

Abstract
Background:
Mitochondrial diseases are a group of multisystem heterogeneous diseases caused by pathologic dysfunction of the mitochondrial respiratory chain. A wide range of clinical expression has been described. However, pulmonary hypertension has rarely been described in association with mitochondrial disease until the past decade, and there is no currently recognized treatment for the pulmonary hypertension complicated with mitochondrial disorder.
Patient concerns:
We reported the case of a 15-year-old boy who presented with shortness of breath and exercise limitation after a cold, and the diagnosis of pulmonary hypertension was confirmed by right heart catheter. Other examinations, such as blood tests, high- resolution chest computed tomography scan, and pulmonary function test, excluded other associated diseases as causes of pulmonary hypertension.
Diagnoses and Outcomes:
The initial diagnosis was idiopathic pulmonary arterial hypertension and an injection of vasodilator (Treprostinil) was given. However, the dyspnea and fatigue subsequently got worsened. Tracing back his family history, together with the electromyography, nerve conduction studies, and the result of muscle biopsy, mitochondrial disease was confirmed. After treatment with vitamin E, vitamin B2, ATP, and coenzyme Q10, the patient's condition improved.
Conclusion:
Pulmonary hypertension should be considered as another potential manifestation of mitochondrial disease. Both mechanism and treatment for pulmonary hypertension complicated with mitochondrial disease are unclear. Further study is necessary.
Keywords: combination therapies, literature review, mitochondrial disease, pulmonary hypertension
1. Introduction
Mitochondrial diseases are a group of multisystem diseases caused by pathologic dysfunction of the mitochondrial respiratory chain.[1] These diseases are a clinically heterogeneous group of disorders, and may present at any age.[2] Clinical symptoms originating from all organ systems have been described, predominantly affecting highly oxidative tissue such as heart, muscle, and brain.[3] Cardiorespiratory complications have previously been described in association with mitochondrial respiratory chain defects. Hypertrophic cardiomyopathy and cardiac conduction defects are the commonest cardiac abnormalities associated with mitochondrial disease.[2–5]
Pulmonary hypertension as a manifestation of mitochondrial disease has been considered rare until the past decade. Most reports are cases not large clinical research, and the mechanism of pulmonary hypertension complicated with mitochondrial disorder remains unclear. A causal relation could be explained by cellular proliferation from mitochondrial dysfunction, and decreased production of reactive oxygen species.[6]
The clinical presentation of mitochondrial disorders is extremely heterogeneous[3] and combined with the large number of causal genetic and environmental factors, which makes their diagnosis and therapy challenging. A multifaceted approach including histopatholgical and electron-microscopic examination, enzymatic assays, and, where relevant, identification of specific mtDNA (mitochondrial DNA) or nuclear mutations should be required to diagnosis of respiratory chain defects.[7,8] As a result of their genetic and biochemical heterogeneity, mitochondrial disorders present therapeutic challenges. The treatment of pulmonary hypertension complicated with mitochondrial disorder is unknown. We reported a case of pulmonary hypertension complicated with mitochondrial disorder.
2. Case report
A 15-year-old boy who had a history of normal birth, was admitted to our clinic for progressive dyspnea, generalized muscle weakness for 5 days after a cold. He complained of shortness of breath and exercise limitation. Physical examination revealed him to be emaciated, with a weight of 36 kg and a height of 162 cm; body mass index was 13.7. His pulse was 105 beats/min and blood pressure 100/68 mm Hg. He had an accentuated second pulmonic heart sound. Neurological examination revealed bilateral lower limbs weakness, decreased muscle tone, and tendon reflexes. Babinski signs were negative. The blood gases analysis indicated that PaO2 was 88 mm Hg and PaCO2 was 57 mm Hg in breathing room air. The lung function damage is very severe limitation ventilation dysfunction. The pro-brain natriuretic peptide level was 3311 pg/mL (normal <300 pg/mL). The results of the thyroid functional tests, autoimmune antibodies parameters, and antineutrophil cytoplasmic antibody tests were within the normal range. The transthoracic echocardiography showed right atrial enlargement, mild tricuspid regurgitation, and mild pulmonary hypertension with the right ventricular systolic pressure (RVSP) 62 mm Hg. The chest computed tomography (CT) scan images showed bilateral pleural effusion and pulmonary artery widened (Fig. 1). The computer CT pulmonary angiography result was negative for pulmonary embolism. Right heart catheter showed pulmonary hypertension with a mean pulmonary artery pressure at 41 mm Hg and pulmonary artery wedge pressure of 16 mm Hg.
Figure 1.
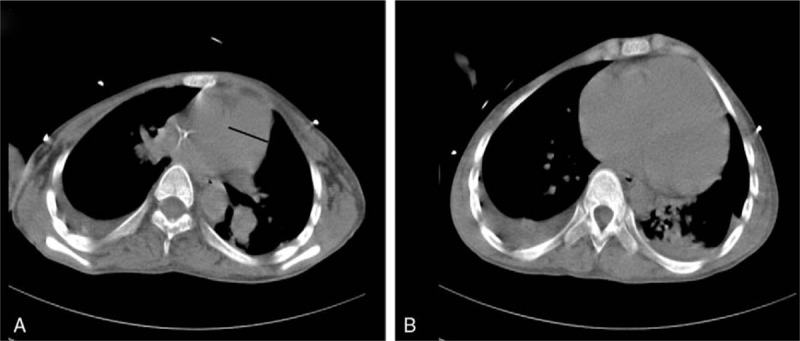
(A) Pulmonary artery widened observed in the chest CT scan. (B) Bilateral few pleural effusions displayed in the chest CT scan. CT = computed tomography.
The initial diagnosis was idiopathic pulmonary arterial hypertension. Because of carbon dioxide retention, the patient was treated with noninvasive positive pressure ventilation. In terms of drug therapies, he was received digoxin, furosemide, and an injection of vasodilator (Treprostinil), which is a stable, long-acting prostacyclin analog, and has been shown to improve clinical state, functional class, exercise capacity, and quality of life in patients with pulmonary arterial hypertension.[9] After treatment, blood gas analysis indicated the partial pressure of carbon dioxide improved. However, the clinical symptom and signs of the patient did not show any response, and the dyspnea and fatigue worsened. Why using targeted therapy for pulmonary hypertension had poor effects in this patient, and weather he had some underlying condition? Stopping Treprostinil injections and tracing back his history, the patient had short stature, was underweight, and had poor physical fitness at an early age, which was similar to his mother, who had mitochondrial diseases carrying A3243G mutation. So, further examination was carried out for him. The serum creatine kinase level was 604 U/L (normal 50–310 U/L). The lactate in serum was 13.9 mmol/L (normal 0.90–1.70 mmol/L). Electromyography and nerve conduction studies revealed unusual results. Muscle biopsy was conducted for him, and the results (Fig. 2) showed many ragged red fibers by modified gomori trichrome staining, which conformed to mitochondrial disease. Finally, the diagnosis of mitochondrial disease was confirmed, and the patient was further treated with vitamin E (100 mg once daily for 14 days), Vitamin B2 (500ug once daily for 14 days), ATP (20 mg twice daily for 14 days) and coenzyme Q10 (30 mg 3 times daily for 14 days). A month and a half later, he achieved a significant clinical and echocardiographic improvement (RVSP 36 mm Hg).
Figure 2.
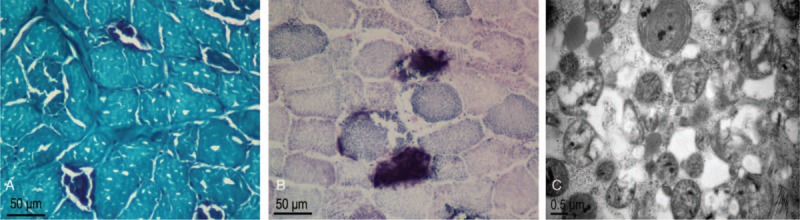
(A) MGT staining showed many ragged red fibers (original magnification 400×, scale bar 50 μm). (B) SDH activity staining showed one strongly SDH-reactive blood vessel. (original magnification 400×, scale bar 50 μm). (C) An electron microscopy image showed an increased number of mitochondria with variable sizes and shapes (scale bar 0.5 μm). MGT = modified gomori trichrome, SDH = succinic dehydrogenase
This study was approved by the Ethics Committee of Sir Run Run Shaw Hospital. Written informed consent was obtained from the patient.
3. Discussion
Mitochondrial disorders are caused by pathologic dysfunction of the mitochondrial respiratory chain. Clinical symptoms originating from all organ systems have been described, but pulmonary hypertension as a manifestation of mitochondrial disease has been considered rare until the past decade. Among them, progressive dyspnea and exercise limitation were presented as main clinical manifestations in this type of patients, which were similar to the patients with idiopathic pulmonary arterial hypertension, leading to misdiagnosis between these 2 conditions.
As we know, pulmonary hypertension is distinguished by increased pulmonary arterial pressure and secondary right ventricular failure. The gold standard for diagnosis of pulmonary arterial hypertension is right heart catheter.[10] Based on etiology, the World Health Organization classified pulmonary hypertension into 5 groups[11]: group 1—pulmonary arterial hypertension; group 2—pulmonary hypertension owing to left heart disease; group 3—pulmonary hypertension owing to lung diseases or hypoxemia; group 4—chronic thromboembolic pulmonary hypertension; and group 5—pulmonary hypertension with unclear multifactorial mechanisms.
Nowadays, the mechanisms of pulmonary hypertension complicated with mitochondrial disorder are unknown. It may be related to the damage of small pulmonary arteries and hypoxia-induced vasoconstriction. The former was similar to the pathological changes of idiopathic pulmonary arterial hypertension, and the latter was similar to group 3 which failed to respond to target therapy, and that may be the reason why the Treprostinil did not improve the clinical symptoms at all of the patient in our case.
Mitochondrial disorder is a multisystem heterogeneous disease that has both genetic and acquired factors. Because of unique genetic mechanism and self-organized DNA, mitochondria are different from other cellular compartments playing the role of energy conversion transformation inside eukaryotic cells. With the development of genetic diagnosis, the mutation of mtDNA has been considered to be related with various mitochondria diseases.[12,13] Among them, A3243G mutation in mtDNA is the most common pathogenic point mutation, especially in mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS). Similar to the case like the patient's mother, although with negative A3243G mutation, the patient showed the same clinical symptoms of mitochondrial diseases such as shortness of breath, exercise limitation dyspnea, and lower body weight, but denied typical features of MELAS including stroke-like episodes, dementia, epilepsy, and vision loss. These results support the maternal inheritance pattern of A3243G mutation, consistent with previous reports,[14,15] which meanwhile show the diversity of the phenotype. However, the mechanisms for phenotypic differences are not yet fully understood, and it is recognized to be related to both the proportion of mutant and wild-type mtDNA and the oxygen consumption in the tissue.[16,17]
As a result of their genetic and biochemical heterogeneity, mitochondrial disorders present therapeutic challenges. Many experimental therapies have been explored, with varying success.[18] In several previous clinical trials, therapy with coenzyme Q10 and multiple vitamins has shown only subjective or no benefit at all.[19–21] However, several case reports using a “cocktail” of supplements have shown clinical and biochemical improvements after treatment.[22–24] For pulmonary hypertension complicated with mitochondrial disorder, rare relevant treatment research has been reported. In our case, the target therapy of pulmonary hypertension was ineffective. Finally, the patient has shown improvement after treatment with vitamin E, vitamin B2, ATP, and coenzyme Q10. What is the optimal treatment strategy for this kind of patient? This is a problem which needs to be resolved.
Although the case report has shown transient improvement after treatment, the clinical effects remains to be seen, and the follow-up schedule is necessary and useful for us to assess the clinical benefits more objectively. Apart from this, because of some privacy concerns and economic reasons, the case lacks a detailed family history and a full-genome scan. Moreover, we wish that large number of case studies and clinical trials can be carried out in the future.
4. Conclusions
According to the guideline,[11] mitochondrial respiratory chain defects was not include as a known cause of pulmonary hypertension. Here, we reported a case of pulmonary hypertension complicated with mitochondrial disease. Pulmonary hypertension maybe a potential manifestation of mitochondrial disease. Though the mechanism and treatment for pulmonary hypertension which complicated with mitochondrial disease are unclear. Further study would be necessary.
Footnotes
Abbreviations: CT = computed tomography, MELAS = mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes, RVSP = right ventricular systolic pressure.
Funding: This study was supported by research grant 81570043 from the National Natural Science Foundation of China, and grant 2017207446 from the Zhejiang Province Medical Science and Technology Planning Projects.
The authors report no conflicts of interest.
References
- [1].Dahl HH, Thorburn DR. Mitochondrial diseases: beyond the magic circle. Am J Med Genet 2001;106:1–3. [DOI] [PubMed] [Google Scholar]
- [2].Lee SR, Han J. Mitochondrial mutations in cardiac disorders. Adv Exp Med Biol 2017;982:81–111. [DOI] [PubMed] [Google Scholar]
- [3].Mahoney DJ, Parise G, Tarnopolsky MA. Nutritional and exercise-based therapies in the treatment of mitochondrial disease. Curr Opin Clin Nutr Metab Care 2002;5:619–29. [DOI] [PubMed] [Google Scholar]
- [4].Gambardella J, Sorriento D, Ciccarelli M, et al. Functional role of mitochondria in arrhythmogenesis. Adv Exp Med Biol 2017;982:191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Venditti CP, Harris MC, Huff D, et al. Congenital cardiomyopathy and pulmonary hypertension: another fatal variant of cytochrome-c oxidase deficiency. J Inherit Metab Dis 2004;27:735–9. [DOI] [PubMed] [Google Scholar]
- [6].Waypa GB, Schumacker PT. Role for mitochondrial reactive oxygen species in hypoxic pulmonary vasoconstriction. Novartis Found Symp 2006;272:176–92. [PubMed] [Google Scholar]
- [7].Chinnery PF, Turnbull DM. Epidemiology and treatment of mitochondrial disorders. Am J Med Genet 2001;106:94–101. [DOI] [PubMed] [Google Scholar]
- [8].Poulton J, Brown GK. Investigation of mitochondrial disease. Arch Dis Child 1995;73:94–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vachiéry JL, Naeije R. Treprostinil for pulmonary hypertension. Expert Rev Cardiovasc Ther 2004;2:183–91. [DOI] [PubMed] [Google Scholar]
- [10].Rubin LJ. Primary pulmonary hypertension. Chest 1993;104:236–50. [DOI] [PubMed] [Google Scholar]
- [11].Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2016;37:67–119. [DOI] [PubMed] [Google Scholar]
- [12].Clark J, Reddy S, Zheng K, et al. Association of PGC-1alpha polymorphisms with age of onset and risk of Parkinson's disease. BMC Med Genet 2011;12:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Prasad M, Narayan B, Prasad AN, et al. MELAS: a multigenerational impact of the MTTL1 A3243G MELAS mutation. Can J Neurol Sci 2014;41:210–9. [DOI] [PubMed] [Google Scholar]
- [14].Lembke A, Gomez R, Tenakoon L, et al. The mineralocorticoid receptor agonist, fludrocortisone, differentially inhibits pituitary-adrenal activity in humans with psychotic major depression. Psychoneuroendocrinology 2013;38:115–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pohjoismaki JL, Holmes JB, Wood SR, et al. Mammalian mitochondrial DNA replication intermediates are essentially duplex but contain extensive tracts of RNA/DNA hybrid. J Mol Biol 2010;397:1144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gropman A. The neurological presentations of childhood and adult mitochondrial disease: established syndromes and phenotypic variations. Mitochondrion 2004;4:503–20. [DOI] [PubMed] [Google Scholar]
- [17].Schapira A. Mitochondrial disease. Lancet 2006;368:70–82. [DOI] [PubMed] [Google Scholar]
- [18].Gold DR, Cohen BH. Treatment of mitochondrial cytopathies. Semin Neurol 2001;21:309–25. [DOI] [PubMed] [Google Scholar]
- [19].Peterson PL. The treatment of mitochondrial myopathies and encephalomyopathies. Biochim Biophys Acta 1995;1271:275–80. [DOI] [PubMed] [Google Scholar]
- [20].Bakker HD, Scholte HR, Jeneson JA, et al. Vitamin-responsive complex I deficiency in a myopathic patient with increased activity of the terminal respiratory chain and lactic acidosis. J Inherit Metab Dis 1994;17:196–204. [DOI] [PubMed] [Google Scholar]
- [21].Matthews PM, Ford B, Dandurand RJ, et al. Coenzyme Q10 with multiple vitamins is generally ineffective in treatment of mitochondrial disease. Neurology 1993;43:884–90. [DOI] [PubMed] [Google Scholar]
- [22].Napolitano A, Salvetti S, Vista M, et al. Long-term treatment with idebenone and riboflavin in a patient with MELAS. Neurol Sci 2000;21:S981–2. [DOI] [PubMed] [Google Scholar]
- [23].Tanaka J, Nagai T, Arai H, et al. Treatment of mitochondrial encephalomyopathy with a combination of cytochrome C and vitamins B1 and B2. Brain Dev 1997;19:262–7. [DOI] [PubMed] [Google Scholar]
- [24].Kuroda Y, Ito M, Naito E, et al. Concomitant administration of sodium dichloroacetate and vitamin B1 for lactic acidemia in children with MELAS syndrome. J Pediatr 1997;131:450–2. [DOI] [PubMed] [Google Scholar]