

Summary
The p110β isoform of PI3K is preferentially activated in many tumors deficient in the phosphatase and tensin homolog (PTEN). However, the mechanism(s) linking PTEN loss to p110β activation remain(s) mysterious. Here we identify CRKL as a member of the class of PI3Kβ interacting proteins. Silencing CRKL expression in PTEN-null human cancer cells leads to a decrease in p110β-dependent PI3K signaling and cell proliferation. In contrast, CRKL depletion does not impair p110α-mediated signaling. Further study showed that CRKL binds to tyrosine phosphorylated p130Cas in PTEN-null cancer cells. Since Src family kinases are known both to be regulated by PTEN and to phosphorylate and activate p130Cas, we tested and found that Src inhibition cooperated with p110β inhibition to suppress the growth of PTEN-null cells. These data suggest both a potential mechanism linking PTEN loss to p110β activation and the possible benefit of dual inhibition of Src and PI3K for PTEN-null tumors.
In Brief
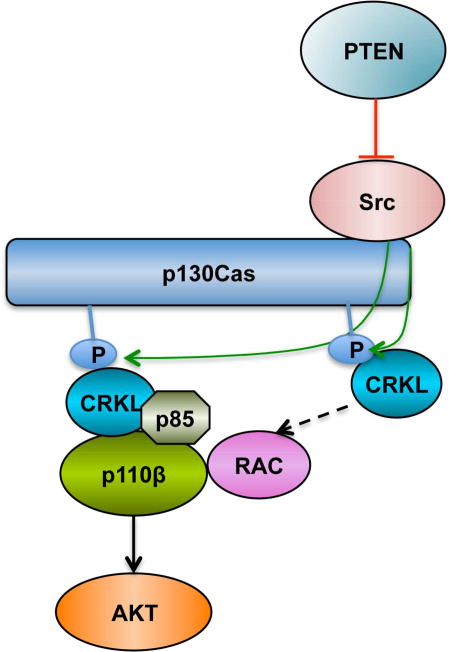
Zhang et al. find a role for CRKL in associating with and regulating p110β-dependent PI3K activity in PTEN-null cancer cells. A PTEN/FAK/Src/p130Cas axis may activate CRKL/p110β in PTEN-null cancer cells. Src inhibition cooperates with PI3K or p110βinhibition to suppress the growth of PTEN-null tumor cells.
Introduction
Class I phosphatidylinositol 3-kinases (PI3Ks) are key regulators of cell survival, proliferation, adhesion and motility. In response to extracellular signals mediated by receptor tyrosine kinases (RTKs), G protein-coupled receptors (GPCRs), or many oncogenes, class I PI3Ks are recruited to plasma membrane and phosphorylate phosphatidylinositol-4,5-bisphosphate (PIP2) to phophstidylinositol-3,4,5-trisphosphate (PIP3), which activates multiple effectors including the Ser/Thr kinase AKT. The PI3K pathway is negatively regulated by the tumor suppressor PTEN, which dephosphorylates PIP3 to PIP2. Class I PI3Ks are obligate heterodimers and further divided into class IA PI3Ks, which consist of a p85 regulatory subunit (p85α, p85β, or p55γ) bound to a p110 catalytic subunit (p110α, p110β, or p110δ) and class IB PI3Ks that feature a p101 regulatory subunit and a p110γ catalytic subunit. Notably, p110α and p110β are ubiquitously expressed, whereas p110δ and p110γ are largely confined to immune system (Thorpe et al., 2015).
Despite a high degree of homology, p110α and p110β have distinct functions in both normal physiology and disease. While p110α plays a dominant role in RTK signaling, p110β is the major effector for GPCRs and has important kinase-independent functions as well (Thorpe et al., 2015). In mice, p110α and p110β have distinct roles in insulin metabolic action in liver (Sopasakis et al., 2010, Jia et al., 2008, Ciraolo et al., 2008) and hypothalamic energy regulation (Donato et al., 2010). p110α is critical for tumor formation induced by RAS, polyoma middle T and HER2/Neu as reviewed by Thorpe et al. (Thorpe et al., 2015). Surprisingly, deletion or inactivation of p110β inhibits prostate, ovarian and haematologic tumor formation driven by PTEN loss in mice or murine cells (Jia et al., 2008, Schmit et al., 2014, Yuzugullu et al., 2015). p110β is the primary PI3K isoform involved in many cases of tumorigenesis driven by PTEN loss in human cells (Torbett et al., 2008, Wee et al., 2008, Ni et al., 2012) though the mechanism underlying this specificity is currently unknown.
CRKL is an adaptor protein consisting of an N-terminal Src homology 2 (SH2) domain followed by two SH3 domains. CRKL is activated when its SH2 domain binds to a phosphorylated Y-x-x-P motif found in docking proteins, such as p130Cas, paxillin, and GAB; the SH3N domain then mediates interaction with effector proteins via a proline-rich P-x-x-P-x-K motif (Birge et al., 2009). Overexpression of CRKL promotes anchorage-independent growth in Rat-1 fibroblasts (Bell and Park, 2012) and transforms human airway epithelial cells by activating SOS1–RAS–RAF–ERK and Src–C3G–RAP1 pathways (Cheung et al., 2011). Elevated expression of CRKL has been found in human non-small cell lung cancer (NSCLC), ovarian cancer and head neck squamous cell carcinoma (Bell and Park, 2012). Overexpression of CRKL renders NSCLC cells with EGFR mutations resistant to EGFR inhibitors by activating MAPK and AKT signaling (Cheung et al., 2011).
In this study, we demonstrate that CRKL preferentially binds p110β and regulates PI3K signaling in p110β-dependent PTEN-deficient tumor cells. Phospho-p130Cas is the major tyrosine phosphorylated protein associated with CRKL in PTEN-deficient tumor cells. Combined inhibition of PI3K and Src antagonizes cell proliferation in those cells, suggesting a promising strategy of treating tumors featuring PTEN loss.
Results
CRKL preferentially associates with p110β
To elucidate the mechanism by which p110β is activated in PTEN null tumors, we first searched for proteins that were preferentially bound by p110β. Both p110α and p110β were expressed at comparable levels in 293FT cells transduced with BacMam viruses (Figure S1). Protein complexes containing each p110 isoform were purified by tandem FLAG and StrepTactin purification, and analyzed by mass spectrometry across two independent biological replicates (Figure 1A). As expected, p85 subunits PIK3R1, PIK3R2 and PIK3R3 were detected with similar stoichiometry in both p110α and p110β complexes (Figure 1A). Notably, CRKL was among the most abundant proteins exclusively detected in the p110β-containing complexes.
Figure 1. Identification of CRKL as a selective p110β interacting protein.

(A) p110α and p110β differentially associate with binding partners. Proteins reproducibly identified across two independent TAP experiments for p110α and p110β were quantified based on the average intensity of extracted ion chromatograms for the top three most abundant constituent peptides. The color intensity is proportional to the abundance of a protein relative to a given p110 isoform. Grey: proteins which were detected but not reliably quantified. White: proteins which were not detected.
(B) 293FT cells were transduced with BacMam viruses containing His-GST-p85NI, Strep (II)-FLAG-p110α/His-GST-p85NI, or Strep (II)-FLAG-p110β/His-GST-p85NI, followed by immunoprecipitation with anti-FLAG M2 agarose and immunoblot analysis with indicated antibodies.
(C) Endogenous CRKL was immunoprecipitated with anti-CRKL antibody from 293FT cell lysates. Rabbit immunoglobulin G (IgG) was used as a control. Cell lysates and immunoprecipitates were analyzed by immunoblotting with indicated antibodies.
(D) 293FT cells were cotransfected with FLAG-tagged bovine p85 and HA-tagged wild-type (p110β-WT) or mutant (p110β-Δp85) p110β. Cell lysates were prepared 2 d post-transfection followed by immunoprecipitation with anti-CRKL antibody and immunoblot analysis with indicated antibodies.
(E) CRKL requires its SH3N domain to efficiently interact with p110β. Top: Schematic illustration of wild-type and mutant CRKL. Bottom: Cell lysates from BT549 cells stably expressing LacZ control, V5-tagged wild-type or mutant human CRKL were immunoprecipitated with anti-V5 antibody and immunoblotted with indicated antibodies.
See also Figure S1.
To validate the mass spectrometry result, we overexpressed Strep (II)-FLAG-tagged p110α/His-GST-p85NI, Strep (II)-FLAG-tagged p110β/His-GST-p85NI, or His-GST-p85NI in 293FT cells followed by anti-FLAG immunoprecipitation. With similar amounts of p110α and p110β immunoprecipitated, CRKL is present at much higher levels in the p110β-containing complex (Figure 1B), suggesting that CRKL preferentially associates with p110β at least under conditions of p110 overexpression. This result is consistent with our TAP/MS analysis. We next performed anti-CRKL immunoprecipitation in 293FT cells and found that CRKL coprecipitated with p110β with no detectable p110α in the immunoprecipitates (Figure 1C), demonstrating that CRKL and p110β interact physically at endogenous expression levels.
Previous studies suggested that CRKL can bind to proline-rich regions of p85 through its SH3N domain (Sattler et al., 1997). To determine if the CRKL-p110β interaction requires p85 binding, we expressed HA-tagged wild-type p110β and a mutant p110β lacking the p85-binding domain (p110β-Δp85) together with FLAG-tagged bovine p85 in 293FT cells. We found that CRKL coprecipitates with wild-type p110β, whereas binding of CRKL to p110β-Δp85 mutant is greatly reduced (Figure 1D). To investigate which domain on CRKL is essential for its interaction with p110β, we expressed V5-tagged wild-type CRKL or a CRKL mutant with an amino acid substitution known to disrupt the function of SH3N domain (W160L) (Cheung et al., 2011) in the breast cancer cell line BT549. As shown in Figure 1E, p110β coprecipitates with wild-type CRKL. The SH3N mutant CRKLW160L no longer binds to p85 and shows little association with p110β.
CRKL knockdown suppresses PI3K signaling and growth of PTEN-deficient cancer cells
To investigate the importance of CRKL-p110β interaction in tumor cells, we selected a panel of human cancer cell lines to represent various genetic alterations seen in primary breast and prostate tumors such as PTEN mutation/loss, HER2 overexpression, and PIK3CA mutation (Table S2). As previously reported (Wee et al., 2008, Ni et al., 2012), the p110β-selective inhibitor TGX221 strongly impaired PI3K signaling in PTEN-deficient cancer cell lines, whereas the p110α-selective inhibitor A66 significantly inhibited AKT phosphorylation in cancer cell lines with wild-type PTEN (data not shown). The MCF-10A cell line was used for comparative purposes as a non-tumorigenic, karyotypically normal cell line. Consistent with results in 293FT, CRKL and p110β coprecipitated in all PTEN-null and PTEN-replete cancer cells, whereas no detectable p110α associated with CRKL in either genetic background (Figure 2A and S2A). It is noteworthy that CRKL also interacted with p110β in serum-deprived cells (Figure 2A and S2A), suggesting a constitutive association between the two proteins. This is an important point, since both CRKL and the p110/p85 complex contain SH2 domains and, thus, could bind to a common receptor after growth factor stimulation.
Figure 2. CRKL knockdown suppresses PI3K signaling and growth of PTEN-deficient cancer cells.

(A) PC3 and BT549 cells were cultured under full (10%) serum condition or under serum starvation overnight. Cell lysates were immunoprecipitated with anti-CRKL antibody or rabbit IgG and immunoblotted with indicated antibodies.
(B–C) BT549 (B) and PC3 (C) cells stably transduced with inducible control or CRKL targeting shRNA were cultured with or without Dox (100 ng/ml 72 hr) under full (10%) serum condition. Alternatively similarly treated cells were under serum starvation overnight before harvesting cells. Cell lysates were analyzed by immunoblotting with CRKL, phospho-AKT (S473), phospho-AKT substrate (RXXS*/T*) and tubulin antibodies. The level of pAKT473 was normalized to tubulin. In each experiment the pAKT473/tubulin ratio in cell line expressing control shRNA in the absence of Dox was set to 1. Bar graphics reflect the mean ± SEM; n=3; * p<0.05 (t-test). Dox = doxycycline.
(D) Stable PC3 cells containing Dox-inducible CRKL shRNA were transduced with LacZ control, shRNA-resistant wild-type (CRKLsilent mutant) or SH3N domain mutant (CRKLW160L) CRKL cDNAs. Stable cells expressing the respective rescue constructs were cultured with or without Dox (100 ng/ml 72 hr) under full (10%) serum condition. Cell lysates were analyzed by immunoblotting with indicated antibodies.
(E) Proliferation of stable PC3 cells containing inducible control or CRKL targeting shRNA was visualized by crystal violet staining 3 d after Dox (50 ng/ml) induction. Data represent mean ± SEM; n=4; * p<0.05 by comparing cells expressing control shRNA and shCRKL (t-test).
See also Figure S2 and Table S2.
We next knocked down CRKL in PC3 and BT549 cells using a doxycycline-inducible lentiviral shRNA system (Figure 2B and 2C). No measurable off-target effect on the other closely related CRK family member CRKII was observed (Figure S2B). We found that downregulation of CRKL was accompanied by a substantial reduction in phosphorylation of AKT and downstream AKT substrates in BT549 (Figure 2B) and PC3 (Figure 2C) cells. Similar observations were made when cells were cultured in serum-free medium, suggesting that CRKL also regulates basal PI3K activity in these cells (Figure 2B and 2C).
To confirm that the observed effects of the shRNAs on PI3K signaling were specific for CRKL, we transduced shCRKL-expressing PC3 and BT549 cells with an shRNA-resistant CRKL mutant (CRKLsilent mutant) containing multiple nucleotide substitutions to the shCRKL#1 targeting sequence that did not change the amino acid sequence of CRKL. Expression of the CRKLsilent mutant, but not a control vector, restored PI3K signaling in shCRKL-expressing PC3 (Figure 2D) and BT549 (Figure S2C) cells to a level comparable to that of the parental cell line. Furthermore, the CRKLW160L mutant that ablates interaction with the p85/p110β complex failed to rescue AKT phosphorylation in BT549 expressing shCRKL#2 that targets the 3’UTR of CRKL mRNA, even though the mutant CRKLW160L was expressed at similar levels as the CRKLsilent mutant (Figure S2C). This result suggests that the SH3N domain of CRKL and the association of CRKL with p110β are essential for CRKL function in PI3K signaling in PTEN-deficient cancer cells.
Because of the high frequency of PTEN loss found in prostate cancer and the importance of p110β in prostate tumorigenesis (Jia et al., 2008, Lee et al., 2010), we focused on PC3 cells, which specifically require p110β for cell proliferation (Wee et al., 2008), to further examine the phenotypic consequences of CRKL suppression. We found that the proliferation of PC3 cells was inhibited by CRKL depletion under full (10%) serum conditions (Figure 2E).
Downregulation of CRKL does not impair PI3K signaling in HER2 amplified and PIK3CA mutant breast cancer cell lines or affect insulin signaling in PC3 and MCF10A cells
Since CRKL also associates with p110β in PTEN-replete cancer cells that rely on p110α for PI3K signaling, we examined the effect of CRKL suppression on PI3K signaling in two human breast cancer cell lines with HER2 amplification and activating mutations in PIK3CA: MDA-MB-361 and MDA-MB-453 (Table S2). Despite the reduction in CRKL protein levels, phosphorylation of AKT was not inhibited in cells grown either in the presence or absence of serum (Figure S2D and S2E), indicating that CRKL is dispensable for the maintenance of PI3K signaling in tumors with co-existing PIK3CA mutations and HER2 amplification.
Prior work has linked CRKL function with RTK signaling (Cheung et al., 2011, Sattler et al., 1997), so we examined whether CRKL suppression affects insulin-stimulated AKT phosphorylation. As shown in Figure 3A, the basal PI3K activity in PC3 cells (under starvation) was sensitive to p110β inhibition but not p110α. In response to insulin stimulation, phospho-AKT was increased to similar level regardless of the levels of CRKL expression. Furthermore, insulin-stimulated AKT activation was suppressed by a p110α-selective inhibitor BYL719 but not by a p110β inhibitor TGX221. These data suggest that p110α is required for PI3K signaling in response to RTK stimulation by insulin, which is independent of CRKL in PC3 cells. Consistent with our observation in PC3 cells, insulin- and EGF-stimulated AKT phosphorylation was not reduced by CRKL knockdown in non-tumorigenic MCF10A (Figure 3B).
Figure 3. Downregulation of CRKL does not impair p110α-dependent PI3K signaling.

(A) PC3 cells stably transduced with inducible control or CRKL targeting shRNA were cultured in the presence of Dox (100 ng/ml 72 hr). Cells were starved in serum-free medium overnight and treated with indicated inhibitors 1 hr prior to insulin stimulation (4 µg/ml 10 min). Cell lysates were analyzed by immunoblotting with indicated antibodies. The level of pAKT473 was normalized to tubulin. For each experiment the pAKT473/tubulin ratio in cell line expressing control shRNA in the presence of DMSO was set to 1. Bar graphics reflect the mean ± SEM; n=3; * p<0.05 by comparing cells expressing control shRNA and shCRKL (t-test).
(B) MCF10A cells stably transduced with inducible control or CRKL targeting shRNA were cultured with or without Dox (100 ng/ml 72 hr). Cells were starved in serumfree medium overnight prior to insulin (4 µg/ml 10 min) or EGF (20 ng/ml 10 min) stimulation. Cell lysates were analyzed by immunoblotting with indicated antibodies. The level of pAKT473 was normalized to tubulin. For each experiment the pAKT473/tubulin ratio in cell line expressing control shRNA in the absence of Dox and stimulated with insulin was set to 1. Bar graphics reflect the mean ± SEM; n=3. No significant difference was found by comparing cells expressing control shRNA and shCRKL (t-test).
A PTEN/FAK/Src/p130Cas axis may activate CRKL/p110β in PTEN-deficient cancer cells
To investigate signaling upstream of the CRKL-p110β complex, we searched for the phospho-tyrosine containing proteins that bind to CRKL in PTEN-null cancer cells by immunoprecipitating CRKL and then blotting with an anti-phosphotyrosine antibody. The major bands were ~130Kd, the size of the CRKL binding protein p130Cas. This identity was confirmed by a phospho-p130Cas antibody (Figure 4A). Intriguingly, tyrosine phosphorylation levels of p130Cas were much higher in PTEN null cells than in PTEN expressing tumor cells (Figure 4A), particularly in serum starved cells.
Figure 4. Inhibition of PI3K and Src reduces cell proliferation in PTEN-deficient cancer cells.

(A) PC3, BT549, MDA-MB-361 and MDA-MB-453 cells were cultured under full (10%) serum condition or starved in serum-free medium overnight. Cell lysates were immunoprecipitated with anti-CRKL antibody or rabbit IgG and immunoblotted with 4G10, phospho-p130Cas, CRKL, and vinculin antibodies.
(B) PC3 and BT549 cells grown under full (10%) serum condition or low (0.1%) serum condition overnight were treated with PF573228 or Dasatinib at indicated concentrations for 10 hr. Cell lysates were immunoblotted with indicated antibodies.
(C) PC3 and BT549 cells were cultured under full (10%) serum condition or low (0.1%) serum condition in the presence of Dasatinib and GDC0491 at indicated concentrations. Cell proliferation was measured by MTS assay after 3 d. Data represent mean ± SD (top left: n=5; top right: n=6; bottom left: n=8; bottom right: n=8); * p<0.05 (t-test).
(D) Model representing PI3K signaling under condition of PTEN loss. In cells lacking PTEN, the active FAK-Src complex recruits and phosphorylates the scaffolding protein p130Cas. The phosphorylation of p130Cas creates docking sites for CRKL, thereby recruiting PI3Kβ. The other effectors of CRKL stimulate RAC activity, both a known activator of p110β and a known target downstream of PI3K via PIP3 activated RAC GEFs, forming a potential positive feedback loop.
p130Cas is a nonenzymatic scaffolding protein with repetitive Y-x-x-P motifs, the preferred binding site for the SH2 domain of CRKL. The tyrosine residues in these motifs are usually phosphorylated by Src (Sharma and Mayer, 2008) with subsequent recruitment of CRKL (Birge et al., 2009, Li et al., 2003). It has been reported that pp60c-Src can be phosphorylated at Tyr416 and hence activated in PTEN null cells (Zhang et al., 2011a, Dey et al., 2008). It has also long been known that activated Src preferentially phosphorylates proteins in focal adhesions including p130Cas (Wozniak et al., 2004). Since ligation of the SH2 domain of CRKL is known to activate it for downstream signaling via interactions of its SH3 domain with effector molecules, our finding suggests the presence in PTEN null cells of a signaling cascade beginning with Src and continuing through p130Cas to CRKL and finally to p110β. To analyze the potential role of FAK, Src, and other Src family kinases in the activation of PI3K in PTEN null cells, we treated PC3 and BT549 cells grown in full serum medium or serum-free medium with FAK or Src inhibitors. Intriguingly, treatment with the Src inhibitor Dasatinib resulted in a marked decline in both p130Cas and AKT phosphorylation. However, FAK inhibition did not significantly suppress AKT phosphorylation (Figure 4B).
Src has been shown to mediate cellular oncogenic transformation independent of PI3Ks (Jia et al., 2008, Rodriguez-Viciana et al., 1997, Zhao et al., 2006). To analyze a possible cooperation between the Src and PI3K pathways in PTEN null cells, we treated PC3 and BT549 cells under full serum condition or starvation with the Src inhibitor Dasatinib and pan-PI3K inhibitor GDC0491. The combination of Dasatinib and GDC0491 yielded more substantial suppression of cell proliferation than either individual agent (Figure 4C), suggesting that Src inhibition cooperates with PI3K or p110β inhibition to suppress the growth of PTEN-null tumor cells.
Discussion
The present data only consider CRKL and not the closely related protein CRKII. It has been shown that the SH3N domain of CRKII interacts with the proline-rich region of p85 (Gelkop et al., 2001); hence, it is thus reasonable to assume CRKII might also play a role in regulating PI3K signaling in PTEN-null tumors. Technical difficulties prevented us from obtaining definitive results on any potential role of CRKII in p110β activation, but we do note that expression of a dominant negative allele of CRKL is more effective than CRKL knockdown in blocking AKT activation in PTEN null cells (Figure 2D and S2C).
One intriguing and surprising finding in our study was the significant preference for CRKL in binding to p110β over p110α, even though CRKL binds p110β at least in large part via p85. Although all tested cell lines seemed to have higher endogenous levels of p110β than p110α, CRKL associated preferentially with overexpressed p110β even when p110α and p110β were overexpressed at comparable levels as judged by immunoprecipitation via epitope tags, suggesting that relative expression of p110s is not the cause. A second possibility is that p110α and p110β may selectively bind different p85 isoforms, which in turn affect their association with CRKL. Each p85 isoform can bind each p110 isoform under overexpression conditions (Thorpe et al., 2015); but little is known about the selectivity of endogenous proteins. There is evidence of selective pairing between p110 and p85 isoforms, such as selective binding of p110β to p85β in the nucleus (Kumar et al., 2011). Whether CRKL-p110β interaction exploits specific p85 isoform awaits further investigation. A third explanation is that the p110β/p85 complex may differ from the p110α/p85 complex structurally in a way favoring CRKL binding. Indeed, crystallographic data suggested that the structure of the p110β/p85 complex is different from that of p110α/p85 complex in the p110/p85 interaction interface (Huang et al., 2007, Zhang et al., 2011b). Unfortunately the proline-rich regions of p85 where CRKL binds are not present in the published structures. Further structural data will be needed to clarify this possibility. Alternatively the CRKL-p110β association may involve additional domains of the two proteins or other bridging proteins that specifically bind p110β. Indeed, we observed some residual interaction between CRKL and p110β when CRKL/p85 binding was abrogated.
A number of hypotheses have been raised to explain the selective activation of p110β in PTEN null cells. One hypothesis is that p110β has a modest level of constitutive activity which is counteracted by PTEN molecules specifically associating with p110β/p85 complex under normal growth conditions. Upon PTEN loss, this basal activity of p110β is no longer controlled by PTEN and, thus, downstream signaling becomes constitutively activated. Supporting this argument, it has been found that PTEN associates with p110β/p85 complex through interaction with p85, which enhances PTEN phosphatase activity (Rabinovsky et al., 2009, Chagpar et al., 2010). Although it remains to be proven whether the p110β-dependency in PTEN-null tumors can be explained solely by the loss of interaction of PTEN with p110β/p85, CRKL seems to provide another link between p110β and PTEN, potentially at focal adhesions or in other distinct membrane microdomains. Cell attachment to extracellular matrix leads to activation of FAK, which in turn recruits proteins including p85. p85 binds to Tyr397 on FAK, a site that PTEN has been proposed to compete for (Tamura et al., 1999). CRKL has been found to translocate to focal adhesions in integrin signaling (Li et al., 2003), and p110β has also been implicated in integrin signaling in platelets (Schoenwaelder et al., 2010). Our results indicate that CRKL binds phospho-p130Cas, a nodal scaffolding protein phosphorylated by Src at focal adhesions, in PTEN-null cancer cells. In line with this idea, knockdown of p130Cas has been found to reduce AKT phosphorylation in BT549 cells (Cunningham-Edmondson and Hanks, 2009).
Our results might also help to explain certain clinical findings in PTEN null tumors. Recent clinical trials indicated that many PTEN-null tumors don't respond well to PI3K inhibitors as monotherapy (Yap et al., 2015). Although it is possible that the pan-PI3K inhibitors currently in trials are less potent towards p110β than p110α, it is also possible that PTEN loss activates more than the PI3K pathway and renders tumors featuring PTEN loss intrinsically more resistant to PI3K inhibition than are tumors driven by p110α activation. We note that PTEN loss activates both Src and p110β. Notably Src has been shown to mediate cellular oncogenic transformation independent of PI3Ks (Jia et al., 2008, Rodriguez-Viciana et al., 1997, Zhao et al., 2006). One example of this type of PI3K independent oncogenic signaling is manifest in STAT pathway activation known to be elicited by Src (Silva, 2004). Similarly, activation of p130Cas by Src in PTEN null tumors also activates PI3K independent oncogenic signaling via RAC (Birge et al., 2009, Zhang et al., 2011a, Dey et al., 2008). The latter may be particularly important in that RAC is both a known activator of p110β (Fritsch et al., 2013) and a known target downstream of PI3K via PIP3 activated RAC GEFs such as PREX and TIAM (Welch et al., 2003). Thus PTEN loss could set up a positive feedback loop mediated by Src, p130Cas and RAC. Since RAC effectors activate multiple pathways including ERKs, a PTEN/Src/p130Cas/RAC cascade may also contribute to PI3K resistance in PTEN null cells. Indeed, we showed that Src inhibition cooperated with PI3K inhibition to suppress the growth of PTEN-null tumors suggesting a combination of Src and PI3K inhibitors may achieve growth inhibition in tumors with PTEN deficiency in clinical settings. Notably it has also been shown that activated Src can transform independently of p110α (Zhao et al., 2006) so it is quite possible that Src inhibitors would also cooperate with p110α inhibition.
We have recently published data showing that RAC activation is necessary to localize p110β to lipid rafts, an essential component of cell growth signaling in PTEN null cells that rely on p110β for PI3K signaling (Cizmecioglu et al., 2016). We showed that a second signal was also required for full p110β activation. In the case of GPCR signaling, this second signal comes from a direct interaction of the Gβγ subunit of the relevant G protein that is activated by the GPCR. In the case of PTEN null cells the origin of this second signal is not known. Notably the Src/p130Cas/CRKL pathway described here could conceivably supply both the activated RAC signal via other effectors of CRKL and the Gβγ like signal, perhaps via the CRKL/p85/p110β interaction itself. More work is clearly required to test this hypothesis which is diagrammed in Figure 4D.
In conclusion, our study identified CRKL as an important regulator of PI3K activity in PTEN-deficient tumor cells through its association with p110β/p85. Although the importance of CRKL in PTEN loss induced tumorigenesis remains to be tested in larger cancer cell panels, this finding provides further insight to the understanding of isoform selectivity of p110α and p110β, defines a specific role for CRKL in regulating p110β kinase activity, and suggests an alternative combination therapy to treat PTEN loss tumors.
Experimental Procedures
Immunoprecipitation
Cell lysates were collected by scraping cells in FLAG-IP buffer. Protein extracts were incubated with 2 µg anti-CRKL antibody (sc-319, Santa Cruz) or normal rabbit IgG (Millipore) at 4°C overnight followed by incubation with 100 µl Protein A/G-PLUS agarose beads (sc-2003, Santa Cruz) for 2 hr at 4°C. Beads were collected by centrifugation at 2,000 rpm for 3 min at 4°C and resuspended in 1 ml FLAG-IP buffer for washing. After repeated washing and centrifugation for 4 times, beads were boiled for 5 min in SDS loading/lysis buffer (4% sodium dodecyl sulfate, 20% glycerol, 120 mM Tris pH 6.8, and bromophenol blue).
Western blot analysis
Cells were lysed in ice-cold RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS) containing complete protease inhibitors (Roche) and phosphatase inhibitor cocktail II and III (Sigma) and clarified by centrifugation. Alternatively, cells were lysed directly on tissue culture dish in SDS loading/lysis buffer. Equal amounts of lysates were resolved on SDS-PAGE and transferred to nitrocellulose membranes. After incubation with blocking buffer (50% LI-COR blocking buffer in phosphate-buffered saline) for 1 hr, the membranes were incubated with primary antibody overnight at 4°C and then incubated with fluorescently labeled secondary antibodies for 1 hr at room temperature. The protein signal was detected using an Odyssey scanner (LI-COR). The quantification of western blots was performed with ImageStudio version 3.1.4.
Please refer to Supplemental Experimental Procedures for a detailed description of other experiments.
Supplementary Material
Highlights.
CRKL preferentially associates with p110β.
CRKL knockdown inhibits p110β-dependent PI3K signaling in PTEN-null cancer cells.
A PTEN/FAK/Src/p130Cas axis may activate CRKL/p110β in PTEN-null cancer cells.
Inhibitors of Src and PI3K cooperate to inhibit growth of PTEN-null cancer cells.
Acknowledgments
We thank Dr. William C. Hahn (Dana-Farber Cancer Institute, Boston, MA) for CRKL expression constructs and CRKL-targeting shRNAs. This work was supported by NIH Grants R01 CA172461 (T.M.R. and J.J.Z.), R01 CA187918 (T.M.R. and J.J.Z.), P50 CA168504 (T.M.R. and J.J.Z.), P50 CA165962 (T.M.R. and J.J.Z.), P01 CA142536 (J.J.Z), GM110352 (M.J.E.) and Dana-Farber Strategic Research Initiative, NINDS P01NS047572 (J.A.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
T.M.R. and J.J.Z. supervised the research. J.Z. designed and performed the majority of experiments. F.S. and J.Z. generated BacMam viruses under the guidance of T.M.R. and M.J.E. G.A. performed LC-MS/MS analysis under the guidance of J.A.M. X.G. assisted with western blot analysis in Figure 4B and data not shown. J.Z., G.A., T.M.R. and J.J.Z. wrote the manuscript with input from all co-authors.
Accession Numbers
Raw mass spectrometry data files are available for free download from: ftp://massive.ucsd.edu/MSV000081148.
References
- Bell ES, Park M. Models of crk adaptor proteins in cancer. Genes Cancer. 2012;3:341–52. doi: 10.1177/1947601912459951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birge RB, Kalodimos C, Inagaki F, Tanaka S. Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun Signal. 2009;7:13. doi: 10.1186/1478-811X-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagpar RB, Links PH, Pastor MC, Furber LA, Hawrysh AD, Chamberlain MD, Anderson DH. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 2010;107:5471–6. doi: 10.1073/pnas.0908899107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung HW, Du J, Boehm JS, He F, Weir BA, Wang X, Butaney M, Sequist LV, Luo B, Engelman JA, Root DE, Meyerson M, Golub TR, Janne PA, Hahn WC. Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non-small cell lung cancers. Cancer Discov. 2011;1:608–25. doi: 10.1158/2159-8290.CD-11-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciraolo E, Iezzi M, Marone R, Marengo S, Curcio C, Costa C, Azzolino O, Gonella C, Rubinetto C, Wu H, Dastru W, Martin EL, Silengo L, Altruda F, Turco E, Lanzetti L, Musiani P, Ruckle T, Rommel C, Backer JM, Forni G, Wymann MP, Hirsch E. Phosphoinositide 3-kinase p110beta activity: key role in metabolism and mammary gland cancer but not development. Sci Signal. 2008;1:ra3. doi: 10.1126/scisignal.1161577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cizmecioglu O, Ni J, Xie S, Zhao JJ, Roberts TM. Rac1-mediated membrane raft localization of PI3K/p110beta is required for its activation by GPCRs or PTEN loss. Elife. 2016:5. doi: 10.7554/eLife.17635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham-Edmondson AC, Hanks SK. p130Cas substrate domain signaling promotes migration, invasion, and survival of estrogen receptor-negative breast cancer cells. Breast Cancer (London) 2009;2009:39–52. doi: 10.2147/BCTT.S6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey N, Crosswell HE, De P, Parsons R, Peng Q, Su JD, Durden DL. The protein phosphatase activity of PTEN regulates SRC family kinases and controls glioma migration. Cancer Res. 2008;68:1862–71. doi: 10.1158/0008-5472.CAN-07-1182. [DOI] [PubMed] [Google Scholar]
- Donato J, Jr, Frazao R, Elias CF. The PI3K signaling pathway mediates the biological effects of leptin. Arq Bras Endocrinol Metabol. 2010;54:591–602. doi: 10.1590/s0004-27302010000700002. [DOI] [PubMed] [Google Scholar]
- Fritsch R, de Krijger I, Fritsch K, George R, Reason B, Kumar MS, Diefenbacher M, Stamp G, Downward J. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell. 2013;153:1050–63. doi: 10.1016/j.cell.2013.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelkop S, Babichev Y, Isakov N. T cell activation induces direct binding of the Crk adapter protein to the regulatory subunit of phosphatidylinositol 3-kinase (p85) via a complex mechanism involving the Cbl protein. J Biol Chem. 2001;276:36174–82. doi: 10.1074/jbc.M100731200. [DOI] [PubMed] [Google Scholar]
- Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, Amzel LM. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318:1744–8. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, Zhao JJ. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–9. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Redondo-Munoz J, Perez-Garcia V, Cortes I, Chagoyen M, Carrera AC. Nuclear but not cytosolic phosphoinositide 3-kinase beta has an essential function in cell survival. Mol Cell Biol. 2011;31:2122–33. doi: 10.1128/MCB.01313-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Poulogiannis G, Pyne S, Jia S, Zou L, Signoretti S, Loda M, Cantley LC, Roberts TM. A constitutively activated form of the p110beta isoform of PI3-kinase induces prostatic intraepithelial neoplasia in mice. Proc Natl Acad Sci U S A. 2010;107:11002–7. doi: 10.1073/pnas.1005642107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Guris DL, Okura M, Imamoto A. Translocation of CrkL to focal adhesions mediates integrin-induced migration downstream of Src family kinases. Mol Cell Biol. 2003;23:2883–92. doi: 10.1128/MCB.23.8.2883-2892.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J, Liu Q, Xie S, Carlson C, Von T, Vogel K, Riddle S, Benes C, Eck M, Roberts T, Gray N, Zhao J. Functional characterization of an isoformselective inhibitor of PI3K-p110beta as a potential anticancer agent. Cancer Discov. 2012;2:425–33. doi: 10.1158/2159-8290.CD-12-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovsky R, Pochanard P, McNear C, Brachmann SM, Duke-Cohan JS, Garraway LA, Sellers WR. p85 Associates with unphosphorylated PTEN and the PTEN-associated complex. Mol Cell Biol. 2009;29:5377–88. doi: 10.1128/MCB.01649-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–67. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- Sattler M, Salgia R, Shrikhande G, Verma S, Pisick E, Prasad KV, Griffin JD. Steel factor induces tyrosine phosphorylation of CRKL and binding of CRKL to a complex containing c-kit, phosphatidylinositol 3-kinase, and p120(CBL) J Biol Chem. 1997;272:10248–53. doi: 10.1074/jbc.272.15.10248. [DOI] [PubMed] [Google Scholar]
- Schmit F, Utermark T, Zhang S, Wang Q, Von T, Roberts TM, Zhao JJ. PI3K isoform dependence of PTEN-deficient tumors can be altered by the genetic context. Proc Natl Acad Sci U S A. 2014;111:6395–400. doi: 10.1073/pnas.1323004111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenwaelder SM, Ono A, Nesbitt WS, Lim J, Jarman K, Jackson SP. Phosphoinositide 3-kinase p110 beta regulates integrin alpha IIb beta 3 avidity and the cellular transmission of contractile forces. J Biol Chem. 2010;285:2886–96. doi: 10.1074/jbc.M109.029132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Mayer BJ. Phosphorylation of p130Cas initiates Rac activation and membrane ruffling. BMC Cell Biol. 2008;9:50. doi: 10.1186/1471-2121-9-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva CM. Role of STATs as downstream signal transducers in Src family kinasemediated tumorigenesis. Oncogene. 2004;23:8017–23. doi: 10.1038/sj.onc.1208159. [DOI] [PubMed] [Google Scholar]
- Sopasakis VR, Liu P, Suzuki R, Kondo T, Winnay J, Tran TT, Asano T, Smyth G, Sajan MP, Farese RV, Kahn CR, Zhao JJ. Specific roles of the p110alpha isoform of phosphatidylinsositol 3-kinase in hepatic insulin signaling and metabolic regulation. Cell Metab. 2010;11:220–30. doi: 10.1016/j.cmet.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura M, Gu J, Danen EH, Takino T, Miyamoto S, Yamada KM. PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem. 1999;274:20693–703. doi: 10.1074/jbc.274.29.20693. [DOI] [PubMed] [Google Scholar]
- Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15:7–24. doi: 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torbett NE, Luna-Moran A, Knight ZA, Houk A, Moasser M, Weiss W, Shokat KM, Stokoe D. A chemical screen in diverse breast cancer cell lines reveals genetic enhancers and suppressors of sensitivity to PI3K isoformselective inhibition. Biochem J. 2008;415:97–110. doi: 10.1042/BJ20080639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A. 2008;105:13057–62. doi: 10.1073/pnas.0802655105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch HC, Coadwell WJ, Stephens LR, Hawkins PT. Phosphoinositide 3-kinase-dependent activation of Rac. FEBS Lett. 2003;546:93–7. doi: 10.1016/s0014-5793(03)00454-x. [DOI] [PubMed] [Google Scholar]
- Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004;1692:103–19. doi: 10.1016/j.bbamcr.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Yap TA, Bjerke L, Clarke PA, Workman P. Drugging PI3K in cancer: refining targets and therapeutic strategies. Curr Opin Pharmacol. 2015;23:98–107. doi: 10.1016/j.coph.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzugullu H, Baitsch L, Von T, Steiner A, Tong H, Ni J, Clayton LK, Bronson R, Roberts TM, Gritsman K, Zhao JJ. A PI3K p110beta-Rac signalling loop mediates Pten-loss-induced perturbation of haematopoiesis and leukaemogenesis. Nat Commun. 2015;6:8501. doi: 10.1038/ncomms9501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, Xiong Y, Tseng LM, Li SH, Ding Z, Sahin AA, Esteva FJ, Hortobagyi GN, Yu D. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med. 2011a;17:461–9. doi: 10.1038/nm.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Vadas O, Perisic O, Anderson KE, Clark J, Hawkins PT, Stephens LR, Williams RL. Structure of lipid kinase p110beta/p85beta elucidates an unusual SH2-domain-mediated inhibitory mechanism. Mol Cell. 2011b;41:567–78. doi: 10.1016/j.molcel.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JJ, Cheng H, Jia S, Wang L, Gjoerup OV, Mikami A, Roberts TM. The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci U S A. 2006;103:16296–300. doi: 10.1073/pnas.0607899103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.