

Abstract
In the last years, the explosion of high throughput sequencing technologies has enabled epigenome-wide analyses, allowing a more comprehensive overview of the oropharyngeal squamous cell carcinoma (OPSCC) epigenetic landscape. In this setting, the cellular pathways contributing to the neoplastic phenotype, including cell cycle regulation, cell signaling, DNA repair, and apoptosis have been demonstrated to be potential targets of epigenetic alterations in OPSCC. Of note, it has becoming increasingly clear that HPV infection and OPSCC lifestyle risk factors differently drive the epigenetic machinery in cancer cells. Epigenetic changes, including DNA methylation, histone modifications, and non-coding RNA expression, can be used as powerful and reliable tools for early diagnosis of OPSCC patients and improve prognostication. Since epigenetic changes are dynamic and reversible, epigenetic enzymes may also represent suitable targets for the development of more effective OPSCC therapeutic strategies. Thus, this review will focus on the main known epigenetic modifications that can occur in OPSCC and their exploitation as potential biomarkers and therapeutic targets. Furthermore, we will address epigenetic alterations to OPSCC risk factors, with a particular focus on HPV infection, tobacco exposure, and heavy alcohol consumption.
Keywords: Oropharyngeal cancer, Epigenetics, Human papillomavirus, Environmental risk factors
Background
Head and neck squamous cell carcinoma (HNSCC) is a frequently lethal cancer that mainly develops in the mucosal epithelial lining of oral cavity, hypopharynx, oropharynx, or larynx. In 2020, HNSCC is expected to affect approximately 833,000 new patients worldwide and 151,000 in Europe, thus representing about 5 and 4% of all new cancers, respectively [1]. Generally, HNSCCs are more common in men than in women and in people aged 60 years than in younger persons [1]. Tobacco use and excessive alcohol intake represent the main risk factors for HNSCC development, and they can act synergistically to increase the risk of this malignancy [2, 3]. To date, oropharyngeal SCC (OPSCC)—which comprises SCC of the base of tongue, tonsillar region, soft palate, and the posterior wall of the pharynx between nasopharynx and the hypopharynx—represents a significantly higher proportion of HNSCC [4]. Among alcohol abstainers, tobacco smokers reported a twofold increased risk of OPSCC compared to never smokers, with a dose-response relationship for both intensity and duration [5]. Similarly, regular alcohol drinking was associated to increased OPSCC risk among never smokers, with risks peaking in people drinking ≥ 5 drink/day [5]. In the last decades, a decrease in the incidence of HNSCC from non-oropharyngeal sites has been observed as the results of preventive strategies to reduce tobacco smoking [6]. Conversely, in economically developed countries, incidence of OPSCC did not show such a decline, despite the reduction of tobacco smoking [7]. This suggests that the control of tobacco smoking epidemic has brought out the burden of OPSCC cases associated with high-risk human papillomaviruses (HPV) infection [8]. HPV-driven OPSCC are rapidly increasing in several Western countries [9], mainly topographically restricted to the oropharynx [10], and exhibit a survival benefit compared to HPV-unrelated tumors [10].
Besides genetic alterations, the accumulation of aberrant epigenetic events deeply influence OPSCC biology and may contribute, at least in part, to the differences between HPV-driven and non-HPV-driven OPSCC [11]. To date, the most extensively characterized mediators of epigenetic modifications are DNA methylation and the post-translational modifications of histone proteins [12]. Despite not yet having been extensively characterized, also non-coding RNAs (ncRNAs) are emerging as important factors in the epigenetic determination of gene expression [12–16]. Rather than acting separately, these epigenetic regulators are dynamically connected to each other in the regulation of gene expression. Disruption of this complex epigenetic control mechanism can affect the structure and the integrity of the genome and alter the expression of genes critically involved in tumorigenesis. Of note, it has becoming increasingly clear that environmental and lifestyle risk factors can promote a wide range of epigenetic alterations that are causally involved in cancer development and progression [17]. Based on these considerations, the primary objective of this review is to resume the common epigenetic events in OPSCC and to discuss their potential translational applications for the management of this disease. Any discussion in this review will also relate epigenetic alterations to OPSCC risk factors, with a particular focus on HPV infection, tobacco smoking, and excessive alcohol intake (Fig. 1).
Fig. 1.
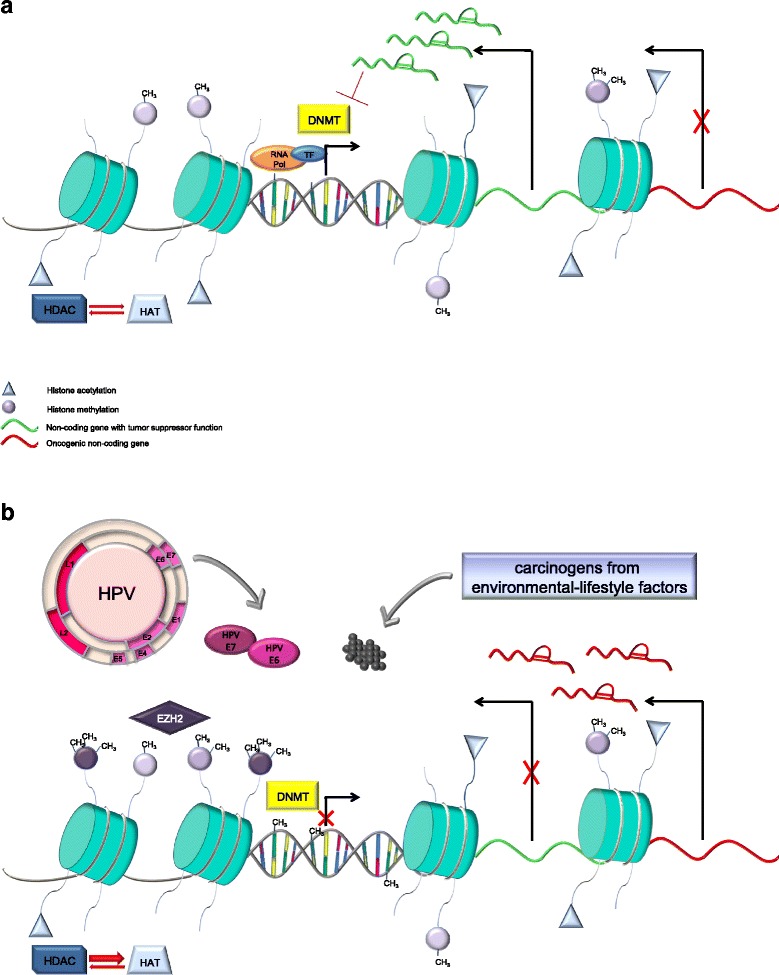
Epigenetic regulation of gene expression involves the crosstalk of DNA methylation, histone modifications, and non-coding RNA (ncRNAs). In normal cells (a), CpG within promoter regions of tumor suppressor genes (TSG) are not methylated and are occupied by complexes including RNA polymerase (RNA pol) and transcription factors (TF), thus allowing gene transcription. Histones undergo several post-translational modifications on their N-terminal tails, including acetylation and methylation. Histone acetylation is the result of the dynamic interplay between histone acetyltransferases (HATs) and histone deacetylases (HDACs), and acetylated histones have been associated with actively expressed genes. Unlike histone acetylation, methylation of histone proteins can result in both repressive or promoting effects on transcription, depending on which residue is modified. NcRNAs, which are involved in almost all major cellular functions, may function either as oncogenes or as TSG. NcRNAs whose expression is increased in tumors may be considered as oncogenes, whereas ncRNAs whose expression is decreased in tumor cells are considered TSG. NcRNAs can also regulate the expression and/or the activity of the epigenetic enzymes, such as DNA methyltransferases (DNMTs). In oropharyngeal squamous cell carcinoma (b), the epigenetic state of cells changes in response to HPV infection and environmental-lifestyle factors (i.e., tobacco smoke and/or excessive alcohol intake). The result is the accumulation of several aberrant epigenetic modifications that lead to inappropriate activation or inhibition of key signaling pathways. For example, E6 and E7 HPV oncoproteins and carcinogens from cigarettes and alcohol have been demonstrated to affect histone acetylation and methylation patterns either by directly interacting with epigenetic enzymes [i.e., DNMT, enhancer of zeste homolog 2 (EZH2)] or by modulating the ncRNA landscape
HPV-driven OPSCC
HPVs are a heterogeneous family consisting of five phylogenetic genera (alpha, beta, gamma, mu, and nu HPVs, encompassing at least 120 genotypes) of small non-enveloped, circular, double-stranded DNA viruses targeting the basal cells of stratified epithelia of the genital and upper respiratory tracts and the skin. Based on their oncogenic potential, the alpha genus (mucosal) HPV types are divided into two groups: low-risk (LR) HPVs, which are mainly associated with benign genital warts, and high-risk (HR) HPVs, which are causative agents of cervical, anogenital, and oropharyngeal cancers (reviewed in [18]). The HPV genome is organized into three regions: a non-coding region, termed the long control region (LCR), regulating gene expression and replication, and two protein-coding regions, the early (E) region coding proteins regulating viral transcription (E2), viral DNA replication (E1, E2), cell proliferation (E5, E6, E7), and viral particle release (E4) and the late (L) region coding for two structural viral capsid proteins (L1 and L2). E5, E6, and E7 are thus viral oncogenes and studies on mucosal HR HPVs have demonstrated that E6 and E7 play a key role in both benign proliferation and malignant transformation, and their continuous expression is critical in maintaining the cancer phenotype in infected cells (reviewed in [19]). In the last two decades, HR alpha HPVs, and in particular HPV type 16 (HPV16), have been causally related to a subset of OPSCC arising from the crypt epithelium of the palatine tonsils and base of tongue [20, 21] as well as to a substantial fraction of SCCs from unknown primary metastatic to the neck nodes [22] to which they confer a more favorable prognosis [23–27]. HPV-driven OPSCC is now considered a rising sexually transmitted disease showing distinctive epidemiological, clinical, and molecular features [4]. Patients with HPV-driven OPSCC are more likely to be younger, without a history of smoking and alcohol abuse, and have a higher socio-economic status and better performance status than those with non-HPV-driven OPSCC [28]. While non-HPV-driven OPSCC show molecular aberrations similar to those observed in SCC of the lung, HPV-driven tumors share similarities with cervical cancer. TP53 and CDKN2A/RB1 axes are the most frequently deregulated signaling pathways in both HPV-driven and non-HPV-driven HNSCC [29]. Most environmental-induced cancers harbor inactivating mutations in the TP53 gene leading to the loss of tumor suppression activity [30]. Furthermore, the p16INK4a-cyclin D1-RB axis is mainly deregulated by deletion or promoter hypermethylation of the CDKN2A gene encoding p16INK4a [31] and/or by CCND1 amplification [32], which encodes cyclin D1, with both leading to a decrease in the growth-suppressive hypo-phosphorylated RB form. Conversely from environmental-related HNSCC and consistently with HPV-mediated carcinogenesis, cells from HPV-driven OPSCC rarely contain loss-of-function TP53 mutations or CDKN2A inactivation and show less genomic instability [33]. In this subset of cancers, the p53 and RB pathways are both inactivated as a result of sequestration by binding viral oncoproteins. The E6 protein drives cell proliferation by stimulating ubiquitination and proteasome-dependent degradation of the p53 protein tumor suppressor protein [34]. E7 viral oncoprotein disrupts the RB/E2F complex, resulting in the dissociation of E2F transcription factors from RB-family proteins, thus inducing S-phase entry [35]. Furthermore, viral integration into host genome may contribute to neoplastic transformations by deregulation of key cellular genes and induction of genome instability [36].
DNA methylation
DNA methylation, catalyzed by DNA methyltransferases (DNMTs), usually occurs at the 5′ position of the cytosine ring within the cytosine-guanine dinucleotides (CpG). Although five members of the DNMT family have been identified, only DNMT1, DNMT3A, and DNMT3B have functional enzymatic activity in mammals. DNMT1 has been called “maintenance” DNMTs since it has a substrate preference for hemi-methylated substrate after DNA replication. Conversely, DNMT3A and DNMT3B are regarded as de novo DNMTs since they create new methylation patterns during embryogenesis and germ-cell development by methylating CpG dinucleotides previously unmethylated on both strands. DNA methylation is associated with repression of genes involved in development and plays a crucial function in genomic imprinting and in X-chromosome inactivation. Besides its role in gene regulation, DNA methylation prevents chromosomal instability by silencing endogenous retroviral and parasitic repetitive sequences (reviewed in [37]). Alterations in DNA methylation patterns have been extensively documented in cancer and appear to deeply contribute to its biology. DNA hypermethylation acts as an alternate and/or complementary mechanism to gene mutation or deletion, resulting in the inactivation of specific gene expression and function of tumor suppressor genes (TSGs) that promote the acquisition of tumorigenic behaviors, such as increased proliferation, enhanced invasiveness, and escape from apoptosis. Besides DNA hypermethylation, the genome of cancer cells undergoes an overall decrease in the level of 5-methylcytosine. This genome-wide hypomethylation affects intergenic and intronic regions of the DNA, particularly repeat sequences and transposable elements, and is believed to facilitate chromosomal instability, loss of imprinting, and reactivation of endogenous parasitic sequences [38].
Impact of aberrant DNA methylation in HPV-positive and HPV-negative OPSCC
The list of genes that are silenced by DNA methylation in OPSCC is growing rapidly and includes genes involved in several pathways, including apoptosis, cell cycle, DNA repair, and WNT signaling. A selection of the most frequently hypermethylated genes in OPSCC is given in Table 1. Notably, differences in DNA methylation profiles between HPV-positive and HPV-negative OPSCC have been frequently observed in several studies. Overall, while HPV-negative cancers are mainly characterized by genome-wide hypomethylation, the HPV-positive counterpart displays higher levels of promoter methylation (Table 1).
Table 1.
Genes hypermethylated in OPSCC
| Pathway | Gene | Name | Hypermethylated in | Region analyzed | Reference |
|---|---|---|---|---|---|
| Apoptosis | DAPK | Death-associated protein kinase 1 | HPV-negative/positive | Promoter region | [39] |
| RASSF1 | Ras association domain-containing protein 1 | HPV-negative | − 244 from TSSa | [50] | |
| Promoter region | [45, 51] | ||||
| STAT5 | Signal transducer and activator of transcription 5 | HPV-negative | + 42 from TSS | [50] | |
| Cell cycle | CCNA1 | Cyclin A1 | HPV-positive | Promoter region | [51] |
| + 7 from TSS | [50] | ||||
| CDKN2A | Cyclin-dependent kinase inhibitor-2A | HPV-negative | Promoter region | [43–45, 51] | |
| HPV-positive | Three loci within the CpG island of CDKN2A gene | [165] | |||
| CHFRb | Checkpoint with forkhead and ring finger domains | HPV-negative | Promoter region | [39] | |
| TP73 | Tumor protein p73 | HPV-negative/positive | Promoter region | [39] | |
| Cell fate determination | APC | Adenomatous polyposis coli | HPV-negative/positive | Promoter region | [39] |
| DNA repair | MGMT | O6-methylguanine-DNA methyltransferases | HPV-negative | − 272 from TSS | [50, 51] |
| Protein glycosylation | TUSC3 | Tumor suppressor candidate 3 | HPV-positive | + 29 from TSS | [50] |
| Inflammation | JAK3 | Janus kinase 3 | HPV-positive | + 64 from TSS | [50] |
| Invasion and metastasis | CADM1 | Cell Adhesion Molecule 1 | HPV-positive | Promoter region | [39] |
| CDH11 | Cadherin 11 | HPV-positive | − 354 from TSS | [50] | |
| CDH13a | Cadherin 13 | HPV-negative/positive | Promoter region | [39] | |
| IGSF4 | Immunoglobulin superfamily member 4 | HPV-positive | Promoter region | [166] | |
| SPDEF | SAM pointed domain-containing Ets transcription factor | HPV-negative | + 116 from TSS | [50] | |
| TIMP3 | TIMP metallopeptidase inhibitor 3 | HPV-positive | Promoter region | [39, 51] | |
| SYBL1 | Synaptobrevin-like 1 | HPV-positive | − 349 from TSS | [50] | |
| Signaling | ESR1 | Estrogen receptor 2 | HPV-negative/positive | Promoter region | [39] |
| ESR2 | Estrogen receptor 2 | HPV-negative | + 66 from TSS | [50] | |
| GALR1 | Galanin receptor type 1/2 | HPV-positive | Two loci within the CpG island of the GALR1 gene | [165] | |
| GRB7 | Growth factor receptor-bound protein 7 | HPV-positive | − 160 from TSS | [50] | |
| RARβa | Retinoic acid receptor β | HPV-negative/positive | Promoter region | [39] | |
| Transcription | RUNX1T1 | RUNX1 translocation partner 1 | HPV-positive | + 145 from TSS | [50] |
| TCF21 | Transcription factor 21 | HPV-positive | Promoter region | [167] | |
| WNT signaling | SFRP1 | Soluble frizzled receptor protein 1 | Drinkers | Promoter region | [56] |
| SFRP4 | Soluble frizzled receptor protein 4 | HPV-positive | Promoter region | [56] | |
| WIF1 | WNT inhibitory factor 1 | NA | Promoter region | [54] |
NA not applicable
a TSS transcription start site
bHypermethylation of these genes is associated with development of radioresistance in other tumor types
Apoptosis
Defects in the apoptotic pathways are essential for cancer development and progression, but also for resistance to chemotherapy and radiotherapy. Thus, identification of genes related to apoptosis in OPSCC may offer newer therapeutic modalities. The pro-apoptotic gene death-associated protein kinase (DAPK) is commonly hypermethylated in at least 20% of OPSCC independent of HPV status, indicating it is involved in both HPV-positive and HPV-negative OPSCC carcinogenesis [39]. DAPK gene encodes for a calcium/calmodulin-regulated serine/threonine kinase that is required for apoptosis induced by interferon-gamma [40].
Cell cycle
Cell cycle regulation depends on the appropriate expression of cyclin-dependent kinases (CDKs), their binding partners, and the inhibitory molecules such as cyclin-dependent kinase inhibitor-2A (CDKN2A). By using different first exons, CDKN2A encodes two overlapping, but very disparate proteins, p16INK4a and p14ARF, which are both involved in negatively regulating cell cycle progression through the pRB and the p53 pathways, respectively [12]. The CDKN2A locus, which is frequently hypermethylated in tumors, is often overexpressed in HPV-positive OPSCC [41]. Consistently, immunohistochemical staining of p16INK4A protein is commonly used as a surrogate marker for HPV infection in OPSCC [42]. To date, any correlation was observed between HPV status and CDKN2A promoter methylation [43–45], thus suggesting that all of the CDKN2A promoters of HPV-positive tumors may not be methylated. However, Schlecht et al. have recently identified four hypermethylated CDKN2A loci downstream of the p16INK4A and p14ARF transcription start sites. Interestingly, the hypermethylation of this region was associated with p16INK4A protein expression and correlated with an increased expression of p14ARF in OPSCC. Although the involvement of HPV proteins in the methylation of the CpG loci downstream of the CDKN2A gene promoter remains unclear, this may represent a potential mechanism for p16INK4A overexpression in HPV-positive OPSCC [46].
DNA repair
The maintenance of cellular integrity depends on the efficiency of multiple specific DNA repair pathways (reviewed in [47]) which are crucial in protecting against genomic instability, a characteristic of tumor development [48]. Genotoxic exposure to carcinogens such as tobacco often results in DNA damage that represents an important mechanism of OPSCC etiology [49]. Aberrant DNA methylation affecting the O6-methylguanine-DNA methyltransferases (MGMT) gene has been revealed in HPV-negative OPSCC [50, 51], but also in lung cancer [52], suggesting the loss of the repair function of MGMT may reduce the ability of cells to repair DNA damage in smoking-induced tumors.
WNT signaling
WNT proteins belong to a large family of secreted glycoproteins activating several pathways, including the best-known Wnt/β-catenin or canonical pathway [53]. Aberrant methylation of the negative regulators of this pathway has been observed in HNSCC, including OPSCC [54]. Downregulation of the soluble frizzled receptor protein (SFRP) genes, encoding soluble antagonists of WNT protein receptors, has been described as an alternative mechanism of stabilization and activation of β-catenin [55]. The promoter methylation status of these genes was examined in 350 patients with HNSCC, of which 25% derived from the oropharynx. Of note, SFRP1 aberrant methylation occurred at a higher prevalence in both heavy and light drinkers, whereas SFRP4 promoter methylation was detected more frequently in never and former smokers and was also associated with HPV16 infection [56].
Radiotherapy resistance
Some genes, which are associated with the development of tumor radioresistance, are frequently hypermethylated in OPSCC [11, 57] (Table 1). However, the molecular mechanism by which their inactivation may contribute to radiotherapy resistance in OPSCC is yet to be determined. Among these, checkpoint with forkhead and RING finger domains protein (CHFR) was hypermethylated in 25% of HPV-negative OPSCC patients, while no promoter methylation of this gene was observed in HPV-positive group [11, 57]. CHFR silencing was associated to the upregulation of PARP1, a gene coding for a DNA repair enzyme involved in radiotherapy resistance in HNSCC [58, 59].
In contrast to gene-specific hypermethylation, which usually occurs in HPV-positive OPSCC, genome-wide and global hypomethylation are more frequently observed in HPV-negative tumors [60], likely leading to chromosomal instability. Although the exact mechanism of global genomic DNA hypomethylation in HNSCC has not fully elucidated yet, differences in the expression and/or activity of DNMTs may explain HPV-related differences among OPSCC [46, 61]. Consistently, methylation levels of the long interspersed nucleotide element-1 (LINE-1) repetitive elements, a widely accepted surrogate of overall genomic DNA methylation content, were shown to be higher in HPV16-positive than in negative HNSCC [60, 62]. This finding suggests that HPV16-infected cells may attempt to silence the virus by DNA methylation, which can result in increased methylation of LINE-1 repetitive elements [62].
Methylation of HPV genome in HPV-associated OPSCC
HPV genome harbors CpG dinucleotides within conserved palindromic sequences [63]; thus, its DNA strands may be potentially targeted by covalent alterations such as methylation. As HPV genome does not encode any proteins with methyltransferase activity, the methylation of HPV-DNA is supposed to be under the control of the human host cell DNMT. It was shown that HPV-E7 is capable to edit DNA methylation by forming a complex with DNMT1 [46, 64]. In addition, both DNMT1 [65] and DNMT3a [61] were found to be more highly expressed in cells from HPV-positive HNSCC than in those from HPV-negative tumors.
Overall, the analysis of the methylation pattern of the integrate HPV16 and human host genome in cultured cells from HNSCC, including SCC from the base of the tongue, revealed that the methylation status of HPV16 is dramatically affected by the methylation status of the host DNA flanking the integration site with HPV16-DNA being highly methylated when integrated into intergenic highly methylated host genome sites, while remaining largely unmethylated when incorporated into poorly methylated intergenic regions [66]. This observation would argue for a bystander role of the virus methylome rather than an important phenomenon in HPV-driven carcinogenesis.
Different changes of the HPV methylome were observed in relation to squamous epithelial differentiation suggesting that HPV-DNA methylation is a more dynamic phenomenon both in the context of the viral life cycle and progression towards transforming infection modes [67]. The HPV genome includes a non-coding LCR, or upstream regulatory region, which contains a large number of cis-responsive elements governing HPV gene expression and replication. The p97 promoter at the E6 proximal part of the LCR regulates the transcription of E6 and E7 viral oncogenes [68]. HPV16 polymorphisms in the LCR may alter the oncogenic potential of the virus by enhancing p97 promoter activity [69]. Furthermore, numerous mutations were uncovered in the LCRs from oral cancer cells and HPV-immortalized oral epithelial cells which increase the expression of HPV-transforming proteins [70]. Therefore, the methylation status of LCR was intensively investigated both in cervical cancer and OPSCC. The functional significance of CpG methylation in the LCR may be indeed an attempt by the host cell to silence the expression of viral genes or a virus-induced strategy to shift from the productive stage of the viral life cycle towards the transforming phase of HPV infection [67]. Unexpectedly, two research groups observed an unmethylated status of the CpG sites within LCR in the majority of OPSCC samples with integrated HPV16 genome [71, 72]. On the other hand, consistently with previous findings derived from cervical cancer, they found a CpG methylation enrichment at the boundary of the L1 and L2 viral gene. In cervical cancer, the rate of hypermethylation at the L1 and L2 sites was observed to rise progressively with the increasing severity of the lesions [73], and HPV16 CpG methylation at L1 and L2 sites was a reliable biomarker of pre-cancer that can potentially stratify the risk in HPV-positive women [74]. Regardless of the oncological consequences of L1/L2 methylation which remains unknown, the authors suggest its potential utility as diagnostic biomarker of HPV-driven OPSCC [72].
The level of viral oncogene E6/E7 expression is regulated by binding of E2 to E2-binding sites (E2BSs). In particular, at high concentration, E2 binds low-affinity E2BS3 and E2BS4 resulting in inhibition of the p97 promoter and thereby maintaining low levels of E6 and E7 (reviewed in [75]). Thus, the transition towards a transforming HPV infection requires the inactivation of E2. The main mechanism by which this is achieved is the linearization of the viral DNA within the E2 open reading frame [76]. However, in about 60% of OPSCC HPV16 integration appears not necessary for viral transformation [36, 77]. In addition, viral integration in host genome may result in head-to-tail concatemers of full-length HPV16 genomes [78]. In the above two contexts, other mechanisms may contribute to the inhibition of E2 functions. Interestingly, tumor samples from OPSCC patients harboring intact E2 sequences (episomal state or integrated head-to-tail concatemers) displayed intermediated to complete methylation of E2BS3 and E2BS4 and high methylation levels at these sites were closely associated with the highest E6 and E7 expression levels and worse prognosis [79, 80]. Methylation status of E2BSs was critical in maintaining the transformed phenotype in oral SCC cells, as demethylation of HPV16 LCR by 5-aza-2′-deoxycytidine (5-AZA-CdR) caused repression of E6 and E7 expression followed by cell cycle arrest at G2/M [80]. Thus, in the presence of an intact E2 open reading frame, methylation at E2BSs in the LCR of E6 and E7 appears to positively regulate their expression. On the contrary, in a context of E2 gene disruption, the selective pressure on cellular clones harboring methylated LCR site is lost.
Impact of lifestyle risk factors in OPSCC aberrant DNA methylation
Tobacco smoke has been associated with both TSG promoter hypermethylation and genome-wide hypomethylation, especially in long-term tobacco users, along with prolonged alcohol consumption [81–83]. Tobacco and its metabolites influence the methylation profiles in cancers by impairing DNMT1 and DNMT3 expression, both at transcript and protein level, and by altering its enzymatic activity (reviewed in [84]). The genotoxic exposure to cigarette smoke condensate and heavy metals that are present in tobacco smoke has been also associated with global DNA hypomethylation [85]. Cigarette smoke decreases acid folic and vitamin B12 levels [86], which are required for the maintenance of methylation patterns in DNA [87]. Consistently, a reduced concentration of folate was found in the buccal mucosal cells of tobacco smokers [88]. Although these evidences indicated that deficiencies of folate and vitamin B12 may be associated with increased risk of developing OPSCC, several CpG sites were found to be differentially methylated in HPV-negative OPSCC patients with the highest levels of both vitamin A and vitamin B12 intake. Interestingly, vitamin B12 intake was positively correlated with RARβ hypomethylation in HPV-positive OPSCC patients, thus emphasizing the differences in tumor biology between HPV-positive and HPV-negative OPSCC [89]. Excessive alcohol intake has been reported to affect DNA methylation by altering folate metabolism and transmethylation reactions (for reviews see [90, 91]). Alcohol can also reduce or increase DNMT activity [92, 93], consistent with the evidence that within the nucleus accumbens core chronic low-level drinking promoted DNA hypomethylation, whereas high levels of drinking resulted in CpG hypermethylation [94]. Despite the limited number of studies that have looked for associations between DNA methylation and alcohol consumption, an analysis on cancers of the upper aerodigestive tract revealed increased levels of CDKN2A promoter methylation among alcohol drinkers [95].
Histone modifications
Histone (H) 2A, H2B, H3, and H4 core histones represent abundant nuclear proteins involved in the chromatin architecture since they enter into the constitution of nucleosomes. Core histones display N-terminal tails that protrude from the nucleosome and are subject to combinations of covalent modifications including acetylation, methylation, phosphorylation, sumoylation, and ubiquitination. These modifications determine how tightly the chromatin is compacted, playing a decisive role in modulating gene expression, as well as serve as docking stations for protein recognition modules which recruit specific functional complexes (reviewed in [96]). Histone acetylation and methylation are most commonly associated with carcinogenesis [37].
Histone acetylation is the result of the dynamic interplay between histone acetyltransferases (HATs) and histone deacetylases (HDACs). In general, transcriptional activators recruit HATs, which alter nucleosomal conformation to produce an open chromatin structure where transcription factors and co-activators can bind to turn on gene transcription, whereas transcriptional repressor associates with HDACs, which promote a more condensed and inactive chromatin state. HATs and HDACs target not only histone tails, but also non-chromatin proteins [97]. Histone methylation, catalyzed by histone methyltransferases (HMTs), occurs at both arginine and lysine residues on the tails of histone proteins H3 and H4. Similar to acetylation/deacetylation, histone methylation is reversible, and demethylation is catalyzed by histone demethylases (HDMs). Unlike histone acetylation, methylation of histone proteins can result in either activation or repression, depending on which residue is affected. Indeed, trimethylation of histone H3 lysine 9 (H3K9), 27 (H3K27), and histone H4 lysine 20 (H4K20) is associated with silent chromatin and transcriptionally inactive genes. Conversely, methylation of lysines 4, 36, and 79 on histone H3 (H3K4, H3K36, and H3K79) can experience various methylated states, including monomethylated, dimethylated, and trimethylated which is closely linked with active transcription [98, 99]. Myeloid mixed-lineage leukemia (MLL) is a HMT that targets several lysine residues on histones, including H3K4, and usually acts as a positive transcriptional regulator. A whole-exome sequencing analysis identified inactivating mutations in MLL2 and MLL3 genes in both HPV-positive and HPV-negative OPSCC [100], indicating a tumor suppressor function. In the same study, the mutational spectrum of HPV-negative tumors resulted very similar to those observed in lung and esophageal squamous cell carcinomas and included mutations of the HMT nuclear-receptor-binding SET-domain-containing 1 (NSD1), which preferentially targets H3K36 methylation [100]. By analyzing public genomic and epigenomic data sets from HNSCC, Papillon-Cavanagh et al. have recently identified a DNA methylation cluster that exclusively contained samples carrying NSD1 mutations or H3K36 alterations. Results were further validated in an independent cohort of OPSCC samples, in which the presence of H3K36 alterations associated to a drastic decrease in H3K36 methylation levels [101]. Altogether, these findings suggest that NSD1 mutations and/or H3K36 alterations may be associated with a genome-wide hypomethylation phenotype in OPSCC.
With the exception of H4, all “canonical” histone proteins in mammals have several variants with different sequences [102]. The “canonical” histones are expressed at high levels during the S-phase of the cell cycle, whereas replication-independent histone “variants” are expressed and incorporated into chromatin throughout the cell cycle (for a review see [103]). Among histone “variants,” phosphorylated H2A.X (γH2A.X) variant represents a useful marker of DNA integrity and repair, because of its ability in recruiting DNA repair proteins at the site of the dysplastic tissue. In response to double-strand breaks (DSB), the serine/threonine kinase ataxia-telangiectasia mutated is activated and rapidly phosphorylates the histone variant H2A.X on S139, forming γH2A.X [104]. Upon DSB induction, γH2A.X appears as subnuclear foci within minutes. The release of γH2A.X foci and the subsequent repair of damage DNA depend on γH2A.X acetylation and subsequent ubiquitination. [105]. Residual γH2A.X foci which are detectable 24 h after damage likely indicate misrepaired or incompletely repaired DSB [106]. Interestingly, Park et al. reported that HPV E7 oncoprotein was able to increase retention of γH2AX nuclear foci following radiation-induced DNA damage. Consequently, the normal kinetics of DNA damage repair was impaired in HPV-positive OPSCC cells both in vitro and in vivo [25]. These findings may explain why HPV-positive tumors are more sensitive to radiotherapy.
HPV infection and histone modifications in OPSCC
E6 and E7 HR-HPV oncoproteins have been demonstrated to affect histone acetylation and methylation pattern by interacting with HATs, HDACs, HMTs, and HDMs (reviewed in [107]), thus affecting the chromatin landscape of cancer cells. In HPV-driven cancers, histone modifications on targeted genes can mediate bidirectional effects on gene transcription. The interaction between E6 and E7 HPV oncoproteins and the HAT p300/CBP is, in fact, mainly direct towards non-histone targets and aimed to enhance the deregulation of TP53 and CDKN2A/RB1 pathways. Independent of its ability to induce p53 degradation, E6 inhibits p300-mediated p53 acetylation, leading to repression of p53-targeted gene activation [108]. By recruiting p300/CBP and pRb, E7 brings the histone acetyltransferase domain of p300/CBP into proximity to pRb and promotes its acetylation, leading to cell cycle deregulation [109]. The C-terminal zinc-binding domain of E7 interacts with HDAC1 and 2 through Mi2β protein, a component of the nucleosome remodeling deacetylase complex, thus inhibiting histone deacetylase activity [110, 111]. Of interest, human keratinocyte-expressing HPV16 E7 show an increased histone H3K9 acetylation on E2F-responsive promoters, which depend on E7 binding with both pRb and HDAC. In addition, methylation of H3K4, which is associated with transcriptional activation, was also increased [112]. This results in the weakening of the histone-DNA interaction at E2F-responsive sites and may promote the transcription of cell cycle progression genes. Enhancer of zeste homolog 2 (EZH2), the functional enzymatic component of the polycomb repressive complex 2 (PRC2), is a HMT catalyzing the addition of methyl groups to H3K27 and, eventually, contributing to formation of a repressive chromatin state [113]. Thus, PRC2 marks transcriptionally silenced genes. Polycomb silencers mediate the repression of key tumor-suppressor pathways and play a crucial role in suppressing genes required for differentiation and maintaining a cancer stem cell phenotype (reviewed in [114]). In vitro studies demonstrated that EZH2 promoter can be activated by HPV E7 oncoprotein via the release of E2F factors from growth-inhibitory pocket proteins [115]. HPV-positive OPSCC have genome-wide elevation in the repressive H3K27me3 histone modification [116], thus confirming HPV-driven carcinogenesis and EZH2 overexpression are closely related. Furthermore, double immunofluorescence quantification of histone lysine methylation revealed that p16-positive OPSCC had global elevation of H3K27me3 and H4K20me1 that are both involved in generating a repressive gene environment through the formation of facultative heterochromatin [117]. Intriguingly, in HPV-positive HNSCC, several PRC2 target genes were found to undergo hypermethylation including members of the cadherin superfamily whose deregulation is implicated in several tumor progression and metastasis processes such as epithelial to mesenchymal transition [118]. Furthermore, a significant enrichment of highly methylated promoter regions of PRC2 targets together with a higher expression of DNMT3a was observed in cell lines from HPV-positive OPSCC compared to HPV-negative ones [61]. In contrast to the abovementioned studies, HPV E7 oncoprotein was shown to induce the expression of the lysine demethylases KDM6A and KDM6B causing epigenetic reprogramming mainly by removing the repressive H3K27me3 marks [119]. While in environmental-related carcinomas the cyclin-dependent kinase inhibitor p16INK4a is usually downregulated mainly by gene mutation or deletion, it is frequently overexpressed in HPV-driven tumor and thus, it is considered a surrogate marker for active HPV involvement in OPSCC carcinogenesis. p16INK4a upregulation was considered the effect of transcriptional activation by E2F transcription factor released after E7-mediated disruption of pRb/E2F complexes. But detailed mechanistic investigations suggested that p16INK4a is overexpressed upon HPV E7 oncoprotein signaling via induction of the demethylase KDM6B that removes repressive H3K27me3 marks from the p16INK4a -encoding CDKN2A promoter region [119, 120].
Tobacco smoke may induce chromatin histone modifications in HPV-negative OPSCC
To date, the effect of excessive alcohol exposure and tobacco consumption on histone modifications has not been investigated in OPSCC. However, cigarette smoking was shown to induce specific post-translational modifications in H3 and H4 lysine and arginine during the pathogenesis of smoking-related diseases [121]. In lung cancer, mutations and deregulations of histone-modifying enzymes have been described in association with tobacco smoke condensate [101, 122], and smoke-induced modifications in histone patterns have linked to aberrant gene expression in immune cells [123]. Given that HPV-negative OPSCC most closely resemble lung SCC [124], deregulation of histone-modifying enzymes and chromatin structure may also play a role in tobacco smoke-induced OPSCC.
NcRNA involvement in OPSCC
NcRNAs are of increasing biologic and therapeutic relevance due to their role in modulating gene expression [12–16]. Generally, ncRNAs less than 200 bp are known as small ncRNAs (sncRNAs) and included small interfering RNAs, micro RNAs (miRNAs), and PIWI-interacting RNAs (piRNAs), while all larger transcripts are defined as long non-coding RNAs (lncRNAs) [125]. NcRNAs can interact with histone-modifying complexes and/or DNMTs, being also targets of these epigenetic mediators [12, 16]. At present, the list of ncRNAs involved in OPSCC includes a large number of sncRNAs (mainly miRNAs) and few lncRNAs (Table 2).
Table 2.
NcRNAs altered in OPSCC
| MiRNA | Up-/downregulation or polymorphism | HPV-associated | Environmental factors associated | Epigenetic regulation | References |
| Let-7c | Down | No | [127] | ||
| MiR-9 | Down | No | Promoter methylation | [131] | |
| Up | Yes | [168, 169, 140] | |||
| MiR-18a | Down | Yes | [168] | ||
| Up | No | [127] | |||
| MiR-20a | Up | No | [127] | ||
| MiR-20b | Up | Yes | [169, 140] | ||
| MiR-21 | Up | No | [127, 151] | ||
| MiR-26b | Down | Yes | [142] | ||
| MiR-30a | Up | No | Alcohol | [150] | |
| MiR-30d | Up | No | [127] | ||
| MiR-31 | Down | Yes | [168] | ||
| MiR-93 | Up | Yes | [140] | ||
| MiR-101 | Down | Yes | [142] | ||
| Up | No | Alcohol | [150] | ||
| MiR-103 | Up | No | [170] | ||
| MiR-106b | Up | Yes | [140] | ||
| MiR-125a | Down | Yes | [142] | ||
| MiR-126 | Down | Yes | [140, 142] | ||
| MiR-127 | Down | Yes | [142] | ||
| MiR-130a | Up | No | [127] | ||
| MiR-137 | Down | No | Promoter methylation | [171] | |
| MiR-143 | Down | Yes | [140, 142] | ||
| MiR-145 | Down | Yes | [140, 142] | ||
| MiR-146 | Polymorphism | Yes | [129] | ||
| MiR-149 | Polymorphism | Yes | [129] | ||
| MiR-155 | Up | No | [170] | ||
| Yes | [168] | ||||
| MiR-181b/d | Up | No | [170] | ||
| MiR-191 | Up | No | [170] | ||
| MiR-195 | Up | Yes | [142] | ||
| MiR-196 | Polymorphism | Yes | [129] | ||
| MiR-198 | Down | No | [127] | ||
| MiR-199a/b | Down | Yes | [140, 142] | ||
| MiR-200c | Up | No | [127] | ||
| MiR-222 | Up | Yes | [140] | ||
| MiR-223 | Down | Yes | [168] | ||
| MiR-320 | Up | Yes | [140] | ||
| MiR-363 | Up | Yes | [140, 142] | ||
| MiR-372 | Up | No | [127] | ||
| MiR-375 | Up | Alcohol | [151] | ||
| MiR-379 | Down | Yes | [142] | ||
| MiR-381 | Down | Yes | [142] | ||
| MiR-409 | Down | Yes | [142] | ||
| Up | No | [127] | |||
| MiR-432 | Down | Yes | [142] | ||
| MiR-433 | Down | Yes | [142] | ||
| MiR-499 | Up | No | [127] | ||
| Polymorphism | Yes | [129] | |||
| MiR-517 | Down | Yes | [142] | ||
| MiR-675 | Up | No | Alcohol | [150] | |
| MiR-934 | Up | No | Alcohol | [150] | |
| MiR-1201 | Down | Yes | [142] | ||
| MiR-1266 | Up | No | Alcohol | [150] | |
| MiR-3164 | Up | No | Alcohol | [150] | |
| MiR-3178 | Up | No | Alcohol | [150] | |
| MiR-3690 | Up | No | Alcohol | [150] | |
| LncRNA | Up-/downregulation | HPV-associated | Environmental factors associated | Epigenetic regulation | References |
| CDKN2B-AS | Up | Yes | [135] | ||
| EGOT | Up | Yes | [135] | ||
| LINC00152 | Down | Yes | [135] | ||
| NCRNA00185 | Down | Yes | [135] | ||
| PRINS | Up | Yes | [135] | ||
| TTTY14 | Up | Yes | [135] | ||
| TTTY15 | Up | Yes | [135] | ||
| XIST | Up | Yes | [135] |
MiRNAs represent important mediators of epigenetic regulation of gene expression. MiRNAs can direct endonucleolytic cleavage of the targeted mRNAs or inhibit translation through perfect or nearly perfect complementarity to targeted mRNAs at the 3′ untranslated [12]. To date, limited information is available regarding mechanisms by which miRNA alterations may contribute to OPSCC carcinogenesis and progression. MiRNAs are transcribed in the nucleus by RNA polymerase II into long primary transcripts, which are further processed by two RNase-III enzymes, Drosha and Dicer [126]. Of note, increased expression levels of Drosha, Dicer, and other components of the miRNAs machinery were detected in tonsil SCC [127]. Consistent with these data, a number of miRNAs were upregulated, with the exception of miR-198 and let-7 [127], which has been previously shown to negatively regulate Dicer expression [128]. Among upregulated miRNAs, miR-21 and miR-499 were found to suppress the programmed cell death protein 4 (PDCD4), a tumor suppressor protein that is lost in the majority of tonsil SCC [127]. Apart from miRNA machinery, single nucleotide polymorphisms in miRNA precursors may influence their maturation, and thereby modulate their expression as reported for miR-146, miR-149, miR-196, and miR-499. Polymorphisms in the immature form of these miRNAs were found to significantly increase the risk of HPV16-associated OPSCC, particularly in never smokers [129].
Interestingly, miRNAs have been reported to be epigenetically silenced in laryngeal SCC, oral cavity SCC (OSCC), and OPSCC. In particular, treatment of HNSCC cell lines with 5-AZA-CdR has proven to be effective in restoring the expression of miR-9, one of the best characterized miRNA regulated by DNA methylation in cancer [130]. Furthermore, miR-9 ectopic expression led to PTEN upregulation, and a significant repression of HNSCC proliferation [131]. Thus, miR-9 could represent an important negative regulator of OPSCC cells growth. Other than being regulated by epigenetic mechanisms, miRNAs can in turn modulate the expression of the epigenetic enzymes. Along this line, miR-874 silencing by aberrant CpG promoter methylation has been frequently described in laryngeal SCC, OSCC, and OPSCC. Performing an in silico database analysis, miR-874 was found to negatively regulate the expression of HDAC1. Accordingly, luciferase reporter assay demonstrated that miR-874 directly regulated HDAC1 in HNSCC cells, thus creating a complicated network of reciprocal interconnections [132].
As described above, lncRNAs are defined as RNA transcripts longer than 200 nucleotides that lack protein-coding potential. After transcription via RNA polymerase, lncRNAs are processed are subject to 5′-capping, polyadenylation, and intron splicing. Most lncRNAs are retained in the nucleus, but in some cases, they can also be exported to the cytoplasm. LncRNAs represent potent cis- and trans-regulators of gene transcription and act as scaffolds for chromatin-modifying complexes (reviewed in [133]). At present, the knowledge of lncRNA involvement in carcinogenesis is still in its infancy, largely due to the novelty of these molecules. The putative role of lncRNAs in OPSCC further confirm this, as only few lncRNAs have been studied in detail, and have been directly linked to HPV oncogenic protein activity [134, 135].
NcRNA deregulation in HPV-positive OPSCC
Although the involvement of ncRNAs in HNSCC is well recognized, only few studies have focused specifically on ncRNAs profile in HPV-positive OPSCC (Table 2) and investigated how HPV modulates their expression [136, 137], since both E6 and E7 HPV oncoproteins were shown to modulate the ncRNA landscape in cancer cells [138, 139]. Using different cohorts of OPSCC, a miRNA panel that differentiates HPV-positive OPSCC from HPV-negative tumors has been recently identified [140]. Interestingly, strong upregulation of miR-9 has been observed in HPV-positive OPSCC but not in HPV-negative tumors [140], in which miR-9 expression was found silenced by promoter methylation [131]. Consistent with this data, increased miR-9 expression associated with HPV activity has been reported in cervical cancer [141]. However, the mechanism by which HPV promotes miR-9 expression is yet to be discovered. In a study of Lajer et al., HPV-positive OPSCC showed a considerable upregulation of miR-363 expression [142], consistent with another report of Wald et al., in which HPV16 E6 knockdown was accompanied by a reduction of miR-363 levels in HNSCC [143], thus suggesting a possible role for miR-363 in HPV-positive OPSCC. MiRNAs can also directly target HPV E6/E7 mRNA, as it has been demonstrated via the ectopic expression of miR-375 mimic in OPSCC and cervical cancer cell lines [144]. Notably, Liu et al. has recently showed that E6 oncoprotein promoted miR-375 epigenetic silencing through overexpression of DNMT1 in HPV16 positive cervical cancer cells [145]. Furthermore, miR-375 was found to negatively regulate the metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) lncRNA [145], which is frequently overexpressed in cancers, and has extensively involved in oncogenic processes [146]. In a previous study of Jang et al., MALAT1 expression increased in oral keratinocytes transfected with HPV16 E6 and HPV E6/E7 oncoproteins, indicating HPV16 may promote cell proliferation by promoting MALAT1 upregulation [134]. A recent study performed on the extensive data set available on The Cancer Genome Atlas portal showed ncRNAs as a significantly upregulated transcriptional RNA population in HPV16-positive HNSCC with the most prominent differentially expressed ncRNAs between HPV16-positive and HPV-negative being associated with protein-coding “targets” involved in the cell cycle and cell-cell signaling [147]. Altogether, these findings suggest the role of ncRNAs in HPV-driven OPSCC warrants future investigation.
Environmental regulation of ncRNAs in OPSCC
NcRNA expression changes following exposure to environmental carcinogens have been documented in HNSCC. Unfortunately, results were obtained using heterogeneous tumor tissues or cell lines from different sites of HNSCC, so they are not exclusively specific for OPSCC. Overexpression of miR-23a was described in HNSCC from areca-nut chewing patients and correlated with an increase of the DNA damage marker γH2A.X and a reduction of DBS repair [148]. A recent study reported that the downregulation of miR-145 in oral fibroblasts exposed to cigarette smoke condensates promoted pro-tumorigenic stromal-epithelial interactions [149]. A number of aberrantly expressed miRNAs were identified in HNSCC patients, who were subdivided into non-drinkers, light, and heavy drinkers. Among these, miR-30a and miR-934 were the most highly upregulated in drinkers, and their overexpression was associated to the induction of the anti-apoptotic gene BCL-2 and to increased levels of cell proliferation [150]. MiR-375 was also demonstrated to increase with alcohol consumption in OPSCC [151], but the pathway involved between this miRNA and the excessive alcohol use has not yet been elucidated.
Epigenetic alterations as diagnostic biomarkers in OPSCC
Epigenetic alterations share some characteristics that render them particularly attractive as clinically applicable biomarkers, since they are characterized by high stability in biologic samples, and they can be easily detected in body fluids (e.g., serum, plasma, saliva, urine). Furthermore, the possibility to amplify them in a cost-effective manner represents a further advantage for routine analyses [152]. The increasingly recognized role of aberrant epigenetic modifications in OPSCC biology strongly suggests for the opportunity to test epigenetic markers as potential indicators of disease prognosis and response to therapy. The ability to determine epigenetic alterations in premalignant lesions, serum, and saliva may also provide valuable biomarkers for the early detection of OPSCC and for monitoring its recurrence.
Consistent with the increasing role of aberrant DNA methylation in HNSCC biology, different studies have reported the methylation of single genes/loci to have a potential in predicting OPSCC clinical outcome. However, a number of them have been conducted on study populations consisted of OPSCC and other HNSCC (Table 3). Taioli et al. studied the methylation of CDKN2A, MGMT, and RASSF1 in correlation with OS and tumor recurrence in OSCC and OPSCC. Results demonstrated that MGMT promoter methylation was significantly associated with poorer outcome, consistent with the critical role of MGMT in DNA repair [45]. In the last years, a number of studies have sought to establish a correlation between promoter methylation and improved survival rate in HPV-positive OPSCC. Along this line, Gubanova et al. provided the evidence that the downregulation of the serine/threonine-protein kinase SMG-1 by promoter hypermethylation correlated with HPV-positive status and improved OPSCC patient survival, and also with enhanced response to radiotherapy in HPV-positive HNSCC cell lines [153]. Subsequently, Kostareli et al. described an HPV-related promoter methylation signature of five genes (ALDH1A2, GATA4, GFR4, IRX4, and OSR2) with strong correlation and predictive power for clinical outcome of OPSCC patients [154]. A more recent study has investigated the influence of the overall level of genomic DNA methylation on early OPSCC relapse risk. Results reported that OPSCC smokers who relapsed within 24 months exhibited significantly reduced methylation levels of the LINE-1 repetitive elements. Interestingly, the association between smoking habits and LINE-1 hypomethylation was stronger in HPV16-negative OPSCC cases [60].
Table 3.
Association of aberrant DNA methylation and clinical outcome in OPSCC patients
| Gene | Hyper-/hypomethylation | Cohort | Clinical outcome | References |
|---|---|---|---|---|
| MGMT | Hyper | 88 OPSCC | Poorer PFS and OS | [45] |
| ALDH3A1 | Hyper | 76 HNSCC, including 15 OPSCC | Decreased OS | [172] |
| TAP1 | Hyper | |||
| SMG1 | Hypo | 40 OPSCC | Improved OS in HPV-positive OPSCC | [153] |
| ALDH1A2 | Hypo | 170 OPSCC (3 independent cohorts) | Improved clinical outcome in HPV-positive OPSCC | [154] |
| GATA4 | Hyper | |||
| GRIA4 | Hyper | |||
| IRX4 | Hyper | |||
| OSR2 | Hypo | |||
| DAPK1 | Hyper | 70 HNSCC, including 9 OPSCC | Lymph node metastasis | [173] |
| MGMT | Hyper | |||
| WIF1 | Hyper | 43 HNSCC, including 19 OPSCC | Decreased OS | [54] |
| GALR1/2 | Hyper | 202 HNSCC, including 58 OPSCC | Poor survival with the highest association in HPV-negative OPSCC | [174] |
| LINE-1 | Hypo | 110 OPSCC (2 independent cohorts) | Increased risk of early relapse in HPV-negative OPSCC | [60] |
ALDH aldehyde dehydrogenase, DAPK death-associated protein kinase, GALR galanin receptor, GATA GATA binding protein, GRIA glutamate receptor, IRX iroquois homeobox, LINE long interspersed nuclear element, MGMT O-6-methylguanine-DNA methyltransferases, OSR odd-skipped-related, SMG nonsense mediated mRNA decay associated PI3K-related kinase, TAP transporter associated with antigen processing, WIFI WNT inhibitory factor
Due to their stability as well as the potential role of their dysregulation at different stages of carcinogenesis, ncRNA may represent very promising non-invasive biomarkers for OPSCC. Based on their size and greater stability with respect to mRNA, miRNA represent an attractive target for salivary-based diagnostic [155]. Consistently, several groups have already published miRNA profiles correlated to clinical outcome of OPSCC patients (Table 4). In most cases, the causal relationship has not been completely elucidated. Since the expression of several miRNA is specifically modulated in HPV-driven tumors, miRNA profiles may also be useful for identifying HPV-positive OPSCC patients in the early stage of the disease. The clinical importance of another class of small ncRNA, called piRNAs, has emerged from a study of Firmino et al. in which piRNA expression was assessed in 498 non-malignant and HNSCC tissues, including OPSCC. Data obtained revealed 87 piRNAs that were exclusively expressed in HNSCC, with 41 piRNA clearly associated to HPV status. Among these, 11 piRNAs were significantly downregulated in HPV16/18 tumors compared to other HPV types. Based on these data, authors defined an expression signature of five piRNAs that correlated with OS exclusively in HPV-positive patients, indicating the potential utility of piRNAs in assessing HNSCC patient outcome [156] (Table 4).
Table 4.
Association of ncRNAs profiles and clinical outcome in OPSCC patients
| NcRNA | Conclusion | Cohort | References |
|---|---|---|---|
| MiR-221 | MiR-21 expression correlated with poor prognosis in HNSCC patients | 147 HNSCC, including 31 OPSCC | [151] |
| Let-7d | Reduced expression of let-7d and miR-205 is a significant predictor of HNSCC progression independent of anatomical site, tumor stage, treatment, or HPV | 104 HNSCC, including 32 OPSCC | [175] |
| MiR-205 | |||
| MiR-107 | Associated with overall survival (OS), independent of HPV status | 88 OPSCC | [169] |
| MiR-151 | |||
| MiR-492 | |||
| MiR-20b | Associated with disease-free survival, independent of HPV status | ||
| MiR-107 | |||
| MiR-151 | |||
| MiR-182 | |||
| MiR-361 | |||
| MiR-151 | Associated with distant metastasis, independent of HPV status | ||
| MiR-152 | |||
| MiR-324-5p | |||
| MiR-361 | |||
| MiR-492 | |||
| MiR-9 | Associated with OS in HPV-positive patients | 150 OPSCC | [168] |
| MiR-18a | |||
| MiR-31 | |||
| MiR-155 | |||
| MiR-223 | |||
| MiR-146 | Single nucleotide polymorphisms in these miRNAs were associated with reduced and increased risk of OPSCC recurrence, respectively | 1008 OPSCC | [176] |
| MiR-196 | |||
| MiR-193b-3p | Positively associated with OS | 81 OPSCC obtained from “The Cancer Genome Atlas”, and 95 OPSCC patients included for validation | [177] |
| MiR-455-5p | |||
| MiR-92a-3p | Negatively associated with OS | ||
| MiR-497-5p | |||
| PiR-30506 | Associated with OS in HPV-positive patients | 498 non-malignant and HNSCC tissues, including 66 OPSCC | [156] |
| PiR-35953 | |||
| PiR-36715 | |||
| PiR-36984 | |||
| PiR-39592 |
Epigenome-modifying enzymes as potential therapeutic targets in OPSCC
Enzymes that maintain and modify the epigenome seem to play a crucial role in OPSCC. In this context, epigenetic drugs might represent an important therapeutic modality for the clinical management of OPSCC patients.
Being regulated by multiple oncogenic pathways and affecting OPSCC phenotype, DNMTs constitute a potential anti-cancer target. So far, the most widely studied DNMT inhibitors (DNMTi) 5-azacytidine (azacitidine, Vidaza) and 5-AZA-CdR (Decitabine, Dacogen) have undergone intensive clinical development that led to their Food and Drug Administration (FDA) approval for patients affected by hematological malignancies [37]. As described above, treatment with 5-AZA-CdR has proven to be effective in restoring the expression of miRNA with tumor suppressor function in HNSCC [131], but also in HPV-transformed cell lines [157]. Considering that HPV-positive OPSCC have been found to have higher levels of TSG promoter methylation, DNMTi might represent an additional treatment option for these patients. Furthermore, an immunomodulatory activity of 5-AZA-CdR, which may ensure efficient therapeutic anti-tumor effects in HPV-positive malignancies, has been also shown in mice vaccinated with HPV DNA vaccines. In fact, 5-AZA-CdR co-delivered with a DNA vaccine encoding calreticulin (CRT) linked to E7 antigen (CRT/E7) was able to increase CRT/E7 DNA expression and to enhance E7-specific CD8+ T cell immune responses [158].
Although the biological consequences of HDAC deregulation in OPSCC are still largely unknown, in vitro studies have reported that HDAC inhibitors (HDACi) could induce cell cycle arrest [159] and promote apoptosis in HNSCC cell lines [160]. Epigenetic drugs have also been explored in combination with chemotherapeutics, indicating they may sensitize HNSCC cells to chemotherapy-induced apoptosis. For example, the combinations of 5-AZA-CdR or HDACi with cisplatin enhanced the cytotoxic effectiveness of this well-established chemotherapeutic in HNSCC treatment [161, 162]. DNMTi also demonstrated some synergistic effect with radiation by reducing HNSCC cell survival compared to the single treatments and by increasing radiation-induced apoptosis [163]. Furthermore, in vivo HDACi administration promoted DNA repair and survival in normal cells after radiation, indicating that HDACi could protect normal tissue from radiation-induced side effects [164]. Based on these promising pre-clinical data, OPSCC clinical trials involving HDACi have been carried out or are on-going (NCT01064921, NCT01695122, NCT01249443, available at www.clinicaltrials.gov).
Unlike DNMTi and HDACi, only a few HMT inhibitors are currently known, and most of them were discovered through random screening approaches [37]. In this context, epigenetic inhibitors targeting the EZH2 pathway have recently shown effectiveness in suppressing OPSCC growth and survival, with a major effect in HPV-positive cell lines [116]. Despite these promising results, no significant decrease in EZH2 and its substrate H3K27 was observed [116], thus indicating the mechanisms of these HMTi need to be further elucidated in OPSCC.
Conclusions
The incidence of OPSCC is rising rapidly with about 60% of patients presenting with loco-regionally advanced disease at diagnosis and requiring combined modality treatment strategies. Thus, improving survival rate and reducing treatment morbidity are both pressing issues. Epigenetic alterations, including DNA methylation, histone modification, and ncRNAs, clearly impact on key pathways that are involved in OPSCC biology. Epigenetic events occurring in OPSCC should be considered as the consequence of a network of interactions between epigenetic enzymes, on one side, and HPV infection and environmental-lifestyle factors on the other. HPV-positive and HPV-negative OPSCC have singular epigenetic drivers which may impact on different clinical behaviors and treatment response and strengthen the concept that HPV-driven OPSCC are biologically distinct from non-HPV-driven tumors. Expanding our understanding on how epigenetic modifications contribute to OPSCC and enlightening the convergent crosstalk existing between DNA methylation, histone modification, and ncRNA networks may improve the knowledge of its pathogenesis and provide new novel biomarkers for diagnosis or prediction of disease outcome and/or response to therapy. Furthermore, the development of next-generation epigenetic drugs may offer the tools necessary for promising therapeutic treatment of both HPV-positive and HPV-negative OPSCC patients.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the Centro di Riferimento Oncologico, IRCCS-National Cancer Institute (5x1000 Institutional Grant to EF).
Availability of data and materials
Not applicable.
Abbreviations
- 5-AZA-CdR
5-aza-2′-deoxycytidine
- CDKs
Cyclin-dependent kinases
- CHFR
Forkhead and RING finger domains protein
- CpG
Cytosine-guanine dinucleotides
- DAPK
Death-associated protein kinase
- DNMTs
DNA methyltransferases
- DSB
Double-strand breaks
- E
Early
- E2BS
E2-binding site
- EZH2
Enhancer of zeste homolog 2
- H
Histone
- HAT
Histone acetyltransferase
- HDAC
Histone deacetylases
- HDACi
HDAC inhibitors
- HDM
Histone demethylase
- HMT
Histone methyltransferase
- HNSCC
Head and neck squamous cell carcinoma
- HPV
Human papillomavirus
- HPV16
HPV type 16
- HR
High risk
- K
Lysine
- L
Late
- LCR
Long control region
- LINE-1
Long interspersed nucleotide element-1
- lncRNA
Long non-coding RNA
- LR
Low-risk
- MALAT1
Metastasis-associated lung adenocarcinoma transcript 1
- MGMT
O6-Methylguanine-DNA methyltransferases
- miRNA
Micro RNA
- MLL
Myeloid mixed-lineage leukemia
- ncRNA
Non-coding RNA
- NSD1
Nuclear-receptor-binding SET-domain-containing 1
- OPSCC
Oropharyngeal squamous cell carcinoma
- OSCC
Oral cavity squamous cell carcinoma
- PDCD4
Programmed cell death protein 4
- piRNA
PIWI-interacting RNA
- PRC2
Polycomb repressive complex 2
- SFRP
Soluble frizzled receptor protein
- sncRNA
Small non-coding RNA
- TSG
Tumor suppressor gene
Authors’ contributions
EF was a major contributor, composing and writing the manuscript. PBR largely contributed to the writing of this review and was involved in critically revising the manuscript. CF, VL, and JP helped during the initial stages of manuscript preparation and were involved in drafting, correcting, and critically revising the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D, Bray F. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49(6):1374–1403. doi: 10.1016/j.ejca.2012.12.027. [DOI] [PubMed] [Google Scholar]
- 2.Wright G, Morgan MY. Alcohol and tobacco misuse: reducing aerodigestive cancer risk. World J Hepatol. 2013;5(8):452–457. doi: 10.4254/wjh.v5.i8.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hashibe M, Brennan P, Chuang SC, Boccia S, Castellsague X, Chen C, Curado MP, Dal Maso L, Daudt AW, Fabianova E, et al. Interaction between tobacco and alcohol use and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol Biomark Prev. 2009;18(2):541–550. doi: 10.1158/1055-9965.EPI-08-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boscolo-Rizzo P, Del Mistro A, Bussu F, Lupato V, Baboci L, Almadori G, Da Mosto MC, Paludetti G. New insights into human papillomavirus-associated head and neck squamous cell carcinoma. Acta Otorhinolaryngol Ital. 2013;33(2):77–87. [PMC free article] [PubMed] [Google Scholar]
- 5.Hashibe M, Brennan P, Benhamou S, Castellsague X, Chen C, Curado MP, Dal Maso L, Daudt AW, Fabianova E, Fernandez L, et al. Alcohol drinking in never users of tobacco, cigarette smoking in never drinkers, and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. J Natl Cancer Inst. 2007;99(10):777–789. doi: 10.1093/jnci/djk179. [DOI] [PubMed] [Google Scholar]
- 6.Underwood JM, Lakhani N, Rohan E, Moore A, Stewart SL. An evaluation of cancer survivorship activities across national comprehensive cancer control programs. J Cancer Surviv. 2015;9(3):554–559. doi: 10.1007/s11764-015-0432-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaturvedi AK, Anderson WF, Lortet-Tieulent J, Curado MP, Ferlay J, Franceschi S, Rosenberg PS, Bray F, Gillison ML. Worldwide trends in incidence rates for oral cavity and oropharyngeal cancers. J Clin Oncol. 2013;31(36):4550–4559. doi: 10.1200/JCO.2013.50.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hashibe M, Sturgis EM. Epidemiology of oral-cavity and oropharyngeal carcinomas: controlling a tobacco epidemic while a human papillomavirus epidemic emerges. Otolaryngol Clin N Am. 2013;46(4):507–520. doi: 10.1016/j.otc.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Hussein AA, Helder MN, de Visscher JG, Leemans CR, Braakhuis BJ, de Vet HCW, Forouzanfar T. Global incidence of oral and oropharynx cancer in patients younger than 45 years versus older patients: a systematic review. Eur J Cancer. 2017;82:115–127. doi: 10.1016/j.ejca.2017.05.026. [DOI] [PubMed] [Google Scholar]
- 10.Secretan B, Straif K, Baan R, Grosse Y, El Ghissassi F, Bouvard V, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, et al. A review of human carcinogens—part E: tobacco, areca nut, alcohol, coal smoke, and salted fish. The Lancet Oncology. 2009;10(11):1033–1034. doi: 10.1016/s1470-2045(09)70326-2. [DOI] [PubMed] [Google Scholar]
- 11.van Kempen PM, Noorlag R, Braunius WW, Stegeman I, Willems SM, Grolman W. Differences in methylation profiles between HPV-positive and HPV-negative oropharynx squamous cell carcinoma: a systematic review. Epigenetics. 2014;9(2):194–203. doi: 10.4161/epi.26881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fratta E, Sigalotti L, Covre A, Parisi G, Coral S, Maio M. Epigenetics of melanoma: implications for immune-based therapies. Immunotherapy. 2013;5(10):1103–1116. doi: 10.2217/imt.13.108. [DOI] [PubMed] [Google Scholar]
- 13.Santosh B, Varshney A, Yadava PK. Non-coding RNAs: biological functions and applications. Cell Biochem Funct. 2015;33(1):14–22. doi: 10.1002/cbf.3079. [DOI] [PubMed] [Google Scholar]
- 14.Sun M, Liu X-H, Wang K-M, F-q N, Kong R, J-s Y, Xia R, Xu T-P, Jin F-Y, Liu Z-J, et al. Downregulation of BRAF activated non-coding RNA is associated with poor prognosis for non-small cell lung cancer and promotes metastasis by affecting epithelial-mesenchymal transition. Mol Cancer. 2014;13:68. doi: 10.1186/1476-4598-13-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang L, Lin C, Jin C, Yang JC, Tanasa B, Li W, Merkurjev D, Ohgi KA, Da M, Zhang J, et al. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature. 2013;500(7464):598–602. doi: 10.1038/nature12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 17.Singh N, Senapati S, Bose K. Insights into the mechanism of human papillomavirus E2-induced procaspase-8 activation and cell death. Sci Rep. 2016;6:21408. doi: 10.1038/srep21408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2(5):342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 19.Tommasino M. The human papillomavirus family and its role in carcinogenesis. Semin Cancer Biol. 2014;26:13–21. doi: 10.1016/j.semcancer.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Castellsague X, Alemany L, Quer M, Halec G, Quiros B, Tous S, Clavero O, Alos L, Biegner T, Szafarowski T, et al. HPV involvement in head and neck cancers: comprehensive assessment of biomarkers in 3680 patients. J Natl Cancer Inst. 2016;108(6):djv403. doi: 10.1093/jnci/djv403. [DOI] [PubMed] [Google Scholar]
- 21.Huber MA, Tantiwongkosi B. Oral and oropharyngeal cancer. Med Clin North Am. 2014;98(6):1299–1321. doi: 10.1016/j.mcna.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 22.Boscolo-Rizzo P, Schroeder L, Romeo S, Pawlita M. The prevalence of human papillomavirus in squamous cell carcinoma of unknown primary site metastatic to neck lymph nodes: a systematic review. Clin Exp Metastasis. 2015;32(8):835–845. doi: 10.1007/s10585-015-9744-z. [DOI] [PubMed] [Google Scholar]
- 23.Kimple RJ, Smith MA, Blitzer GC, Torres AD, Martin JA, Yang RZ, Peet CR, Lorenz LD, Nickel KP, Klingelhutz AJ, et al. Enhanced radiation sensitivity in HPV-positive head and neck cancer. Cancer Res. 2013;73(15):4791–4800. doi: 10.1158/0008-5472.CAN-13-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O'Rorke MA, Ellison MV, Murray LJ, Moran M, James J, Anderson LA. Human papillomavirus related head and neck cancer survival: a systematic review and meta-analysis. Oral Oncol. 2012;48(12):1191–1201. doi: 10.1016/j.oraloncology.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 25.Park JW, Nickel KP, Torres AD, Lee D, Lambert PF, Kimple RJ. Human papillomavirus type 16 E7 oncoprotein causes a delay in repair of DNA damage. Radiother Oncol. 2014;113(3):337–344. doi: 10.1016/j.radonc.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rieckmann T, Tribius S, Grob TJ, Meyer F, Busch CJ, Petersen C, Dikomey E, Kriegs M. HNSCC cell lines positive for HPV and p16 possess higher cellular radiosensitivity due to an impaired DSB repair capacity. Radiother Oncol. 2013;107(2):242–246. doi: 10.1016/j.radonc.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 27.Schroeder L, Boscolo-Rizzo P, Dal Cin E, Romeo S, Baboci L, Dyckhoff G, Hess J, Lucena-Porcel C, Byl A, Becker N, et al. Human papillomavirus as prognostic marker with rising prevalence in neck squamous cell carcinoma of unknown primary: a retrospective multicentre study. Eur J Cancer. 2017;74:73–81. doi: 10.1016/j.ejca.2016.12.020. [DOI] [PubMed] [Google Scholar]
- 28.D'Souza G, Kreimer AR, Viscidi R, Pawlita M, Fakhry C, Koch WM, Westra WH, Gillison ML. Case-control study of human papillomavirus and oropharyngeal cancer. N Engl J Med. 2007;356(19):1944–1956. doi: 10.1056/NEJMoa065497. [DOI] [PubMed] [Google Scholar]
- 29.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wichmann G, Rosolowski M, Krohn K, Kreuz M, Boehm A, Reiche A, Scharrer U, Halama D, Bertolini J, Bauer U, et al. The role of HPV RNA transcription, immune response-related gene expression and disruptive TP53 mutations in diagnostic and prognostic profiling of head and neck cancer. Int J Cancer. 2015;137(12):2846–2857. doi: 10.1002/ijc.29649. [DOI] [PubMed] [Google Scholar]
- 31.Shi H, Chen X, Lu C, Gu C, Jiang H, Meng R, Niu X, Huang Y, Lu M. Association between P16INK4a promoter methylation and HNSCC: a meta-analysis of 21 published studies. PLoS One. 2015;10(4):e0122302. doi: 10.1371/journal.pone.0122302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanken H, Grobe A, Cachovan G, Smeets R, Simon R, Sauter G, Heiland M, Blessmann M. CCND1 amplification and cyclin D1 immunohistochemical expression in head and neck squamous cell carcinomas. Clin Oral Investig. 2014;18(1):269–276. doi: 10.1007/s00784-013-0967-6. [DOI] [PubMed] [Google Scholar]
- 33.Cancer Genome Atlas N Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu X, Dakic A, Zhang Y, Dai Y, Chen R, Schlegel R. HPV E6 protein interacts physically and functionally with the cellular telomerase complex. Proc Natl Acad Sci U S A. 2009;106(44):18780–18785. doi: 10.1073/pnas.0906357106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghittoni R, Accardi R, Hasan U, Gheit T, Sylla B, Tommasino M. The biological properties of E6 and E7 oncoproteins from human papillomaviruses. Virus Genes. 2010;40(1):1–13. doi: 10.1007/s11262-009-0412-8. [DOI] [PubMed] [Google Scholar]
- 36.Parfenov M, Pedamallu CS, Gehlenborg N, Freeman SS, Danilova L, Bristow CA, Lee S, Hadjipanayis AG, Ivanova EV, Wilkerson MD, et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc Natl Acad Sci U S A. 2014;111(43):15544–15549. doi: 10.1073/pnas.1416074111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fratta E, Montico B, Rizzo A, Colizzi F, Sigalotti L, Dolcetti R. Epimutational profile of hematologic malignancies as attractive target for new epigenetic therapies. Oncotarget. 2016;7(35):57327–57350. doi: 10.18632/oncotarget.10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1(2):239–259. doi: 10.2217/epi.09.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Kempen PM, van Bockel L, Braunius WW, Moelans CB, van Olst M, de Jong R, Stegeman I, van Diest PJ, Grolman W, Willems SM. HPV-positive oropharyngeal squamous cell carcinoma is associated with TIMP3 and CADM1 promoter hypermethylation. Cancer Med. 2014;3(5):1185–1196. doi: 10.1002/cam4.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michie AM, McCaig AM, Nakagawa R, Vukovic M. Death-associated protein kinase (DAPK) and signal transduction: regulation in cancer. FEBS J. 2010;277(1):74–80. doi: 10.1111/j.1742-4658.2009.07414.x. [DOI] [PubMed] [Google Scholar]
- 41.Paschos K, Allday MJ. Epigenetic reprogramming of host genes in viral and microbial pathogenesis. Trends Microbiol. 2010;18(10):439–447. doi: 10.1016/j.tim.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boscolo-Rizzo P, Pawlita M, Holzinger D. From HPV-positive towards HPV-driven oropharyngeal squamous cell carcinomas. Cancer Treat Rev. 2016;42:24–29. doi: 10.1016/j.ctrv.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 43.de Freitas C-SM, Stur E, Agostini LP, de Podesta JR, de Oliveira JC, Soares MS, Mendonca EF, Gouvea SA, Von Zeidler SV, Louro ID. Promoter hypermethylation in primary squamous cell carcinoma of the oral cavity and oropharynx: a study of a Brazilian cohort. Mol Biol Rep. 2012;39(12):10111–10119. doi: 10.1007/s11033-012-1885-4. [DOI] [PubMed] [Google Scholar]
- 44.O'Regan EM, Toner ME, Finn SP, Fan CY, Ring M, Hagmar B, Timon C, Smyth P, Cahill S, Flavin R, et al. p16(INK4A) genetic and epigenetic profiles differ in relation to age and site in head and neck squamous cell carcinomas. Hum Pathol. 2008;39(3):452–458. doi: 10.1016/j.humpath.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 45.Taioli E, Ragin C, Wang XH, Chen J, Langevin SM, Brown AR, Gollin SM, Garte S, Sobol RW. Recurrence in oral and pharyngeal cancer is associated with quantitative MGMT promoter methylation. BMC Cancer. 2009;9:354. doi: 10.1186/1471-2407-9-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schlecht NF, Ben-Dayan M, Anayannis N, Lleras RA, Thomas C, Wang Y, Smith RV, Burk RD, Harris TM, Childs G, et al. Epigenetic changes in the CDKN2A locus are associated with differential expression of P16INK4A and P14ARF in HPV-positive oropharyngeal squamous cell carcinoma. Cancer Med. 2015;4(3):342–353. doi: 10.1002/cam4.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Julsing JR, Peters GJ. Methylation of DNA repair genes and the efficacy of DNA targeted anticancer treatment. Oncology Discovery. 2014;2(1):3. [Google Scholar]
- 48.Yao Y, Dai W. Genomic instability and cancer. J Carcinog Mutagen. 2014;5:1000165. doi: 10.4172/2157-2518.1000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reiter M, Baumeister P, Jaiser S, Reiss A, Schwenk-Zieger S, Kleinsasser N, Harreus U. DNA repair and mutagen sensitivity of epithelial cells and lymphocytes in oropharyngeal cancer. Oncol Lett. 2012;3(1):100–106. doi: 10.3892/ol.2011.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Colacino JA, Dolinoy DC, Duffy SA, Sartor MA, Chepeha DB, Bradford CR, McHugh JB, Patel DA, Virani S, Walline HM, et al. Comprehensive analysis of DNA methylation in head and neck squamous cell carcinoma indicates differences by survival and clinicopathologic characteristics. PLoS One. 2013;8(1):e54742. doi: 10.1371/journal.pone.0054742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weiss D, Basel T, Sachse F, Braeuninger A, Rudack C. Promoter methylation of cyclin A1 is associated with human papillomavirus 16 induced head and neck squamous cell carcinoma independently of p53 mutation. Mol Carcinog. 2011;50(9):680–688. doi: 10.1002/mc.20798. [DOI] [PubMed] [Google Scholar]
- 52.Huang T, Chen X, Hong Q, Deng Z, Ma H, Xin Y, Fang Y, Ye H, Wang R, Zhang C, et al. Meta-analyses of gene methylation and smoking behavior in non-small cell lung cancer patients. Sci Rep. 2015;5:8897. doi: 10.1038/srep08897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gordon MD, Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem. 2006;281(32):22429–22433. doi: 10.1074/jbc.R600015200. [DOI] [PubMed] [Google Scholar]
- 54.Paluszczak J, Sarbak J, Kostrzewska-Poczekaj M, Kiwerska K, Jarmuz-Szymczak M, Grenman R, Mielcarek-Kuchta D, Baer-Dubowska W. The negative regulators of Wnt pathway-DACH1, DKK1, and WIF1 are methylated in oral and oropharyngeal cancer and WIF1 methylation predicts shorter survival. Tumour Biol. 2015;36(4):2855–2861. doi: 10.1007/s13277-014-2913-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caldwell GM, Jones C, Gensberg K, Jan S, Hardy RG, Byrd P, Chughtai S, Wallis Y, Matthews GM, Morton DG. The Wnt antagonist sFRP1 in colorectal tumorigenesis. Cancer Res. 2004;64(3):883–888. doi: 10.1158/0008-5472.can-03-1346. [DOI] [PubMed] [Google Scholar]
- 56.Marsit CJ, McClean MD, Furniss CS, Kelsey KT. Epigenetic inactivation of the SFRP genes is associated with drinking, smoking and HPV in head and neck squamous cell carcinoma. Int J Cancer. 2006;119(8):1761–1766. doi: 10.1002/ijc.22051. [DOI] [PubMed] [Google Scholar]
- 57.Noorlag R, van Kempen PM, Moelans CB, de Jong R, Blok LE, Koole R, Grolman W, van Diest PJ, van Es RJ, Willems SM. Promoter hypermethylation using 24-gene array in early head and neck cancer: better outcome in oral than in oropharyngeal cancer. Epigenetics. 2014;9(9):1220–1227. doi: 10.4161/epi.29785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khan K, Araki K, Wang D, Li G, Li X, Zhang J, Xu W, Hoover RK, Lauter S, O’Malley B, Jr, et al. Head and neck cancer radiosensitization by the novel poly(ADP-ribose) polymerase inhibitor GPI-15427. Head Neck. 2010;32(3):381–391. doi: 10.1002/hed.21195. [DOI] [PubMed] [Google Scholar]
- 59.Chow JP, Man WY, Mao M, Chen H, Cheung F, Nicholls J, Tsao SW, Li Lung M, Poon RY. PARP1 is overexpressed in nasopharyngeal carcinoma and its inhibition enhances radiotherapy. Mol Cancer Ther. 2013;12(11):2517–2528. doi: 10.1158/1535-7163.MCT-13-0010. [DOI] [PubMed] [Google Scholar]
- 60.Furlan C, Polesel J, Barzan L, Franchin G, Sulfaro S, Romeo S, Colizzi F, Rizzo A, Baggio V, Giacomarra V, et al. Prognostic significance of LINE-1 hypomethylation in oropharyngeal squamous cell carcinoma. Clin Epigenetics. 2017;9:58. doi: 10.1186/s13148-017-0357-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sartor MA, Dolinoy DC, Jones TR, Colacino JA, Prince ME, Carey TE, Rozek LS. Genome-wide methylation and expression differences in HPV(+) and HPV(−) squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis. Epigenetics. 2011;6(6):777–787. doi: 10.4161/epi.6.6.16216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Richards KL, Zhang B, Baggerly KA, Colella S, Lang JC, Schuller DE, Krahe R. Genome-wide hypomethylation in head and neck cancer is more pronounced in HPV-negative tumors and is associated with genomic instability. PLoS One. 2009;4(3):e4941. doi: 10.1371/journal.pone.0004941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Galvan SC, Martinez-Salazar M, Galvan VM, Mendez R, Diaz-Contreras GT, Alvarado-Hermida M, Alcantara-Silva R, Garcia-Carranca A. Analysis of CpG methylation sites and CGI among human papillomavirus DNA genomes. BMC Genomics. 2011;12:580. doi: 10.1186/1471-2164-12-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laurson J, Khan S, Chung R, Cross K, Raj K. Epigenetic repression of E-cadherin by human papillomavirus 16 E7 protein. Carcinogenesis. 2010;31(5):918–926. doi: 10.1093/carcin/bgq027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martinez I, Wang J, Hobson KF, Ferris RL, Khan SA. Identification of differentially expressed genes in HPV-positive and HPV-negative oropharyngeal squamous cell carcinomas. Eur J Cancer. 2007;43(2):415–432. doi: 10.1016/j.ejca.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hatano T, Sano D, Takahashi H, Hyakusoku H, Isono Y, Shimada S, Sawakuma K, Takada K, Oikawa R, Watanabe Y, et al. Identification of human papillomavirus (HPV) 16 DNA integration and the ensuing patterns of methylation in HPV-associated head and neck squamous cell carcinoma cell lines. Int J Cancer. 2017;140(7):1571–1580. doi: 10.1002/ijc.30589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vinokurova S, von Knebel DM. Differential methylation of the HPV 16 upstream regulatory region during epithelial differentiation and neoplastic transformation. PLoS One. 2011;6(9):e24451. doi: 10.1371/journal.pone.0024451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chong T, Apt D, Gloss B, Isa M, Bernard HU. The enhancer of human papillomavirus type 16: binding sites for the ubiquitous transcription factors oct-1, NFA, TEF-2, NF1, and AP-1 participate in epithelial cell-specific transcription. J Virol. 1991;65(11):5933–5943. doi: 10.1128/jvi.65.11.5933-5943.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pientong C, Wongwarissara P, Ekalaksananan T, Swangphon P, Kleebkaow P, Kongyingyoes B, Siriaunkgul S, Tungsinmunkong K, Suthipintawong C. Association of human papillomavirus type 16 long control region mutation and cervical cancer. Virol J. 2013;10(1):30. doi: 10.1186/1743-422X-10-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen Z, Storthz KA, Shillitoe EJ. Mutations in the long control region of human papillomavirus DNA in oral cancer cells, and their functional consequences. Cancer Res. 1997;57(8):1614–1619. [PubMed] [Google Scholar]
- 71.Park IS, Chang X, Loyo M, Wu G, Chuang A, Kim MS, Chae YK, Lyford-Pike S, Westra WH, Saunders JR, et al. Characterization of the methylation patterns in human papillomavirus type 16 viral DNA in head and neck cancers. Cancer Prev Res (Phila) 2011;4(2):207–217. doi: 10.1158/1940-6207.CAPR-10-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wilson GA, Lechner M, Koferle A, Caren H, Butcher LM, Feber A, Fenton T, Jay A, Boshoff C, Beck S. Integrated virus-host methylome analysis in head and neck squamous cell carcinoma. Epigenetics. 2013;8(9):953–961. doi: 10.4161/epi.25614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fernandez AF, Rosales C, Lopez-Nieva P, Grana O, Ballestar E, Ropero S, Espada J, Melo SA, Lujambio A, Fraga MF, et al. The dynamic DNA methylomes of double-stranded DNA viruses associated with human cancer. Genome Res. 2009;19(3):438–451. doi: 10.1101/gr.083550.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mirabello L, Frimer M, Harari A, McAndrew T, Smith B, Chen Z, Wentzensen N, Wacholder S, Castle PE, Raine-Bennett T, et al. HPV16 methyl-haplotypes determined by a novel next-generation sequencing method are associated with cervical precancer. Int J Cancer. 2015;136(4):E146–E153. doi: 10.1002/ijc.29119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Doeberitz M, Vinokurova S. Host factors in HPV-related carcinogenesis: cellular mechanisms controlling HPV infections. Arch Med Res. 2009;40(6):435–442. doi: 10.1016/j.arcmed.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 76.Choo KB, Pan CC, Han SH. Integration of human papillomavirus type 16 into cellular DNA of cervical carcinoma: preferential deletion of the E2 gene and invariable retention of the long control region and the E6/E7 open reading frames. Virology. 1987;161(1):259–261. doi: 10.1016/0042-6822(87)90195-4. [DOI] [PubMed] [Google Scholar]
- 77.Olthof NC, Speel EJ, Kolligs J, Haesevoets A, Henfling M, Ramaekers FC, Preuss SF, Drebber U, Wieland U, Silling S, et al. Comprehensive analysis of HPV16 integration in OSCC reveals no significant impact of physical status on viral oncogene and virally disrupted human gene expression. PLoS One. 2014;9(2):e88718. doi: 10.1371/journal.pone.0088718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xu F, Cao M, Shi Q, Chen H, Wang Y, Li X. Integration of the full-length HPV16 genome in cervical cancer and Caski and Siha cell lines and the possible ways of HPV integration. Virus Genes. 2015;50(2):210–220. doi: 10.1007/s11262-014-1164-7. [DOI] [PubMed] [Google Scholar]
- 79.Reuschenbach M, Huebbers CU, Prigge ES, Bermejo JL, Kalteis MS, Preuss SF, Seuthe IM, Kolligs J, Speel EJ, Olthof N, et al. Methylation status of HPV16 E2-binding sites classifies subtypes of HPV-associated oropharyngeal cancers. Cancer. 2015;121(12):1966–1976. doi: 10.1002/cncr.29315. [DOI] [PubMed] [Google Scholar]
- 80.Zhang C, Deng Z, Pan X, Uehara T, Suzuki M, Xie M. Effects of methylation status of CpG sites within the HPV16 long control region on HPV16-positive head and neck cancer cells. PLoS One. 2015;10(10):e0141245. doi: 10.1371/journal.pone.0141245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ishida E, Nakamura M, Ikuta M, Shimada K, Matsuyoshi S, Kirita T, Konishi N. Promotor hypermethylation of p14ARF is a key alteration for progression of oral squamous cell carcinoma. Oral Oncol. 2005;41(6):614–622. doi: 10.1016/j.oraloncology.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 82.Saatci C, Caglayan AO, Ozkul Y, Tahiri S, Turhan AB, Dundar M. Detection of p16 promotor hypermethylation in “Maras powder” and tobacco users. Cancer Epidemiol. 2009;33(1):47–50. doi: 10.1016/j.canep.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 83.Zeilinger S, Kuhnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, Weidinger S, Lattka E, Adamski J, Peters A, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013;8(5):e63812. doi: 10.1371/journal.pone.0063812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee KW, Pausova Z. Cigarette smoking and DNA methylation. Front Genet. 2013;4:132. doi: 10.3389/fgene.2013.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wangsri S, Subbalekha K, Kitkumthorn N, Mutirangura A. Patterns and possible roles of LINE-1 methylation changes in smoke-exposed epithelia. PLoS One. 2012;7(9):e45292. doi: 10.1371/journal.pone.0045292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Warad S, Kalburgi NB, Manak M, Kalburgi VC, Koregol AC, Patanashetti J, Rao S, Kokatnur MV. Determining the effect of gutkha on serum levels of vitamin B12 and folic acid as compared to smoking among chronic periodontitis subjects: a cross-sectional study. J Clin Diagn Res. 2014;8(12):ZC85–ZC89. doi: 10.7860/JCDR/2014/10575.5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Crider KS, Yang TP, Berry RJ, Bailey LB. Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate's role. Adv Nutr. 2012;3(1):21–38. doi: 10.3945/an.111.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gabriel HE, Crott JW, Ghandour H, Dallal GE, Choi S-W, Keyes MK, Jang H, Liu Z, Nadeau M, Johnston A, et al. Chronic cigarette smoking is associated with diminished folate status, altered folate form distribution, and increased genetic damage in the buccal mucosa of healthy adults. Am J Clin Nutr. 2006;83(4):835–841. doi: 10.1093/ajcn/83.4.835. [DOI] [PubMed] [Google Scholar]
- 89.Colacino JA, Arthur AE, Dolinoy DC, Sartor MA, Duffy SA, Chepeha DB, Bradford CR, Walline HM, McHugh JB, D'Silva N, et al. Pretreatment dietary intake is associated with tumor suppressor DNA methylation in head and neck squamous cell carcinomas. Epigenetics. 2012;7(8):883–891. doi: 10.4161/epi.21038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Varela-Rey M, Woodhoo A, Martinez-Chantar ML, Mato JM, Lu SC. Alcohol, DNA methylation, and cancer. Alcohol Res. 2013;35(1):25–35. [PMC free article] [PubMed] [Google Scholar]
- 91.Zakhari S. Alcohol metabolism and epigenetics changes. Alcohol Res. 2013;35(1):6–16. [PMC free article] [PubMed] [Google Scholar]
- 92.Bonsch D, Lenz B, Fiszer R, Frieling H, Kornhuber J, Bleich S. Lowered DNA methyltransferase (DNMT-3b) mRNA expression is associated with genomic DNA hypermethylation in patients with chronic alcoholism. J Neural Transm (Vienna) 2006;113(9):1299–1304. doi: 10.1007/s00702-005-0413-2. [DOI] [PubMed] [Google Scholar]
- 93.Mukhopadhyay P, Rezzoug F, Kaikaus J, Greene RM, Pisano MM. Alcohol modulates expression of DNA methyltranferases and methyl CpG-/CpG domain-binding proteins in murine embryonic fibroblasts. Reprod Toxicol. 2013;37:40–48. doi: 10.1016/j.reprotox.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cervera-Juanes R, Wilhelm LJ, Park B, Grant KA, Ferguson B. Genome-wide analysis of the nucleus accumbens identifies DNA methylation signals differentiating low/binge from heavy alcohol drinking. Alcohol. 2017;60:103–113. doi: 10.1016/j.alcohol.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mani S, Szymanska K, Cuenin C, Zaridze D, Balassiano K, Lima SC, Matos E, Daudt A, Koifman S, Filho VW, et al. DNA methylation changes associated with risk factors in tumors of the upper aerodigestive tract. Epigenetics. 2012;7(3):270–277. doi: 10.4161/epi.7.3.19306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta. 2014;1839(8):627–643. doi: 10.1016/j.bbagrm.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell. 2008;31(4):449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cheung P, Lau P. Epigenetic regulation by histone methylation and histone variants 97. Mol Endocrinol. 2005;19(3):563–573. doi: 10.1210/me.2004-0496. [DOI] [PubMed] [Google Scholar]
- 99.Vakoc CR, Mandat SA, Olenchock BA, Blobel GA. Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol Cell. 2005;19(3):381–391. doi: 10.1016/j.molcel.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 100.Seiwert TY, Zuo Z, Keck MK, Khattri A, Pedamallu CS, Stricker T, Brown C, Pugh TJ, Stojanov P, Cho J, et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res. 2015;21(3):632–641. doi: 10.1158/1078-0432.CCR-13-3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Papillon-Cavanagh S, Lu C, Gayden T, Mikael LG, Bechet D, Karamboulas C, Ailles L, Karamchandani J, Marchione DM, Garcia BA, et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat Genet. 2017;49(2):180–185. doi: 10.1038/ng.3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Marzluff WF, Duronio RJ. Histone mRNA expression: multiple levels of cell cycle regulation and important developmental consequences. Curr Opin Cell Biol. 2002;14(6):692–699. doi: 10.1016/s0955-0674(02)00387-3. [DOI] [PubMed] [Google Scholar]
- 103.Talbert PB, Henikoff S. Histone variants—ancient wrap artists of the epigenome. Nat Rev Mol Cell Bio. 2010;11(4):264–275. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- 104.Bakkenist CJ, Kastan MB. Chromatin perturbations during the DNA damage response in higher eukaryotes. DNA Repair (Amst) 2015;36:8–12. doi: 10.1016/j.dnarep.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yamagata K, Kitabayashi I. Sirt1 physically interacts with Tip60 and negatively regulates Tip60-mediated acetylation of H2AX. Biochem Biophys Res Commun. 2009;390(4):1355–1360. doi: 10.1016/j.bbrc.2009.10.156. [DOI] [PubMed] [Google Scholar]
- 106.Klokov D, MacPhail SM, Banath JP, Byrne JP, Olive PL. Phosphorylated histone H2AX in relation to cell survival in tumor cells and xenografts exposed to single and fractionated doses of X-rays. Radiother Oncol. 2006;80(2):223–229. doi: 10.1016/j.radonc.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 107.Durzynska J, Lesniewicz K, Poreba E. Human papillomaviruses in epigenetic regulations. Mutat Res. 2017;772:36–50. doi: 10.1016/j.mrrev.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 108.Thomas MC, Chiang CM. E6 oncoprotein represses p53-dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol Cell. 2005;17(2):251–264. doi: 10.1016/j.molcel.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 109.Jansma AL, Martinez-Yamout MA, Liao R, Sun P, Dyson HJ, Wright PE. The high-risk HPV16 E7 oncoprotein mediates interaction between the transcriptional coactivator CBP and the retinoblastoma protein pRb. J Mol Biol. 2014;426(24):4030–4048. doi: 10.1016/j.jmb.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brehm A, Nielsen SJ, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 1999;18(9):2449–2458. doi: 10.1093/emboj/18.9.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Longworth MS, Wilson R, Laimins LA. HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs. EMBO J. 2005;24(10):1821–1830. doi: 10.1038/sj.emboj.7600651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang B, Laribee RN, Klemsz MJ, Roman A. Human papillomavirus type 16 E7 protein increases acetylation of histone H3 in human foreskin keratinocytes. Virology. 2004;329(1):189–198. doi: 10.1016/j.virol.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 113.Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439(7078):871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 114.Sauvageau M, Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010;7(3):299–313. doi: 10.1016/j.stem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Holland D, Hoppe-Seyler K, Schuller B, Lohrey C, Maroldt J, Durst M, Hoppe-Seyler F. Activation of the enhancer of zeste homologue 2 gene by the human papillomavirus E7 oncoprotein. Cancer Res. 2008;68(23):9964–9972. doi: 10.1158/0008-5472.CAN-08-1134. [DOI] [PubMed] [Google Scholar]
- 116.Idris S, Lindsay C, Kostiuk M, Andrews C, Cote DW, O'Connell DA, Harris J, Seikaly H, Biron VL. Investigation of EZH2 pathways for novel epigenetic treatment strategies in oropharyngeal cancer. J Otolaryngol Head Neck Surg. 2016;45(1):54. doi: 10.1186/s40463-016-0168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Biron VL, Mohamed A, Hendzel MJ, Alan Underhill D, Seikaly H. Epigenetic differences between human papillomavirus-positive and -negative oropharyngeal squamous cell carcinomas. J Otolaryngol Head Neck Surg. 2012;41(Suppl 1):S65–S70. [PubMed] [Google Scholar]
- 118.Lechner M, Fenton T, West J, Wilson G, Feber A, Henderson S, Thirlwell C, Dibra HK, Jay A, Butcher L, et al. Identification and functional validation of HPV-mediated hypermethylation in head and neck squamous cell carcinoma. Genome Med. 2013;5(2):15. doi: 10.1186/gm419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.McLaughlin-Drubin ME, Crum CP, Munger K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc Natl Acad Sci U S A. 2011;108(5):2130–2135. doi: 10.1073/pnas.1009933108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Barradas M, Anderton E, Acosta JC, Li S, Banito A, Rodriguez-Niedenfuhr M, Maertens G, Banck M, Zhou MM, Walsh MJ, et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009;23(10):1177–1182. doi: 10.1101/gad.511109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sundar IK, Nevid MZ, Friedman AE, Rahman I. Cigarette smoke induces distinct histone modifications in lung cells: implications for the pathogenesis of COPD and lung cancer. J Proteome Res. 2014;13(2):982–996. doi: 10.1021/pr400998n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hussain M, Rao M, Humphries AE, Hong JA, Liu F, Yang M, Caragacianu D, Schrump DS. Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer Res. 2009;69(8):3570–3578. doi: 10.1158/0008-5472.CAN-08-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Qiu F, Liang CL, Liu H, Zeng YQ, Hou S, Huang S, Lai X, Dai Z. Impacts of cigarette smoking on immune responsiveness: up and down or upside down? Oncotarget. 2017;8(1):268–284. doi: 10.18632/oncotarget.13613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dillon MT, Harrington KJ. Human papillomavirus-negative pharyngeal cancer. J Clin Oncol. 2015;33(29):3251–3261. doi: 10.1200/JCO.2015.60.7804. [DOI] [PubMed] [Google Scholar]
- 125.Gomes AQ, Nolasco S, Soares H. Non-coding RNAs: multi-tasking molecules in the cell. Int J Mol Sci. 2013;14(8):16010–16039. doi: 10.3390/ijms140816010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 2002;21(17):4663–4670. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang X, Gee H, Rose B, Lee CS, Clark J, Elliott M, Gamble JR, Cairns MJ, Harris A, Khoury S, et al. Regulation of the tumour suppressor PDCD4 by miR-499 and miR-21 in oropharyngeal cancers. BMC Cancer. 2016;16:86. doi: 10.1186/s12885-016-2109-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jakymiw A, Patel RS, Deming N, Bhattacharyya I, Shah P, Lamont RJ, Stewart CM, Cohen DM, Chan EK. Overexpression of dicer as a result of reduced let-7 MicroRNA levels contributes to increased cell proliferation of oral cancer cells. Genes Chromosomes Cancer. 2010;49(6):549–559. doi: 10.1002/gcc.20765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Manasa VG, Kannan S. Impact of microRNA dynamics on cancer hallmarks: an oral cancer scenario. Tumour Biol. 2017;39(3):1010428317695920. doi: 10.1177/1010428317695920. [DOI] [PubMed] [Google Scholar]
- 130.Li Y, Xu Z, Li B, Zhang Z, Luo H, Wang Y, Lu Z, Wu X. Epigenetic silencing of miRNA-9 is correlated with promoter-proximal CpG island hypermethylation in gastric cancer in vitro and in vivo. Int J Oncol. 2014;45(6):2576–2586. doi: 10.3892/ijo.2014.2667. [DOI] [PubMed] [Google Scholar]
- 131.Minor J, Wang X, Zhang F, Song J, Jimeno A, Wang XJ, Lu X, Gross N, Kulesz-Martin M, Wang D, et al. Methylation of microRNA-9 is a specific and sensitive biomarker for oral and oropharyngeal squamous cell carcinomas. Oral Oncol. 2012;48(1):73–78. doi: 10.1016/j.oraloncology.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nohata N, Hanazawa T, Kinoshita T, Inamine A, Kikkawa N, Itesako T, Yoshino H, Enokida H, Nakagawa M, Okamoto Y, et al. Tumour-suppressive microRNA-874 contributes to cell proliferation through targeting of histone deacetylase 1 in head and neck squamous cell carcinoma. Br J Cancer. 2013;108(8):1648–1658. doi: 10.1038/bjc.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Forrest ME, Khalil AM. Review: regulation of the cancer epigenome by long non-coding RNAs. Cancer Lett. 2017;407:106-112. [DOI] [PubMed]
- 134.Jiang Y, Li Y, Fang S, Jiang B, Qin C, Xie P, Zhou G, Li G. The role of MALAT1 correlates with HPV in cervical cancer. Oncol Lett. 2014;7(6):2135–2141. doi: 10.3892/ol.2014.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tomar S, Graves CA, Altomare D, Kowli S, Kassler S, Sutkowski N, Gillespie MB, Creek KE, Pirisi L. Human papillomavirus status and gene expression profiles of oropharyngeal and oral cancers from European American and African American patients. Head Neck. 2016;38 Suppl 1(Suppl 1):E694–E704. doi: 10.1002/hed.24072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gunasekharan V, Laimins LA. Human papillomaviruses modulate microRNA 145 expression to directly control genome amplification. J Virol. 2013;87(10):6037–6043. doi: 10.1128/JVI.00153-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang X, Wang HK, Li Y, Hafner M, Banerjee NS, Tang S, Briskin D, Meyers C, Chow LT, Xie X, et al. microRNAs are biomarkers of oncogenic human papillomavirus infections. Proc Natl Acad Sci U S A. 2014;111(11):4262–4267. doi: 10.1073/pnas.1401430111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Melar-New M, Laimins LA. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J Virol. 2010;84(10):5212–5221. doi: 10.1128/JVI.00078-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chiantore MV, Mangino G, Iuliano M, Zangrillo MS, De Lillis I, Vaccari G, Accardi R, Tommasino M, Columba Cabezas S, Federico M, et al. Human papillomavirus E6 and E7 oncoproteins affect the expression of cancer-related microRNAs: additional evidence in HPV-induced tumorigenesis. J Cancer Res Clin Oncol. 2016;142(8):1751–1763. doi: 10.1007/s00432-016-2189-1. [DOI] [PubMed] [Google Scholar]
- 140.Miller DL, Davis JW, Taylor KH, Johnson J, Shi Z, Williams R, Atasoy U, Lewis JS, Jr, Stack MS. Identification of a human papillomavirus-associated oncogenic miRNA panel in human oropharyngeal squamous cell carcinoma validated by bioinformatics analysis of the Cancer Genome Atlas. Am J Pathol. 2015;185(3):679–692. doi: 10.1016/j.ajpath.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Liu W, Gao G, Hu X, Wang Y, Schwarz JK, Chen JJ, Grigsby PW, Wang X. Activation of miR-9 by human papillomavirus in cervical cancer. Oncotarget. 2014;5(22):11620–11630. doi: 10.18632/oncotarget.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lajer CB, Nielsen FC, Friis-Hansen L, Norrild B, Borup R, Garnaes E, Rossing M, Specht L, Therkildsen MH, Nauntofte B, et al. Different miRNA signatures of oral and pharyngeal squamous cell carcinomas: a prospective translational study. Br J Cancer. 2011;104(5):830–840. doi: 10.1038/bjc.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wald AI, Hoskins EE, Wells SI, Ferris RL, Khan SA. Alteration of microRNA profiles in squamous cell carcinoma of the head and neck cell lines by human papillomavirus. Head Neck. 2011;33(4):504–512. doi: 10.1002/hed.21475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jung HM, Phillips BL, Chan EK. miR-375 activates p21 and suppresses telomerase activity by coordinately regulating HPV E6/E7, E6AP, CIP2A, and 14-3-3zeta. Mol Cancer. 2014;13:80. doi: 10.1186/1476-4598-13-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Liu S, Song L, Yao H, Zhang L, Xu D, Gao F, Li Q. MiR-375 is epigenetically downregulated by HPV-16 E6 mediated DNMT1 upregulation and modulates EMT of cervical cancer cells by suppressing lncRNA MALAT1. PLoS One. 2016;11(9):e0163460. doi: 10.1371/journal.pone.0163460. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 146.Gutschner T, Hammerle M, Diederichs S. MALAT1—a paradigm for long noncoding RNA function in cancer. J Mol Med (Berl) 2013;91(7):791–801. doi: 10.1007/s00109-013-1028-y. [DOI] [PubMed] [Google Scholar]
- 147.Salyakina D, Tsinoremas NF. Non-coding RNAs profiling in head and neck cancers. npj Genomic Medicine. 2016;1:15004. [DOI] [PMC free article] [PubMed]
- 148.Tsai YS, Lin CS, Chiang SL, Lee CH, Lee KW, Ko YC. Areca nut induces miR-23a and inhibits repair of DNA double-strand breaks by targeting FANCG. Toxicol Sci. 2011;123(2):480–490. doi: 10.1093/toxsci/kfr182. [DOI] [PubMed] [Google Scholar]
- 149.Pal A, Melling G, Hinsley EE, Kabir TD, Colley HE, Murdoch C, Lambert DW. Cigarette smoke condensate promotes pro-tumourigenic stromal-epithelial interactions by suppressing miR-145. J Oral Pathol Med. 2013;42(4):309–314. doi: 10.1111/jop.12017. [DOI] [PubMed] [Google Scholar]
- 150.Saad MA, Kuo SZ, Rahimy E, Zou AE, Korrapati A, Rahimy M, Kim E, Zheng H, Yu MA, Wang-Rodriguez J, et al. Alcohol-dysregulated miR-30a and miR-934 in head and neck squamous cell carcinoma. Mol Cancer. 2015;14(1):181. doi: 10.1186/s12943-015-0452-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Avissar M, McClean MD, Kelsey KT, Marsit CJ. MicroRNA expression in head and neck cancer associates with alcohol consumption and survival. Carcinogenesis. 2009;30(12):2059–2063. doi: 10.1093/carcin/bgp277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sigalotti L, Fratta E, Parisi G, Coral S, Maio M. Epigenetic markers of prognosis in melanoma. In: Thurin M, Marincola FM, editors. Molecular diagnostics for melanoma: methods and protocols. Totowa: Humana Press; 2014. pp. 481–499. [DOI] [PubMed] [Google Scholar]
- 153.Gubanova E, Brown B, Ivanov SV, Helleday T, Mills GB, Yarbrough WG, Issaeva N. Downregulation of SMG-1 in HPV-positive head and neck squamous cell carcinoma due to promoter hypermethylation correlates with improved survival. Clin Cancer Res. 2012;18(5):1257–1267. doi: 10.1158/1078-0432.CCR-11-2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kostareli E, Holzinger D, Bogatyrova O, Hielscher T, Wichmann G, Keck M, Lahrmann B, Grabe N, Flechtenmacher C, Schmidt CR, et al. HPV-related methylation signature predicts survival in oropharyngeal squamous cell carcinomas. J Clin Invest. 2013;123(6):2488–2501. doi: 10.1172/JCI67010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Park NJ, Zhou H, Elashoff D, Henson BS, Kastratovic DA, Abemayor E, Wong DT. Salivary microRNA: discovery, characterization, and clinical utility for oral cancer detection. Clin Cancer Res. 2009;15(17):5473–5477. doi: 10.1158/1078-0432.CCR-09-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Firmino N, Martinez VD, Rowbotham DA, Enfield KSS, Bennewith KL, Lam WL. HPV status is associated with altered PIWI-interacting RNA expression pattern in head and neck cancer. Oral Oncol. 2016;55:43–48. doi: 10.1016/j.oraloncology.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Stich M, Ganss L, Puschhof J, Prigge ES, Reuschenbach M, Guiterrez A, Vinokurova S, von Knebel DM. 5-aza-2′-deoxycytidine (DAC) treatment downregulates the HPV E6 and E7 oncogene expression and blocks neoplastic growth of HPV-associated cancer cells. Oncotarget. 2017;8(32):52104–52117. doi: 10.18632/oncotarget.10631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lu D, Hoory T, Monie A, Wu A, Wang MC, Hung CF. Treatment with demethylating agent, 5-aza-2′-deoxycytidine enhances therapeutic HPV DNA vaccine potency. Vaccine. 2009;27(32):4363–4369. doi: 10.1016/j.vaccine.2009.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Miki Y, Mukae S, Murakami M, Ishikawa Y, Ishii T, Ohki H, Matsumoto M, Komiyama K. Butyrate inhibits oral cancer cell proliferation and regulates expression of secretory phospholipase A2-X and COX-2. Anticancer Res. 2007;27(3B):1493–1502. [PubMed] [Google Scholar]
- 160.Gasche JA, Goel A. Epigenetic mechanisms in oral carcinogenesis. Future Oncol. 2012;8(11):1407–1425. doi: 10.2217/fon.12.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Viet CT, Dang D, Achdjian S, Ye Y, Katz SG, Schmidt BL. Decitabine rescues cisplatin resistance in head and neck squamous cell carcinoma. PLoS One. 2014;9(11):e112880. doi: 10.1371/journal.pone.0112880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Diyabalanage HV, Granda ML, Hooker JM. Combination therapy: histone deacetylase inhibitors and platinum-based chemotherapeutics for cancer. Cancer Lett. 2013;329(1):1–8. doi: 10.1016/j.canlet.2012.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Brieger J, Mann SA, Pongsapich W, Koutsimpelas D, Fruth K, Mann WJ. Pharmacological genome demethylation increases radiosensitivity of head and neck squamous carcinoma cells. Int J Mol Med. 2012;29(3):505–509. doi: 10.3892/ijmm.2011.843. [DOI] [PubMed] [Google Scholar]
- 164.Chung YL, Lee MY, Pui NN. Epigenetic therapy using the histone deacetylase inhibitor for increasing therapeutic gain in oral cancer: prevention of radiation-induced oral mucositis and inhibition of chemical-induced oral carcinogenesis. Carcinogenesis. 2009;30(8):1387–1397. doi: 10.1093/carcin/bgp079. [DOI] [PubMed] [Google Scholar]
- 165.Lleras RA, Smith RV, Adrien LR, Schlecht NF, Burk RD, Harris TM, Childs G, Prystowsky MB, Belbin TJ. Unique DNA methylation loci distinguish anatomic site and HPV status in head and neck squamous cell carcinoma. Clin Cancer Res. 2013;19(19):5444–5455. doi: 10.1158/1078-0432.CCR-12-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chen KM, Stephen JK, Havard S, Mahan M, Divine G, Worsham MJ. IGSF4 methylation as an independent marker of human papillomavirus-positive oropharyngeal squamous cell carcinoma. JAMA otolaryngology. 2015;141(3):257–263. doi: 10.1001/jamaoto.2014.3393. [DOI] [PubMed] [Google Scholar]
- 167.Weiss D, Stockmann C, Schrodter K, Rudack C. Protein expression and promoter methylation of the candidate biomarker TCF21 in head and neck squamous cell carcinoma. Cell Oncol (Dordr) 2013;36(3):213–224. doi: 10.1007/s13402-013-0129-5. [DOI] [PubMed] [Google Scholar]
- 168.Gao G, Gay HA, Chernock RD, Zhang TR, Luo J, Thorstad WL, Lewis JS, Jr, Wang X. A microRNA expression signature for the prognosis of oropharyngeal squamous cell carcinoma. Cancer. 2013;119(1):72–80. doi: 10.1002/cncr.27696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hui AB, Lin A, Xu W, Waldron L, Perez-Ordonez B, Weinreb I, Shi W, Bruce J, Huang SH, O'Sullivan B, et al. Potentially prognostic miRNAs in HPV-associated oropharyngeal carcinoma. Clin Cancer Res. 2013;19(8):2154–2162. doi: 10.1158/1078-0432.CCR-12-3572. [DOI] [PubMed] [Google Scholar]
- 170.Barker EV, Cervigne NK, Reis PP, Goswami RS, Xu W, Weinreb I, Irish JC, Kamel-Reid S. microRNA evaluation of unknown primary lesions in the head and neck. Mol Cancer. 2009;8(1):127. doi: 10.1186/1476-4598-8-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kozaki K, Imoto I, Mogi S, Omura K, Inazawa J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008;68(7):2094–2105. doi: 10.1158/0008-5472.CAN-07-5194. [DOI] [PubMed] [Google Scholar]
- 172.Poage GM, Butler RA, Houseman EA, McClean MD, Nelson HH, Christensen BC, Marsit CJ, Kelsey KT. Identification of an epigenetic profile classifier that is associated with survival in head and neck cancer. Cancer Res. 2012;72(11):2728–2737. doi: 10.1158/0008-5472.CAN-11-4121-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Melchers LJ, Clausen MJ, Mastik MF, Slagter-Menkema L, van der Wal JE, Wisman GB, Roodenburg JL, Schuuring E. Identification of methylation markers for the prediction of nodal metastasis in oral and oropharyngeal squamous cell carcinoma. Epigenetics. 2015;10(9):850–860. doi: 10.1080/15592294.2015.1075689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Misawa K, Mochizuki D, Endo S, Mima M, Misawa Y, Imai A, Shinmura K, Kanazawa T, Carey TE, Mineta H. Site-specific methylation patterns of the GAL and GALR1/2 genes in head and neck cancer: potential utility as biomarkers for prognosis. Mol Carcinog. 2017;56(3):1107–1116. doi: 10.1002/mc.22577. [DOI] [PubMed] [Google Scholar]
- 175.Childs G, Fazzari M, Kung G, Kawachi N, Brandwein-Gensler M, McLemore M, Chen Q, Burk RD, Smith RV, Prystowsky MB, et al. Low-level expression of microRNAs let-7d and miR-205 are prognostic markers of head and neck squamous cell carcinoma. Am J Pathol. 2009;174(3):736–745. doi: 10.2353/ajpath.2009.080731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chen X, Sturgis EM, Wang C, Cao X, Li Y, Wei Q, Li G. Significance of microRNA-related variants in susceptibility to recurrence of oropharyngeal cancer patients after definitive radiotherapy. Oncotarget. 2016;7(23):35015–35025. doi: 10.18632/oncotarget.9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wong N, Khwaja SS, Baker CM, Gay HA, Thorstad WL, Daly MD, Lewis JS, Jr, Wang X. Prognostic microRNA signatures derived from The Cancer Genome Atlas for head and neck squamous cell carcinomas. Cancer Med. 2016;5(7):1619–1628. doi: 10.1002/cam4.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.