

Abstract
Introduction
Type 1 diabetic patients can develop skeletal muscle weakness and atrophy by molecular mechanisms that are not well understood. Alternative splicing (AS) is critical for gene expression in the skeletal muscle, and its dysregulation is implicated in muscle weakness and atrophy. Therefore, we investigated whether AS patterns are affected in type 1 diabetic skeletal muscle contributing to skeletal muscle defects.
Methods
AS patterns were determined by reverse transcription–polymerase chain reaction and levels of RNA binding proteins were assessed by Western blot in type 1 diabetic mouse skeletal muscle and during normal mouse skeletal muscle development.
Results
Five genes with critical functions in the skeletal muscle are misspliced in type 1 diabetic skeletal muscle, resembling their AS patterns at embryonic stages. AS of these genes undergoes dramatic transitions during skeletal muscle development, correlating with changes in specific RNA binding proteins.
Conclusion
Embryonic spliced variants are inappropriately expressed in type 1 diabetic skeletal muscle.
Keywords: alternative splicing, muscle development, RNA binding proteins, skeletal muscle, type 1 diabetes
Type 1 diabetic patients exhibit multisystemic complications, such as nephropathy, retinopathy, neuropathy, myopathy, and cardiovascular disease.1–9 Diabetic myopathy is characterized by skeletal muscle weakness and atrophy.5 Under diabetic conditions, skeletal muscle function, growth, development, and repair are compromised.5,10–12 In type 1 diabetic patients, muscle mass and myofiber size are reduced, and muscle metabolism switches to a glycolytic pathway.11–14 In addition to growth and function, repair capacity of skeletal muscle is blunted in type 1 diabetes due to the adverse effects of hyperglycemia on muscle progenitor (satellite) cells.11,15–18 However, molecular mechanisms responsible for diabetic myopathy are not well understood.
Alternative splicing (AS) is critical for muscle growth, development, and function via directly regulating gene expression.17–21 AS is a complex process controlled by RNA binding proteins that bind to specific motifs in the pre-mRNA.22 The abundance and activity of these regulators are important for AS decisions.22 RNA binding protein Fox-1 homolog (RBFOX), Muscleblind-like (MBNL), and CUG-binding protein Elav-like family (CELF) of RNA binding proteins are linked to AS regulation during heart and skeletal muscle development.23–27 Dysregulation of AS is linked to skeletal muscle and heart pathophysiology of myotonic dystrophy.28–33
We have recently shown that global changes in AS occur in type 1 diabetic mouse heart due to changes in RNA binding proteins, negatively affecting gene expression and contributing to diabetic cardiomyopathy.27,34 Therefore, we hypothesized that type 1 diabetic skeletal muscle may also display abnormal AS signatures mediated by RNA binding proteins.
METHODS
Type 1 Diabetic Mice
A well-established type 1 diabetic mouse model, the non-obese diabetic (NOD) mouse, which can closely mimic human type 1 diabetes,10,35,36 was used in our experiments. In this mouse model, females develop diabetes around 30 weeks of age due to autoimmune-mediated destruction of β islet cells.10,35,36 NOD mice (≥5 months of age) were purchased (NOD/ShiLtj, Stock #001976; Jackson Laboratory, Bar Harbor, Maine). Fasting blood glucose levels of both NOD and control mice were tested weekly using tail vein blood by a OneTouch glucometer. Female NOD mice that exhibited fasting glucose levels >400 mg/dl for 3 weeks were used in our experiments because female mice develop diabetes at an earlier time-point and more consistently than males.27 Age-matched ICR (ICR/HaJ, Stock #009122; Jackson Laboratory) female mice were used as non-diabetic controls. ICR mice have the same major histocompatibility complex (MHC) haplotype as NOD mice, but they do not develop insulitis or diabetes. In addition, NOD mice descend from an ICR strain, making them ideal controls. NOD and ICR mice were euthanized and the gastrocnemius muscle was isolated for RNA and protein extraction.
Mouse Development
Friend virus B (FVB/NJ, Stock #001800) wild-type mice were purchased (Jackson Laboratory). Timed matings were conducted. Embryonic day 18 (E18) and newborn (NB) mice were euthanized and hindlimbs were obtained. Quadricep muscles were isolated from 6-month-old wild-type FVB (adult) mice.
All mouse experiments were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and approved by the institutional animal care and use committee of the University of Texas Medical Branch (#1101001).
RNA Extraction
RNA was extracted from mouse tissues using TRIzol (15596018; Thermo Fisher Scientific/Invitrogen, Waltham, Massachusetts) following the manufacturer’s protocol. RNA concentrations were measured using a microplate spectrometer (Epoch; BioTek, Winooski, Vermont).
Quantitative Reverse Transcription–Polymerase Chain Reaction and Statistical Analysis
Quantitative reverse transcription–polymerase chain reaction (qRT-PCR) and statistical analysis were performed as described elsewhere.27,34 Briefly, a cDNA library was generated by annealing 125 ng of oligo(dT) (S1316S; New England BioLabs, Ipswich, Massachusetts) to the poly(A) tail of total RNA (1 μg) at 65° C for 10 min and then incubated with 15 units/μg Avian Myeloblastosis Virus Reverse Transcriptase (AMV-RT) (AMV 007-1; Life Sciences Advance Technologies, St. Petersburg, Florida) and 10 μmol/L deoxynucleotide triphosphates (Thermo Fisher Scientific/Invitrogen) at 42° C for 1 hour. For the PCR reaction, primer sequences were designed on exons upstream and downstream of alternative exons. Genes and primer sequences used for this study are included in the Supplementary Material (available online).
PCR amplification was carried out in a 20-μl reaction using 5 μl of cDNA, 15 units/μg Biolase Taq polymerase (21066; Bioline, London, UK) and 200 ng of each primer under the following conditions: 95° C, 45 s; 59° C, 45 s; and 75° C, 1 min, for 25 cycles. PCR products were resolved with 5% non-denaturing polyacrylamide gels and stained with ethidium bromide. DNA bands were imaged using Gel Doc XR + (Bio-Rad, Hercules, California) and quantified by normalizing the ethidium bromide signal to the size of the DNA bands using Bio-Rad Image Lab software.27 Percent inclusion of alternative exons was calculated using the formula as we have described elsewhere.27 The Student’s t-test was used to determine statistical significance between 2 groups and one-way analysis of variance (ANOVA) for more than 2 groups using GraphPad Prism (GraphPad, Inc., La Jolla, California).
Western Blotting
Thirty to fifty μg of protein was separated on 10% sodium dodecylsulfate–polyacrylamide gel electrophoresis gels, and transferred to Immobilon-P polyvinylidene fluoride membranes (EMD IPVH00010; EMD Millipore, Temecula, California). Membranes were stained with Ponceau S (P7170-1L; Sigma Co. St. Louis, Missouri) to assess transfer quality and equal protein loading. Membranes were blocked using 5% dry fat-free milk solution in phosphate-buffered saline with 0.1% Tween 20 (PBST) for 1 hour, followed by overnight incubation with the specified primary antibody at 4° C. Membranes were subsequently washed 3 times for 15 minutes each with PBST followed by incubation with the relevant horseradish peroxidase (HRP)–labeled secondary antibody for 3 hours at room temperature. HRP activity was determined using Immobilon Western Chemiluminescent (WBKLS0500; EMD Millipore) or SuperSignal West Femto Chemiluminescent (34096; Thermo Fisher Scientific/Pierce) followed by exposure to GeneMate X-ray film (F-9024-8x10; Bioexpress, Kaysville, Utah). Protein levels were quantified by measuring protein band intensity and normalizing it to loading controls using the Bio-Rad Image Lab software as described elsewhere.34,37 Primary antibodies used in WB analyses were as follows: rabbit anti-RBFOX1 (HPA040809; Sigma); mouse anti-CELF1 (ab9549; Abcam, Cambridge, Massachusetts); mouse anti-MBNL1 (M3320; Sigma), rabbit anti-MYF5 (ab125301; Abcam); mouse anti-MYOD1 (ab16148; Abcam); and mouse anti-histone H3 (ab10799; Abcam). Secondary antibodies used in WB analyses were as follows: goat anti-mouse light-chain IgG-HRP (Jackson ImmunoResearch Laboratories, West Grove, Pennsylvania) and goat anti-rabbit IgG-HRP (EMD Millipore).
RESULTS
Alternative Splicing Is Dysregulated in Type 1 Diabetic Mice
To determine whether AS changes occur in diabetic skeletal muscle and contribute to diabetic myopathy, we tested AS patterns of 5 genes involved in muscle contraction, growth, and metabolism. Using non-obese diabetic (NOD) mice, we assessed AS regulation of calcineurin (Ppp3ca), plasma membrane calcium-transporting ATPase 1 (Atp2b1), fragile X mental retardation syndrome–related protein 1 (Fxr1), myotubularin-related phosphatase 3 (Mtmr3), and glutathione peroxidase 8 (Gpx8).
We checked in NOD mouse skeletal muscle for splicing of Ppp3ca exon 13 that encodes for the auto-inhibitory domain of the protein and found that exon 13 was more excluded in type 1 diabetic mice (Fig. 1A and B). Next, we examined AS of Atp2b1 exon 21 that is within the calmodulin-binding domain. We found that exon 21 (corresponds to exon 22 in human) of Atbp2b1 was less included in the NOD mouse gastrocnemius muscle in comparison to control mice (Fig. 1C). AS of Fxr1 exons 15 and 16 was also altered in type 1 diabetic mouse skeletal muscle (Fig. 1D). AS of Mtmr3 exon 16 was affected modestly in NOD mice (Fig. 1E). Finally, we checked splicing of Gpx8 exon 2 that encodes for the thioredoxin superfamily domain essential for antioxidant properties of the enzyme. We found that exon 2 of Gpx8 was more excluded in the skeletal muscle of NOD mice (Fig. 1F). Overall, all 5 genes exhibited altered AS patterns in type 1 diabetic mouse skeletal muscle when compared with controls (Fig. 1).
FIGURE 1.
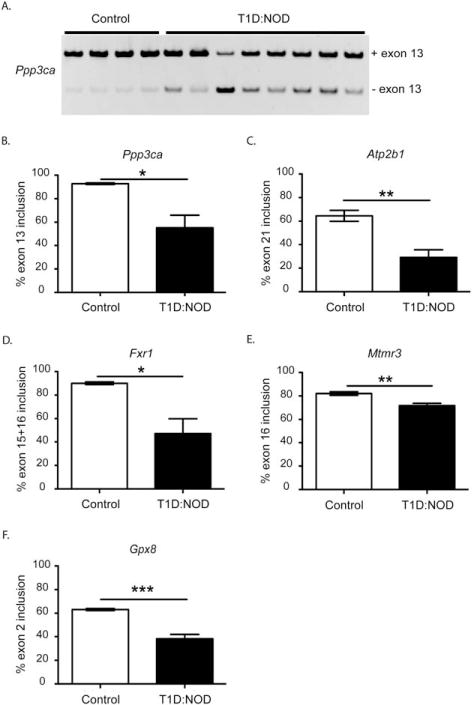
Alternative splicing is dysregulated in skeletal muscle of non-obese diabetic mice. Alternative splicing analysis of (A) and (B), Ppp3ca exon 13, (C) Atp2b1 exon 21, (D) Fxr1 exon 15 + 16, (E) Mtmr3 exon 16, and (F) Gpx8 exon 2 in ICR/HaJ (control) (white bars) or type 1 non-obese diabetic (T1D:NOD) (black bars) mouse gastrocnemius muscle, as determined by qRT-PCR (n ≥ 4; *P < 0.05, **P < 0.01, ***P < 0.005).
Alternative Splicing Events Altered in Diabetic Skeletal Muscle Are Developmentally Regulated
To determine whether AS events affected in diabetic skeletal muscle are developmentally regulated, we tested AS of these genes in vivo during normal mouse skeletal muscle development. We examined AS events in wild-type mouse skeletal muscle at embryonic day 18 (E18), newborn, and adult (6-month-old) stages. AS of Ppp3ca (Fig. 2A), Atp2b1 (Fig. 2B), Fxr1 (Fig. 2C), and Mtmr3 (Fig. 2D) transitioned in developing skeletal muscle such that alternative exons were included in adult, but excluded in newborn and embryonic skeletal muscle. Gpx8 exon 2 was included similarly in embryonic and adult skeletal muscle, but it was included less in newborn skeletal muscle (Fig. 2E). AS events altered in type 1 diabetic skeletal muscle underwent embryonic-to-postnatal transitions during normal skeletal muscle development in mice.
FIGURE 2.
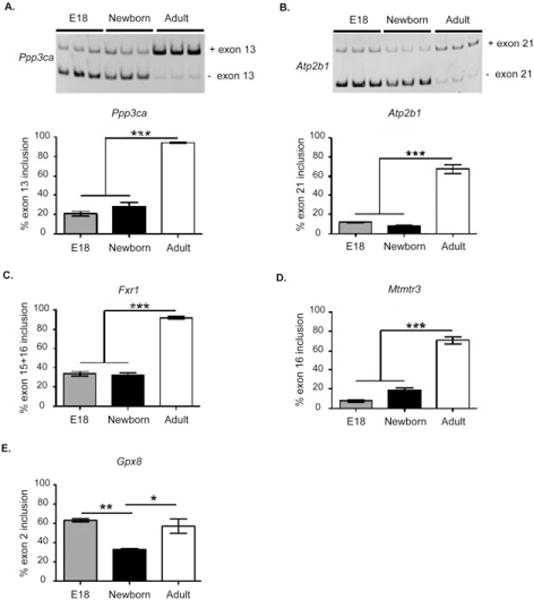
Alternative splicing events affected in type 1 diabetic skeletal muscle undergo changes during mouse skeletal muscle development. Representative gel images of (A) Ppp3ca and (B) Atp2b1 AS in normal FVB mouse skeletal muscle during development. Quantification of alternative exon inclusions of (A) Ppp3ca, (B) Atp2b1, (C) Fxr1, (D) Mtmr3, and (E) Gpx8 in embryonic day 18 (E18) limb, newborn limb, and adult mouse quadriceps (n = 3; *P < 0.05, **P < 0.01, ***P < 0.005).
RNA Binding Proteins that Regulate Alternative Splicing Undergo Dynamic Changes during Mouse Skeletal Muscle Development
To determine the protein levels of RNA binding proteins that regulate AS during skeletal muscle development, we examined steady-state levels of CELF1, MBNL1, and RBFOX1 in E18 and newborn limb and adult quadriceps by Western blot (WB). Histone H3 WB and Ponceau S stainings of membranes were used as loading controls. CELF1 protein levels were high at E18 and newborn stages, but were dramatically decreased in adult skeletal muscle relative to embryonic skeletal muscle (Fig. 3). RBFOX1 protein levels increased as the muscle developed. Although 2 isoforms (~48 kDa and ~37–40 kDa) of MBNL1 expressed in skeletal muscle were abundant at embryonic and newborn stages, they were severely downregulated in adult mice relative to embryonic and newborn mice (Fig. 3). In summary, CELF1, RBFOX1, and MBNL1 protein levels were dynamically controlled at embryonic, newborn, and adult stages in skeletal muscle.
FIGURE 3.
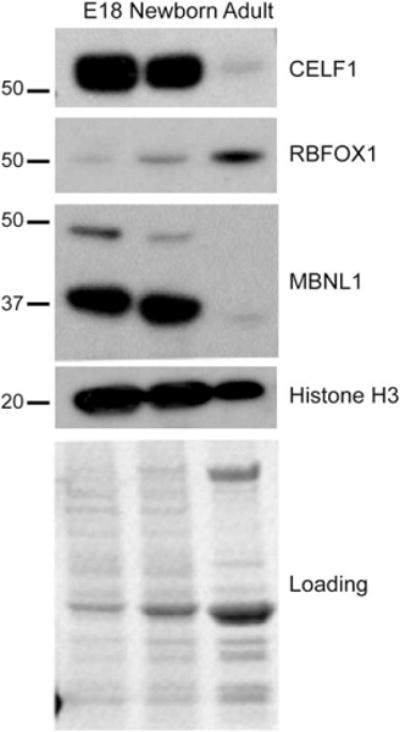
Expression of splicing regulators is dynamically controlled during skeletal muscle development. Western blot (WB) analysis of CELF1, RBFOX1, MBNL1, and Histone H3 in wild-type E18 embryonic mouse limb, newborn mouse limb, and adult mouse quadriceps. Ponceau S staining and Histone H3 WB were used to monitor protein loading.
RNA Binding Protein Levels Are Altered in Type 1 Diabetic Skeletal Muscle
To determine whether expression of embryonic spliced variants in type 1 diabetic skeletal muscle correlates with changes in RBFOX1, CELF1, and MBNL1, protein levels of these RNA binding proteins were determined by WB analysis using gastrocnemius muscle from control ICR or NOD mice. RBFOX1 protein was increased by 2.65-fold (Fig. 4A). There was no statistically significant change in MBNL1 protein levels in NOD mice skeletal muscle (Fig. 4A). CELF1 protein levels were upregulated by 2.78-fold in the skeletal muscle of NOD mice in comparison to control mice (Fig. 4B). These results show that RBFOX1 and CELF1 proteins are modulated in skeletal muscle under type 1 diabetic conditions. Notably, upregulation of CELF1 protein levels in diabetic skeletal muscle mimicked its protein levels at embryonic and newborn stages in muscle, consistent with more embryonic/newborn-like AS patterns of Atp2b1, Fxr1, and Mtmr3 in diabetic skeletal muscle (Fig. 1C–E vs. Fig. 2B–D).
FIGURE 4.
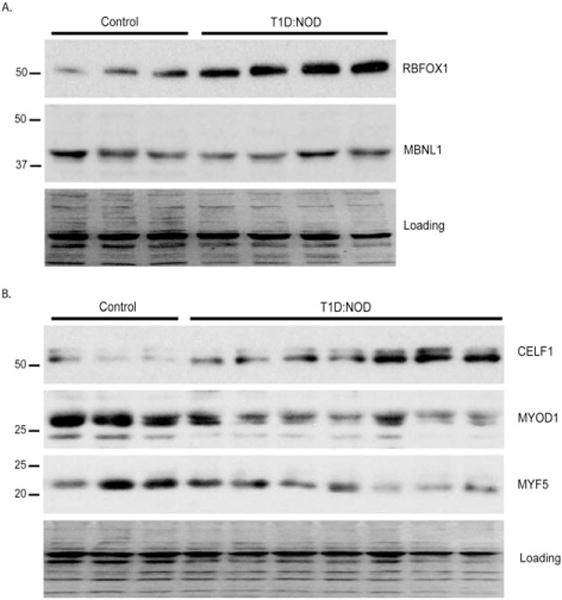
Protein levels of splicing regulators are modulated in non-obese diabetic skeletal muscle. Western blot analysis of (A) RBFOX1 and MBNL1 in ICR (control) or non-obese diabetic (T1D:NOD) gastrocnemius muscle of mice. (B) CELF1, MYOD1, and MYF5 in control (n = 3) and NOD (n = 7) mice gastrocnemius muscle. Ponceau S staining was used to monitor protein loading. Fold-change in each protein in NOD mice skeletal muscle was quantified after normalizing to the loading control and compared with control ICR mice (*P < 0.05, **P < 0.01, ***P < 0.005, NS = not significant). Fold changes in protein levels were as follows: RBFOX1 protein 2.65 ± 0.15 (P < 0.01); MBNL1 protein 1.1 ± 0.1 (not significant); CELF1 protein 2.78 ± 0.36 (P < 0.05); MYOD1 protein −2.17 ± 0.06 (P < 0.005); and MYF5 protein −1.79 ± 0.09 (P < 0.05).
Next, we tested whether CELF1 increase in diabetic skeletal muscle is a primary event or is secondary due to muscle regeneration. Therefore, we assessed protein levels of MYF5 and MYOD1 transcription factors that are upregulated in regenerating muscle. Figure 4B shows that CELF1 protein levels were elevated in NOD mice where MYOD1 and MYF5 levels were significantly downregulated – 2.17-fold and –1.79-fold, respectively (Fig. 4B). Overall, we found that increases in CELF1 protein levels correlated with expression of embryonic spliced variants in type 1 skeletal muscle and this increase was not due to muscle regeneration.
DISCUSSION
We have previously identified genome-wide AS changes in diabetic hearts.27,34 In this study we examined whether AS defects occur in type 1 diabetic skeletal muscle. We found that 5 genes relevant to skeletal muscle physiology were misspliced in type 1 diabetic skeletal muscle. Because all the alternative exons of these genes are located within a functional domain of the protein, and these exons are excluded in diabetic skeletal muscle, AS changes likely affect their functions/activities under diabetic conditions. We found that AS patterns of these genes are regulated dramatically during mouse skeletal muscle development. Expression of “fetal-like” spliced variants is implicated in skeletal muscle pathogenesis of myotonic dystrophy, in which there is severe muscle wasting and atrophy.28–33 Although diabetic myopathy displays less severe muscle atrophy and weakness, expression of embryonic spliced variants may contribute to myopathy in diabetic patients. In this study we tested only 5 genes, but it is possible that there is a global switch to embryonic AS programs in diabetic skeletal muscle. Future studies are needed to test this possibility. Several other studies, including ours, have shown that embryonic splicing patterns are also activated in failing and diabetic hearts.27,38,39 Therefore, a defect in developmental regulation of AS patterns may be a common phenomenon in striated muscle under pathological conditions.
We found that CELF1 and RBFOX1 protein levels are elevated in type 1 diabetic skeletal muscle (Fig. 4). Importantly, expression of embryonic spliced variants was in agreement with upregulation of CELF1 protein levels. Increase in CELF1 protein levels and expression of embryonic-like spliced variants in diabetic skeletal muscle supports our previous results showing that high CELF1 protein level correlates with subsequent reactivation of embryonic splicing programs in diabetic hearts.27 In support of our findings, it has been shown that AS of Fxr1, Mtmr3, and Atp2b1 revert back to an embryonic pattern in CELF1 heart-specific transgenic mice, suggesting that CELF1 controls AS of these genes.23 In addition, our results show that CELF1 upregulation is not a secondary effect from muscle regeneration and supports previous findings that muscle growth, development, and regeneration are diminished under diabetic conditions.5,10–12,40
RBFOX1 protein levels are also increased in type 1 diabetic skeletal muscle (Fig. 4A). Interestingly, we did not see a switch to an embryonic expression pattern of RBFOX1 in diabetic skeletal muscle. RBFOX1 protein levels were low at embryonic stages but high at adult stages in skeletal muscle (Fig. 3). In diabetic skeletal muscle protein levels increased further. The mechanism for this increase and its consequences on alternative splicing patterns are unknown.
In our study, Fxr1 exons 15 + 16 were mostly excluded in type 1 diabetic mouse skeletal muscle. FXR1 regulates mRNA translation of muscle structural genes such as Talin2 and Desmoplakin.41 Fxr1 ablation in mice leads to defects in formation of limb musculature and reduced Fxr1 expression in zebrafish induces muscle dystrophy.42,43 Fxr1 exon 15 and 16 splicing is regulated by both CELF and RBFOX proteins.23,34 Exons 15 and 16 are critical for binding to the muscle-specific target mRNAs of FXR1. A naturally occurring constitutively active cal-cineurin is generated via exclusion of Ppp3ca exons 13 and 14 that encode for the autoinhibitory domain, and this calcineurin isoform is thought to affect muscle regeneration.44–46 Therefore, the change in AS pattern of Ppp3ca (calcineurin) in diabetic mice generates a constitutively active isoform that may play a role in the muscle fiber changes and regeneration defects observed in type 1 diabetic skeletal muscle.13 Atp2b1 exon 21 is mostly excluded in type 1 diabetic mouse skeletal muscle, similar to its AS pattern in myotonic dystrophy patient myoblasts.47 Exclusion of the same exon, which is important for calmodulin interactions, may contribute to defects in calcium homeostasis in type 1 diabetic skeletal muscle. Atp2b1 exon 21 inclusion was shown to be controlled in an RBFOX and CELF-dependent manner.23,34 Gpx8 has 3 exons, exon 2 is skipped more in type 1 diabetic mouse skeletal muscle. This exon encodes for the thioredoxin-like superfamily domain found in antioxidant proteins.48,49 Exclusion of this exon may impact the antioxidant activity of GPX8 in type 1 diabetic skeletal muscle, but it is still unclear what regulates inclusion/exclusion of this exon under diabetic conditions.
Identifying biologically relevant AS changes in type 1 diabetic skeletal muscle may provide new targets for potential treatment options in patients. In the future, the use of modified oligonucleotides that are currently being tested for treatment of other muscle diseases50–52 may provide novel tools to correct AS changes in diabetic patients.
Supplementary Material
Acknowledgments
This work was supported in part by the March of Dimes (Award 5FY12-21), American Heart Association (Grant 15GRNT22830010), UTMB IHII Mini Center (pilot award), University of Texas Medical Branch startup funds, BMB (pilot award), the National Institutes of Health/National Heart Lung, and Blood Institute (Grant R01HL135031 to M.N.K.-M), and the University of Texas Medical Branch (Kempner Fellowship to C.A.N.).
The authors thank Dr. Heather Lander for editing of the manuscript.
Abbreviations
- ANOVA
analysis of variance
- AS
alternative splicing
- CELF1
CUG-binding protein Elav-like family member 1
- E18
embryonic day 18
- FVB
Friend virus B
- HRP
horseradish peroxidase
- MHC
histocompatibility complex
- MBNL1
muscleblind-like splicing regulator 1
- NB
newborn
- NOD
non-obese diabetic
- qRT-PCR
quantitative reverse transcript–polymerase chain reaction
- RBFOX1
RNA binding protein Fox-1 homolog 1
- T1D
type 1 diabetes
- WB
Western blot
Footnotes
Additional supporting information may be found in the online version of this article.
Publisher's Disclaimer: Disclaimer: The authors claim sole responsibility for the study’s content, which may not necessarily represent views of the National Institutes of Health.
References
- 1.Aiello LP, Gardner TW, King GL, Blankenship G, Cavallerano JD, Ferris FL, 3rd, et al. Diabetic retinopathy. Diabetes Care. 1998;21:143–156. doi: 10.2337/diacare.21.1.143. [DOI] [PubMed] [Google Scholar]
- 2.Huynh K, Bernardo BC, McMullen JR, Ritchie RH. Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol Ther. 2014;142:375–415. doi: 10.1016/j.pharmthera.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Miki T, Yuda S, Kouzu H, Miura T. Diabetic cardiomyopathy: pathophysiology and clinical features. Heart Fail Rev. 2013;18:149–166. doi: 10.1007/s10741-012-9313-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coppini DV. Enigma of painful diabetic neuropathy: can we use the basic science, research outcomes and real-world data to help improve patient care and outcomes? Diabet Med. 2016;33:1477–1482. doi: 10.1111/dme.13089. [DOI] [PubMed] [Google Scholar]
- 5.Hernandez-Ochoa EO, Vanegas C. Diabetic myopathy and mechanisms of disease. Biochem Pharmacol (Los Angel) 2015;4 doi: 10.4172/2167-0501.1000e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conserva F, Gesualdo L, Papale M. A systems biology overview on human diabetic nephropathy: from genetic susceptibility to post-transcriptional and post-translational modifications. J Diabetes Res. 2016;2016:7934504. doi: 10.1155/2016/7934504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan GC, Tang SC. Diabetic nephropathy: landmark clinical trials and tribulations. Nephrol Dial Transplant. 2016;31:359–368. doi: 10.1093/ndt/gfu411. [DOI] [PubMed] [Google Scholar]
- 8.Schneider AL, Kalyani RR, Golden S, Stearns SC, Wruck L, Yeh HC, et al. Diabetes and prediabetes and risk of hospitalization: The Atherosclerosis Risk in Communities (ARIC) study. Diabetes Care. 2016;39:772–779. doi: 10.2337/dc15-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El-Beblawy NM, Andrawes NG, Ismail EA, Enany BE, Abou El-Seoud HS, Erfan MA. Serum and urinary orosomucoid in young patients with type 1 diabetes: a link between inflammation, microvascular complications, and subclinical atherosclerosis. Clin Appl Thromb Hemost. 2016;22:718–726. doi: 10.1177/1076029616637185. [DOI] [PubMed] [Google Scholar]
- 10.Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. 2014;383:69–82. doi: 10.1016/S0140-6736(13)60591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Souza DM, Al-Sajee D, Hawke TJ. Diabetic myopathy: impact of diabetes mellitus on skeletal muscle progenitor cells. Front Physiol. 2013;4:379. doi: 10.3389/fphys.2013.00379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krause MP, Al-Sajee D, D’Souza DM, Rebalka IA, Moradi J, Riddell MC, et al. Impaired macrophage and satellite cell infiltration occurs in a muscle-specific fashion following injury in diabetic skeletal muscle. PLoS One. 2013;8:e70971. doi: 10.1371/journal.pone.0070971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fritzsche K, Bluher M, Schering S, Buchwalow IB, Kern M, Linke A, et al. Metabolic profile and nitric oxide synthase expression of skeletal muscle fibers are altered in patients with type 1 diabetes. Exp Clin Endocrinol Diabetes. 2008;116:606–613. doi: 10.1055/s-2008-1073126. [DOI] [PubMed] [Google Scholar]
- 14.Andersen H, Gadeberg PC, Brock B, Jakobsen J. Muscular atrophy in diabetic neuropathy: a stereological magnetic resonance imaging study. Diabetologia. 1997;40:1062–1069. doi: 10.1007/s001250050788. [DOI] [PubMed] [Google Scholar]
- 15.Brack AS, Rando TA. Tissue-specific stem cells: lessons from the skeletal muscle satellite cell. Cell Stem Cell. 2012;10:504–514. doi: 10.1016/j.stem.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dhawan J, Rando TA. Stem cells in postnatal myogenesis: molecular mechanisms of satellite cell quiescence, activation and replenishment. Trends Cell Biol. 2005;15:666–673. doi: 10.1016/j.tcb.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 17.Babij P. Tissue-specific and developmentally regulated alternative splicing of a visceral isoform of smooth muscle myosin heavy chain. Nucl Acids Res. 1993;21:1467–1471. doi: 10.1093/nar/21.6.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo W, Mulligan GJ, Wormsley S, Helfman DM. Alternative splicing of beta-tropomyosin pre-mRNA: cis-acting elements and cellular factors that block the use of a skeletal muscle exon in nonmuscle cells. Genes Dev. 1991;5:2096–2107. doi: 10.1101/gad.5.11.2096. [DOI] [PubMed] [Google Scholar]
- 19.Bland CS, Cooper TA. Micromanaging alternative splicing during muscle differentiation. Dev Cell. 2007;12:171–172. doi: 10.1016/j.devcel.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 20.Cooper TA. Alternative splicing regulation impacts heart development. Cell. 2005;120:1–2. doi: 10.1016/j.cell.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 21.Cooper TA, Ordahl CP. A single troponin T gene regulated by different programs in cardiac and skeletal muscle development. Science. 1984;226:979–982. doi: 10.1126/science.6095446. [DOI] [PubMed] [Google Scholar]
- 22.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Ann Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 23.Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, et al. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci USA. 2008;105:20333–20338. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ladd AN, Charlet N, Cooper TA. The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Mol Cell Biol. 2001;21:1285–1296. doi: 10.1128/MCB.21.4.1285-1296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blech-Hermoni Y, Stillwagon SJ, Ladd AN. Diversity and conservation of CELF1 and CELF2 RNA and protein expression patterns during embryonic development. Dev Dyn. 2013;242:767–777. doi: 10.1002/dvdy.23959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blech-Hermoni Y, Ladd AN. RNA binding proteins in the regulation of heart development. Int J Biochem Cell Biol. 2013;45:2467–2478. doi: 10.1016/j.biocel.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verma SK, Deshmukh V, Liu P, Nutter CA, Espejo R, Hung ML, et al. Reactivation of fetal splicing programs in diabetic hearts is mediated by protein kinase C signaling. J Biol Chem. 2013;288:35372–35386. doi: 10.1074/jbc.M113.507426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuyumcu-Martinez NM, Cooper TA. Misregulation of alternative splicing causes pathogenesis in myotonic dystrophy. Prog Mol Subcell Biol. 2006;44:133–159. doi: 10.1007/978-3-540-34449-0_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–741. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- 30.Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 31.Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- 32.Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 33.Freyermuth F, Rau F, Kokunai Y, Linke T, Sellier C, Nakamori M, et al. Splicing misregulation of SCN5A contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nat Commun. 2016;7:11067. doi: 10.1038/ncomms11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nutter CA, Jaworski EA, Verma SK, Deshmukh V, Wang Q, Botvinnik OB, et al. Dysregulation of RBFOX2 Is an early event in cardiac pathogenesis of diabetes. Cell Rep. 2016;15:2200–2213. doi: 10.1016/j.celrep.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.King AJ. The use of animal models in diabetes research. Br J Pharmacol. 2012;166:877–894. doi: 10.1111/j.1476-5381.2012.01911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leiter EH, Schile A. Genetic and pharmacologic models for type 1 diabetes. Curr Protoc Mouse Biol. 2013;3:9–19. doi: 10.1002/9780470942390.mo120154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verma SK, Deshmukh V, Nutter CA, Jaworski E, Jin W, Wadhwa L, et al. Rbfox2 function in RNA metabolism is impaired in hypoplastic left heart syndrome patient hearts. Sci Rep. 2016;6:30896. doi: 10.1038/srep30896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao C, Ren S, Lee JH, Qiu J, Chapski DJ, Rau CD, et al. RBFox1-mediated RNA splicing regulates cardiac hypertrophy and heart failure. J Clin Invest. 2016;126:195–206. doi: 10.1172/JCI84015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei C, Qiu J, Zhou Y, Xue Y, Hu J, Ouyang K, et al. Repression of the central splicing regulator RBFox2 is functionally linked to pressure overload-induced heart failure. Cell Rep. 2015;10:1521–1533. doi: 10.1016/j.celrep.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aragno M, Mastrocola R, Catalano MG, Brignardello E, Danni O, Boccuzzi G. Oxidative stress impairs skeletal muscle repair in diabetic rats. Diabetes. 2004;53:1082–1088. doi: 10.2337/diabetes.53.4.1082. [DOI] [PubMed] [Google Scholar]
- 41.Whitman SA, Cover C, Yu L, Nelson DL, Zarnescu DC, Gregorio CC. Desmoplakin and talin2 are novel mRNA targets of fragile X-related protein-1 in cardiac muscle. Circ Res. 2011;109:262–271. doi: 10.1161/CIRCRESAHA.111.244244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van’t Padje S, Chaudhry B, Severijnen LA, van der Linde HC, Mientjes EJ, Oostra BA, et al. Reduction in fragile X related 1 protein causes cardiomyopathy and muscular dystrophy in zebrafish. J Exp Biol. 2009;212:2564–2570. doi: 10.1242/jeb.032532. [DOI] [PubMed] [Google Scholar]
- 43.Mientjes EJ, Willemsen R, Kirkpatrick LL, Nieuwenhuizen IM, Hoogeveen-Westerveld M, Verweij M, et al. Fxr1 knockout mice show a striated muscle phenotype: implications for Fxr1p function in vivo. Hum Mol Genet. 2004;13:1291–1302. doi: 10.1093/hmg/ddh150. [DOI] [PubMed] [Google Scholar]
- 44.Lara-Pezzi E, Winn N, Paul A, McCullagh K, Slominsky E, Santini MP, et al. A naturally occurring calcineurin variant inhibits FoxO activity and enhances skeletal muscle regeneration. J Cell Biol. 2007;179:1205–1218. doi: 10.1083/jcb.200704179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev. 2000;80:1483–1521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- 46.Reuter A, Mi J, Sehrsam I, Ludolph AC, Volkel H. A novel calcineurin splice variant that modifies calcineurin activity. Eur J Biochem. 2001;268:5955–5960. doi: 10.1046/j.0014-2956.2001.02551.x. [DOI] [PubMed] [Google Scholar]
- 47.Ketley A, Chen CZ, Li X, Arya S, Robinson TE, Granados-Riveron J, et al. High-content screening identifies small molecules that remove nuclear foci, affect MBNL distribution and CELF1 protein levels via a PKC-independent pathway in myotonic dystrophy cell lines. Hum Mol Genet. 2014;23:1551–1562. doi: 10.1093/hmg/ddt542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bosello-Travain V, Forman HJ, Roveri A, Toppo S, Ursini F, Venerando R, et al. Glutathione peroxidase 8 is transcriptionally regulated by HIFalpha and modulates growth factor signaling in HeLa cells. Free Radic Biol Med. 2015;81:58–68. doi: 10.1016/j.freeradbiomed.2014.12.020. [DOI] [PubMed] [Google Scholar]
- 49.Brigelius-Flohe R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830:3289–3303. doi: 10.1016/j.bbagen.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 50.Wheeler TM. Myotonic dystrophy: therapeutic strategies for the future. Neurotherapeutics. 2008;5:592–600. doi: 10.1016/j.nurt.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao Z, Cooper TA. Antisense oligonucleotides: rising stars in eliminating RNA toxicity in myotonic dystrophy. Hum Gene Ther. 2013;24:499–507. doi: 10.1089/hum.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muir LA, Chamberlain JS. Emerging strategies for cell and gene therapy of the muscular dystrophies. Expert Rev Mol Med. 2009;11:e18. doi: 10.1017/S1462399409001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.