

Supplemental Digital Content is available in the text.
Keywords: exercise, glycolysis, hypertrophy, metabolomics, mitochondria, ventricular remodeling
Abstract
Background:
Exercise promotes metabolic remodeling in the heart, which is associated with physiological cardiac growth; however, it is not known whether or how physical activity–induced changes in cardiac metabolism cause myocardial remodeling. In this study, we tested whether exercise-mediated changes in cardiomyocyte glucose metabolism are important for physiological cardiac growth.
Methods:
We used radiometric, immunologic, metabolomic, and biochemical assays to measure changes in myocardial glucose metabolism in mice subjected to acute and chronic treadmill exercise. To assess the relevance of changes in glycolytic activity, we determined how cardiac-specific expression of mutant forms of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase affect cardiac structure, function, metabolism, and gene programs relevant to cardiac remodeling. Metabolomic and transcriptomic screenings were used to identify metabolic pathways and gene sets regulated by glycolytic activity in the heart.
Results:
Exercise acutely decreased glucose utilization via glycolysis by modulating circulating substrates and reducing phosphofructokinase activity; however, in the recovered state following exercise adaptation, there was an increase in myocardial phosphofructokinase activity and glycolysis. In mice, cardiac-specific expression of a kinase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase transgene (GlycoLo mice) lowered glycolytic rate and regulated the expression of genes known to promote cardiac growth. Hearts of GlycoLo mice had larger myocytes, enhanced cardiac function, and higher capillary-to-myocyte ratios. Expression of phosphatase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase in the heart (GlycoHi mice) increased glucose utilization and promoted a more pathological form of hypertrophy devoid of transcriptional activation of the physiological cardiac growth program. Modulation of phosphofructokinase activity was sufficient to regulate the glucose–fatty acid cycle in the heart; however, metabolic inflexibility caused by invariantly low or high phosphofructokinase activity caused modest mitochondrial damage. Transcriptomic analyses showed that glycolysis regulates the expression of key genes involved in cardiac metabolism and remodeling.
Conclusions:
Exercise-induced decreases in glycolytic activity stimulate physiological cardiac remodeling, and metabolic flexibility is important for maintaining mitochondrial health in the heart.
Editorial, see p 2158
Clinical Perspective.
What Is New?
Episodic, exercise-induced changes in cardiac metabolism appear to be required for healthy, physiological growth of the heart.
In the heart, glucose utilization via glycolysis is reduced during exercise and in the early recovery period after exercise.
Low rates of myocardial glycolysis are sufficient to activate gene programs that instigate physiological cardiac growth.
Metabolic inflexibility of the heart, such as occurs in heart failure and diabetes mellitus, is sufficient to diminish mitochondrial function.
Phosphofructokinase-mediated changes in metabolism appear to regulate genes involved in processes critical for metabolic remodeling, transcription, cell division, differentiation, cell proliferation, and contraction.
What Are the Clinical Implications?
Regular exercise diminishes cardiovascular disease risk. Preclinical studies identifying the mechanisms by which exercise-induced metabolic changes in the heart regulate cardiac growth and health could be used to promote healthy cardiac adaptation and delay disease progression.
Identification of genes regulated by metabolism could lead to new therapeutic targets to mitigate maladaptive cardiac remodeling and heart failure.
The finding that loss of cardiac metabolic flexibility promotes mitochondrial dysfunction could be important for predicting mitochondrial derangements in patients and for devising personalized treatment strategies to improve the metabolic flexibility of the heart.
Exercise promotes cardiovascular wellness,1 augments musculoskeletal function,2 and lengthens lifespan.3 These salutary effects of exercise are dependent on the ability of the body to adapt to recurrent bouts of physical activity. Cumulative adaptation of the circulatory, pulmonary, and musculoskeletal systems augments exercise capacity,4 which is the strongest indicator of cardiovascular health and a robust predictor of mortality.5 Cardiovascular adaptations to exercise are particularly critical to enable adequate oxygen and nutrient delivery to peripheral tissues, and they help preserve circulatory system health with age.6,7 Physiological cardiac growth attributable to exercise training is a key component of these cardiovascular adaptations and contributes not only to exercise capacity, but also to cardioprotection.8 Nevertheless, the mechanisms by which regular exercise triggers and sustains cardiac adaptation remain unclear. Understanding how exercise promotes healthy cardiac remodeling could provide the knowledge required to delay the progression of heart disease or reverse tissue pathology.
Previous studies have identified signaling pathways and gene programs essential for cardiac adaptation.9,10 For example, C/EBPβ and CITED4 are known to be critical for regulating the transcriptional programs that instigate cardiac growth11–13; however, the mechanisms by which such transcriptional programs are activated with exercise are unclear. In tissues such as skeletal muscle, adaptive changes in gene expression are thought to be initiated by the repetitive bouts of metabolic stress triggered by strenuous physical activity.2 Although it follows that the heart should conform to this paradigm, this has not been tested and it remains unclear which metabolic pathways and flux control steps may be important to provoke tissue adaptation to exercise.
In this study, we examined exercise-induced changes in glucose metabolism and their relationship to cardiac remodeling. We provide evidence that exercise regulates the activity of myocardial phosphofructokinase 1 (PFK1) and that the resulting changes in metabolism are sufficient to activate a transcriptional program influencing exercise-induced cardiac growth. Nonetheless, our data also indicate that chronically low or high relative levels of glycolysis, such as occurs in type 2 diabetes mellitus or heart failure, are damaging to mitochondria. These findings suggest that changes in myocardial glucose metabolism, brought about by regular exercise, regulate transcriptional programming and physiological remodeling of the heart and that maintenance of metabolic flexibility is required to preserve mitochondrial health.
Methods
Experimental Animals
All procedures were approved by the University of Louisville Institutional Animal Care and Use Committee. Transgenic mice expressing a kinase-deficient (ie, GlycoLo mice) or phosphatase-deficient (ie, GlycoHi mice) form of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK2) under the control of the α-myosin heavy chain promoter and wild-type (WT) littermates on the FVB/NJ background were used for this study.14,15 Adult, male mice were used for all studies, with the exception of fructose 2,6,-bisphosphate (F-2,6-P2) measurements in isolated cardiomyocytes, where both male and female mice were used. At the conclusion of animal experiments, and after a 6-hour fast, mice were anaesthetized with sodium pentobarbital (150 mg/kg, intraperitoneally) and euthanized.
Exercise Capacity Testing and Training
We performed exercise familiarization, capacity testing, and training as we previously outlined.16
Echocardiographic Assessment
Cardiac function was measured by transthoracic echocardiography, as described.17
Measurements of Myocardial Glucose Utilization
Hearts were excised and perfused in nonrecirculating Langendorff mode with Krebs-Henseleit buffer (37°C; 5% CO2/95% O2; containing in mmol/L: 118.5 NaCl, 25 NaHCO3, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 2.5 CaCl2, and 5 glucose; pH 7.4). Glycolytic flux was assessed by measuring the amount of 3H2O released through the metabolism of exogenous [5-3H]glucose (Perkin Elmer), as described.14,15 Glucose utilization was then normalized to total heart weight.
Blood and Plasma Measurements
Blood glucose and blood lactate were measured via tail clip using the Accu-Check Aviva (Roche) and Lactate Plus meter (Nova Biomedical), respectively. Free fatty acids were quantified using a colorimetric enzymatic assay (Sigma-Aldrich), as instructed by the manufacturer. Insulin-like growth factor-1 was quantified using a colorimetric assay (R&D Systems), per the manufacturer.
Glycogen Assay
Cardiac glycogen content was determined using a colorimetric assay, as described by the manufacturer (Biovision).
Measurement of F-2,6-P2 Concentration
Metabolite levels of F-2,6-P2 were measured in isolated ventricular myocytes as outlined previously.14,15
Histology
After euthanasia, the heart tissue was excised, flushed with 1 mol/L KCl solution, and fixed in 10% formalin, embedded in paraffin, and sectioned at 4 µm. Heart cross sections were stained with 4′,6-diamidino-2-phenylindole (Invitrogen) and wheat germ agglutinin (ThermoFisher) for quantification of cardiomyocyte cross-sectional area. In addition, sections were stained with isolectin B4 (Vector Laboratories) to assess capillary density and with Sirius red for fibrosis. Quantitative measurements were determined using Nikon Elements software.
Mitochondrial Function
Heart mitochondria were isolated and subjected to respiratory function assays using the Seahorse XF24, similar to that described.18
Protein and mRNA Analysis
Western blotting and quantitative reverse transcription polymerase chain reaction were performed as described in the online-only Data Supplement. Lists of antibodies and primers are found in the online-only Data Supplement.
Analysis of Cardiac Ultrastructure
Left ventricular tissue was dissected into 3-mm3 pieces and fixed in 2.5% glutaraldehyde overnight. The samples were then postfixed in 2% aqueous osmium tetroxide, dehydrated in ascending concentrations of ethanol, embedded in Araldite 502 (Electron Microscopy Sciences), sectioned and mounted on 200-mesh copper grids, and doubly stained with uranyl acetate and lead citrate. Ultrastructure was examined by using a Phillips CM-10 electron microscope with a Laboratory 6 cathode (Phillips Electronic Instruments Co) operating at 80 kV.
Metabolomic Analyses
After a 6-hour fast, hearts were freeze-clamped by using Wollenberger forceps ultracooled in liquid nitrogen. Samples were stored frozen (–80°C) until metabolite extraction by Metabolon, Inc., and subjected to metabolic profiling by ultrahigh-performance liquid chromatography/tandem mass spectrometry.17 Detailed methods are provided in the online-only Data Supplement.
Transcriptomic Analyses
Hearts of male WT, GlycoLo, and GlycoHi mice (15–16 weeks of age) were subjected to transcriptomic analysis by using the GeneChip Mouse Genome 430 2.0 Array (Affymetrix, Inc). Detailed methods on analysis and software used can be found in the online-only Data Supplement.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 7, Metaboanalyst, and the R program. Statistical parameters including the value of n (number of mice), the definition of center, dispersion and precision measures (mean±SEM or SD), and statistical significance is reported in the figures and figure legends. A P value of ≤0.05 was considered statistically significant. For direct comparisons, statistical significance was calculated by unpaired or paired Student t test. Multiple comparisons were assessed by 1-way or 2-way ANOVA followed by Bonferroni or Sidak multiple comparison tests, as appropriate. Details of the statistical analysis can be found in the online-only Data Supplement.
Results
Exercise Promotes Physiological Cardiac Growth
Wild-type mice subjected to 4 weeks of treadmill training showed a 48% increase in distance run and a 77% increase in work accomplished (Figure IA and IB in the online-only Data Supplement); successful exercise testing was validated by elevated blood lactate levels, indicating that the mice ran to exhaustion (Figure IC in the online-only Data Supplement). In comparison with untrained mice, trained mice had 15% higher levels of circulating plasma insulin-like growth factor-1 (Figure ID in the online-only Data Supplement), further demonstrating systemic adaptation to the training regimen. Exercise-adapted mice displayed physiological cardiac growth characterized by a 10% increase in heart weight to tibia length (Figure 1A), a 24% increase in cardiomyocyte cross-sectional area, and a 35% increase in capillary-to-myocyte ratio (Figure 1B through 1D). Furthermore, exercise increased chamber diameter and wall thicknesses, improved indices of cardiac function (end-diastolic volume, stroke volume, and cardiac output; Table I in the online-only Data Supplement), and activated the prohypertrophic signaling intermediate protein kinase B (AKT)19–23 (Figure 1E). Exercise training downregulated Cebpb and upregulated Cited4, an observation consistent with previous findings showing necessary involvement of these transcriptional mediators in regulating physiological growth.8,10–13 Moreover, the expression of Nfatc2, the deletion of which ameliorates pathological hypertrophy and heart failure but does not prevent physiological hypertrophy,24 was markedly lower in hearts from exercise-adapted mice (Figure 1F). Collectively, these results indicate that exercise training elicits robust cardiac adaptation in mice.
Figure 1.
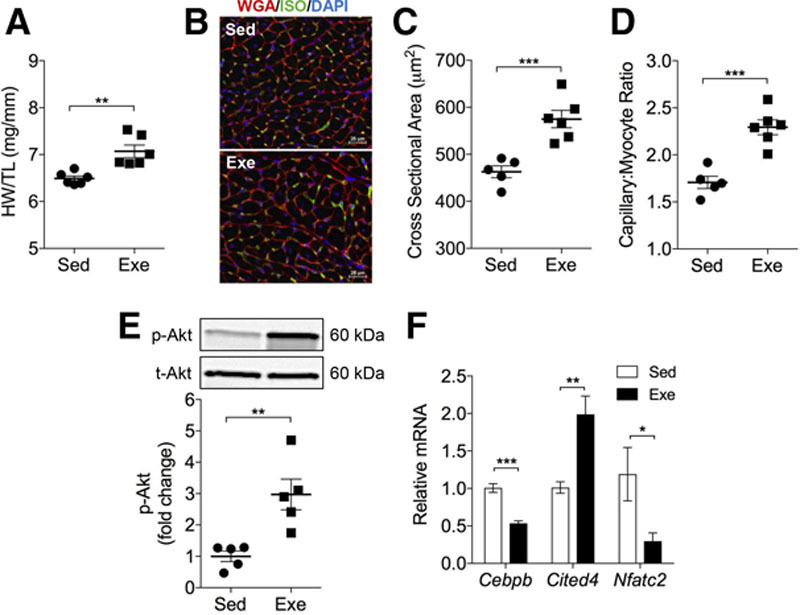
Exercise training promotes physiological cardiac growth. Effect of treadmill training on the exercise-induced cardiac growth program. A, Gravimetric measurements of cardiac size (HW/TL, heart weight/tibia length). B, Representative myocardial sections stained with wheat germ agglutinin (red) and isolectin B4 (green). Nuclei are stained with DAPI (blue). C, Cardiomyocyte cross-sectional area. D, Capillary-to-myocyte ratio. E, Phosphorylation of AKT (Ser473) 24 hours after the last training session. F, Relative mRNA expression of Cebpb, Cited4, and Nfatc2. Data are represented as mean±SEM. *P<0.05. **P<0.01. ***P<0.001. n=5–6 per group. DAPI indicates 4′,6-diamidino-2-phenylindole; Exe, exercise; ISO, isolectin B4; Sed, sedentary; and WGA, wheat germ agglutinin.
Metabolomic Changes in the Exercise-Adapted Heart at Steady State
To examine global metabolic changes in exercise-adapted hearts, we first compared the relative abundance of 424 metabolites in hearts isolated from sedentary and exercise-adapted mice. To capture the metabolic state of the adapted heart as accurately as possible, the hearts were freeze-clamped in situ 24 hours after the final exercise bout (Figure IIA in the online-only Data Supplement). Principal component analysis showed little separation between the groups (Figure IIB in the online-only Data Supplement), and only 20 metabolites (of 424 measured) were found to be significantly different (Figure IIC in the online-only Data Supplement). Of these, only 2 metabolites had a q value <0.05 (Table II in the online-only Data Supplement). These findings demonstrate that the steady-state abundance of most metabolites in the exercise-adapted heart is unchanged in comparison with that of sedentary mice.
Exercise Induces Periodicity in Glucose Metabolism
Because the steady-state abundance of biochemical pathway intermediates may not be reflective of pathway flux,17,25 and because several studies show that exercise changes cardiac metabolism (eg,22,26), we next measured the metabolism of [5-3H]glucose in hearts from sedentary and exercise-adapted mice. Radiometric assays of perfused hearts showed a nearly 2-fold increase in glucose utilization in exercise-adapted hearts in comparison with hearts from sedentary mice (Figure 2A). Glycolytic enzymes were found in similar abundance in exercise-adapted hearts relative to hearts from sedentary mice (Figure III in online-only Data Supplement). These data indicate that changes in enzyme abundance are unlikely to contribute to the increase in glycolysis that occurs in the exercise-adapted heart.
Figure 2.
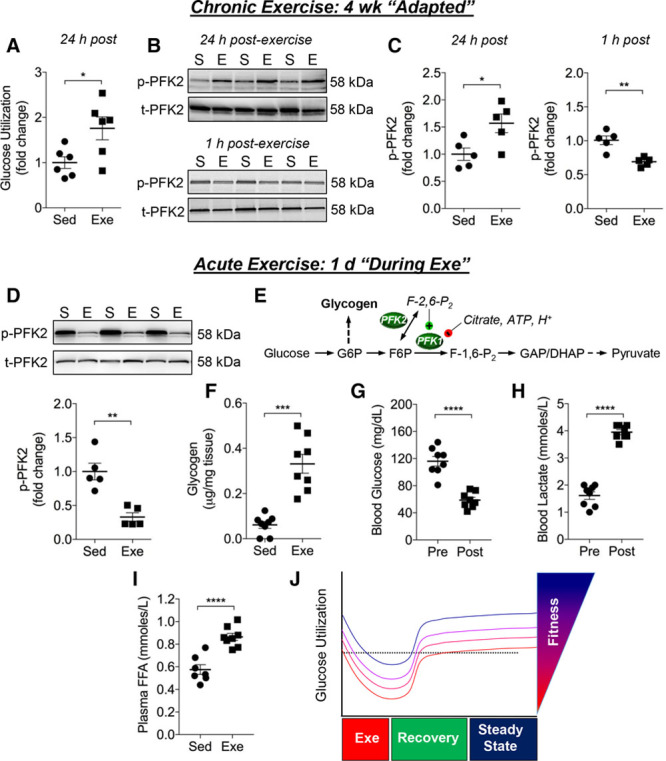
Exercise dynamically regulates glucose utilization in the heart. Measurements of glucose metabolism in the heart. A, Measurement of relative myocardial glucose utilization in isolated perfused hearts from mice that remained sedentary (Sed) or that were exercised for 4 weeks (Exe). Measurements were performed on hearts excised 24 hours after the last exercise session. B, Representative immunoblots from a separate cohort of mice of S483 phosphorylation of the cardiac isoform of PFK2 (PFKFB2) in the sedentary (S) and the exercise-adapted (E) state: Upper, levels of PFK2 phosphorylation 24 hours after the last bout of exercise; Lower, PFK2 phosphorylation occurring 1 hour after the last exercise bout. C, Quantification of PFK2 phosphorylation from blots represented in B. D, Representative immunoblots of S483 phosphorylation of PFK2 in hearts of mice exercised for 1 day; these hearts were excised immediately after exercise, which would appear to be indicative of the state of PFK2 occurring during or immediately after exercise. E, Schematic representation of glycolysis showing relationship of PFK activity with glycogen storage. F, Myocardial glycogen content in hearts of Sed mice and in hearts excised immediately after 1 bout of exercise. Circulating levels of blood glucose (G), blood lactate (H), and plasma free fatty acids (FFA) (I) in mice subjected to a single bout of exercise. J, Schematic illustrating changes in glucose utilization occurring with acute and chronic exercise, ie, exercise-induced metabolic periodicity. Data are represented as mean±SEM. *P<0.05. **P<0.01. ****P<0.0001. n=5– 8 per group. DHAP indicates dihydroxyacetone phosphate; F-1,6-P2, fructose 1,6-bisphosphate; F6P, fructose 6-phosphate; G6P, glucose 6-phosphate; and GAP, glyceraldehyde-3-phosphate.
Because glycolysis is influenced by rate-limiting steps, such as occurs at the phosphofructokinase reaction, we next examined factors that regulate PFK1—the rate-limiting and committed step of glycolysis.27, 28 The PFK1 reaction exerts more control on glycolysis than any other step29 and is dependent on several factors including: the levels of fructose-2,6,-bisphosphate (F-2,6-P2), substrate availability, energy charge, the levels of citrate and ATP, and pH. Because PFK2 regulates the levels of F-2,6-P2, a potent activator of PFK1, we examined the phosphorylation status of the myocardial isoform of PFK2, PFKFB2. Phosphorylation of PFKFB2 at S483 can increase F-2,6-P2 levels and is known to be under the influence of insulin-like growth factor-1/AKT signaling.20, 30-32 Notably, both cardiac AKT phosphorylation and plasma insulin-like growth factor-1 were elevated in exercise-adapted mice (Figure 1E and Figure ID in the online-only Data Supplement). In agreement with radiometric findings, S483 phosphorylation of PFKFB2 (p-PFK2) was 50% higher in exercise-adapted hearts compared with hearts of sedentary mice (Figure 2B, upper panel and Figure 2C, left panel). Taken together, the radiometric and immunologic data provide convergent evidence that glycolysis is increased in the exercise-adapted heart and indicate that this increase in glycolytic activity may be attributable at least in part to PFK activation.
Because exercise is a dynamic challenge that leads to rapid changes in metabolism, we next examined glycolytic changes that occur early after exercise, ie, in the trained state during recovery. Because ex vivo measurements of flux in the recovery period after an exercise session are unlikely to provide meaningful data (eg, because of the lack of a metabolic steady state), we measured levels of p-PFK2 as a possible indicator of phosphofructokinase activity and glucose utilization through glycolysis. In comparison with corresponding sedentary controls, hearts excised from exercise-adapted mice 1 hour after the last exercise session showed significantly diminished PFK2 phosphorylation (Figure 2B, lower, and Figure 2C, right).
To determine whether these changes in metabolism precede cardiac adaptation, we eliminated the variable of extended training and examined PFK2 activation immediately after a single bout of exercise. For this, a separate group of mice was subjected to a single bout of exercise for 40 minutes. Immediately after this exercise bout, mice were euthanized and the hearts were excised for biochemical analysis. We found that p-PFK2 levels in these hearts were remarkably lower than those in sedentary controls (Figure 2D), suggesting that a bout of exercise might acutely decrease the rate of glycolysis in the heart. Because measurement of glycolytic rate ex vivo would likely obfuscate the metabolic changes that occur during or shortly after a bout of exercise (because of the loss of hemodynamic, metabolic, and paracrine [eg, catecholamine] influence), we chose to measure glycogen content as an additional surrogate of PFK activity and glycolysis. Thus, we hypothesized that, during exercise, PFK-mediated decreases in glycolytic rate should promote glycogen accumulation (Figure 2E), which is consistent with the notion that decreases in PFK activity promote glycogenesis.33 Indeed, glycogen content was elevated >5-fold in hearts collected immediately after a single bout of exercise (Figure 2F). It is noteworthy that this elevation in cardiac glycogen is not related to the short (6-hour) fasting period occurring in sedentary and exercised mice, because independent experiments showed that fasting mice for this duration does not affect myocardial glycogen content (unfasted, 0.17±0.03 µg/mg tissue; 6 hours fasted, 0.18±0.05 µg/mg tissue; n=7–8 per group).
Because substrate availability and delivery are key determinants of myocardial substrate preference and utilization,34–37 we measured the circulating levels of glucose, lactate, and free fatty acids in mice immediately after a bout of exercise. In comparison with preexercise levels, mice subjected to 40 minutes of treadmill exercise showed a 50% decrease in blood glucose and a 2.5-fold increase in blood lactate (Figure 2G and 2H). Moreover, plasma free fatty acids were significantly elevated (Figure 2I), which along with the changes in blood glucose and lactate, would stimulate lactate and fat oxidation and diminish overall glucose utilization. These data are concordant with our biochemical measurements and provide convergent evidence suggesting that, during and immediately after exercise, the rate of myocardial glycolysis is diminished; however, on adaptation and full recovery, the steady-state rates of glycolysis appear to be increased in the heart (Figure 3J).
Figure 3.
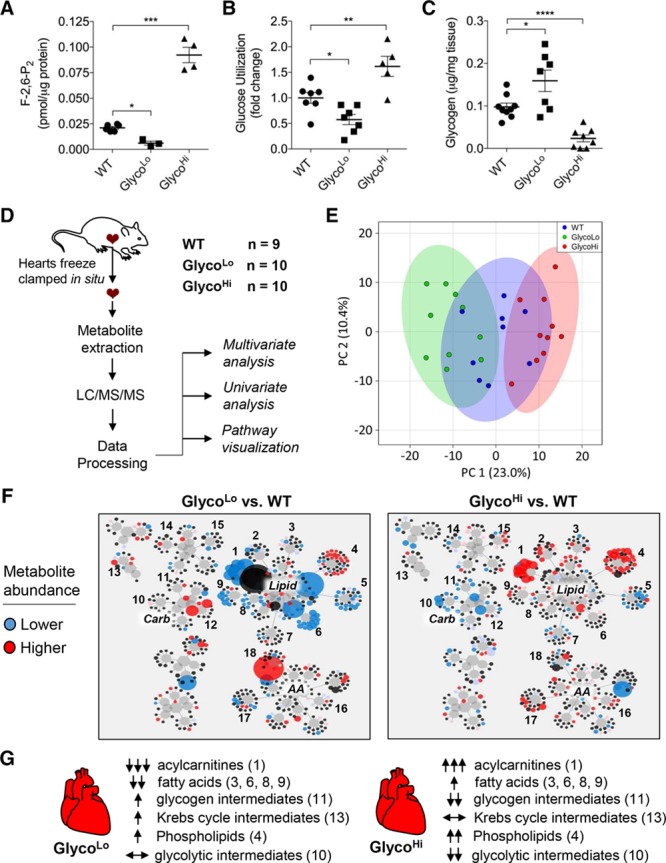
Cardiac F-2,6-P2 regulates myocardial glucose and lipid metabolism. Metabolic phenotyping of GlycoLo and GlycoHi mice. A, Levels of F-2,6-P2 in cardiomyocytes isolated from WT, GlycoLo, and GlycoHi mice. n=3 to 6 per group. B, Relative myocardial glucose utilization in isolated perfused hearts. n=5 to 7 per group. C, Myocardial glycogen content. n=7 to 9 per group. D, Schematic of metabolomic analysis of WT, GlycoLo, and GlycoHi hearts. E, 2D PCA analysis. F, Metabolic network visualization of metabolites in GlycoLo and GlycoHi hearts in comparison with corresponding WT hearts. Circle size is indicative of the magnitude of change (blue and red circles indicate significantly decreased or increased metabolite abundance in comparison with WT controls, respectively). Numbers represent metabolites in the following subpathways: 1, acylcarnitines; 2, monacylglycerols; 3, monohydroxy fatty acids; 4, phospholipid metabolism; 5, lysolipids; 6, polyunsaturated fatty acids; 7, sphingolipids; 8, dicarboxylic fatty acids; 9, long-chain fatty acids; 10, glycolysis; 11, fructose, mannose, galactose metabolism; 12, pentose metabolism; 13, TCA cycle; 14, purine metabolism, (hypo)xanthine/inosine; 15, purine metabolism, adenine; 16, Gly, Ser, Thr metabolism; 17, BCAA metabolism; 18, Met, Cys, SAM, Tau metabolism. G, Overview of changes in major metabolic subpathways in GlycoLo and GlycoHi hearts (F). *P<0.05. **P<0.01. ***P<0.001. ****P<0.0001 versus the indicated group. BCAA indicates branched-chain amino acid; F-2,6-P2, fructose 2,6,-bisphosphate; LC/MS/MS, liquid chromatography/tandem mass spectrometry; PCA, principal component analysis; SAM, S-adenosyl methionine; TCA, tricarboxylic acid; and WT, wild type.
Phosphofructokinase Is a Strong Regulator of the Glucose–Fatty Acid Cycle in the Heart
We next examined whether exercise-induced periodicity in glucose metabolism might be essential for structural and functional cardiac adaptation. This hypothesis is consistent with the notion that repetitive bouts of exercise are a trigger for the transcriptional adaptation in skeletal muscle.2 As tools to examine the relationship between changes in myocardial glucose metabolism and cardiac remodeling, we used genetically modified mice expressing kinase-deficient (GlycoLo) or phosphatase-deficient (GlycoHi) point-mutant isoforms of PFK2 under the control of the α-myosin heavy chain promoter, which constitutively decrease or increase PFK1 activity and myocardial glycolytic rate.14,15 In comparison with cardiomyocytes isolated from WT mice, the abundance of F-2,6-P2 was >2-fold lower in GlycoLo cardiomyocytes and >5-fold higher in GlycoHi cardiomyocytes (Figure 3A). The rate of glycolysis in perfused hearts was in accordance with transgene-induced alterations in F-2-6-P2 (Figure 3B). Consistent with the notion that PFK activity regulates glycogen storage,33 GlycoLo hearts showed 1.6-fold higher myocardial glycogen content, and GlycoHi hearts demonstrated 4-fold lower levels of glycogen (Figure 3C).
To determine how glycolysis affects myocardial metabolism, we measured the steady-state abundance of myocardial metabolites in both GlycoLo and GlycoHi hearts (Figure 3D). Principal component analysis showed that the relative abundances of metabolites could discriminate between the genotypes (Figure 3E). Volcano plot analysis demonstrated that, of the 424 metabolites measured, 108 metabolites in GlycoLo hearts (Figure IVA and Table III in the online-only data supplement) and 109 metabolites in GlycoHi hearts (Figure IVB and Table IV in the online-only Data Supplement) were significantly different from WT hearts. Visualization of changes in the metabolic network demonstrated the greatest impact on the lipid superfamily, with acylcarnitines (subfamily 1) showing robust, diametrically opposite changes in the GlycoLo and GlycoHi hearts (Figure 3F and 3G). Z-score analyses illustrate remarkable increases in medium- and long-chain acylcarnitines in GlycoHi hearts; the corresponding species were lower in abundance in GlycoLo hearts (Figure VA and Tables III and IV in the online-only Data Supplement). In addition, long-chain fatty acids (saturated and unsaturated) were lower in GlycoLo mice, whereas many of the same species were elevated in GlycoHi mice (Figure 3F and 3G, Figure VA and Tables III and IV in the online-only Data Supplement). It is interesting to note that GlycoLo hearts demonstrated elevated levels of aconitate, homocitrate, and α-ketoglutarate (Figure 3F and 3G and Table III in the online-only Data Supplement), suggesting that high rates of lipid oxidation in these hearts might flood the Krebs cycle. Glycolytic intermediates were lower in GlycoHi mice, whereas most glycolytic intermediates were unchanged in abundance in GlycoLo mice (Figure 3F and 3G and Figure VB in the online-only Data Supplement). Changes in the steady-state abundances of glycolytic and lipid metabolites are most consistent with increased propensity for glucose oxidation in GlycoHi hearts and increased lipid oxidation in GlycoLo hearts. These changes would be sufficient to explain the accumulation of acylcarnitines and diminished levels of glycolytic intermediates occurring in GlycoHi hearts, and the lower levels of acylcarnitines in GlycoLo hearts. Polarization of myocardial metabolism also resulted in modest changes in amino acid metabolites (see Figure VC and Tables III and IV in the online-only Data Supplement). Collectively, these results show the high degree to which phosphofructokinase regulates the glucose–fatty acid cycle in the heart.
Constitutively Low Glycolytic Activity Promotes Structural and Transcriptional Changes Consistent With the Exercise-Adapted Heart
To examine whether PFK activity regulates cardiac growth, we assessed the structure and function of GlycoLo and GlycoHi hearts. Both the GlycoLo and GlycoHi hearts showed a mild increase in cardiac size, which was associated with increased cardiomyocyte cross-sectional area (Figure 4A through 4C, 4E, and 4F); however, significantly increased capillary-to-myocyte ratio was observed only in GlycoLo hearts (Figure 4A and 4D). Picrosirius red staining showed no noteworthy myocardial fibrosis in either strain (Figure 4A). It is interesting to note that GlycoLo hearts showed elevated end-diastolic volume and slightly increased wall thicknesses in comparison with WT mice (Table V in the online-only Data Supplement), which is similar to that found in WT, exercise-adapted mice (see Table I in the online-only Data Supplement). In contrast, GlycoHi hearts had elevated end-systolic volume, lower ejection fraction, and larger chamber diameters, suggesting a more dilated cardiac phenotype (Table V in the online-only Data Supplement).
Figure 4.
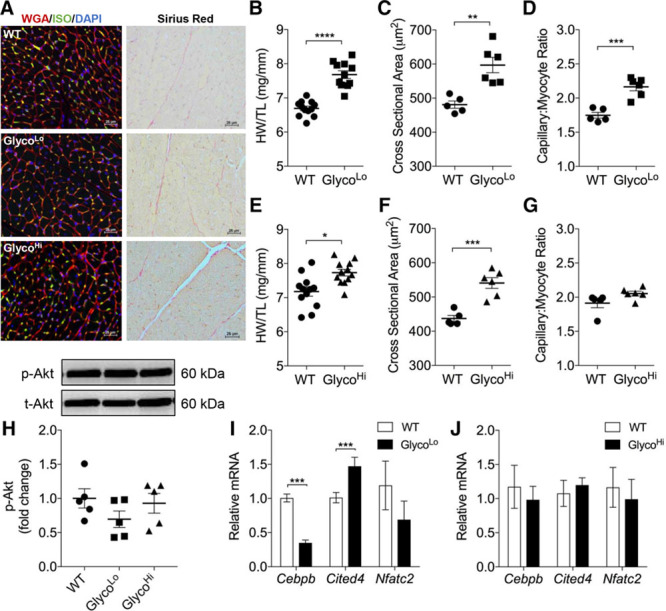
Constitutive changes in glycolysis promote cardiac growth and hypertrophy. Structural and molecular indices of cardiac growth/hypertrophy. A, Representative myocardial sections stained for: Left, wheat germ agglutinin (red), isolectin B4 (green), and DAPI (blue); and Right, Picrosirius red. B and E, Gravimetric measurement of cardiac size (HW/TL, heart weight/tibia length). C and F, Cardiomyocyte cross-sectional area. D and G, Capillary-to-myocyte ratio. H, Basal phosphorylation of AKT (Ser473). I and J, Relative mRNA expression of genes associated with the physiological growth program. Data are represented as mean±SEM. *P<0.05. **P<0.01. ***P<0.001. ****P<0.0001. n=6 to 12 per group. DAPI indicates 4′,6-diamidino-2-phenylindole; ISO, isolectin B4; WGA, wheat germ agglutinin; and WT, wild type.
To determine whether the signaling mediators and transcriptional programs underlying exercise-induced cardiac growth10–12,38 are affected by constitutive changes in glucose utilization, we next examined the phosphorylation of AKT and the relative expression of Cebpb and Cited4. Although no differences in basal p-AKT (S473) were observed in either strain (Figure 4H), Cebpb expression was lower and Cited4 expression was higher in GlycoLo hearts than in WT counterparts (Figure 4I). Expression of these genes was unchanged in GlycoHi hearts (Figure 4J). Although Nfatc2 showed a trend toward lower expression in GlycoLo hearts, this did not reach statistical significance. These data suggest that constitutively low or high rates of glycolysis cause mild cardiac hypertrophy and that a low rate of glycolysis, in the absence of AKT activation, is sufficient to promote a structural, functional, and transcriptional phenotype resembling the exercise-adapted heart.
Metabolic Inflexibility Causes Mitochondrial Structural and Functional Abnormalities
Because constitutive polarization of metabolism toward either glucose or fat oxidation could have deleterious effects on mitochondrial structure and function, we examined mitochondrial ultrastructure by transmission electron microscopy and measured respiration in mitochondria isolated from the hearts of both GlycoLo and GlycoHi mice. As shown in Figure 5A, qualitative analysis of electron micrographs showed disorganization of mitochondrial cristae and diminished cristae density in comparison with WT littermates. Consistent with this observation, mitochondria from both GlycoLo and GlycoHi hearts showed lower respiration in the presence of pyruvate+malate or succinate (Figure 5B and 5C). In addition, respiratory control ratios were found to be lower in cardiac mitochondria from both transgenic strains, suggesting diminished coupling of mitochondrial electron transport to ATP synthesis (Figure 5D). Thus, although constitutively low glycolysis imparts some structural, functional, and transcriptional features characteristic of the exercise-adapted heart, the mitochondrial compartment is damaged by metabolic inflexibility.
Figure 5.
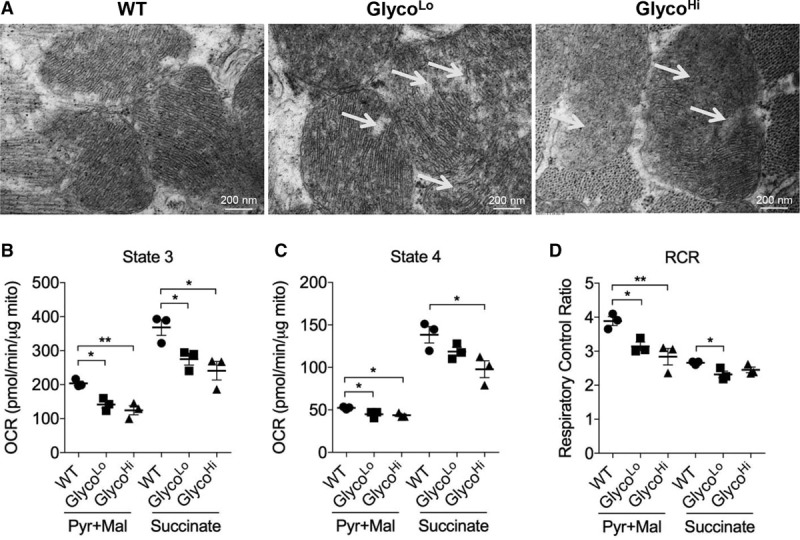
Metabolic inflexibility disrupts cristae structure and causes mitochondrial dysfunction. A, Electron micrographs of WT, GlycoLo, and GlycoHi hearts. Arrows indicate areas of decreased cristae organization and density in comparison with WT controls. State 3 (B) and State 4 (C) respiration in isolated cardiac mitochondria measured by using complex I (pyruvate+malate)– and complex II (succinate)–specific substrates. D, Respiratory control ratios. Data are represented as mean±SEM. *P<0.05. **P<0.01. n=3 per group. OCR indicates oxygen consumption rate; RCR, respiratory control ratio; and WT, wild type
Constitutively Low Glycolysis Is Sufficient for Maximal Cardiac Growth
With the observation that GlycoLo hearts have a cardiac phenotype partially resembling the exercise-adapted heart, we next asked whether these mice are capable of performing more work or adapting to a greater degree than their WT counterparts. To address this question, we subjected both WT and GlycoLo mice to exercise testing before and after 4 weeks of treadmill training. Although GlycoLo mice were able to adapt systemically to the training regimen, they were not able to run farther or perform more work than WT mice (Figure 6A and 6B). Unlike WT mice, which showed a ≈15% increase in cardiac size, GlycoLo mice showed no further increase in cardiac growth after training (Figure 6C). Similarly, myocardial expression of Cebpb and Cited4 were not found to be different in exercised GlycoLo mice than in sedentary GlycoLo mice (Figure 6D), suggesting that their baseline cardiac phenotype is sufficient to meet the increased cardiovascular demands brought about by exercise. These results suggest that a constitutively low glycolytic rate is sufficient to maximally activate physiological cardiac growth.
Figure 6.
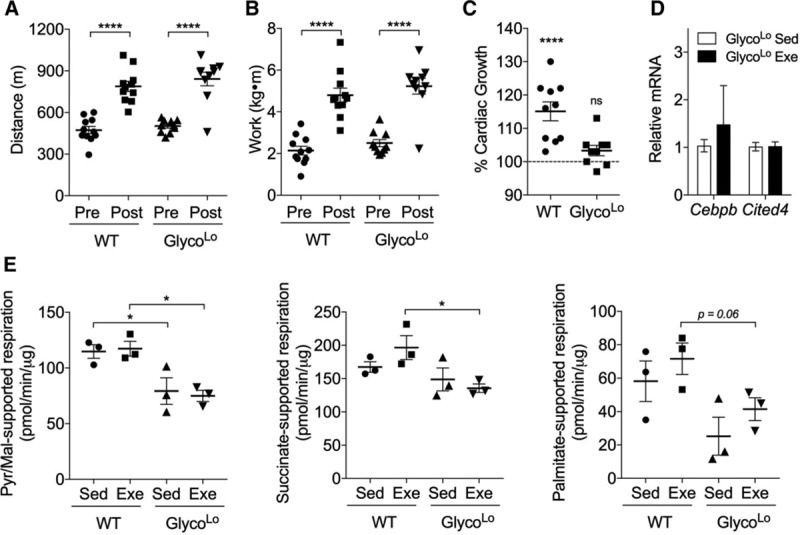
Low myocardial glycolytic rates are sufficient for maximal cardiac growth. Effects of exercise training on exercise capacity, cardiac growth, and mitochondrial function in WT and GlycoLo mice. Pre- and posttraining distance (A) and work (B). C, Percent cardiac growth in exercise-adapted (Exe) mice in comparison with sedentary (Sed) controls. n=10 to 11 per group. D, Relative mRNA expression of genes associated with the physiological growth program. n=5 per group. E, State 3 respiration in isolated cardiac mitochondria using complex I (pyruvate+malate)–, complex II (succinate)–, and fatty acid oxidation (palmitoylcarnitine+malate)–specific substrates. n=3 per group. Data are represented as mean±SEM. *P<0.05. ****P<0.0001. WT indicates wild type.
Because exercise could have beneficial effects on mitochondrial function,23 we next posited that it might restore mitochondrial function in GlycoLo hearts. In WT mice, exercise did not affect state 3 respiration in the presence of any substrate tested, and, in GlycoLo mice, exercise did not reverse mitochondrial dysfunction caused by their metabolic phenotype (Figure 6E).
Glycolysis Regulates Gene Expression in the Heart
To gain further insight into the extent to which glycolysis might regulate myocardial gene expression, we assessed relative transcript abundance in GlycoLo and GlycoHi hearts. Results of Genechip arrays showed that the expression of 619 genes were significantly different in GlycoLo and GlycoHi hearts in comparison with hearts of corresponding WT mice (Up: GlycoLo, 7; GlycoHi, 309; Down: GlycoLo, 22; GlycoHi, 295 genes; 14 differentially expressed genes overlapped in GlycoLo and GlycoHi hearts). As shown in Figure VIA in the online-only Data Supplement, the top 100 genes include genes critical for intermediary metabolism, neurotransmission and ion channel activity/transport, DNA damage/repair, cell proliferation/differentiation, cell signaling, gene transcription, inflammation and stress responses, detoxification and xenobiotic metabolism, and cytoskeletal/contractile function. In particular, we were interested in genes that are differentially regulated by glycolysis, ie, affected in diametric opposition with respect to low or high glycolytic activity (Figure VIB in the online-only Data Supplement). In addition to Cited4, we found that glycolysis regulates the expression of genes involved in one-carbon metabolism and downstream methylation reactions (Pah, Phgdh, Mtr), lipid metabolism (Insg1, Angptl3), gluconeogenic activity (Fbp2), cell division (Cenpf), osmoregulation (Cenpf, Aqp4), transcription (Tox3), G-protein signaling (Ramp1), calcium-related processes (Rcan1, Cacna1g). Gene ontology analysis showed that low rates of glycolysis had a strong influence on genes involved in oxidation-reduction processes, lipid metabolism, and responses to cAMP (Figure VIC in the online-only Data Supplement). High glycolytic rates also influenced genes involved in metabolism, responses to cAMP, responses to hormonal stimuli, and transcription. In addition, genes involved in angiogenesis and differentiation, showed highly significant changes (Figure VID in the online-only Data Supplement). Collectively, the findings of this study indicate that the activity of phosphofructokinase has pervasive effects on the metabolic, transcriptional, structural, and functional characteristics of the adult heart.
Discussion
In this study, we provide evidence suggesting that exercise dynamically regulates myocardial glycolytic activity at the level of phosphofructokinase. To assess the importance of exercise-induced periodicity in glucose metabolism, we used transgenic mice expressing mutant transgenes that constitutively increase or decrease phosphofructokinase activity in the heart. Expression of a dominant-negative form of PFK2 (GlycoLo) in the heart decreased glycolytic rate and was sufficient to modulate the Cebpb/Cited4 transcriptional program that regulates physiological cardiac growth.11–13 Mice in which myocardial glycolytic rate is constitutively high also showed slight hypertrophy, but these hearts did not engage the physiological growth gene program. The corollary of these findings is that exercise-induced changes in phosphofructokinase activity are necessary to activate transcriptional programs dictating cardiac growth and hypertrophy. This is supported by our transcriptomics data from both GlycoLo mice and GlycoHi mice, which revealed coordinated regulation of not only the Cited4 gene, but also genes important for epigenetic regulation, lipid metabolism, gluconeogenic activity, contractility, and angiogenesis. Nevertheless, constitutive changes in myocardial metabolism incited damage to the mitochondrial compartment. Collectively, these results suggest that episodic decreases in glycolytic activity caused by exercise are required for physiological cardiac remodeling and that metabolic flexibility is important to maintain mitochondrial health in the heart.
Our findings in the heart are reminiscent of exercise-induced changes in skeletal muscle, where transient and repetitive changes in gene expression, likely initiated by acute disruption of metabolic equanimity during exercise, stimulate adaptation.2 Although testing the role of metabolic periodicity in the heart is difficult, the posttranslational regulation of PFK2 that we found to occur with exercise provided a unique opportunity to test not only how glucose utilization affects the cardiac phenotype, but also how restricting fuel flexibility regulates cardiac structure, function, and gene expression. The dominant-negative approach to overcome the activity of endogenous myocardial PFK2, ie, expression of the kinase-deficient form of PFK2, was sufficient to trigger a physiological cardiac growth phenotype: GlycoLo hearts showed lower Cebpb and higher Cited4 expression, which mirrored the transcriptional changes found in the exercise-adapted heart. This transcriptional signature was complemented by a cardiac growth phenotype replete with enlarged myocytes and increased capillary density. In addition, the chamber volumes, wall thicknesses, and functional values of GlycoLo hearts were nearly identical to that found in exercise-adapted hearts. GlycoLo hearts appear to have a maximal functional and structural physiological growth phenotype because they could not further activate the physiological growth gene program or undergo additional cardiac growth in response to the exercise regimen. It is important to note that the phenotype of GlycoLo hearts developed in the absence of AKT activation, which has been suggested to underlie exercise-induced downregulation of Cebpb and upregulation of Cited4 and the general cardiac growth phenotype.9,10 Thus, these data help form a model in which exercise-induced modulation of glucose metabolism is a proximal regulator of physiological cardiac growth.
Several factors appear to contribute to acute, exercise-induced decreases in glycolysis and phosphofructokinase activity. The lower circulating blood glucose levels and increased levels of blood lactate and free fatty acids that we found to occur immediately after a single bout of exercise suggest diminished glycolytic activity occurring in the heart during and in the early recovery period after exercise. Indeed, lactate and fatty acids are preferred substrates of the heart that suppress glucose utilization both in vivo34,39,40 and ex vivo.36,37 Moreover, previous studies in humans and animal models suggest that changes in these substrates during exercise contribute to diminished exercise oxygen extraction ratios for glucose and lower glucose uptake and utilization.41,42 Not only do increases in circulating lactate and fatty acids with exercise appear to favor lower rates of glucose utilization, they also divert glucose from glycolysis to glycogen synthesis, which is thought to be caused in part by diminished phosphofructokinase activity.43–45 Consistent with this idea, our results show diminished phosphorylation of S483 of PFK2 and increased levels of glycogen, which support the hypothesis that PFK1 activity and glycolysis are diminished with prolonged exercise or during the early recovery period. In the context of glucose utilization, changes in PFK1 are particularly important because this node of metabolism represents the major control point of glycolysis, exerting 65% control over flux through the pathway.29
Although constitutively low glycolytic rates appeared sufficient to drive physiological growth, metabolic inflexibility in both the GlycoHi and GlycoLo hearts appeared sufficient to cause some pathology as well. Although GlycoHi hearts were enlarged, they did not demonstrate significantly increased capillary-to-myocyte ratios, and, functionally, they presented with a mildly diminished ejection fraction, significantly higher end-systolic volume, and larger chamber diameters. These findings are in agreement with previous studies15 and suggest that chronically high rates of glycolysis promote a more pathological form of hypertrophy. Furthermore, that mitochondria isolated from both GlycoHi and GlycoLo hearts showed mitochondrial dysfunction suggests that deleterious changes in mitochondria occurring in the heart in the context of type 2 diabetes mellitus or heart failure, conditions associated with excessive myocardial reliance on fatty acids or glucose, respectively,46,47 could in part be caused by severe metabolic inflexibility. Although genetic downregulation of Cebpb and upregulation of Cited4 protect against pathological insults,11,12 mice with constitutively low glycolysis subjected to pressure overload display exacerbated pathological remodeling,48 which we suggest may be attributable to poor mitochondrial health caused by metabolic inflexibility.
Although at steady state the exercise-adapted heart showed higher glycolytic activity, we found relatively few changes in metabolite abundance. We speculate that higher myocardial metabolic flux in the absence of changes in the steady-state abundance of metabolites may be an indication of a physiological form of metabolic remodeling that occurs in the exercise-adapted heart. However, it should be noted that our results contrast somewhat with a previous finding that showed modestly lower abundances of acylcarnitines and organic acids in hearts of female mice adapted to 2 months of voluntary wheel running.49 The discrepancy between these 2 studies could be attributable to sex differences, strain differences, differences in the training modality, and intensity. Moreover, given the high energy utilization of the heart, even slightly different methods of handling the samples prior to freezing may in part explain differences in metabolomics results.
Although our data support the idea that PFK-mediated modulation of metabolism influences transcriptional programs relating to cardiac growth and remodeling, it is possible that fat utilization or lactate oxidation, which are regulated in concert with glucose metabolism, are also important triggers of cardiac adaptation. Future studies to determine how exercise acutely changes the cardiac metabolome could help answer whether changes in metabolites in each pathway during or shortly after exercise promote biofeedback that coordinates transcriptional programming. Moreover, identifying how exercise intensity and duration regulate cardiac metabolism and gene transcription would be important for optimizing cardiac adaptations and for further understanding links between metabolism and cardiac growth. Particularly intriguing is the question of how such changes in metabolism may regulate epigenetic modifications. In our study, we found that glycolysis regulated Phgdh, Mthfd2, and Mtr, which code for proteins critical for serine biosynthesis, 1-carbon metabolism, and methylation reactions. This could be significant because Cited4 is epigenetically regulated: hypermethylation of Cited4-associated CpG islands causes lower expression of Cited4 in oligodendroglial tumors.50 Although our data do not address this mechanism, they suggest that low glycolytic rates could prevent such methylation events, thereby permitting Cited4 transcription, especially when appropriate stimuli are present.
In summary, our results indicate that low rates of glycolysis during or shortly after exercise regulate gene programs important for cardiac growth and that invariantly high rates of glycolysis promote a more pathological form of hypertrophy. Although metabolic inflexibility is damaging to mitochondria, changes in glucose utilization attributable to regular exercise appear to be important to maintain mitochondrial health and to derive maximal cardiac benefits from physical activity. In addition, by integrating metabolomic and transcriptomic data, these studies provide new insights into how PFK-mediated changes in metabolism regulate gene expression in the heart. Additional work is needed to further disclose how exercise-mediated changes in metabolism regulate gene transcription and tissue remodeling and how these changes could be targeted to maximize the beneficial cardiovascular effects of exercise.
Acknowledgments
The authors acknowledge the assistance of Don Mosley, the Animal Core, and the Imaging and Physiology Core of the Diabetes and Obesity Center.
Sources of Funding
This work was supported in part by grants from the National Institutes of Health (HL122580 [to Dr Hill], HL130174 [to Dr Hill], HL131647 [to Dr Jones], GM103492 [to Dr Bhatnagar], HL78825 [to Drs Bhatnagar and Jones]), a Predoctoral Fellowship from the American Heart Association (16PRE31010022 [Mr Gibb]), and the American Diabetes Association Pathway to Stop Diabetes Grant (1-16-JDF-041 [to Dr Hill]).
Disclosures
None.
Supplementary Material
Footnotes
Sources of Funding, see page 2155
The online-only Data Supplement is available with this article at http://circ.ahajournals.org/lookup/suppl/doi:10.1161/CIRCULATIONAHA.117.028274/-/DC1.
Circulation is available at http://circ.ahajournals.org.
References
- 1.Vega RB, Konhilas JP, Kelly DP, Leinwand LA. Molecular mechanisms underlying cardiac adaptation to exercise. Cell Metab. 2017;25:1012–1026. doi: 10.1016/j.cmet.2017.04.025. doi: 10.1016/j.cmet.2017.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013;17:162–184. doi: 10.1016/j.cmet.2012.12.012. doi: 10.1016/j.cmet.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 3.López-Otín C, Galluzzi L, Freije JM, Madeo F, Kroemer G. Metabolic control of longevity. Cell. 2016;166:802–821. doi: 10.1016/j.cell.2016.07.031. doi: 10.1016/j.cell.2016.07.031. [DOI] [PubMed] [Google Scholar]
- 4.Bassett DR, Jr, Howley ET. Limiting factors for maximum oxygen uptake and determinants of endurance performance. Med Sci Sports Exerc. 2000;32:70–84. doi: 10.1097/00005768-200001000-00012. [DOI] [PubMed] [Google Scholar]
- 5.Wilson MG, Ellison GM, Cable NT. Basic science behind the cardiovascular benefits of exercise. Br J Sports Med. 2016;50:93–99. doi: 10.1136/bjsports-2014-306596rep. doi: 10.1136/bjsports-2014-306596rep. [DOI] [PubMed] [Google Scholar]
- 6.Roh J, Rhee J, Chaudhari V, Rosenzweig A. The role of exercise in cardiac aging: from physiology to molecular mechanisms. Circ Res. 2016;118:279–295. doi: 10.1161/CIRCRESAHA.115.305250. doi: 10.1161/CIRCRESAHA.115.305250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seals DR. Edward F. Adolph Distinguished Lecture: the remarkable anti-aging effects of aerobic exercise on systemic arteries. J Appl Physiol (1985) 2014;117:425–439. doi: 10.1152/japplphysiol.00362.2014. doi: 10.1152/japplphysiol.00362.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mann N, Rosenzweig A. Can exercise teach us how to treat heart disease? Circulation. 2012;126:2625–2635. doi: 10.1161/CIRCULATIONAHA.111.060376. doi: 10.1161/CIRCULATIONAHA.111.060376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14:38–48. doi: 10.1038/nrm3495. doi: 10.1038/nrm3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lerchenmüller C, Rosenzweig A. Mechanisms of exercise-induced cardiac growth. Drug Discov Today. 2014;19:1003–1009. doi: 10.1016/j.drudis.2014.03.010. doi: 10.1016/j.drudis.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 11.Boström P, Mann N, Wu J, Quintero PA, Plovie ER, Panáková D, Gupta RK, Xiao C, MacRae CA, Rosenzweig A, Spiegelman BM. C/EBPβ controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010;143:1072–1083. doi: 10.1016/j.cell.2010.11.036. doi: 10.1016/j.cell.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bezzerides VJ, Platt C, Lerchenmuller C, Paruchuri K, Oh NL, Xiao C, Cao Y, Mann N, Spiegelman BM, Rosenzweig A. CITED4 induces physiologic hypertrophy and promotes functional recovery after ischemic injury. JCI Insight. 2016;1(9):e85904. doi: 10.1172/jci.insight.85904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryall KA, Bezzerides VJ, Rosenzweig A, Saucerman JJ. Phenotypic screen quantifying differential regulation of cardiac myocyte hypertrophy identifies CITED4 regulation of myocyte elongation. J Mol Cell Cardiol. 2014;72:74–84. doi: 10.1016/j.yjmcc.2014.02.013. doi: 10.1016/j.yjmcc.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donthi RV, Ye G, Wu C, McClain DA, Lange AJ, Epstein PN. Cardiac expression of kinase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase inhibits glycolysis, promotes hypertrophy, impairs myocyte function, and reduces insulin sensitivity. J Biol Chem. 2004;279:48085–48090. doi: 10.1074/jbc.M405510200. doi: 10.1074/jbc.M405510200. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q, Donthi RV, Wang J, Lange AJ, Watson LJ, Jones SP, Epstein PN. Cardiac phosphatase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase increases glycolysis, hypertrophy, and myocyte resistance to hypoxia. Am J Physiol Heart Circ Physiol. 2008;294:H2889–H2897. doi: 10.1152/ajpheart.91501.2007. doi: 10.1152/ajpheart.91501.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gibb AA, McNally LA, Riggs DW, Conklin DJ, Bhatnagar A, Hill BG. FVB/NJ mice are a useful model for examining cardiac adaptations to treadmill exercise. Front Physiol. 2016;7:636. doi: 10.3389/fphys.2016.00636. doi: 10.3389/fphys.2016.00636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sansbury BE, DeMartino AM, Xie Z, Brooks AC, Brainard RE, Watson LJ, DeFilippis AP, Cummins TD, Harbeson MA, Brittian KR, Prabhu SD, Bhatnagar A, Jones SP, Hill BG. Metabolomic analysis of pressure-overloaded and infarcted mouse hearts. Circ Heart Fail. 2014;7:634–642. doi: 10.1161/CIRCHEARTFAILURE.114.001151. doi: 10.1161/CIRCHEARTFAILURE.114.001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato S, Ogura Y, Mishra V, Shin J, Bhatnagar S, Hill BG, Kumar A. TWEAK promotes exercise intolerance by decreasing skeletal muscle oxidative phosphorylation capacity. Skelet Muscle. 2013;3:18. doi: 10.1186/2044-5040-3-18. doi: 10.1186/2044-5040-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shiojima I, Yefremashvili M, Luo Z, Kureishi Y, Takahashi A, Tao J, Rosenzweig A, Kahn CR, Abel ED, Walsh K. Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. J Biol Chem. 2002;277:37670–37677. doi: 10.1074/jbc.M204572200. [DOI] [PubMed] [Google Scholar]
- 20.Kim J, Wende AR, Sena S, Theobald HA, Soto J, Sloan C, Wayment BE, Litwin SE, Holzenberger M, LeRoith D, Abel ED. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol Endocrinol. 2008;22:2531–2543. doi: 10.1210/me.2008-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wende AR, O’Neill BT, Bugger H, Riehle C, Tuinei J, Buchanan J, Tsushima K, Wang L, Caro P, Guo A, Sloan C, Kim BJ, Wang X, Pereira RO, McCrory MA, Nye BG, Benavides GA, Darley-Usmar VM, Shioi T, Weimer BC, Abel ED. Enhanced cardiac Akt/protein kinase B signaling contributes to pathological cardiac hypertrophy in part by impairing mitochondrial function via transcriptional repression of mitochondrion-targeted nuclear genes. Mol Cell Biol. 2015;35:831–846. doi: 10.1128/MCB.01109-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riehle C, Wende AR, Zhu Y, Oliveira KJ, Pereira RO, Jaishy BP, Bevins J, Valdez S, Noh J, Kim BJ, Moreira AB, Weatherford ET, Manivel R, Rawlings TA, Rech M, White MF, Abel ED. Insulin receptor substrates are essential for the bioenergetic and hypertrophic response of the heart to exercise training. Mol Cell Biol. 2014;34:3450–3460. doi: 10.1128/MCB.00426-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noh J, Wende AR, Olsen CD, Kim B, Bevins J, Zhu Y, Zhang QJ, Riehle C, Abel ED. Phosphoinositide dependent protein kinase 1 is required for exercise-induced cardiac hypertrophy but not the associated mitochondrial adaptations. J Mol Cell Cardiol. 2015;89:297–305. doi: 10.1016/j.yjmcc.2015.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bourajjaj M, Armand AS, da Costa Martins PA, Weijts B, van der Nagel R, Heeneman S, Wehrens XH, De Windt LJ. NFATc2 is a necessary mediator of calcineurin-dependent cardiac hypertrophy and heart failure. J Biol Chem. 2008;283:22295–22303. doi: 10.1074/jbc.M801296200. doi: 10.1074/jbc.M801296200. [DOI] [PubMed] [Google Scholar]
- 25.Brown GC. Control of respiration and ATP synthesis in mammalian mitochondria and cells. Biochem J. 1992;284 (pt 1):1–13. doi: 10.1042/bj2840001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burelle Y, Wambolt RB, Grist M, Parsons HL, Chow JC, Antler C, Bonen A, Keller A, Dunaway GA, Popov KM, Hochachka PW, Allard MF. Regular exercise is associated with a protective metabolic phenotype in the rat heart. Am J Physiol Heart Circ Physiol. 2004;287:H1055–H1063. doi: 10.1152/ajpheart.00925.2003. doi: 10.1152/ajpheart.00925.2003. [DOI] [PubMed] [Google Scholar]
- 27.Mor I, Cheung EC, Vousden KH. Control of glycolysis through regulation of PFK1: old friends and recent additions. Cold Spring Harb Symp Quant Biol. 2011;76:211–216. doi: 10.1101/sqb.2011.76.010868. doi: 10.1101/sqb.2011.76.010868. [DOI] [PubMed] [Google Scholar]
- 28.Yalcin A, Telang S, Clem B, Chesney J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp Mol Pathol. 2009;86:174–179. doi: 10.1016/j.yexmp.2009.01.003. doi: 10.1016/j.yexmp.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 29.Cortassa S, Caceres V, Bell LN, O’Rourke B, Paolocci N, Aon MA. From metabolomics to fluxomics: a computational procedure to translate metabolite profiles into metabolic fluxes. Biophys J. 2015;108:163–172. doi: 10.1016/j.bpj.2014.11.1857. doi: 10.1016/j.bpj.2014.11.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem. 1997;272:17269–17275. doi: 10.1074/jbc.272.28.17269. [DOI] [PubMed] [Google Scholar]
- 31.Pozuelo Rubio M, Peggie M, Wong BH, Morrice N, MacKintosh C. 14-3-3s regulate fructose-2,6-bisphosphate levels by binding to PKB-phosphorylated cardiac fructose-2,6-bisphosphate kinase/phosphatase. EMBO J. 2003;22:3514–3523. doi: 10.1093/emboj/cdg363. doi: 10.1093/emboj/cdg363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 33.Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab. 2009;297:E578–E591. doi: 10.1152/ajpendo.00093.2009. doi: 10.1152/ajpendo.00093.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lassers BW, Kaijser L, Wahlqvist ML, Carlson LA. Relationship in man between plasma free fatty acids and myocardial metabolism of carbohydrate substrates. Lancet. 1971;2:448–450. doi: 10.1016/s0140-6736(71)92624-9. [DOI] [PubMed] [Google Scholar]
- 35.Drake AJ, Haines JR, Noble MI. Preferential uptake of lactate by the normal myocardium in dogs. Cardiovasc Res. 1980;14:65–72. doi: 10.1093/cvr/14.2.65. [DOI] [PubMed] [Google Scholar]
- 36.Schönekess BO. Competition between lactate and fatty acids as sources of ATP in the isolated working rat heart. J Mol Cell Cardiol. 1997;29:2725–2733. doi: 10.1006/jmcc.1997.0504. doi: 10.1006/jmcc.1997.0504. [DOI] [PubMed] [Google Scholar]
- 37.Goodwin GW, Taegtmeyer H. Improved energy homeostasis of the heart in the metabolic state of exercise. Am J Physiol Heart Circ Physiol. 2000;279:H1490–H1501. doi: 10.1152/ajpheart.2000.279.4.H1490. [DOI] [PubMed] [Google Scholar]
- 38.DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, Muslin AJ. Akt1 is required for physiological cardiac growth. Circulation. 2006;113:2097–2104. doi: 10.1161/CIRCULATIONAHA.105.595231. doi: 10.1161/CIRCULATIONAHA.105.595231. [DOI] [PubMed] [Google Scholar]
- 39.Wisneski JA, Gertz EW, Neese RA, Gruenke LD, Morris DL, Craig JC. Metabolic fate of extracted glucose in normal human myocardium. J Clin Invest. 1985;76:1819–1827. doi: 10.1172/JCI112174. doi: 10.1172/JCI112174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gertz EW, Wisneski JA, Stanley WC, Neese RA. Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. J Clin Invest. 1988;82:2017–2025. doi: 10.1172/JCI113822. doi: 10.1172/JCI113822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lassers BW, Kaijser L, Wahlqvist M, Carlson LA. Effect of prolonged exercise on myocardial metabolism in man. Br Heart J. 1971;33:609. [PubMed] [Google Scholar]
- 42.Takala TE, Ruskoaho HJ, Hassinen IE. Transmural distribution of cardiac glucose uptake in rat during physical exercise. Am J Physiol. 1983;244:H131–H137. doi: 10.1152/ajpheart.1983.244.1.H131. [DOI] [PubMed] [Google Scholar]
- 43.Kemppainen J, Fujimoto T, Kalliokoski KK, Viljanen T, Nuutila P, Knuuti J. Myocardial and skeletal muscle glucose uptake during exercise in humans. J Physiol. 2002;542(pt 2):403–412. doi: 10.1113/jphysiol.2002.018135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Depre C, Ponchaut S, Deprez J, Maisin L, Hue L. Cyclic AMP suppresses the inhibition of glycolysis by alternative oxidizable substrates in the heart. J Clin Invest. 1998;101:390–397. doi: 10.1172/JCI1168. doi: 10.1172/JCI1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Depre C, Rider MH, Veitch K, Hue L. Role of fructose 2,6-bisphosphate in the control of heart glycolysis. J Biol Chem. 1993;268:13274–13279. [PubMed] [Google Scholar]
- 46.Fillmore N, Mori J, Lopaschuk GD. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br J Pharmacol. 2014;171:2080–2090. doi: 10.1111/bph.12475. doi: 10.1111/bph.12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 48.Wang J, Xu J, Wang Q, Brainard RE, Watson LJ, Jones SP, Epstein PN. Reduced cardiac fructose 2,6 bisphosphate increases hypertrophy and decreases glycolysis following aortic constriction. PLoS One. 2013;8:e53951. doi: 10.1371/journal.pone.0053951. doi: 10.1371/journal.pone.0053951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, Kapoor K, Koves TR, Stevens R, Ilkayeva OR, Vega RB, Attie AD, Muoio DM, Kelly DP. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail. 2014;7:1022–1031. doi: 10.1161/CIRCHEARTFAILURE.114.001469. doi: 10.1161/CIRCHEARTFAILURE.114.001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tews B, Roerig P, Hartmann C, Hahn M, Felsberg J, Blaschke B, Sabel M, Kunitz A, Toedt G, Neben K, Benner A, von Deimling A, Reifenberger G, Lichter P. Hypermethylation and transcriptional downregulation of the CITED4 gene at 1p34.2 in oligodendroglial tumours with allelic losses on 1p and 19q. Oncogene. 2007;26:5010–5016. doi: 10.1038/sj.onc.1210297. doi: 10.1038/sj.onc.1210297. [DOI] [PubMed] [Google Scholar]