

Abstract
The voltage-gated L-type Ca2+ channel CaV1.2 is crucial for initiating heartbeat and control of a number of neuronal functions such as neuronal excitability and long-term potentiation. Mutations of CaV1.2 subunits result in serious health problems including arrhythmia, autism spectrum disorders, immunodeficiency, and hypoglycemia. Thus precise control of CaV1.2 surface expression and localization is essential. We previously reported that α-actinin associates and colocalizes with neuronal CaV1.2 channels and that shRNA-mediated depletion of α-actinin significantly reduces localization of endogenous CaV1.2 in dendritic spines in hippocampal neurons. Here we investigated the hypothesis that direct binding of α-actinin to CaV1.2 supports its surface expression. Using two-hybrid screens and pull-down assays we identified three point mutations (K1647A, Y1649A, and I1654A) in the central, pore-forming α11.2 subunit of CaV1.2 that individually impaired α-actinin binding. Surface biotinylation and flow cytometry assays revealed that CaV1.2 channels composed of the corresponding α-actinin-binding-deficient mutants results in a 35–40% reduction in surface expression compared to wild-type channels. Moreover, the mutant CaV1.2 channels expressed in HEK293 cells exhibit a 60–75% reduction in current density. The larger decrease in current density as compared to surface expression imparted by these α11.2 subunit mutations hints at the possibility that α-actinin not only stabilizes surface localization of CaV1.2 but also augments its ion conducting activity.
Keywords: alpha-actinin, calcium channel, cell surface, electrophysiology, flow cytometry, plasma membrane, protein-protein interaction, site-directed mutagenesis, surface biotinylation, yeast two-hybrid
Graphical Abstract
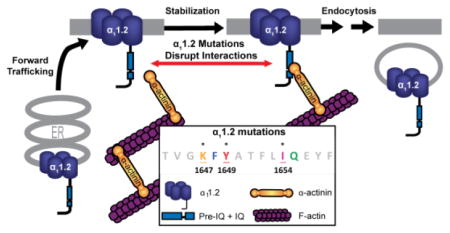
INTRODUCTION
Ca2+ is a potent secondary messenger involved in triggering a number of key biochemical signaling pathways that regulate a multitude of physiological processes. Voltage-dependent calcium channels (VDCCs) mediate Ca2+ influx into cells upon changes in membrane potential. Thus VDCCs act as cellular transistors, converting electrical to biochemical signals. The four dihydropyridine-sensitive L-type Ca2+ channels CaV1.1– CaV1.4 are each composed of distinct pore-forming α1 subunits (α11.1 – α11.4) encoded by different genes. In addition to this large transmembrane α1 subunit, VDCCs also require associated auxiliary β and α2δ and, in case of CaV1.1, γ subunits, for full activity (1–3). These subunits facilitate release of α1 subunits from the endoplasmic reticulum (ER), inhibit ubiquitin-mediated degradation of VDCCs, influence electrophysiological properties of Ca2+ channels, such as activation and inactivation, and play diverse roles in the regulation of these channels (1–5).
CaV1.2 is expressed in a variety of cell types including neurons, cardiomyocytes, endocrine cells, and immune cells. It is the primary voltage-gated L-type channel of the central nervous system (6, 7) and heart (8). CaV1.2 is critical for numerous neuronal functions, including synaptic plasticity (9–12), learning, memory (10), and gene expression (13–17). Mutations in CaV1.2 have been linked to arrhythmia, autism spectrum disease, immunodeficiency, hypoglycemia, webbing fingers, as observed in the multi-syndromic disease called Timothy syndrome (18). Recent genome-wide association studies suggest that genetic variations in the α11.2 encoding gene CACNA1C are involved in many psychiatric diseases including schizophrenia, depression, bipolar and autism spectrum disease (19, 20). Thus it is no surprise that knockout of CACNA1C in mice is embryonically lethal (8).
In neurons, CaV1.2 is enriched at postsynaptic sites (21–25), while in cardiomyocytes, it is highly enriched in the T-tubules where, in synchrony with ryanodine receptors, it triggers excitation-contraction coupling (26). In neurons such precisely defined subcellular localization of CaV1.2 allows for Ca2+ enriched nanodomains that can more accurately control excitability (27–29) and long-term potentiation (9–12). In this study, we investigate the molecular mechanisms inherent to CaV1.2 and its interactions with binding partners that ultimately are critical for its functional distribution to the plasma membrane.
Surface expression and internalization of CaV1.2 is activity-controlled in transfected cell lines and primary cortical and hippocampal neurons (30–32). Disruption of F-actin networks diminishes CaV1.2 surface expression and L-type currents in neurons (33), cardiomyocytes (34), vascular smooth muscle cells (35), and retinal neurons (36). The ~100kDa ubiquitous actin-binding protein α-actinin forms anti-parallel dimers, which facilitate F-actin cross-linking and linkage to integral membrane proteins and ion channels. Like CaV1.2 (21–25), α-actinin is enriched in postsynaptic dendritic spines (37, 38), where it colocalizes with CaV1.2 (32). We recently reported that α-actinin binds to an 86 amino acid (AA) region (residues 1,584 to 1,670) in the proximal C-terminus of rabbit α11.2 (32). Previous work of others found that the deletion of 43 AAs within this region (AAs 1,623–1,666) reduces surface expression of CaV1.2 (4, 39, 40). This region encompasses the IQ segment (AAs 1,644–1,666), which binds to calmodulin (CaM) and fosters Ca2+-dependent inactivation (CDI) (41–45). CaM has been reported to promote surface expression of CaV1.2 (46) and has been hypothesized to help mask ER retention signals within the quaternary structure of CaV1.2 (47). However, this effect appears to be only engaged in the absence of β subunits and others find no effect of CaM on CaV1.2 plasma membrane targeting in HEK293 cells (4). Previously we showed that α-actinin knockdown (KD) impairs surface expression of CaV1.2 in HEK293T/17 cells and spine accumulation in neurons (32). However, whether these effects were due to loss of direct α-actinin binding to CaV1.2 or due to indirect effects of the α-actinin KD (perhaps by disturbing F-actin) remained unclear. Here we precisely define specific residues within α11.2 that are critical for α-actinin-1 binding. Three different point mutations that disrupt α-actinin binding reduce CaV1.2 surface expression. Unexpectedly they appear to down regulate current density to a larger degree than surface expression.
MATERIALS AND METHODS
Reagents and Antibodies
The FP1 antibody against Loop II/III of α11.2 has been previously described (48). The mouse antibody against the hemagglutinin (HA)-epitope, mono HA.11 16B12 (Lot# 14945101) used for immunoblotting was from Covance, the mouse-anti-α-tubulin antibody (DM1A) from Santa Cruz Biotechnology, the mouse-anti-GAPDH antibody (Lot# 214592) from EMD Millipore, the anti- glutathione S-transferase (GST) antibody from GE Heathcare Life Science, and the anti-Maltose Binding Protein (MBP) antibody from NewEngland Biosciences (NEB). Horseradish peroxidase (HRP)-conjugated goat-anti-mouse and mouse-anti-rabbit secondary antibodies were from Jackson ImmunoResearch. Bovine serum albumin (BSA) used for immunoblotting was from Research Products International Corp (RPI Corp). Polyvinylidene difluoride (PVDF) membranes and HRP-coupled protein A was from Bio-Rad (Life Sciences Group, Hercules CA). Luminata Classico and Crescendo Western HRP Substrate reagents were from Fisher Scientific. For flow cytometry the Alexa Fluor® 647-Conjugated anti-HA-Tag Mouse monoclonal antibody (6E2) was from Cell Signaling Technology, propidium iodide (PI) from Sigma. Isradipine was from Enzo Life Sciences and accutase from MP Biomedicals. EZ-link-sulfo-NHS-LC-biotin and the NeutrAvidin UltraLink Resin were from ThermoFisher. Other reagents were obtained from Fisher Scientific, VWR or similar suppliers.
Yeast Two-Hybrid Plasmids and Methodology
The Matchmaker Gold yeast (GAL4 transcription) two-hybrid system was utilized for testing protein interactions. The cDNAs encoding AAs 1-123, 1506-1610, 1506-1638, and 1639-1871 of the rabbit α11.2 subunit of CaV1.2 (GenBank ID: NM001136522.1) were subcloned in-frame with the Gal4 DNA-binding domain of the NdeI and EcoRI digested ‘bait’ expression vector pGBKT7 (Clontech; for oligonucleotides see Supporting Information, Table S1). The cDNAs encoding full length and various regions of α-actinin-1 were subcloned in frame with the Gal4 activation domain of the NdeI/EcoRI digested ‘prey’ expression vector pGADT7 (Clontech; for oligonucleotides see Supporting Information, Table S2). Pairs of these bait and prey plasmids were cotransformed into Y2H Gold yeast and cells harboring both plasmids selected by growth on double dropout (DDO, Leu−/Trp−/X-α-Gal+) plates to ensure successful cotransformation. Selected colonies were then transferred to the more stringent quadruple dropout (QDO, Leu−/Trp−/His−/Ade−/X-α-Gal+) plates. Yeast colonies that express interacting Gal4 fusion proteins digest the X-α-Gal chromogenic substrate and produce a reporter indigo-blue chromophore. Site-specific mutations in AAs 1639-1871 of α11.2 were generated via PCR using specific oligonucleotide primer sets (see Supporting Information, Tables S3 (triple alanine) and S4 (single alanine)) and the QuikChange II Site-Directed Mutagenesis kit (Stratagene/Agilent Technologies).
Molecular Cloning of other Plasmids
For in vitro pull-down assays, we used the GST-CT1 plasmid, which covers rabbit α11.2 AAs 1513-1733 in the GST-fusion protein expression vector pGEX-4T1 (GE healthcare) as described (32). The site-specific mutations K1647A, Y1649A, and I1654A were generated via QuikChange II as above (see Supporting Information, Table S4). A cDNA fragment encoding AAs 391-892 of human α-actinin-1 was sub-cloned into KpnI/EcoRI digested plasmid DNA of the MBP fusion protein expression vector, pMAL-c2e (NEB). For identification of CaV1.2 expressing HEK293T/17 transfectants in electrophysiological, flow cytometry and surface biotinylation studies, we used a rat α11.2 (GenBank ID: M67515.1) construct that carried an HA tag in the S5-H5 extracellular loop of domain II, which only modestly reduces current density in heterologous expression systems (25, 49). This α11.2 cDNA was subcloned into a SalI/SacII restriction endonuclease digested pECFP-C1 vector (Clontech). The site-specific mutations K1647A, Y1649A, and I1654A in the rat α11.2 IQ domain were again generated via QuikChange II with rat-appropriate oligonucleotide primer sets (see Supporting Information, Table S5).
Production of GST- and MBP-tagged α11.2 Fragments and α-Actinin for In Vitro Binding Studies
Expression, purification, and pull-down assays using the CaV1.2 α11.2 subunit-GST fusion proteins were performed essentially as before (32, 50, 51). Wild Type (WT) and mutant (K1647A, Y1649A, I1654A) rabbit α11.2 AAs 1576-1733 were expressed as GST-fusion proteins using the expression vector pGEX-4T1. Human α-actinin-1 (AAs 391-892)-MBP fusion protein was produced from the expression vector, pMAL-c2e (New England Biolabs, NEB). Expression and purification of MBP-tagged α-actinin-1 was performed according to the manufacturer’s protocol (NEB). For pull-down assays, GST-tagged WT and mutant α11.2 AAs 1576-1733 were expressed in E. coli before extraction and adsorption of 150 μg onto glutathione-Sepharose, washing, and incubation with 100 μg of MBP-purified α-actinin-1. Resins were washed three times in ice-cold TBS plus 0.1% Triton X-100 and extracted with 1.5x SDS-PAGE sample buffer at 90 °C for 5 min. Extracted proteins were loaded onto 7.5% polyacrylamide gels, fractionated by SDS-PAGE and transferred to PVDF membranes for immunoblot detection with anti-GST antibodies to assess relative amounts of immobilized α11.2 bait and anti-MBP antibodies to determine relative amounts of α-actinin pulled down by the resin.
Fluorescence Polarization (FP)
Synthetic peptides spanning α11.2 AAs 1644-1668 with fluorescein covalently linked to their N-terminal amino groups were obtained from ChinaPeptides (Shanghai, China). Peptides were titrated with untagged purified ApoCaM as described (11, 52). First, serial dilutions of CaM were prepared starting with 100 μM CaM and diluting it twofold eight times, i.e., each time 1:1, with FP buffer (50 mM HEPES, 100 mM KCl, 1 mM MgCl2, 0.05 mM EGTA, 5 mM NTA, pH 7.4) in black 384-well polystyrene plates (Corning). Then fluorescein-labeled peptides were added to each well for a final concentration of 1μM. After a minimum of 45 min, FP was determined with a Synergy 2 (Biotek) plate reader using polarization filters to determine parallel and perpendicular fluorescence intensities of exciting (485/20λ) and emitted light (528/20λ). Data were recorded with the Gen5 software. FP was calculated as P = (Iv - g*Ih)/(Iv + g*Ih); Iv and Ih: vertical and horizontal fluorescence intensities, respectively; g: correction factor for fluorescein. Fractional binding was calculated as (FP- FPmin)/(FPmax-FPmin) where FPmax and FPmin were the highest and lowest values obtained, respectively, for each dilution series. To obtain binding curves and Kd values, fractional binding values were fitted in GraphPad Prism 5 to the equation Y = B*X/(Kd + X); B: maximal FP value that would be reached at saturation as determined by extrapolation of the fitted curve (53).
Cell Culture
HEK293T/17 cells were cultured in DMEM-10 [Dulbecco’s modified Eagle’s medium (Life Technologies) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals)] at 37 °C in humidified incubators injected with 5% CO2 and 95% air.
Electrophysiology
HEK293T/17 cells were seeded onto poly-L-lysine-coated coverslips 24 hours prior to transfection with pGWIH-based plasmids encoding rat β2a (54) and rabbit α2δ1 (55) subunits and the HA- and CFP-tagged WT or mutant α11.2 (see above) at a 1:1:1 ratio using Lipofectamine 2000™. Whole-cell Ba2+ currents (IBa) were recorded 48~72 hours after the transfection from CFP positive HEK293T/17 cells. Extracellular solution contained (in mM) 130 N-Methyl-D-Glucamine HCl (NMG-Cl), 5 BaCl2, 1 MgCl2, 10 HEPES, pH 7.4 and 300 mOsm, and pipette solution 125 CsMeSO4, 10 EGTA, 10 Tetraethylammonium Chloride (TEA-Cl), 10 glucose, 3 Mg-ATP, 0.5 Na-GTP, and 1 MgCl2, pH 7.4 and ~290 mOsm. Osmolarity was adjusted with sucrose and pH with NMG or CsOH. Pipette electrodes were made from borosilicate glass capillaries with 1.5~2.5 MΩ resistances by use of a Flaming micropipette puller (Model P-97, Sutter Instruments). Data were obtained with an Axopatch 200B amplifier, with 1440 digitizer and pCLAMP 10 software (Molecular Devices). The sampling rate was 5 kHz and filter rate was 2 kHz. Pipette and whole cell capacitance were manually offset via the amplifier. P/4 leak subtraction was applied through the pClamp software. Cells were held at −70 mV and whole cell currents were triggered by depolarizing pulses ranging from −40 to +80 mV with 10 mV increments. To assess current density, IBa from individual cells were normalized to their whole cell capacitance. The current densities were plotted against the function of depolarization pulse as the current-voltage relationships (I–V curves). The I–V curves were fitted with the Boltzmann IV equation: I(V) = (Gmax*(V – Vrev))/(1 + e((V – V1/2)/k) with Gmax as maximum conductance, Vrev as reversal potential, and V1/2 (Vhalf) the voltage where 50% of the channels are open, and k is the slope function which denotes RT/zF, where R is the gas constant, T is absolute temperature (in K), z is charge number, and F is the Faraday constant. Curve fitting and descriptive statistics were performed and plotted by ORIGIN software. Data were analyzed using one-way analysis of variance (ANOVA) statistical analysis including Tukey and Bonferroni post hoc test and shown as mean +/− S.E.M. All recordings were performed several times in at least three independently transfected cell cultures for each condition.
Cell Surface Biotinylation Assays
HEK293T/17 cells were transfected with cDNA plasmids for α2δ1, β2a, and either WT or mutant CFP-HA-tagged α11.2 at a 1:1:1 ratio using HEPES-buffered (pH 7.11) calcium phosphate DNA precipitation (56, 57). Seven hours after transfection the medium was removed and replaced with fresh DMEM-10 medium (Enzo). Cells were harvested during the log phase of growth at 22 h post-transfection by washing with phosphate-buffered saline (PBS, pH 7.1) followed by incubation with Accutase™ (MP Biomedicals) for 3 min at room temperature (RT) to detach cells from substrate. Resuspended cells were collected and diluted in DMEM-10, triturated to mono-dispersion and held on ice. Cell counts with trypan blue exclusion were performed to assess concentration and viability. Samples with greater than 95% viability (trypan blue-negative) were collected by centrifugation and washed with ice cold HEPES biotinylation buffer (HBB; 140 mM NaCl, 0.5 mM MgCl2, 10 mM HEPES, pH 7.8,). 6×106 washed cells were resuspended in 2 ml of ice cold HBB and treated with 0.4 mg/mL of the cell membrane-impermeant EZ-link-sulfo-NHS-LC-biotin (ThermoFisher) (58) for 30 min on ice as described (32, 59). The labeling reaction was quenched with 4 washes of 20 mM glycine in HEPES buffer (pH 7.8). The cells were lysed in ice-cold RIPA buffer (50 mM Tris-Cl pH 7.4, 150 mM NaCl, 5 mM EGTA, pH 7.4, 10 mM EDTA, 1% NP-40, 10% Glycerol, 0.05% SDS, 0.4% DOC) supplemented with a cocktail of protease inhibitors (1 μg/mL leupeptin (Merck Millipore), 2 μg/mL aprotinin (Merck Millipore), 1 μg/mL pepstatin A (Merck Millipore) and 34 μg/mL phenylmethylsulfonyl fluoride (Sigma)). The solubilized material was cleared of insoluble debris by centrifugation at 200,000 × g for 15 min. Biotinylated constituents in 250 μg cell protein lysate were affinity purified by incubation with 30 μl of NeutrAvidin-conjugated Sepharose beads (ThermoFisher) for 2 hours at 4°C. Bead-bound material was collected by centrifugation, washed 3x with ice cold buffer and immobilized proteins extracted in SDS-sample buffer. Extracted samples were fractionated by SDS-PAGE in 7.5% acrylamide gels and transferred onto PVDF membranes for immunoblot detection using antibodies against the HA tag of α11.2 and against the internal loop II/III FP1 epitope as well as the control cytosolic proteins GAPDH and α-tubulin.
Immunoblot Analysis
Protein-bound PVDF membranes were incubated in blocking buffer (TBS containing 0.10% Tween and 2% bovine serum albumin (BSA, RPI Corp.)) for 1 hour at RT before incubation with primary antibodies (diluted in blocking buffer) for 3 hours at RT. Following primary incubation, membranes were washed for 40 min with > 4 exchanges of TBS+0.10% Tween (TBST) buffer, incubated with HRP-conjugated secondary antibodies (Jackson) for 1 hour at RT, washed again with TBST exchanges for 1.5 hours and bound antibody detected using Luminata Classico or Crescendo Western HRP substrates (Merck Millipore) to generate luminescent signals captured on X-ray film (Denville Scientific Inc.). Multiple exposures over increasing time periods were taken to ensure that all signals were in the linear range (60, 61).
Flow Cytometry
HEK293T/17 cells were transfected with cDNA for α2δ1, β2a, and CFP-HA-α11.2 as above. The medium was changed to DMEM-10 7 h after transfection as above. At 22 h post transfection the cells were washed with PBS, and detached by incubating with Accutase™ as above. Cells held on ice were mono-dispersed and counted using trypan blue exclusion assays as above. 6×106 cells were washed with PBS, resuspended in ice cold PBS containing 0.5% BSA (RPI Corp.) and incubated on ice plus or minus 2.5 μg/mL Alexa Fluor 647-conjugated mouse anti-HA-tag antibodies (either mouse IgG2b clone TANA2 from MBL Int. Corp. or mouse IgG1 clone 16B12 from Biolegend Inc.) for 1 h in the dark. The remaining non-immunostained cells were lysed in ice-cold RIPA buffer as above for immunoblotting of total α11.2 expression. Immunostained cells were washed three times with ice cold PBS before a final resuspension in ice cold PBS containing 0.5% BSA. Resuspended cells were filtered through cell-strainer caps into Falcon 5 mL round bottom polystyrene tubes (Corning) to remove aggregates. To allow for live cell gating, 1 μg/mL of the membrane-impermeant DNA intercalating dye propidium iodide (PI, Sigma) was added before flow cytometry data collection using a Becton-Dickinson LSRII 17 color flow cytometer equipped with FACS DIVA software. The expressed CFP- α11.2 fusion protein and control CFP vector transfected cells was detected by excitation at 405nm using the violet excitation laser line with a 560/40 filter and the fluorescence of PI at 640nm was detected using the 488nm laser line and 670/30 filter. Alexa Fluor 647 (anti-HA-tag immunostained cells) fluorescence positive cells were excited and detected using the 638nm red laser line and 670/30 filter. Emission spectra of all chosen fluorophores were well separated to minimize any possible ambiguity that might arise from spectral overlap. Before data collection for each experimental run, mock- and CFP-alone-transfected HEK293T/17 cells +/− PI were used to establish appropriate instrument settings and thresholds for detection of each single fluorophore. During each experimental run, equal numbers of live cell events (e.g., 50,000 PI-negative cells) from each test sample encompassed within the forward scatter (FSC) by side scatter (SSC) population gate were collected for further analysis of their CFP and Alexa Fluor 647 fluorescence (anti-HA) intensity. Doublets were excluded via FSC height by area gating. Surface HA staining was set as the percent of CFP positive cells exhibiting Alexa Fluor 647 intensity beyond that of the immunostained CFP control gate and normalized to that exhibited by WT α11.2. Data analysis for each experiment was done using FlowJo flow 10.1 cytometry analysis software.
Data Analysis of Surface Expression Assays
Immunoblot signals were quantified using ImageJ (National Institutes of Health) and Adobe Photoshop CS3 (Adobe Systems Incorporated). Statistical analysis was with GraphPad Prism version 4.0 (GraphPad Software, Inc.). Data were obtained from > three independent experimental trials for each condition. Data are shown as mean ± S.E. Analysis of variance (one-way ANOVA with the Tukey and Bonferroni post hoc tests) was used for comparisons of more than two groups. p < 0.05 was considered statistically significant (*).
RESULTS
Yeast Two-hybrid Screening
We previously showed co-immunoprecipitation of α-actinin with α11.2 and determined that a C-terminal region in α11.2 spanning AAs 1588-1670 is required for its interaction with α-actinin (32). This α11.2 region also encompasses the IQ motif (AAs 1644-1670) where calmodulin binds to facilitate Ca2+-dependent inactivation (41, 42). To better define the region in α-actinin required for this interaction and to test whether this interaction is direct, we used the yeast two-hybrid (Y2H) system (62). We designed a series of Y2H expression constructs covering the different polypeptide regions in α-actinin 1 (Figure 1A). Out of eleven α-actinin constructs, only the SR4EF and SR234EF polypeptides consisting of the C-terminal four EF hands preceded by either one (SR4EF) or three (SR234EF) spectrin repeats of α-actinin-1 showed interactions with the α11.2 AAs 1506-1871) (Figure 1B). SR234EF gave consistently strong interaction readouts and was thus used for all subsequent interaction assays.
Figure 1. Defining α-actinin-1 - α11.2 interacting regions by yeast two-hybrid assay.

A, Top: Schematic of α11.2 ‘bait’ encoded by a cDNA for the C-terminal region of α11.2 spanning AAs 1506-1871 (red frame), which was subcloned in frame with the Gal4 DNA-binding domain in the Y2H ‘bait’ expression vector pGBKT7 for initial identification of the α-actinin-1 binding region. Bottom: Schematic of α-actinin-1. The optimal ‘prey’ polypeptide encompassing spectrin repeats 2–4 and EF hands (SR234EF; AAs 391-892) of α-actinin-1 is depicted by the red frame. B) Full length (FL) and 10 different fragments of α-actinin-1 were expressed from the pGADT7 vector as fusion proteins in frame with the Gal4 activation domain. A potent interaction between these prey polypeptides and the α11.2 (1506-1871) bait leads to Gal4 activation and conversion of X-α-Gal to an indigo-blue chromophore that is reflected by the turquois color in the cultures. Blank pGBKT7 plasmid with α-actinin-1 pGADT7 SR234EF (NC) served as negative control (not depicted but see NC2 in Fig. 2A). C) Selected colonies of Y2H Gold yeast expressing different fragments of α11.2 C-terminus grown on double-drop out media (Leu−/Trp−/X-α-Gal+) plates were replated onto agar plates of quadruple dropout (QDO, Leu−/Trp−/His−/Ade−) media plus X-α-Gal. Note α-actinin-1 SR234EF showed interaction only with IQ fragments (IQ, AAs 1639-1871) within the α11.2 C-terminus, but not with pre-IQ (AAs 1506-1638), EF hands (EF, AAs 1506-1610), nor N-terminus (NT, AAs1-123). Co-transformation of WT α11.2-pGBKT7 construct with blank pGADT7 plasmid or blank pGBKT7 plasmid with α-actinin-1 pGADT7 construct served as negative controls (not depicted but see NC1 and NC2, respectively, in Fig. 2A; N=2 independent experiments).
Our earlier peptide binding results suggested that α-actinin-1 could interact with different sites within α11.2 AAs 1588-1670 (32). Here we tested whether α11.2 AAs 1506-1610 (mainly encompasses the single EF hand motif of α11.2), 1506-1638 (includes in addition the so-called pre-IQ motif of α11.2), and 1639-1871 (with the IQ motif region near the N-terminus of this α11.2 segment) would bind the α-actinin-1 SR234EF polypeptide. The Y2H results indicated that α-actinin-1 strongly interacts with the IQ motif-containing AAs 1639-1871 but not with the other constructs containing the EF hand and pre-IQ motif (AAs 1506-1638 and 1506-1610, respectively), nor, as expected, with the negative control N-terminal AAs 1-123 of α11.2 (Fig. 1C).
After establishing that α11.2 interacts with the α-actinin-1 SR234EF region via its IQ motif we sought to define which of the AAs of α11.2 encompassed within the IQ motif are required for this interaction. Here we used alanine scanning mutagenesis of α11.2 in combination with the Y2H interactions assays. We generated overlapping triple alanine (AAA) mutations in the α11.2 IQ sequence region (AAs 1644-1670) and tested the ability of the mutant polypeptides to interact with the α-actinin bait in Y2H screens. We found two out of nine triple alanine mutations, AAA2 (AAs 1647-1649) and AAA4 (AAs 1653-1655), resulted in abrogation of a detectable interaction between the α11.2 IQ polypeptide and α-actinin SR234EF (Fig. 2A). To more precisely define the individual AAs required for this interaction, we generated single alanine replacement point mutations within these two triplets and determined that the K1647A, Y1649A, and I1654A point mutations could individually diminish α-actinin-1 binding (Fig. 2B).
Figure 2. Defining AAs in the α11.2 IQ region required for α-actinin-1 binding.

A) Nine triple alanine mutations spanning AAs 1644-1670 within the IQ domain (see top sequence schematic showing placement of mutations) were engineered into pGBKT7 encoding α11.2 residues 1506-1871. Note that the inability of the two triple alanine mutants (red highlighted) A2 (KFY/AAA) and A4 (LIQ/AAA) to trigger conversion of X-α-Gal to the blue chromophore indicates loss of the α11.2 IQ bait polypeptide interaction with α-actinin-1 SR234EF prey polypeptide. Co-transformation of WT α11.2-pGBKT7 construct with blank pGADT7 plasmid (NC1) or blank pGBKT7 plasmid with α-actinin-1 pGADT7 construct (NC2) served as negative controls. B) The single alanine substitutions K1647A, Y1649A, and I1654A within the original WT α11.2 IQ A2 and A4 segments (highlighted in red in the sequence schematic in A) individually disrupted the interaction with the SR234EF prey (N=2 independent experiments).
Validation of the Role of K1647, Y1649 and I1654 of α11.2 in α-Actinin Binding
To corroborate the findings of our Y2H screen, we performed in vitro pull-down assays. Residues 1576-1733 of the WT and mutant α11.2 C-terminus were subcloned and expressed as GST-tagged proteins in E. coli and the α-actinin-1 SR234EF core (AAs 391-892) was subcloned and expressed as an MBP-tagged fusion protein. Purified GST-tagged WT and mutant α11.2 IQ motifs were immobilized on glutathione-Sepharose and equal amounts of purified MBP-α-actinin-1 were added. In contrast to the ability of immobilized WT α11.2 IQ-GST to pull-down recombinant α-actinin-1, equal amounts of the K1647A, Y1649A and I1654A-GST fusion proteins exhibited only minimal amounts of pull-down of the α-actinin target and GST alone no detectable pull-down of (Fig. 3).
Figure 3. Pull-down assay using glutathione-Sepharose-bound WT and point-mutant α11.2 C-terminal polypeptides as bait to capture purified MBP-α-actinin-1.
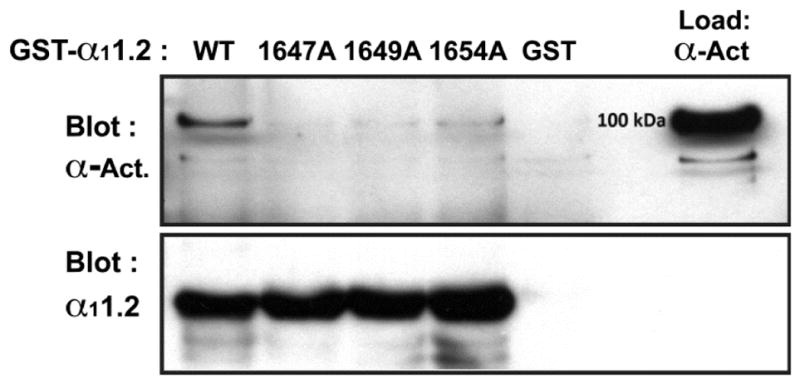
The indicated WT and point-mutant α11.2 C-terminal polypeptides spanning AA1576-1733 were expressed as GST-fusion proteins and immobilized on glutathione-conjugated Sepharose (bottom panel). α-actinin-1 polypeptide covering AAs 391-892 was expressed as a fusion protein with maltose binding protein (Load lane, top panel) from pMAL-c2e. Anti-α-actinin-1 immunoblotting (top panel) shows strong interaction of WT α11.2 and very weak interactions of α11.2 K1647A, Y1649A, or I1654A with α-actinin-1. Bottom panel illustrates that comparable amounts for each the GST - α11.2 fusion polypeptides were present. Similar results were obtained in two other experiments.
Under basal conditions when Ca2+ is low apoCaM pre-associates with the IQ motif (63, 64) and could mediate potential effects of point mutations within the IQ motif. To evaluate whether these mutations affected binding of apoCaM we utilized a fluorescence polarization (FP) assay we had adopted earlier for defining CaM binding (52) to evaluate the relative binding-affinities of the WT and mutant α11.2 IQ domains for apoCaM (Fig. 4; Table 1). Accordingly, only the I1654A but not K1647A or Y1649A mutation affected apoCaM binding, in perfect agreement with earlier work (64). The F1648A and Q1655A also did not affect apoCaM binding, again in full agreement with previous work (64).
Figure 4. Titration of a11.2 IQ domain – derived peptides with apoCaM.
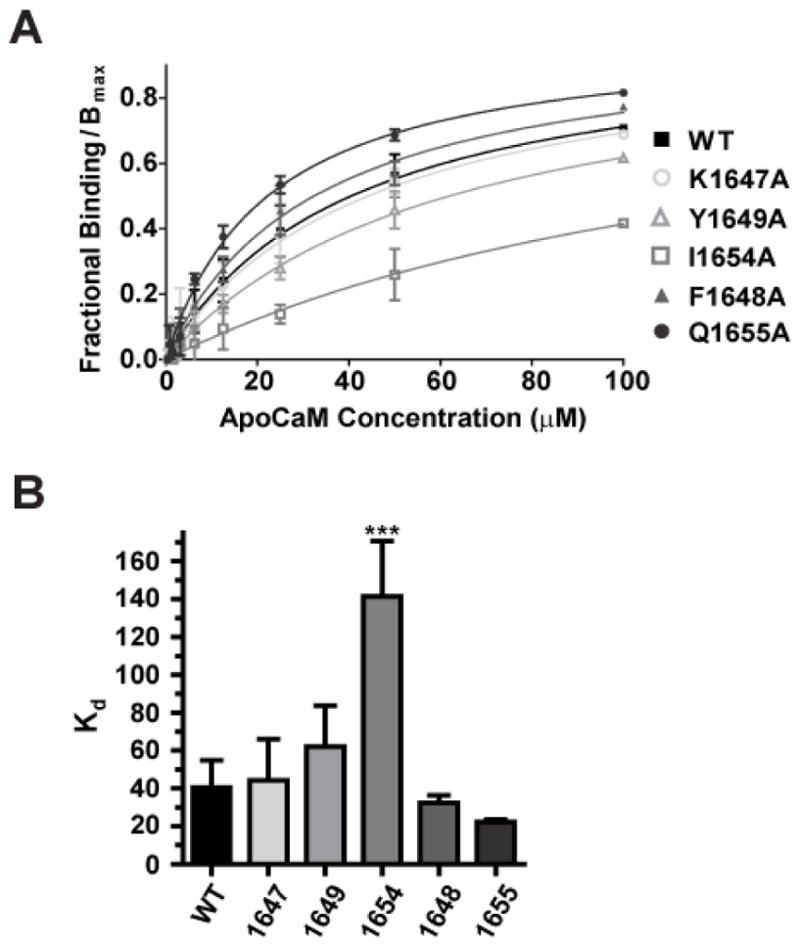
One μM fluorescein-labeled peptides spanning the α11.2 IQ domain (AAs 1644-1668) were incubated with serial dilutions of purified apoCaM. Changes in fluorescence polarization (FP) were measured to determine binding affinities of the individual IQ domain peptides. A) Shown are averages of fractional bindings normalized to Bmax against concentration of apoCaM. Error bars represent standard deviation of the fractional binding for each concentration. Binding curves were calculated by fitting the data to the equation Y = B*X/(Kd + X); B: maximal fractional binding value that would be reached at saturation as determined by extrapolation of the fitted curve. For fractional binding, the maximum and minimum polarization value for each titration curve was set to 1 and 0, respectively. B) Binding affinity Kd values were obtained from curve fitting. Only the I1654A mutant peptide showed reduced binding affinity to apoCaM relative to the WT α11.2 peptide, which was statistically significant (***p<0.001, Tukey post hoc test; N=3–6; see Table 1).
Table 1.
Kd values ± standard deviations for apoCaM binding to IQ domain – derived peptides
| WT | K1647A | F1648A | Y1649A | I1654A | Q1655A | |
|---|---|---|---|---|---|---|
| Kd (μM) | 40.5±14.3 | 44.2±21.8 | 32.4±3.8 | 61.9±21.8 | 141.2±29.5*** | 22.3±1.3 |
Significant differences to WT means were only found for I1654A (***, p<0.001; ANOVA followed by Tukey and Bonferroni post hoc test; N=3–6).
Site-specific Mutational Disruption of α11.2 IQ Domain Binding to α-Actinin Reduces Current Density of CaV1.2
As expression of dominant negative constructs of α-actinin and KD of α-actinin both reduced current density and surface biotinylation of CaV1.2 (32), we hypothesized that the direct interaction of α-actinin with CaV1.2 is important for surface expression of CaV1.2. As the AAs K1647, Y1649 and I1654 in α11.2 are important for efficient binding to α-actinin-1, we tested whether individually mutating these residues in the IQ motif affects current density in transfected HEK293T/17 cells expressing CFP- and HA-tagged full length WT or mutant α11.2 together with rat β2a and rabbit α2δ subunits (54, 55). Barium was used as a charge carrier to minimize CDI of the channel. Peak currents were normalized to whole cell capacitance to calculate current densities. Mutating K1647, Y1649, or I1654 to alanine significantly reduced CaV1.2 current density by 60–75% (Fig. 5, Table 2). Mutations F1648A and Q1655A, which did not detectably affect the interaction with α-actinin (Fig. 2B) or with apoCaM (Fig. 4), only minimally affected CaV1.2 current density relative to the WT channel (Fig. 5, Table 2).
Figure 5. Current density in HEK293T/17 cells transfected with β2a, α2δ1 and WT or mutant α11.2 subunits fused to eCFP for identification of α11.2-expressing cells.

Electrophysiological recordings were ~48 hours post transfection. 300 ms depolarization pulses ranging from −40 mV to +80 mV with 10 mV intervals were given from a −70 mV holding potential. A) Summary I/V curves. Ba2+ currents (pA) are normalized by whole cell capacitance (pF) and are plotted as the function of voltage (pA/pF vs. mV). B) Panels depict sample traces ranging from −40 mV to +30 mV of WT (top) and Y1649 mutant (bottom) in corresponding color gradients (data for all mutants summarized in Table 1). C) Bar graph of mean current density at 20 mV (IBa2+, +/− S.E.M) in cells expressing either WT or the indicated mutant α11.2 CaV1.2 channels. Cells expressing α11.2 point mutants K1647A, Y1649A and I1654A exhibit significantly reduced current density when compared to WT, whereas the mutation F1648A and Q1655A did not show a significant reduction. (***= p<0.001, ANOVA, Tukey and Bonferroni post hoc tests; N=6–7).
Table 2.
Current density and curve fitting.
| pA/pF | V1/2 (mV) | Gmax | Erev (mV) | |
|---|---|---|---|---|
| WT | 17.2±1.0 | −1.1 ± 1.4 | 0.30 ± 0.02 | 72.8 ± 2.3 |
| K1647A | 4.4±1.1*** | 1.1 ± 1.4 | 0.09 ± 0.01 | 69.2 ± 1.3 |
| F1648A | 13.3±1.7 | 3.0 ± 1.5 | 0.26 ± 0.02 | 73.5 ± 1.7 |
| Y1649A | 3.9±0.4*** | 4.1 ± 2.7*** | 0.08 ± 0.01 | 66.5 ± 2.9 |
| I 1654A | 6.7±2.1*** | 11.2 ± 1.5*** | 0.15 ± 0.01 | 75.7 ± 1.7 |
| Q1655A | 12.8±1.1 | −2.6 ± 1.6 | 0.25 ± 0.02 | 67.2 ± 2.7 |
Mean current density values at 20 mV (IBa2+, +/− S.E.M), half-activation voltage (V1/2), maximum conductance (Gmax), and equilibrium potential (Erev) calculated from the Boltzmann IV equation (see Methods and Materials). Significant differences to WT means were found for K1647A, Y1649A, and I1654A (***p<0.001; ANOVA followed by Tukey post hoc test; N=6–7).
Site-specific Mutational Disruption of α11.2 IQ Domain Binding to α-Actinin Reduces Surface Expression of CaV1.2
Although the reduction in current density by the above three mutants is consistent with a reduction in surface expression, it is also possible that the reduction in current density is in part or fully due to a reduction in ion conducting activity of CaV1.2. To differentiate between these two possibilities, we subjected CaV1.2 expressing HEK293T/17 cell cultures of good viability (i.e., > 95% trypan-blue-negative cells) to surface biotinylation. Biotinylated proteins in cell lysates were affinity purified using NeutrAvidin-conjugated (NAv) Sepharose, eluted, and subjected to immunoblot analysis. Probing of membranes with anti-α11.2 (Fig. 6A) and anti-HA-epitope antibody (Fig. 6B) revealed a clear band at the expected molecular mass of ~250 kDa for the α11.2 pore-forming subunit of CaV1.2 in NAv pull-down and total lysate samples. As expected, no signal was detectable in the case of CFP control vector transfected or mock-transfected lysates (Fig. 6A, CFP lanes), nor after control pull-down from WT CaV1.2-expressing cultures that had not undergone the biotinylation reaction, although such a band was detected in lysates of the latter (Fig. 6A, right lanes). The signal intensities of the surface biotinylated (pulled down by NAv) α11.2 subunits were corrected for lysate load differences (Fig. 6A,B, top Load panels) and normalized to the biotinylated WT α11.2 signals. The K1647A, Y1649A, and I1654A mutants showed significant decreases in surface expression relative to WT (Fig. 6C). In contrast, the α11.2 F1648A and Q1655A mutants did not exhibit statistically significant differences in degree of surface biotinylation when compared to WT. That the cells were intact and not permeant for the surface biotinylation label was demonstrated by the lack of anti-α-tubulin and anti-GAPDH signals in any of the NAv pull-down reactions, when both of which were readily detectable in lysate. These results were reproducible in replicate experiments and qualitatively consistent with the observations for differences in current density readings (Fig. 5, Table 2).
Figure 6. Surface biotinylation of HA-tagged WT and mutant α11.2 co-expressed with β2a and α2δ1 in HEK293T/17 cells.

A & B) Transfected HEK293T/17 cells were surface biotinylated and solubilized. Biotinylated proteins were pulled down with NeutrAvidin-conjugated Sepharose beads (NAv PD). NAv PD samples and corresponding lysate samples were fractionated in parallel by SDS-PAGE and immunoblotted using indicated antibodies. Ectopic α11.2 was detected with either the FP1 (A, top rows) or anti-HA antibodies (B, top rows). The same blots were probed with anti-α-tubulin (A, middle rows) and anti-GAPDH (A,B, bottom rows) to verify the lack of biotin label permeability into the cell cytoplasm and comparable sample loading. Thin vertical white lines in B indicate where original blots were trimmed for clarity (all three panels in B are from the same respective blots and film exposures). C) Bar graph depicting level of surface labeling for mutant α11.2 relative to surface labeling of WT α11.2. One-way analysis of variance (ANOVA) of the data via Tukey and Bonferroni post hoc tests verify that the differences are statistically significant (***= p<0.001; N=7–13).
As each of the K1647A, Y1649A, and I1654A mutations reduced current density of CaV1.2 to a substantially larger degree than the surface biotinylation, we employed flow cytometry to further evaluate CaV1.2 surface expression. We took advantage of the HA tag engineered into the extracellular channel loop between transmembrane segments S5 and S6 in domain II of α11.2 to detect surface expression of α11.2 by antibody labeling. HEK293T/17 cells were transfected with α2δ1, β2a and either WT or mutant α11.2 subunits (N-terminally tagged with CFP), or an empty CFP control vector. 22 h post transfection unfixed live cells were incubated with an Alexa 647-conjugated anti-HA-epitope antibody or BSA control solution. Washed mono-dispersed cells in each immunostained sample were incubated with the plasma membrane-impermeant DNA intercalating dye propidium iodide (PI) to allow exclusion of plasma membrane-compromised (PI-positive) cells during analysis. Equal numbers of intact live cells across samples for each experiment (e.g., 50,000 PI-negative events) were collected by flow cytometry for analysis. As expected no Alexa-647 fluorescence was observed for either the mock-transfected HEK293T/17 control or CFP vector control immunostained samples (Fig. 7A, C and data not shown). Similarly, histogram analysis of the PI-negative mock-immunostained (BSA control) cell populations of the WT (grey shaded) and mutant α11.2 transfections showed no detectable Alexa-647 fluorescence, the signal intensity identical to that seen for immunostained mock-transfected control (black dotted line, Fig. 7A and data not shown). In contrast, histogram analysis of live cell populations of immunostained WT and mutant α11.2 transfected cultures showed shifts indicating anti-HA staining levels beyond background (Figure 7A). Importantly, the Alexa Fluor 647 signal intensities for WT and mutant 1655 α11.2 transfected cells are greater than that exhibited by equal numbers of the K1647A, Y1649A and I1654A mutant α11.2 transfected cells (Fig. 7A and data not shown). Gating to restrict histogram analysis to only CFP-expressing populations (with comparably equal number of cells) also showed that the anti-HA Alexa-647 fluorescence signals were greater for the WT and Q1655A-mutant expressors compared to the other α11.2 mutants (Fig. 7B and data not shown). Two-dimensional contour plots of the Alexa-647 signal-positive events exhibited by the CFP-positive populations clearly showed a lack of anti-HA-signal for the CFP vector control and a reduction in the anti-HA surface labeling for the α11.2 mutants K1647A, Y1649A and I1654A relative to the surface labeling exhibited by the WT or Q1655A α11.2 expressing samples (Fig. 7C). Each contour line increases the percent of cells within the demarcated area by 2% of the total number of events. The increase in number and frequency of contour lines expanding along the X-axis to the right of the CFP control outermost boundary reflects a greater number of HA positive events. Thus these results indicate there is more surface expression of HA tagged WT and Q1655A α11.2 containing channels compared to the K1647A, Y1649A and I1654A mutant α11.2 channels. Any differences in surface HA staining seen between the K1647A, Y1649A and I1654A mutants varied from experiment to experiment, however, when compared to WT and Q1655A α11.2, each of these mutants consistently showed an obvious reduction in surface HA expression. Statistical analysis of the percentage of CFP-positive cells in the different α11.2 mutant populations showing anti-HA Alexa-647 fluorescence across > 5 experiments (normalized to the WT α11.2 sample level in each experiment) indicated that the K1647A, Y1649A and I1654A α11.2 mutants exhibit a statistically significant lower percentage of anti-HA and CFP co-positive events than found for WT α11.2 expressing populations (Fig. 7D). Notably, this decrease in surface labeling relative to WT was not observed for the Q1655A α11.2 mutant-expressing populations (Fig. 7D). Although assessing the percent of CFP positive cells that are surface-HA positive should eliminate any variation due to transfection/expression efficiency of the various α11.2 constructs, we also analyzed the relative expression level of α11.2 in the different samples via immunoblot analysis for each experiment (Fig. 7E). No statistical difference in the expression levels for mutant versus WT α11.2 pore forming subunits was found across the experiments analyzed above. Thus, over multiple replicate experiments, statistically significant reductions in surface labeling of the α11.2 HA-epitope in the K1647A, Y1649A, and I1654A point mutants were found (~35% relative to WT channel labeling). In summary, our findings from both surface biotinylation and flow cytometry of anti-HA surface staining indicate that the α-actinin-interacting motif in the IQ domain of α11.2 significantly influences surface deposition of CaV1.2 in the plasma membrane.
Figure 7. Flow cytometric analysis of surface expression of WT and mutant α11.2 co-expressed with β2a and α2δ1.

Surface HA expression of HA- and CFP-tagged WT and mutant α11.2 subunits in intact (PI-negative) HEK293T/17 cells assessed by flow cytometry. A) Representative histogram overlays (see key) of Alexa-fluor 647 signals from total PI-negative populations of anti-HA immunostained mock-transfected HEK293T/17 cells (black dotted line), no-antibody (non-stained) control (grey-shaded) and immunostained (red line) WT α11.2 samples and immunostained α11.2 mutants Q1655A (blue line), I1654A (brown line), Y1649A (gold line) and K1647A (green line). B) Histogram overlays of Alexa-fluor 647 signals from CFP-positive, PI-negative populations of no-antibody (non-stained) control (grey-shaded) and anti-HA immunostained (red line) WT α11.2 samples and immunostained α11.2 mutant Q1655A (blue line), I1654A (brown line), Y1649A (gold line) and K1647A (green line) samples from the same experiment shown in A. C) The amount of surface expressed (anti-HA stained) α11.2 (Alexa-647 fluorescence) relative to total channel (CFP signal intensity) in the transfected cell population is depicted in 2D contour plots. The upper-right plot shows contour plot of immunostained CFP-vector alone transfected control sample used to set the limit for the anti-HA signal. The upper left plot depicts anti-HA immunosignal on CFP-positive cell population of WT-α11.2 expressing cells. Unstained WT-α11.2 sample showed a similar fluorescence profile as the anti-HA antibody-stained CFP control (data not shown). The surface HA-immunosignal exhibited by the indicated α11.2 channel mutants from the same experiment is shown in 2D contour plots directly below. D) Bar graph of experimental results from repeat flow cytometry experiments. Anti-HA positive events as percent of total CFP positive events for each of the mutant CaV1.2 expressors normalized to the percent HA-positive observed for CFP positive population of WT CaV1.2 test samples in the same experiments (**= p<0.01 and ***= p<0.001, NS=p>0.05, Tukey post hoc tests; N=5–7). E) Representative immunoblot (left) and quantitative Bar graph analysis (right) of total α11.2 expression in samples used for flow analysis of α11.2 surface expression (p>0.05, N=5–7).
DISCUSSION
Through expression of dominant-negative α-actinin fragments and α-actinin knock-down we previously showed that α-actinin helps anchor CaV1.2 at the cell surface (32). We also determined that α-actinin binds to a CaM-binding region in the C-terminus of α11.2 and that upon Ca2+ influx from CaV1.2, CaM displaces α-actinin binding coincident with loss of CaV1.2 from the cell surface (32). Although suggestive, these findings did not prove that α-actinin promotes surface localization of CaV1.2 via its direct binding to α11.2. We now disambiguate this earlier finding and provide detailed insight into the precise interaction between α-actinin and CaV1.2. We establish through Y2H and pull-down assays that residues K1647, Y1649 and I1654 within the α11.2 IQ motif are required for α-actinin binding (Figs. 1–3). We show by both surface biotinylation and flow cytometry analysis that separately mutating these α-actinin binding residues reduces surface expression of CaV1.2 (Figs. 6&7). Moreover, we found that each of these mutations greatly decreases CaV1.2 current density, whereas nearby point mutations in α11.2 that do not abrogate α-actinin binding do not diminish current density (Fig. 5). Thus efficient anchoring of active CaV1.2 channels at the cell surface requires the ability of the α11.2 IQ domain residues to bind to α-actinin. The mutational abrogation of the α11.2 - α-actinin interaction in HEK293T/17 transfected cells appears to have a more pronounced effect on CaV1.2 current density than it does on the steady state surface deposition of the channel (Figs. 5–7, Table 2). This observation suggests that the α11.2 - α-actinin interaction mediates additional properties of CaV1.2 function. It might promote channel activity through some mechanism that augments channel open probability or conductance. One potential clue is the observation that all three mutations shift the voltage necessary for half maximal activation (V1/2) for CaV1.2 to the right, i.e., towards more depolarized levels (Fig. 5, Table 2). Although this shift appears to be quite small for K1647A and did not reach statistical significance for this mutation, it is quite pronounced and significant for Y1649A and I1654A. It is thus conceivable that α-actinin binding to CaV1.2 could augment its sensitivity towards depolarization by, e.g., easing the outward movement of the voltage sensors of the channel or coupling of this movement to channel opening potentially by reducing the energy barrier for channel opening.
As the interaction between CaM and the α11.2 IQ motif mediates CDI, binding of α-actinin to this region could also impact channel opening or closing, including CDI. However, our earlier studies determined that neither α-actinin KD nor ectopic expression of dominant-negative α-actinin fragments alter activation or inactivation kinetics of Ca2+ or Ba2+ currents through CaV1.2 but do decrease current density and surface biotinylation levels (32). Further defining how loss of this α11.2 - α-actinin interaction leads to reduced channel activity will require new tools and approaches. Thus this future work is beyond the scope of our current study on defining the role of this interaction on CaV1.2 surface expression.
Auxiliary β and α2δ subunits facilitate the release of Ca2+ channel α1 subunits from the ER and thereby surface expression but there is no evidence that these subunits help localize CaV1.2 to defined subcellular regions (1–5). Similarly, binding of the structural protein densin-180 to the N-terminus of α11.2 promotes secretory trafficking of CaV1.2 but not its surface retention (65). Thus, a clear understanding of how CaV1.2 is anchored at certain locations such as postsynaptic sites in neurons (6, 21) has been elusive until recently. Our findings that the K1647A, Y1649A, and I1654A mutations in α11.2 all reduce α-actinin binding and surface expression strongly supports the hypothesis that α-actinin binding to this site mediates retention of CaV1.2 at the cell surface. In an earlier report, FACS analyses indicated that I1654A (but not F1648A) α11.2 mutation reduces surface staining of CaV1.2 (4). However, this mutation also affects apoCaM binding (Fig. 4) (64). Given that only the I1654A but not the K1647A or Y1649A mutation decreases apoCaM binding but each of the three mutations do impair interaction with α-actinin (Figs. 1–4), our results firmly establish the importance of α-actinin binding to this region of α11.2 for the surface localization of CaV1.2. Aptly, our previous findings indicate that this interaction is dynamically regulated. Prolonged Ca2+ influx leads to displacement of α-actinin from CaV1.2 and consequent internalization of CaV1.2 in HEK293T/17 cells and neurons (32). Furthermore, our results for the α11.2 K1647A, Y1649A and I1654A mutants strongly suggest that the downregulation of CaV1.2 cell surface expression is due to Ca2+/CaM-mediated displacement of α-actinin from the α11.2 IQ motif. Activity-induced reduction in CaV1.2 surface localization occurs over a few minutes and thus constitutes a fairly slow but long lasting negative feedback mechanism that curbs Ca2+ influx via CaV1.2 when it is overly active. This mechanism complements the much faster CDI, which occurs within tens of milliseconds. CDI is mediated by CaM, which pre-associates with the IQ motif in its Ca2+-free apo form and undergoes a conformational change upon Ca2+ influx that induces channel closure by re-arranging the proximal membrane region of α11.2 (41–45, 64). Ca2+-induced conformational change in CaM also leads to displacement of α-actinin, which, if lasting long enough (i.e., tens of seconds), will result in removal of CaV1.2 from the cell surface (32). Consistent with these results, a study investigating the functional coupling between L-type Ca2+ channels and Ca2+-activated K+ channels found that chelating intracellular Ca2+ with BAPTA increased the prevalence of L-type Ca2+ channels at the surface of hippocampal pyramidal neurons (27). This activity-driven endocytosis is at least to a good degree reversible when Ca2+ influx ceases (30).
Activity-dependent downregulation of CaV1.2 ion conduction is important for preventing toxic levels of Ca2+ from entering the cell. Ca2+ overload is detrimental to neuronal function and has been implicated in various neurological diseases (66, 67). Ca2+ influx through L-type Ca2+ channels is chronically increased in animal models of aging (68) and contributes to senile symptoms (69). L-type channel hyperactivity causes autism-related behavioral abnormalities (18, 70). Concordantly, use of the L-type channel inhibitor, nifedipine, or of BayK8644, which augments L-type channel activity, elicits antidepressant and depressant-like behavior, respectively (71, 72). Indeed, analysis of the behavioral and biochemical effects of nifedipine and BayK8644 in knock-in mice harboring the α11.2 Thr1066Tyr mutation that makes CaV1.2 insensitive to these drugs strongly indicates that abnormal CaV1.2 activity can cause neurotoxicity and is a major underlying factor in anxiety disorders, depression, and self-injurious behavior (7). Thus it is not surprising that Ca2+ influx via CaV1.2 channels is regulated at multiple levels. Defining these mechanisms is, therefore, of high physiological and clinical relevance.
CONCLUSION
Collectively our data show that α-actinin promotes CaV1.2 surface expression via its direct binding to the IQ motif of the central α11.2 subunit. Three point mutations in the IQ motif impair α-actinin binding and result in significantly reduced CaV1.2 surface expression and current density. Comparing the effect of impaired α-actinin binding on surface expression with that on channel current density suggests that α-actinin might play an additional role by promoting channel activity of CaV1.2. Further evaluation of the intriguing possibility that α-actinin plays a dual role in CaV1.2 function by both localizing it at the correct subcellular regions and fostering its activity when anchored at these sites is an interesting perspective for future studies requiring new methodology beyond the current work. As CaV1.2 regulates multiple neuronal functions in a highly localized manner, understanding the mechanisms underlying its precise subcellular localization and activity is critical.
Supplementary Material
Acknowledgments
FUNDING SOURCES. This work was supported by AHA 11PRE7240069T32 (PYT), T32 GM113770 (AMC), GM099608 (PBH), AHA 14PRE19900021 (PBH), R01 AG017502 (JWH), R01 NS078792 (JWH), and R01 MH097887 (JWH).
We thank Dr. Nipavan Chiamvimonvat (UC Davis) for plasmids and technical advice on use of the yeast two-hybrid system, Dr. Riccardo Dolmetsch (Stanford University) for HA-tagged rat α11.2 cDNA, Ms. Bridget McLaughlin of the UC Davis NCI funded (P30CA093373) Cancer Center Flow Cytometry Facility for expert technical support, and Ms. Rebecca Rubin for help with the flow cytometry experiments.
ABBREVIATIONS
The abbreviations used are
- α11.2
the pore-forming subunit of CaV1.2
- AA
amino acid
- AAA
triple alanine mutations
- ANOVA
analysis of variance
- BSA
Bovine serum albumin
- CaM
calmodulin
- CaV1.2
voltage-gated L-type Ca2+ channel
- CDI
Ca2+-dependent inactivation
- CFP
cyan fluorescent protein
- DMEM-10
Dulbecco’s Modification of Eagles Medium with 10% FBS
- DMEM-10-10IRP
DMEM-10 medium containing 10 μM isradipine
- ER
endoplasmic reticulum
- FBS
fetal bovine serum
- FP1
antibody against Loop II/III of α11.2
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GST
glutathione S-transferase
- HA
hemagglutinin
- HBB
HEPES biotinylation buffer
- HRP
Horseradish peroxidase
- IBa
whole-cell Ba2+ currents
- I/V
current-voltage relationships
- KD
knockdown
- MBP
maltose binding protein
- NeuAv
NeutrAvidin-conjugated Sepharose
- PBS
phosphate-buffered saline
- PD
pull-down
- PI
propidium iodide
- PVDF
polyvinylidene difluoride
- RT
room temperature
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- S.E
standard error
- S.E.M
standard error of the mean
- TBS
Tris-buffered saline
- TBST
Tris-buffered saline +0.05% Tween
- VDCC
voltage-dependent calcium channels
- WT
wild type
- Y2H
yeast two-hybrid
Footnotes
AUTHOR CONTRIBUTIONS. PYT, PBH, ACH, TP, AC, MWL, CMB and MCH acquired, analyzed and interpreted the experimental data. JWH and MCH developed the research strategy and oversaw the project and interpretation of the experimental data. PYT, PBH, and MCH prepared figures and tables. MCH and JWH wrote the manuscript, PYT and PBH contributed to the preparation of it, and PBH assembled and submitted the final draft. All authors reviewed the results and approved the final version of the manuscript.
CONFLICT OF INTEREST. The authors declare that they have no conflicts of interest with the contents of this article.
SUPPORTING INFORMATION. Supporting information encompasses tables of oligonucleotides used for the generation of α11.2 and α-actinin-1 expression constructs. The α11.2 constructs were used for yeast two-hybrid studies, pull-down assays, electrophysiology and surface expression work. These include oligonucleotides used for PCR subcloning into the indicated constructs and for PCR mutagenesis to create the triple and single alanine point mutations in α11.2. Oligonucleotides designed for PCR subcloning of α-actinin-1 into expression vectors for yeast-two hybrid screens are also described.
References
- 1.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 2.Dai S, Hall DD, Hell JW. Supramolecular Assemblies and Localized Regulation of Voltage-gated Ion Channels. Physiol Rev. 2009;89:411–452. doi: 10.1152/physrev.00029.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zamponi GW, Striessnig J, Koschak A, Dolphin AC. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol Rev. 2015;67:821–870. doi: 10.1124/pr.114.009654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bourdin B, Marger F, Wall-Lacelle S, Schneider T, Klein H, Sauve R, Parent L. Molecular determinants of the CaVbeta-induced plasma membrane targeting of the CaV1.2 channel. J Biol Chem. 2010;285:22853–22863. doi: 10.1074/jbc.M110.111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dolphin AC. Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol. 2016;594:5369–5390. doi: 10.1113/JP272262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel a1 subunits. J Cell Biol. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinnegger-Brauns MJ, Hetzenauer A, Huber IG, Renstrom E, Wietzorrek G, Berjukov S, Cavalli M, Walter D, Koschak A, Waldschutz R, Hering S, Bova S, Rorsman P, Pongs O, Singewald N, Striessnig JJ. Isoform-specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L-type Ca 2+ channels. J Clin Invest. 2004;113:1430–1439. doi: 10.1172/JCI20208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seisenberger C, Specht V, Welling A, Platzer J, Pfeifer A, Kuhbandner S, Striessnig J, Klugbauer N, Feil R, Hofmann F. Functional embryonic cardiomyocytes after disruption of the L-type alpha1C (Cav1.2) calcium channel gene in the mouse. J Biol Chem. 2000;275:39193–39199. doi: 10.1074/jbc.M006467200. [DOI] [PubMed] [Google Scholar]
- 9.Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- 10.Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Muller J, Stiess M, Marais E, Schulla V, Lacinova L, Goebbels S, Nave KA, Storm DR, Hofmann F, Kleppisch T. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci. 2005;25:9883–9892. doi: 10.1523/JNEUROSCI.1531-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patriarchi T, Qian H, Di Biase V, Malik ZA, Chowdhury D, Price JL, Hammes EA, Buonarati OR, Westenbroek RE, Catterall WA, Hofmann F, Xiang YK, Murphy GG, Chen C-Y, Navedo MF, Hell JW. Phosphorylation of Cav1.2 on S1928 Uncouples the L-type Ca2+ Channel from the β2 Adrenergic Receptor. EMBO J. 2016;35:1330–1345. doi: 10.15252/embj.201593409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qian H, Patriarchi T, Price JL, Matt L, Lee B, Nieves-Cintron M, Buonarati OR, Chowdhury D, Nanou E, Nystoriak MA, Catterall WA, Poomvanicha M, Hofmann F, Navedo MF, Hell JW. Phosphorylation of Ser1928 mediates the enhanced activity of the L-type Ca2+ channel Cav1.2 by the beta2-adrenergic receptor in neurons. Sci Signaling. 2017;10:eaaf9659. doi: 10.1126/scisignal.aaf9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- 14.Li H, Pink MD, Murphy JG, Stein A, Dell’Acqua ML, Hogan PG. Balanced interactions of calcineurin with AKAP79 regulate Ca2+-calcineurin-NFAT signaling. Nat Struct Mol Biol. 2012;19:337–345. doi: 10.1038/nsmb.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, Tsien RW. Cav1 and Cav2 Channels engage distinct modes of Ca2+ signaling to control CREB-dependent gene expression. Cell. 2012;149:1112–1124. doi: 10.1016/j.cell.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma H, Groth RD, Cohen SM, Emery JF, Li B, Hoedt E, Zhang G, Neubert TA, Tsien RW. gammaCaMKII Shuttles Ca(2+)/CaM to the Nucleus to Trigger CREB Phosphorylation and Gene Expression. Cell. 2014;159:281–294. doi: 10.1016/j.cell.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy JG, Sanderson JL, Gorski JA, Scott JD, Catterall WA, Sather WA, Dell’Acqua ML. AKAP-anchored PKA maintains neuronal L-type calcium channel activity and NFAT transcriptional signaling. Cell Rep. 2014;7:1577–1588. doi: 10.1016/j.celrep.2014.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 19.Bhat S, Dao DT, Terrillion CE, Arad M, Smith RJ, Soldatov NM, Gould TD. CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog Neurobiol. 2012;99:1–14. doi: 10.1016/j.pneurobio.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smoller JW. Disorders and borders: psychiatric genetics and nosology. Am J Med Genet, Part B. 2013;162B:559–578. doi: 10.1002/ajmg.b.32174. [DOI] [PubMed] [Google Scholar]
- 21.Hell JW, Westenbroek RE, Breeze LJ, Wang KKW, Chavkin C, Catterall WA. N-methyl-D-aspartate receptor-induced proteolytic conversion of postsynaptic class C L-type calcium channels in hippocampal neurons. Proc Natl Acad Sci U S A. 1996;93:3362–3367. doi: 10.1073/pnas.93.8.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW. A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2 [see comments] [erratum appears in Science 2001 Aug 3;293(5531):804] Science. 2001;293:98–101. doi: 10.1126/science.293.5527.98. [DOI] [PubMed] [Google Scholar]
- 23.Obermair GJ, Szabo Z, Bourinet E, Flucher BE. Differential targeting of the L-type Ca2+ channel alpha1C (CaV1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. Eur J Neurosci. 2004;19:2109–2122. doi: 10.1111/j.0953-816X.2004.03272.x. [DOI] [PubMed] [Google Scholar]
- 24.Tippens AL, Pare JF, Langwieser N, Moosmang S, Milner TA, Smith Y, Lee A. Ultrastructural evidence for pre- and postsynaptic localization of Cav1.2 L-type Ca2+ channels in the rat hippocampus. J Comp Neurol. 2008;506:569–583. doi: 10.1002/cne.21567. [DOI] [PubMed] [Google Scholar]
- 25.Di Biase V, Tuluc P, Campiglio M, Obermair GJ, Heine M, Flucher BE. Surface Traffic of Dendritic CaV1.2 Calcium Channels in Hippocampal Neurons. J Neurosci. 2011;31:13682–13694. doi: 10.1523/JNEUROSCI.2300-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franzini-Armstrong C, Protasi F, Ramesh V. Shape, size, and distribution of Ca(2+) release units and couplons in skeletal and cardiac muscles. Biophys J. 1999;77:1528–1539. doi: 10.1016/S0006-3495(99)77000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marrion NV, Tavalin ST. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- 28.Liu G, Shi J, Yang L, Cao L, Park SM, Cui J, Marx SO. Assembly of a Ca(2+)-dependent BK channel signaling complex by binding to beta2 adrenergic receptor. EMBO J. 2004;23:2196–2205. doi: 10.1038/sj.emboj.7600228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berkefeld H, Sailer CA, Bildl W, Rohde V, Thumfart JO, Eble S, Klugbauer N, Reisinger E, Bischofberger J, Oliver D, Knaus HG, Schulte U, Fakler B. BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science. 2006;314:615–620. doi: 10.1126/science.1132915. [DOI] [PubMed] [Google Scholar]
- 30.Liu J, Bangalore R, Rutledge A, Triggle DJ. Modulation of L-type Ca2+ channels in clonal rat pituitary cells by membrane depolarization. Mol Pharmacol. 1994;45:1198–1206. [PubMed] [Google Scholar]
- 31.Green EM, Barrett CF, Bultynck G, Shamah SM, Dolmetsch RE. The tumor suppressor eIF3e mediates calcium-dependent internalization of the L-type calcium channel CaV1.2. Neuron. 2007;55:615–632. doi: 10.1016/j.neuron.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hall DD, Dai S, Tseng PY, Malik ZA, Nguyen M, Matt L, Schnizler K, Shephard A, Mohapatra D, Tsuruta F, Dolmetsch RE, Christel CJ, Lee A, Burette A, Weinberg RJ, Hell JW. Competition Between α-Actinin and Ca2+-Calmodulin Controls Surface Retention of the L-type Ca2+ Channel CaV1.2. Neuron. 2013;78:483–497. doi: 10.1016/j.neuron.2013.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson BD, Byerly L. A cytoskeletal mechanism for Ca2+ channel metabolic dependence and inactivation by intracellular Ca2+ Neuron. 1993;10:797–804. doi: 10.1016/0896-6273(93)90196-x. [DOI] [PubMed] [Google Scholar]
- 34.Lader AS, Kwiatkowski DJ, Cantiello HF. Role of gelsolin in the actin filament regulation of cardiac L-type calcium channels. Am J Physiol. 1999;277:C1277–1283. doi: 10.1152/ajpcell.1999.277.6.C1277. [DOI] [PubMed] [Google Scholar]
- 35.Nakamura M, Sunagawa M, Kosugi T, Sperelakis N. Actin filament disruption inhibits L-type Ca(2+) channel current in cultured vascular smooth muscle cells. Am J Physiol Cell Physiol. 2000;279:C480–487. doi: 10.1152/ajpcell.2000.279.2.C480. [DOI] [PubMed] [Google Scholar]
- 36.Mizuno F, Barabas P, Krizaj D, Akopian A. Glutamate-induced internalization of Ca(v)1.3 L-type Ca(2+) channels protects retinal neurons against excitotoxicity. J Physiol. 2010;588:953–966. doi: 10.1113/jphysiol.2009.181305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wyszynski M, Kharazia V, Shanghvi R, Rao A, Beggs AH, Craig AM, Weinberg R, Sheng M. Differential regional expression and ultrastructural localization of alpha-actinin-2, a putative NMDA receptor-anchoring protein, in rat brain. J Neurosci. 1998;18:1383–1392. doi: 10.1523/JNEUROSCI.18-04-01383.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allison DW, Gelfand VI, Spector I, Craig AM. Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci. 1998;18:2423–2436. doi: 10.1523/JNEUROSCI.18-07-02423.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao T, Bunemann M, Gerhardstein BL, Ma H, Hosey MM. Role of the C terminus of the alpha 1C (CaV1.2) subunit in membrane targeting of cardiac L-type calcium channels. J Biol Chem. 2000;275:25436–25444. doi: 10.1074/jbc.M003465200. [DOI] [PubMed] [Google Scholar]
- 40.Kepplinger KJ, Kahr H, Forstner G, Sonnleitner M, Schindler H, Schmidt T, Groschner K, Soldatov NM, Romanin C. A sequence in the carboxy-terminus of the alpha(1C) subunit important for targeting, conductance and open probability of L-type Ca(2+) channels. FEBS Lett. 2000;477:161–169. doi: 10.1016/s0014-5793(00)01791-9. [DOI] [PubMed] [Google Scholar]
- 41.Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 42.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L- type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 43.Kim J, Ghosh S, Nunziato DA, Pitt GS. Identification of the components controlling inactivation of voltage-gated Ca2+ channels. Neuron. 2004;41:745–754. doi: 10.1016/s0896-6273(04)00081-9. [DOI] [PubMed] [Google Scholar]
- 44.Van Petegem F, Chatelain FC, Minor DL., Jr Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain-Ca2+/calmodulin complex. Nat Struct Mol Biol. 2005;12:1108–1115. doi: 10.1038/nsmb1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fallon JL, Halling DB, Hamilton SL, Quiocho FA. Structure of calmodulin bound to the hydrophobic IQ domain of the cardiac Ca(v)1.2 calcium channel. Structure. 2005;13:1881–1886. doi: 10.1016/j.str.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 46.Wang HG, George MS, Kim J, Wang C, Pitt GS. Ca2+/calmodulin regulates trafficking of Ca(V)1.2 Ca2+ channels in cultured hippocampal neurons. J Neurosci. 2007;27:9086–9093. doi: 10.1523/JNEUROSCI.1720-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ravindran A, Lao QZ, Harry JB, Abrahimi P, Kobrinsky E, Soldatov NM. Calmodulin-dependent gating of Ca(v)1.2 calcium channels in the absence of Ca(v)beta subunits. Proc Natl Acad Sci U S A. 2008;105:8154–8159. doi: 10.1073/pnas.0711624105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davare MA, Horne MC, Hell JW. Protein Phosphatase 2A is associated with class C L-type calcium channels (Cav1.2) and antagonizes channel phosphorylation by cAMP-dependent protein kinase. J Biol Chem. 2000;275:39710–39717. doi: 10.1074/jbc.M005462200. [DOI] [PubMed] [Google Scholar]
- 49.Altier C, Dubel SJ, Barrere C, Jarvis SE, Stotz SC, Spaetgens RL, Scott JD, Cornet V, De Waard M, Zamponi GW, Nargeot J, Bourinet E. Trafficking of L-type calcium channels mediated by the postsynaptic scaffolding protein AKAP79. J Biol Chem. 2002;277:33598–33603. doi: 10.1074/jbc.M202476200. [DOI] [PubMed] [Google Scholar]
- 50.Hall DD, Feekes JA, Arachchige Don AS, Shi M, Hamid J, Chen L, Strack S, Zamponi GW, Horne MC, Hell JW. Binding of protein phosphatase 2A to the L-type calcium channel Cav1.2 next to Ser1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochemistry. 2006;45:3448–3459. doi: 10.1021/bi051593z. [DOI] [PubMed] [Google Scholar]
- 51.Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, McKnight GS, Hell JW. Critical role of cAMP-dependent protein kinase anchoring to the L-type calcium channel Cav1.2 via A-kinase anchor protein 150 in neurons. Biochemistry. 2007;46:1635–1646. doi: 10.1021/bi062217x. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Y, Matt L, Patriarchi T, Malik ZA, Chowdhury D, Park DK, Renieri A, Ames JB, Hell JW. Capping of the N-terminus of PSD-95 by calmodulin triggers its postsynaptic release. EMBO J. 2014;33:1341–1353. doi: 10.1002/embj.201488126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lim IA, Hall DD, Hell JW. Selectivity and promiscuity of the first and second PDZ domains of PSD- 95 and synapse-associated protein 102. J Biol Chem. 2002;277:21697–21711. doi: 10.1074/jbc.M112339200. [DOI] [PubMed] [Google Scholar]
- 54.Perez-Reyes E, Castellano A, Kim HS, Bertrand P, Baggstrom E, Lacerda AE, Wei XY, Birnbaumer L. Cloning and expression of a cardiac/brain beta subunit of the L-type calcium channel. J Biol Chem. 1992;267:1792–1797. [PubMed] [Google Scholar]
- 55.Ellis SB, Williams ME, Ways NR, Brenner R, Sharp AH, Leung AT, Campbell KP, McKenna E, Koch WJ, Hui A, Schwartz A, Harpold MM. Sequence and expression of mRNAs encoding the a1 and a2 subunits of the DHP-sensitive calcium channel. Science. 1988;241:1661–1664. doi: 10.1126/science.2458626. [DOI] [PubMed] [Google Scholar]
- 56.Jordan M, Schallhorn A, Wurm FM. Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res. 1996;24:596–601. doi: 10.1093/nar/24.4.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Senatore A, Boone AN, Spafford JD. Optimized transfection strategy for expression and electrophysiological recording of recombinant voltage-gated ion channels in HEK-293T cells. J Visualized Exp. 2011 doi: 10.3791/2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Elia G. Biotinylation reagents for the study of cell surface proteins. Proteomics. 2008;8:4012–4024. doi: 10.1002/pmic.200800097. [DOI] [PubMed] [Google Scholar]
- 59.Horne MC, Roth PE, DeFranco AL. Assembly of the truncated immunoglobulin heavy chain Dm into antigen receptor-like complexes in pre-B cells but not in B cells. Immunity. 1996;4:145–158. doi: 10.1016/s1074-7613(00)80679-2. [DOI] [PubMed] [Google Scholar]
- 60.Davare MA, Hell JW. Increased phosphorylation of the neuronal L-type Ca(2+) channel Ca(v)1.2 during aging. Proc Natl Acad Sci U S A. 2003;100:16018–16023. doi: 10.1073/pnas.2236970100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. From the Cover: Localization of cardiac L-type Ca2+ channels to a caveolar macromolecular signaling complex is required for beta2-adrenergic regulation. Proc Natl Acad Sci U S A. 2006;103:7500–7505. doi: 10.1073/pnas.0503465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu L, Zhang Q, Timofeyev V, Zhang Z, Young JN, Shin HS, Knowlton AA, Chiamvimonvat N. Molecular coupling of a Ca2+-activated K+ channel to L-type Ca2+ channels via alpha-actinin2. Circ Res. 2007;100:112–120. doi: 10.1161/01.RES.0000253095.44186.72. [DOI] [PubMed] [Google Scholar]
- 63.Ben Johny M, Yang PS, Bazzazi H, Yue DT. Dynamic switching of calmodulin interactions underlies Ca2+ regulation of CaV1.3 channels. Nat Commun. 2013;4:1717. doi: 10.1038/ncomms2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bazzazi H, Ben Johny M, Adams PJ, Soong TW, Yue DT. Continuously Tunable Ca Regulation of RNA-Edited Ca1.3 Channels. Cell Rep. 2013;5:367–377. doi: 10.1016/j.celrep.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang S, Stanika RI, Wang X, Hagen J, Kennedy MB, Obermair GJ, Colbran RJ, Lee A. Densin-180 controls the trafficking and signaling of L-type voltage-gated Cav1.2 Ca2+ channels at excitatory synapses. J Neurosci. 2017;37:4679–4691. doi: 10.1523/JNEUROSCI.2583-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Disterhoft JF, Gispen WH, Traber J, Khachaturian ZS, editors. Calcium Hypothesis of Aging and Dementia. 1994;747 [Google Scholar]
- 67.Lee J-M, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399(Suppl):A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- 68.Thibault O, Landfield PW. Increase in single L-type calcium channels in hippocampal neurons during aging. Science. 1996;272:1017–1020. doi: 10.1126/science.272.5264.1017. [DOI] [PubMed] [Google Scholar]
- 69.Deyo RA, Straube KT, Disterhoft JF. Nimodipine facilitates associative learning in aging rabbits. Science. 1989;243:809–811. doi: 10.1126/science.2916127. [DOI] [PubMed] [Google Scholar]
- 70.Jinnah HA, Yitta S, Drew T, Kim BS, Visser JE, Rothstein JD. Calcium channel activation and self-biting in mice. Proc Natl Acad Sci U S A. 1999;96:15228–15232. doi: 10.1073/pnas.96.26.15228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mogilnicka E, Czyrak A, Maj J. Dihydropyridine calcium channel antagonists reduce immobility in the mouse behavioral despair test; antidepressants facilitate nifedipine action. Eur J Pharmacol. 1987;138:413–416. doi: 10.1016/0014-2999(87)90480-8. [DOI] [PubMed] [Google Scholar]
- 72.Mogilnicka E, Czyrak A, Maj J. BAY K 8644 enhances immobility in the mouse behavioral despair test, an effect blocked by nifedipine. Eur J Pharmacol. 1988;151:307–311. doi: 10.1016/0014-2999(88)90813-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.