxylcytosine (5-CaCyt). Next, transformation to an unmodified Cyt requires the coupled action of thymine DNA glycosylase (TDG) and the base excision repair (BER) system. TET1 and double TET1/TET2 knockdown promote B cell malignancies [32, 33]. First reported in myeloid lineage malignant diseases, 5-hmCyt level reduction was related to mutations in TETs or their regulators (e.g., IDH1/2, WT1) [34]. In CLL, DNA demethylation is observed, but mutations in TETs and IDHs are rare events. However, a reduction in TET1/3 and IDH2 was shown to be associated with a poor prognosis [12]. In this report, we could confirm that patients with low mRNA expression of TET1 and TET2 were predominantly classified into the cytogenetic intermediate/high-risk group, which is in agreement with our observation that both 5-mCyt and 5-hmCyt levels were reduced in the intermediate- and/or high-risk groups. Van Damme et al. observed a decreased expression of TET1 in CLL B cells but did not observe an association with DNA hydroxymethylation, and this may be explained, in part, by the small number of patients studied for hydroxymethylation levels [12]. In another study comparing CLL B cells to peripheral blood B cells, TET2 was reported to be overexpressed and such overexpression was not associated with other clinical and biological markers [35]. The list of the mechanisms controlling TET expression in leukemic cells is growing and involves microenvironmental factors [12], hypoxia, reactive oxygen species (ROS), and miRNAs such as miR-15a and miR-29 [36]. Additional experiments are required to better understand the mechanisms controlling active DNA demethylation in CLL and their consequences.
Relevant limitations of our study include the following: (i) global 5-mCyt and 5-hmCyt analysis is likely to reflect variations of repetitive elements (half of the genome) than gene regulatory sequence, which may render comparison with EWAS studies difficult; (ii) the absence of a well-established control B cell for CLL B cells, making difficult the comparison of the transcriptional analysis between normal and tumor cells; and (iii) a transcriptional analysis for DNMTs and TETs, which does not test enzyme activity.
In conclusion, our study established that loss in the 5-Cyt derivatives, 5-mCyt and, to a lesser extent, 5-hmCyt, during disease progression, better predict the CLL patients’ outcome when associated with the classical cytogenetic analysis. Future work is required to develop routine assays for monitoring DNA methylation during disease course and to test their utility as therapeutic response predictors.
Materials and methods
Cells and sample preparation
Clinical and biological data were retrospectively obtained for 127 untreated patients diagnosed with CLL according to the World Health Organization (WHO) classification [37], and samples were collected for 60 CLL patients and 15 healthy volunteers at the Brest University Hospital. Consent was obtained from all individuals and the protocol approved by the Ethical Board at the Brest University Hospital (OFICE, November 26, 2015; ClinicalTrials.gov: NCT03294980; CRB Brest, collection 2008-214), in accordance with the Declaration of Helsinki. Lymphocytes were isolated from peripheral blood mononuclear cells (PBMC) by Ficoll-Hypaque density gradient centrifugation (Eurobio, Courtaboeuf, France), and B cells were further enriched using the Pan B cell Isolation Kit (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). Cell purity was assessed by fluorescence-activated cell sorting (FACS) analysis and was over 95% for B cells (CD19+). Fluorescence-activated cell sorting (FACS) antibodies (Abs) used were purchased from Beckman Coulter.
DNA sample preparation and global DNA 5-mCyt and 5-hmCyt level assessment by ELISA.
DNA was extracted from purified B cells using Biosprint 15 DNA Blood Kit (Qiagen, Hilden, Germany). Next, DNA was quantified and its purity assessed using the NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA). An ELISA previously developed in the laboratory was used and adapted to measure global 5-mCyt and 5-hmCyt [38]. Briefly, high affinity microplates (Thermo 269620, Thermo Fisher Scientific) were pre-coated 90 min at room temperature (RT) with 100 μl poly-l-lysine 0.01% (Sigma-Aldrich, St. Louis, MI) to attach DNA. Next, DNA samples adjusted at 2 ng/μl in carbonate/bicarbonate buffer 0.1 M pH 9.6 were denatured at 95 °C for 6 min, kept on ice 5–10 min, and then 100 μl dispensed in each well, in duplicates. The plates were next incubated overnight at 4 °C, three washes with phosphate-buffered saline (PBS)-Tween 0.01% were performed, and 200 μl of PBS with bovine serum albumin (BSA) 1% was added in each well as blocking solution. After 1-h incubation at RT and extensive washing, 100 μl of mouse IgG anti-5-mCyt (diluted 1:5000 in PBS-BSA 1%) or mouse IgG anti-5-hmCyt (1:1000) was added and plates were incubated 2 h at RT. Anti-cytosine derivative Abs were purchased from Abcam (Cambridge, UK). After six washes, 100 μl of alkaline phosphate-labeled goat anti-mouse (Jackson Laboratory, Bar Harbor, ME), diluted at 1:5000 in PBS-BSA 1%, was added and the plate was incubated for 1 h at RT. After three washes, color was developed with 100 μl p-nitrophenyl-phosphate (Sigma-Aldrich) diluted in carbonate/bicarbonate buffer 0.1 M pH 9.6. Plates were kept at 37 °C for 4 h, and optical density (OD) determined at 405 nm using a Titertek Multiscan microplate reader (Flow laboratories, Rockville, MD). Each sample was tested in duplicate, and non-specific background OD (duplicate wells without DNA) was subtracted from the corresponding test sample. For normalization, a reference sample (salmon sperm DNA—Sigma-Aldrich) was included on each plate and indexes calculated using the ratio between the patient OD and the reference sample OD at 200 ng/well.
Classic cytogenetic and FISH analysis
As previously described [39], cytogenetic analysis was performed on bone marrow or peripheral blood cells cultured for 72 h with B cell mitogens (DSP30+ interleukin-2). Chromosomes were R-banded and the karyotype described according to the International System for Human Cytogenomic Nomenclature (ISCN) [40]. Metaphase and interphase FISH using the Vysis CLL FISH Probe Kit (Abbott, Rungis, France) was performed for the patients analyzed before the year 2012 and then using P53/ATM probe combination (Cytocell, Cambridge, UK) and XL DLEU/LAMP/12cen probe (Metasystems, Altlussheim, Germany). In all the CLL cases, at least 200 interphase nuclei were counted. Positive cases were defined as having ≥ 5% of nuclei with the investigated anomaly. The patients with del(13q) were also separated into monoallelic and biallelic subgroups. Any patient with two clones, harboring both monoallelic and biallelic deletions, were classified into the biallelic del(13q) subgroup.
Mutational status of IGHV
According to the BIOMED-2 consortium guidelines [41], the IGHV gene mutation status was determined by sequencing after conducting a PCR multiplex amplification. Briefly, for multiplex PCR, 100 ng of genomic DNA, 0.25 μl of Ampli Taq Gold DNA Polymerase (Applied Biosystems, Foster City, CA), 10 pmol of each primer, 0.2 mM dNTP Mix, 1.5 mM MgCl2, and 1× PCR Buffer II were adjusted to 50 μl with DNase/RNase-free ultrapure distilled water. Next, PCR products were visualized on 2% agarose gel and purified with ExoSAP-IT PCR product cleanup kit (Affymetrix, High Wycombe, UK). Finally, amplicons were sequenced with a Big Dye Terminator v3.1 cycle sequencing kit (Applied Biosystems). Results were analyzed with the database IMGT/HighV-Quest (The international ImMunoGeneTics information system, Montpellier) and a homology sequence > 98% defined an UM status [42].
RNA sample preparation, reverse transcription, and RTq-PCR
RNA was extracted from purified B cells using the RNeasy Plus Micro Kit (Qiagen). Quantification and purity were assessed using the NanoDrop 2000 Spectrophotometer. Next, RNA (300 ng) was reverse transcribed into cDNA using the Super Script III enzyme and random primers (Invitrogen Life Sciences, Carlsbad, CA). RTq-PCR was carried out in 20 μl mixtures containing 6 μl of template cDNA diluted 1/12 with DNase/RNase-free ultrapure distilled water, 1× Power SYBR® Green PCR Master Mix (Applied Biosystems), and 250 nM of each primer (Table 4) using Applied Biosystems® QuantStudio™ 7 Flex Real-Time PCR System. The PCR conditions were the same for all genes. All assays included a negative control, which consisted of the reaction mixture with no template. Comparison of cycle thresholds was completed with the 2ΔΔCT method using GAPDH as an endogenous control.
Table 4.
Primers used for real-time quantitative PCR
| Symbol | Gene description | Forward primer | Reverse primer |
|---|---|---|---|
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | TGCCCTCAACGACCACTTT | GGTCCAGGGGTCTTACTCCTT |
| DNMT1 | DNA methyltransferase 1 | CCTGTACCGAGTTGGTGATGGT | CCTTCCGTGGGCGTTTC |
| DNMT3A | DNA methyltransferase 3 alpha | CTCCTGTGGGAGCCTCAATGTTACC | CAGTTCTTGCAGTTTTGGCACATTCC |
| DNMT3B | DNA methyltransferase 3 beta | ACCACCTGCTGAATTACTCACGC | GATGGCATCAATCATCACTGGATT |
| TET1 | tet methylcytosine dioxygenase 1 | AATGGAAGCACTGTGGTTTG | ACATGGAGCTGCTCATCTTG |
| TET2 | tet methylcytosine dioxygenase 2 | AATGGCAGCACATTGGTATG | AGCTTCCACACTCCCAAACT |
| TET3 | tet methylcytosine dioxygenase 3 | TTGCGTCGAACAAATAGTGG | CCCGTGTAGATGACCTTCTC |
Clinical outcome endpoints
PFS was defined as the time from disease discovery to disease progression. Disease progression was considered either as the shift from Binet stage A to Binet stage B/C or as a short LDT of less than 6 months. TFS was defined as the interval between the date of disease discovery and the date of treatment initiation.
Statistical analysis
The profile likelihood method using a Cox regression model of TFS was used in univariate analysis to determine the optimal threshold and stratify patients into two groups. This analysis was computed using the Survival and SurvMisc R packages [43]. LDT, TFS, and PFS analyses were next performed using Kaplan–Meier curves, and prognosis differences between groups were assessed with a log-rank test. Continuous data are described as mean ± standard error of the mean (SEM). Differences among groups were analyzed by the Kruskall–Wallis test and the Dunn test was used for post hoc comparisons, or the Fisher exact test for categorical data. Following normality and equality of variance tests, nominal values were compared to controls using the student t test or alternatively by using a nonparametric test (Mann–Whitney rank sum test). p values under 0.05 were considered significant. Statistical analyses were performed using GraphPad Prism 7.0 (La Jolla, CA).
Acknowledgements
Authors express their thanks to Stephanie Deshayes and Charlotte Laot for the technical assistance, to Dr. Wesley H. Brooks (University of South Florida, USA) for the editorial assistance, and to Simone Forest and Genevieve Michel for the secretarial assistance.
Funding
This study was supported by research funding from the “Association Laurette Fugain” (ALF 2015/03), the “Region Bretagne,” and the “Canceropole Grand Ouest.”
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on a reasonable request.
Authors’ contributions
CBa, MZ, VC, and YR designed the study. CBa performed the research. NDG performed the cytogenetic studies. CBa and AB performed the clinical data gathering. AT, HS, and CBe took care of the patients and validated clinical data accuracy. CBa, CL, and YR analyzed the data. CBa and YR prepared the initial draft. The final manuscript was read and approved by all authors.
Ethics approval and consent to participate
Consent was obtained from all individuals and the protocol approved by the Ethical Board at the Brest University Hospital (OFICE, November 26, 2015; ClinicalTrials.gov: NCT03294980; CRB Brest, collection 2008-214), in accordance with the Declaration of Helsinki.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Cristina Bagacean, Email: cristina.bagacean@univ-brest.fr.
Christelle Le Dantec, Email: christelle.ledantec@univ-brest.fr.
Christian Berthou, Email: cberthou@univ-brest.fr.
Adrian Tempescul, Email: Adrian.tempescul@chu-brest.fr.
Hussam Saad, Email: hussam.saad@chu-brest.fr.
Anne Bordron, Email: anne.bordron@univ-brest.fr.
Mihnea Zdrenghea, Email: mzdrenghea@umfcluj.ro.
Victor Cristea, Email: victor_cristea@yahoo.com.
Nathalie Douet-Guilbert, Email: Nathalie.douet-guilbert@chu-brest.fr.
Yves Renaudineau, Phone: 33-298-22-33-84, Email: yves.renaudineau@univ-brest.fr.
References
- 1.Kipps TJ, Stevenson FK, Wu CJ, Croce CM, Packham G, Wierda WG, et al. Chronic lymphocytic leukaemia. Nat Rev Dis Primers. 2017;3:16096. doi: 10.1038/nrdp.2016.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Puiggros A, Blanco G, Espinet B. Genetic abnormalities in chronic lymphocytic leukemia: where we are and where we go. Biomed Res Int. 2014;2014:435983. doi: 10.1155/2014/435983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Dyke DL, Werner L, Rassenti LZ, Neuberg D, Ghia E, Heerema NA, et al. The Dohner fluorescence in situ hybridization prognostic classification of chronic lymphocytic leukaemia (CLL): the CLL research consortium experience. Br J Haematol. 2016;173(1):105–113. doi: 10.1111/bjh.13933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526(7574):525–530. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Landau DA, Clement K, Ziller MJ, Boyle P, Fan J, Gu H, et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell. 2014;26(6):813–825. doi: 10.1016/j.ccell.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rendeiro AF, Schmidl C, Strefford JC, Walewska R, Davis Z, Farlik M, et al. Chromatin accessibility maps of chronic lymphocytic leukaemia identify subtype-specific epigenome signatures and transcription regulatory networks. Nat Commun. 2016;7:11938. doi: 10.1038/ncomms11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuille MR, Condie A, Stone EM, Wilsher J, Bradshaw PS, Brooks L, et al. TCL1 is activated by chromosomal rearrangement or by hypomethylation. Genes Chromosomes Cancer. 2001;30(4):336–341. doi: 10.1002/gcc.1099. [DOI] [PubMed] [Google Scholar]
- 8.Abreu C, Moreno P, Palacios F, Borge M, Morande P, Landoni AI, et al. Methylation status regulates lipoprotein lipase expression in chronic lymphocytic leukemia. Leuk Lymphoma. 2013;54(8):1844–1848. doi: 10.3109/10428194.2013.796057. [DOI] [PubMed] [Google Scholar]
- 9.Mankai A, Bordron A, Renaudineau Y, Martins-Carvalho C, Takahashi S, Ghedira I, et al. Purine-rich box-1-mediated reduced expression of CD20 alters rituximab-induced lysis of chronic lymphocytic leukemia B cells. Cancer Res. 2008;68(18):7512–7519. doi: 10.1158/0008-5472.CAN-07-6446. [DOI] [PubMed] [Google Scholar]
- 10.Fabris S, Bollati V, Agnelli L, Morabito F, Motta V, Cutrona G, et al. Biological and clinical relevance of quantitative global methylation of repetitive DNA sequences in chronic lymphocytic leukemia. Epigenetics. 2011;6(2):188–194. doi: 10.4161/epi.6.2.13528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garaud S, Le Dantec C, Berthou C, Lydyard PM, Youinou P, Renaudineau Y. Selection of the alternative exon 1 from the cd5 gene down-regulates membrane level of the protein in B lymphocytes. J Immunol. 2008;181(3):2010–2018. doi: 10.4049/jimmunol.181.3.2010. [DOI] [PubMed] [Google Scholar]
- 12.Van Damme M, Crompot E, Meuleman N, Maerevoet M, Mineur P, Bron D, et al. Characterization of TET and IDH gene expression in chronic lymphocytic leukemia: comparison with normal B cells and prognostic significance. Clin Epigenetics. 2016;8:132. doi: 10.1186/s13148-016-0298-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bagacean C, Tempescul A, Le Dantec C, Bordron A, Mohr A, Saad H, et al. Alterations in DNA methylation/demethylation intermediates predict clinical outcome in chronic lymphocytic leukemia. Oncotarget. 2017;8:65699–65716. doi: 10.18632/oncotarget.20081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–1916. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 15.Parker H, Rose-Zerilli MJ, Parker A, Chaplin T, Wade R, Gardiner A, et al. 13q deletion anatomy and disease progression in patients with chronic lymphocytic leukemia. Leukemia. 2011;25(3):489–497. doi: 10.1038/leu.2010.288. [DOI] [PubMed] [Google Scholar]
- 16.Dewald GW, Brockman SR, Paternoster SF, Bone ND, O'Fallon JR, Allmer C, et al. Chromosome anomalies detected by interphase fluorescence in situ hybridization: correlation with significant biological features of B-cell chronic lymphocytic leukaemia. Br J Haematol. 2003;121(2):287–295. doi: 10.1046/j.1365-2141.2003.04265.x. [DOI] [PubMed] [Google Scholar]
- 17.Hernandez JA, Rodriguez AE, Gonzalez M, Benito R, Fontanillo C, Sandoval V, et al. A high number of losses in 13q14 chromosome band is associated with a worse outcome and biological differences in patients with B-cell chronic lymphoid leukemia. Haematologica. 2009;94(3):364–371. doi: 10.3324/haematol.13862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oakes CC, Claus R, Gu L, Assenov Y, Hullein J, Zucknick M, et al. Evolution of DNA methylation is linked to genetic aberrations in chronic lymphocytic leukemia. Cancer Discov. 2014;4(3):348–361. doi: 10.1158/2159-8290.CD-13-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714–726. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garding A, Bhattacharya N, Claus R, Ruppel M, Tschuch C, Filarsky K, et al. Epigenetic upregulation of lncRNAs at 13q14.3 in leukemia is linked to the In Cis downregulation of a gene cluster that targets NF-kB. PLoS Genet. 2013;9(4):e1003373. doi: 10.1371/journal.pgen.1003373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cahill N, Bergh AC, Kanduri M, Goransson-Kultima H, Mansouri L, Isaksson A, et al. 450K-array analysis of chronic lymphocytic leukemia cells reveals global DNA methylation to be relatively stable over time and similar in resting and proliferative compartments. Leukemia. 2013;27(1):150–158. doi: 10.1038/leu.2012.245. [DOI] [PubMed] [Google Scholar]
- 22.Quijada-Alamo M, Hernandez-Sanchez M, Robledo C, Hernandez-Sanchez JM, Benito R, Montano A, et al. Next-generation sequencing and FISH studies reveal the appearance of gene mutations and chromosomal abnormalities in hematopoietic progenitors in chronic lymphocytic leukemia. J Hematol Oncol. 2017;10(1):83. doi: 10.1186/s13045-017-0450-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodriguez AE, Hernandez JA, Benito R, Gutierrez NC, Garcia JL, Hernandez-Sanchez M, et al. Molecular characterization of chronic lymphocytic leukemia patients with a high number of losses in 13q14. PLoS One. 2012;7(11):e48485. doi: 10.1371/journal.pone.0048485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haney SL, Upchurch GM, Opavska J, Klinkebiel D, Appiah AK, Smith LM, et al. Loss of Dnmt3a induces CLL and PTCL with distinct methylomes and transcriptomes in mice. Sci Rep. 2016;6:34222. doi: 10.1038/srep34222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Upchurch GM, Haney SL, Opavsky R. Aberrant promoter hypomethylation in CLL: does it matter for disease development? Front Oncol. 2016;6:182. doi: 10.3389/fonc.2016.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palamarchuk A, Yan PS, Zanesi N, Wang L, Rodrigues B, Murphy M, et al. Tcl1 protein functions as an inhibitor of de novo DNA methylation in B-cell chronic lymphocytic leukemia (CLL) Proc Natl Acad Sci U S A. 2012;109(7):2555–2560. doi: 10.1073/pnas.1200003109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arruga F, Gizdic B, Bologna C, Cignetto S, Buonincontri R, Serra S, et al. Mutations in NOTCH1 PEST domain orchestrate CCL19-driven homing of chronic lymphocytic leukemia cells by modulating the tumor suppressor gene DUSP22. Leukemia. 2017;31(9):1882–893. [DOI] [PubMed]
- 28.Kn H, Bassal S, Tikellis C, El-Osta A. Expression analysis of the epigenetic methyltransferases and methyl-CpG binding protein families in the normal B-cell and B-cell chronic lymphocytic leukemia (CLL) Cancer Biol Ther. 2004;3(10):989–994. doi: 10.4161/cbt.3.10.1137. [DOI] [PubMed] [Google Scholar]
- 29.Lai AY, Mav D, Shah R, Grimm SA, Phadke D, Hatzi K, et al. DNA methylation profiling in human B cells reveals immune regulatory elements and epigenetic plasticity at Alu elements during B-cell activation. Genome Res. 2013;23(12):2030–2041. doi: 10.1101/gr.155473.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulis M, Merkel A, Heath S, Queiros AC, Schuyler RP, Castellano G, et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat Genet. 2015;47(7):746–756. doi: 10.1038/ng.3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caron G, Hussein M, Kulis M, Delaloy C, Chatonnet F, Pignarre A, et al. Cell-cycle-dependent reconfiguration of the DNA methylome during terminal differentiation of human B cells into plasma cells. Cell Rep. 2015;13(5):1059–1071. doi: 10.1016/j.celrep.2015.09.051. [DOI] [PubMed] [Google Scholar]
- 32.Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap YS, Bakogianni S, Yu Y, et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat Immunol. 2015;16(6):653–662. doi: 10.1038/ni.3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao Z, Chen L, Dawlaty MM, Pan F, Weeks O, Zhou Y, et al. Combined loss of Tet1 and Tet2 promotes B cell, but not myeloid malignancies, in mice. Cell Rep. 2015;13(8):1692–1704. doi: 10.1016/j.celrep.2015.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bullinger L, Dohner K, Dohner H. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol. 2017;35(9):934–946. doi: 10.1200/JCO.2016.71.2208. [DOI] [PubMed] [Google Scholar]
- 35.Hernandez-Sanchez M, Rodriguez AE, Kohlmann A, Benito R, Garcia JL, Risueno A, et al. TET2 overexpression in chronic lymphocytic leukemia is unrelated to the presence of TET2 variations. Biomed Res Int. 2014;2014:814294. doi: 10.1155/2014/814294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 2017;18(9):517–34. [DOI] [PubMed]
- 37.Swertlow SH, Harris LN, Jaffe ES, et al. World Health Organization classification of Tumours. Lyon, France: IARC Press. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue; 2008.
- 38.Thabet Y, Le Dantec C, Ghedira I, Devauchelle V, Cornec D, Pers JO, et al. Epigenetic dysregulation in salivary glands from patients with primary Sjogren’s syndrome may be ascribed to infiltrating B cells. J Autoimmun. 2013;41:175–181. doi: 10.1016/j.jaut.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 39.De Braekeleer M, Gueganic N, Tous C, Le Bris MJ, Basinko A, Morel F, et al. Translocations involving 13q14 without associated deletion in chronic lymphoid leukaemia target DLEU2. Br J Haematol. 2016;172(3):467–469. doi: 10.1111/bjh.13495. [DOI] [PubMed] [Google Scholar]
- 40.Simons A, Shaffer LG, Hastings RJ. Cytogenetic nomenclature: changes in the ISCN 2013 compared to the 2009 edition. Cytogenet Genome Res. 2013;141(1):1–6. doi: 10.1159/000353118. [DOI] [PubMed] [Google Scholar]
- 41.van Dongen JJ, Langerak AW, Bruggemann M, Evans PA, Hummel M, Lavender FL, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 concerted action BMH4-CT98-3936. Leukemia. 2003;17(12):2257–2317. doi: 10.1038/sj.leu.2403202. [DOI] [PubMed] [Google Scholar]
- 42.Alamyar E, Duroux P, Lefranc MP, Giudicelli V. IMGT((R)) tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol Biol. 2012;882:569–604. doi: 10.1007/978-1-61779-842-9_32. [DOI] [PubMed] [Google Scholar]
- 43.Cui B, Chen L, Zhang S, Mraz M, Fecteau JF, Yu J, et al. MicroRNA-155 influences B-cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood. 2014;124(4):546–554. doi: 10.1182/blood-2014-03-559690. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on a reasonable request.