

Abstract
Objective
In the liver, a contributing factor in the pathogenesis of non-alcoholic fatty liver disease is oxidative stress leading to the accumulation of highly reactive electrophilic α/β unsaturated aldehydes. The objective of this study was to determine if significant differences were evident when evaluating carbonylation in human end-stage fatty nonalcoholic steatohepatitis (fNASH) compared to end-stage nonfatty NASH (nfNASH).
Methods
Using hepatic tissue obtained from healthy humans and patients diagnosed with end stage nfNASH or fNASH, overall carbonylation was assessed by immunohistochemistry (IHC) and LC-MS/MS followed by bioinformatics.
Results
Picrosirius red staining revealed extensive fibrosis in both fNASH and nfNASH which corresponded with increased reactive aldehyde staining. Although significantly elevated when compared to normal hepatic tissue, no significant differences in overall carbonylation and fibrosis were evident when comparing fNASH with nfNASH. Examining proteins that are critical for anti-oxidant defense revealed elevated expression of thioredoxin, thioredoxin interacting protein, glutathione S-transferase p1 and mitochondrial superoxide dismutase in human NASH. As important, using immunohistochemistry, significant colocalization of the aforementioned proteins occurred in cytokeratin 7 positive cells indicating that they are part of the ductular reaction. Expression of catalase and Hsp70 decreased in both groups when compared to normal human liver. Mass spectrometric analysis revealed a total of 778 carbonylated proteins. Of these, 194 were common to all groups, 124 unique to tissue prepared from healthy individuals, 357 proteins exclusive to NASH, 124 proteins distinct to samples from patients with fNASH and 178 unique to nfNASH. Using functional enrichment analysis of hepatic carbonylated proteins revealed a propensity for increased carbonylation of proteins regulating cholesterol and Huntington’s disease related pathways occurred in nfNASH. Examining fNASH, increased carbonylation was evident in proteins regulating Rho cytoskeletal pathways, nicotinic acetylcholine receptor signaling and chemokine/cytokine inflammatory pathways. Using LC-MS/MS analysis and trypsin digests, sites of carbonylation were identified on peptides isolated from vimentin, endoplasmin and serum albumin in nfNASH and fNASH respectively.
Conclusions
These results indicate that cellular factors regulating mechanisms of protein carbonylation may be different depending on pathological diagnosis of NASH. Furthermore these studies are the first to use LC-MS/MS analysis of carbonylated proteins in human NAFLD and explore possible mechanistic links with end stage cirrhosis due to fatty liver disease and the generation of reactive aldehydes.
Graphical Abstract
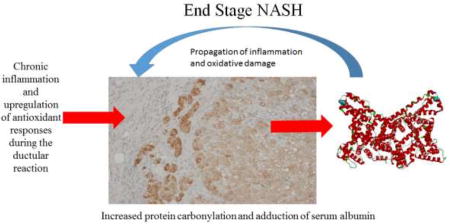
Background
Non-alcoholic fatty liver disease (NAFLD) is a leading cause of liver failure in the United States today. A common phenotype of NAFLD is pronounced hepatic lipid accumulation and enhanced oxidative stress. In a subset of NAFLD patients, symptoms progress to nonalcoholic steatohepatitis (NASH) and in this inflammatory environment a further subset will progress to fibrosis and ultimately cirrhosis1–3.
In NASH, steatosis is frequently regarded as the first “hit” and is hypothesized to be the prerequisite for progression to steatohepatitis. A second, not yet definitively identified, “hit” is required for the progression to steatohepatitis. This second hit has been proposed to include cellular processes such as mitochondrial injury, oxidative stress, innate immunity or proinflammatory cytokines4. Recent data has suggested that inflammation provoked by oxidative stress promotes the production of pro-inflammatory cytokines including tumor necrosis factor-alpha (TNF-α) in Kupffer cells and hepatocytes, leading to stimulation of collagen synthesis by hepatic stellate cells5.
The formation of reactive oxygen species (ROS) during chronic inflammation is central to the progression of chronic liver diseases6,7. ROS are produced by the mitochondrial respiratory chain, the cytochrome P450 system, auto-oxidation of heme proteins, the NADPH oxidase complex, xanthine oxidase, oxidative enzymes and other cellular systems8. While ROS are important in signal transduction, cellular physiology and critical metabolic pathways, a high concentration of ROS can result in hepatocellular damage, apoptosis and necrosis9. Elevated ROS can also lead to a free radical chain reaction with unsaturated fatty acids generating toxic electrophilic α/β unsaturated aldehydes, a process called lipid peroxidation10. Oxidative stress is an important factor in the pathogenesis of NASH and has been proposed contribute to the “second hit”4,11. During NASH progression, markers of oxidative stress including products of lipid peroxidation are elevated and importantly, decrease following bariatric surgery or other types of treatments supporting the relationship between hepatocellular damage and lipid peroxidation12–15.
The best characterized of carbonyl-derivatives are 4-hydroxy-2-nonenal (4-HNE), 4-oxo-2-nonenal (4-ONE), Malondialdehyde (MDA) and acrolein7. Following their formation, these highly reactive lipid peroxides modify nucleophilic Lys, Cys and His residues on proteins exerting pathophysiological inhibitory effects. Lipid peroxidation/protein aldehyde carbonylation is a validated marker of oxidative stress7,16,17. Research in our laboratory has pioneered global proteomic approaches to identify proteins that are post-translationally modified by reactive aldehydes in the liver. In recent murine and human studies in alcoholic liver disease we have identified 829 and 1244 proteins respectively that are carbonylated during conditions of increased hepatocellular stress16,18. In our current study, we utilized LC-MS/MS, immunohistochemistry and Western blotting to determine the impact of human end-stage fatty NASH and nonfatty NASH on protein carbonylation and anti-oxidant defense proteins.
Materials and Methods
Human Tissue
To determine the status of protein carbonylation in human fNASH/nfNASH, paraffin embedded and frozen hepatic tissue from normal and end stage NASH patients (N=8 Non-fatty NASH, 7 Fatty NASH and 7 normal human liver) procured during transplantation (ages 33–70) were obtained from the University of Minnesota Liver Tissue Cell Distribution Center which was funded through NIH Contract #HHSN276201200017C. Whole cell extracts (WCE) of each sample were prepared by dounce homogenization (10×) of tissue resuspended in 50mM tricine pH 8.0, 0.001M NaCl plus phosphatase and protease inhibitors (SIGMA ALDRICH, St Louis, MO) followed by sonication (3×15 seconds@ 4°C). To remove debris, samples were centrifuged at 14,000RPM (16,000g)(4°C) for 10 minutes. Supernatants were drawn off and immediately flash frozen in liquid nitrogen.
Histological Evaluation
To detect fibrosis, formalin fixed slides were stained with Picro-Sirius (PSR). In addition, immunohistochemical staining for 4-HNE, rabbit polyclonal16, 4-oxononenal rabbit polyclonal (4-ONE), acrolein rabbit polyclonal (Cell Sciences, Newburyport, MA), malondialdehyde (MDA) rabbit polyclonal (ABCAM, Billerica, MA), myeloperoxidase (MPO) goat polyclonal (Millipore, Billerica, MA), catalase rabbit polyclonal (SIGMA ALDRICH, St. Louis, MO), GSTπ (rabbit polyclonal MBL Woburn, MA), mitochondrial superoxide dismutase (SOD2) goat polyclonal (ABCAM, Billerica, MA), Thioredoxin (Trx1) rabbit polyclonal (Protein Tech, Rosemont, IL) was completed as previously described17. Histologic images were captured on an Olympus BX51 microscope equipped with a four-megapixel Macrofire digital camera (Optronics; Goleta, CA) using the PictureFrame Application 2.3 (Optronics). All images were cropped and assembled using Photoshop CS2 (Adobe Systems, Inc.; Mountain View, CA). For picrosirius red (PSR) quantification, ten polarized images were made in a “tiling” fashion across each PSR stained slide, then quantitated using the 3I Slidebook program (3I, Denver, Colorado) to arrive at the PSR stained pixels per 100× field for that slide.
Biotin hydrazide purification
Whole cell extracts (500µg) were prepared from age matched hepatic tissue obtained from the same human patients as utilized for both immunohistochemistry and Western blotting (normal (N=4), Fatty NASH (N=4) and Non-Fatty NASH (N=4)). Aldehyde modified proteins from each extract were derivatized using biotin hydrazide (BH; ThermoFisher/Pierce, Waltham, MA; 5mM/2 hrs/RT/dark) followed by NaBH4 reduction (10mM/100mM NaOH 1hr/dark) followed by Streptavidin purification and trypsin digestion as previously described16,18.
LC-MS/MS analysis
For LC-MS/MS analysis, 8µl of each peptide mixture was loaded on a Bruker Amazon Speed LC-MS/MS and a Bruker Maxis IMPACT LC-MS/MS. The instrument was operated using data-dependent collision-induced dissociation (CID) MS/MS or electron transfer dissociation (ETD) with a threshold of 10,000 total ion current (TIC). Data analysis was performed using ProteinScape V3.1.2 (Bruker Daltonics Inc. Billerica, MA) and Mascot (v2.1.04, Matrixscience). First a global search for proteins was conducted with a Mascot cutoff score of 80 with the following variable modifications of carbamidomethyl (C) and oxidized (M). After the initial search a second search was conducted for post-translational modification by reactive aldehydes using the masses previously described16. For this 2nd iteration, peptide significance required a Mascot score higher than 24 and visual validation of spectra. Following protein identification, all data was pooled into each respective group.
Western blotting
Western blotting for E.R. Stress proteins CHOP/GADD153 (rabbit polyclonal Santa Cruz Biotechnology, Santa Clara, CA), Glucose Response Protein 94 (Grp94, Mouse monoclonal, (Millipore, Temecula, CA), Glucose Response Protein 78 (Grp78 Rabbit polyclonal) (ABCAM, Billerica, MA), Heat Shock Protein 70 (Hsp70 Mouse monoclonal), Stressgen/Enzo Lifesciences Farmingdale, NY), Trx1 (Protein Tech, Rosemont, IL), Thioredoxin Reductase 1 (TrxR1), (Protein Tech), Thioredoxin interacting protein (TxNIP) (Invitrogen, Carlsbad, CA), SOD2 (ABCAM), catalase (Sigma, St Louis, MO) and Vimentin (Rabbit polyclonal, Cell Signaling Woburn, MA) was performed from 10µg of liver extracts as previously described17,19–21. GAPDH was used as a loading control with anti-GAPDH (Mouse monoclonal) (ABCAM, Billerica, MA). Quantification of expression of each protein was performed using ImageJ (NIH) and normalized to overall GAPDH expression.
Bioinformatics
Bioinformatics analysis was performed using UniProt IDs from proteins identified in the each sample group. ID’s were initially converted into gene symbols using the UniProt website (UniProt Release 2017_01). Gene symbols were compared to the Panther Pathways database22 available through the Enrichr database23. When the same UniProt ID linked to multiple gene symbols, the multiple gene symbols were collapsed into one in the Panther pathway database prior to enrichment analysis. Enrichment was determined using a one-sided Fisher’s Exact test for 4 mutually exclusive sets of proteins/genes: 1) genes that represent protein carbonylated in the fNASH samples (not nfNASH or normal liver), 2) genes that represent protein carbonylated in the nfNASH samples (not fNASH or normal liver), 3) genes that represent protein carbonylated in the normal liver samples (not nfNASH or fNASH), and 4) genes that represent protein carbonylated in both the fNASH and nfNASH samples (not normal liver). A pathway was significantly enriched if more than one gene/protein from the pathway was in the list of carbonylated proteins and the enrichment p-value was less than 0.05. For visualization, hierarchical clustering was done using Euclidean distance between binary indicators of significance (p<0.05 vs. p>=0.05). Enrichment analyses and the heatmap were generated using R Statistical Software (version 3.3.2).
Molecular modeling
All manipulations were performed using Discovery Studio software (Version 3.1; Accelrys Inc., San Diego, CA). The crystallographic coordinates of the 2.4Å human endoplasmin/Grp94/Hsp90b (PDB code 2olv24), and 1.1 Å human serum albumin (PDB code 4k2c (23)) crystal structures were obtained from the RCSB Protein Data Bank (http://wwww.rcsb.org)25,26.
Statistical Analysis
The data are presented as means ± SE. Comparisons between genotype and diet was accomplished by one-way ANOVA, followed by Student Newman-Keuls post hoc analysis. Comparisons between two groups were accomplished using Student’s T-tests. Statistical significance was set at P<0.05. Prism 5 for Windows (GraphPad Software, San Diego, CA) was used to perform all statistical tests.
Results
Post-translational modification of proteins by products of lipid peroxidation has been implicated as a contributing factor in the progression of chronic liver disease2,3,7. Although significant data have been accumulated using animal and human models of ALD, protein carbonylation in human hepatic tissue in NASH has not been examined16,18. In the present study we have examined the effects of increased oxidative stress with respect to protein carbonylation and relevant anti-oxidant defense systems in human tissue obtained from patients classified as having either end-stage fNASH or end-stage nfNASH.
For this study, fresh frozen human hepatic tissue and formalin fixed tissue from normal and end stage NASH was obtained prior to transplant from the University of Minnesota Liver Tissue cell Distribution Center (NIH Contract #HHSN276201200017C). For each patient relevant hepatic parameters was provided (model for end-stage liver disease (MELD)27, aspartate aminotransferase (AST), international normalized ratio of prothrombin coagulation (INR), serum bilirubin, alkaline phosphatase and albumin). As shown in Table 1, all 15 patients possessed increased MELD scores indicative of severe hepatic dysfunction. MELD scores however were significantly higher in fNASH patients when compared to nfNASH. Examining individual parameters, INR (normal approximately 1.0), total bilirubin (adult normal range 0.1 – 1.3 mg/dL), serum AST (normal range 12 – 39 U/L), alkaline phosphatase (normal range 39 – 117 U/L), were all elevated and serum albumin (normal range 3.5–5.7g/dL) levels suppressed. Comparing fNASH with nfNASH, fNASH patients overall exhibited significant higher INR and bilirubin concentrations. Combined these data suggest that at least for the samples examined, the degree of disease was more severe in individuals diagnosed with fNASH.
Table 1. Serum biochemical parameters of human fNASH and nfNASH patients.
Data was obtained from the University of Minnesota Liver Tissue cell Distribution Center NIH Contract #HHSN276201200017C.
| Biochemical Parameters of human NASH patients | ||||||||
|---|---|---|---|---|---|---|---|---|
| Group | Age | MELD score |
INR | Bilirubin (mg/dl) |
Creatinine (mg/dl) |
AST (U/L) |
Alk. Phos. (U/L) |
Albumin (g/dl) |
| fNASH n=7 | 58.71±2.90 | 31.43±1.72 | 2.44±0.24 | 9.21±2.90 | 2.12±0.50 | 56.00±7.95 | 159.57±21.52 | 2.99±0.24 |
| nfNASH n=8 | 58.38±4.87 | 22.75±2.12** | 1.86±0.14* | 2.68±0.42* | 3.93±1.38 | 50.13±6.85 | 203.00±43.10 | 2.94±0.20 |
MELD (Model for End Stage Liver Disease), INR (International normalized ratio), AST (aspartate aminotransferase), Alk. Phos (Alkaline phosphatase). (N=7 fNASH, 8 nfNASH patients respectively, data are means +/− SEM ***p<0.001)
Cirrhosis is characterized by a marked increase in fibrosis. To verify the extent of fibrosis, tissue sections obtained from human NASH patients were stained with Picrosirius red. As shown in Figure 1, tissue from healthy donors exhibited no evidence of steatosis or Picrosirius red staining characteristic of abnormal fibrotic networks within the periportal and centrilobular regions of the liver (1A). In patients with end stage fNASH, mild steatosis was observed in tissue sections with significant picrosirius staining indicative of bridging fibrosis and cirrhosis (1B). Steatosis was not present in patients diagnosed with nfNASH but all patients exhibited cirrhosis (1C). To further characterize this fibrosis, polarized light images of each picrosirius red stained section (1D–F) were quantified28. As shown in Figure S1, samples analyzed from patients with end-stage f/nf NASH displayed significant increases in picrosirius red intensity as compared to healthy controls but no difference were evident between fNASH and nfNASH.
Figure 1. Pathology of human end-stage NASH.

A. Paraffin embedded formalin fixed tissue sections were stained with picrosirius red (PSR). Top panels; PSR staining (original magnification 100×). Bottom panels; Polarized light exposure of PSR staining.
Protein carbonylation has been proposed to play a significant role in the transition to fibrosis in NASH29. In human NASH patients, protein carbonyls are reported to increase by over 400%13. Pathophysiology of carbonylation has not been examined in end-stage patients diagnosed with fNASH or nfNASH. Tissue sections isolated from human end-stage patients were immunohistochemically probed for post-translational modification by the reactive aldehydes MDA (Figure 2A–C), acrolein (2D–F), 4-ONE (2G–I), and 4-HNE (2J–K). As shown in Figure 2, Panels A, D, G and J, significant staining by any of the reactive aldehydes was not present in normal hepatic tissue. Focusing on the fNASH and nfNASH samples, 4-HNE staining was significantly increased within most hepatocytes in fNASH and nfNASH. Staining by 4-ONE revealed panlobular staining in both fNASH and nfNASH. Examining tissue sections isolated from both nfNASH and fNASH revealed dramatically increased aldehyde staining in hepatocytes. Within fibrotic tissue, both acrolein and 4-ONE staining was increased. Furthermore, staining of all aldehydes increased in cholangiocytes adjacent to fibrosis indicating elevated oxidative stress is occurring during the ductal response. In fNASH and nfNASH, 4-HNE, 4-ONE, MDA and acrolein all exhibited significant staining across the lobules with staining further elevated in areas surround fibrotic tissue. There were no apparent differences in aldehyde staining when comparing nfNASH and fNASH.
Figure 2. Immunohistochemical analysis of protein carbonylation in end-stage fNASH and nfNASH.

Paraffin embedded formalin fixed tissue sections were analyzed immunohistochemically using polyclonal antibodies directed against MDA (A–C), acrolein (D–F), 4-ONE (G–I) and 4-HNE (J–L). Figures are representative of hepatic tissue isolated from three normal, four nfNASH and four fNASH patients respectively.
Neutrophil infiltration and the NADPH oxidase complex is a significant source of reactive species that contribute to the formation of reactive aldehydes. To determine if increased neutrophil infiltration was present in end-stage n/nfNASH, tissue sections were probed for the neutrophil marker myeloperoxidase (MPO). As shown in Figure 3, both nfNASH and fNASH displayed increased neutrophil accumulation within fibrotic tissue as well as in tissue adjacent to fibrotic tissue. Importantly, neutrophil infiltration corresponded to areas where increases in lipid peroxidation are evident. Decreased glutathione S-transferase activity is frequently identified with chronic inflammation and oxidative stress19,30. To determine the effects of fNASH/nfNASH on GST activity, enzyme activity assays were performed using 1-chloronitrobenzene (CDNB) as a substrate. As shown in Figure 3B, global GST activity is suppressed by 30% in end-stage f/nfNASH supporting the presence of increased hepatic oxidative stress. No significant differences were evident between fNASH and nfNASH.
Figure 3. Increased inflammation and oxidative stress in end-stage NASH.

A. Paraffin embedded formalin fixed tissue sections were analyzed immunohistochemically using polyclonal antibodies directed against myeloperoxidase. Figures are representative of hepatic tissue isolated from four normal, 4 nfNASH and 4 fNASH patients respectively. B. Glutathione S-transferase activity in U/mg total protein using whole cell extracts isolated from Normal, fNASH and nfNASH hepatic tissue. Data are means± SEM as analyzed by two-way ANOVA with a Bonferroni post hoc analysis (Normal group compared to nfNASH and fNASH groups), (N=7 patients/group (p<0.05)).
The enzymes catalase and mitochondrial superoxide dismutase play an important role in mitigating reactive intermediates during chronic inflammation. Previous reports examining both mRNA expression and activity, have indicated suppression of catalase in human NASH13,31. Furthermore, during conditions of elevated oxidative stress, by its ability to reduce oxidized cysteine residues, the thioredoxin (Trx1)/thioredoxin reductase system contributes to cellular anti-oxidant defense32. In serum isolated from human NASH patients, levels of Trx1 increase as disease severity increases33,34. To determine the impact of fNASH and nfNASH on hepatic expression of catalase, SOD2, GSTπ and the thioredoxin system (Trx1, TrxR1 and TxNIP), Western blots were performed. As shown in Figure 4A and Table 2, catalase expression decreased in fNASH and nfNASH suggesting that there may be defective anti-oxidant responses whereas SOD2 expression is significantly elevated in both groups. GSTπ has previously been demonstrated to contribute to mitigation of lipid aldehydes. Examining expression of GSTπ, expression is significantly elevated in both NASH groups. Focusing on the antioxidant enzyme Trx1, whereas there was no significant change in TrxR1, Trx1 expression is significantly upregulated in hepatic tissue from both fNASH and exhibited a trend towards an increase in nfNASH patients. Using immunohistochemistry, expression of TxNIP has been reported to be upregulated in NASH patients when compared to patients with NAFLD35. From our Western blotting analysis, TxNIP is significantly upregulated in both fNASH and nfNASH supporting previously reported histology.
Figure 4. Altered expression of oxidative stress responses in end-stage NASH.


(A) Western analysis of overall expression of Trx1, TrxR1, TxNIP, SOD2, Catalase and GSTp1 in 10µg of whole cell extracts prepared from normal, fNASH and nfNASH hepatic tissue. (B) Paraffin embedded formalin fixed tissue sections were analyzed immunohistochemically using polyclonal antibodies directed against catalase and SOD2 respectively followed by confocal microcopy of (Green=SOD2, Red=Catalase, Blue= Hoechst 33342 nuclear staining). Panels A, D, G Normal tissue, B, E, H nfNASH and C, F, I fNASH. Top panels-SOD2 alone, middle panels Catalase alone, bottom panels Catalase and SOD2 expression merged (SOD2 green, catalase Red). Arrows indicate catalase positive cells that are not expressing SOD2.
Table 2. Impact of fNASH and nfNASH on expression of thioredoxin and antioxidant responses.
Quantitative analysis of Western blots presented in Figure 4A.
| Normal fNASH | Thioredoxin signaling | Antioxidant response | ||||
| Trx1 | TrxR1 | TxNIP | GSTπ | SOD2 | catalase | |
|
| ||||||
| 100±10.85 | 100±7.20 | 100±28.56 | 100±30.72 | 100±21.21 | 100±4.66 | |
|
| ||||||
| 183.74±16.60a | 88.31±8.97 | 375.33±61.25a | 7260.08±2157.12a | 217.42±15.09a | 82.09±4.67a | |
|
| ||||||
| Normal nfNASH | Trx1 | TrxR1 | TxNIP | GSTπ | SOD2 | catalase |
|
| ||||||
| 100±18.25 | 100±12.74 | 100±24.11 | 100±53.93 | 100±23.63 | 100±4.85 | |
|
| ||||||
| 128.57±24.19 | 84.38±11.28 | 1407.73±490.01a | 1996.23±336.93a | 216.58±26.29a | 77.03±5.16a | |
Each band was quantified using NIH ImageJ and normalized against total GADPH expression. N=6 normal, 6=fNASH, 6=nfNASH, data are means +/− SEM,
= statistically significant change when compared to expression in normal tissue.
To further support our Western expression data, tissue sections from normal, fNASH and nfNASH groups were probed for catalase, SOD2 using fluorescent immunohistochemistry. As shown in Figure 4C, expression of SOD2 (Green, panels A–C) is clearly elevated in f/nfNASH. Catalase (Red, Panels D–F) however, did not exhibit a similar elevation. Concurrently, there is a significant lack of colocalization of cells expressing catalase and SOD2 (Merge, Panels G–I, yellow arrows). Furthermore, increased SOD2 expression is in the same hepatic region as neutrophil infiltration and post-translational modification by reactive aldehydes supporting enhanced levels of oxidative stress in NASH.
From Western data, both Trx1 and GSTπ are significantly upregulated in both NASH groups. To further explore the impact of NASH on Trx1 and GSTπ expression, an immunohistochemical approach was utilized (Figure 5). Examining Trx1 (Panels A–C), in normal tissue, Trx1 staining was very weak which is in agreement with previously published reports36,37. In nfNASH, Trx1 expression was elevated in cholangiocytes (arrows) and hepatocytes immediately adjacent to fibrotic regions as well as in cells within fibrotic tissue. Comparing nfNASH to fNASH tissue, fNASH tissue exhibited elevated Trx1 expression in hepatocytes adjacent to fibrotic regions but also presented elevated Trx1 expression in areas surrounding steatosis. Examining GSTπ (Panels D–F), in normal tissue, GSTπ expression was localized within hepatocytes surrounding the central vein, in monocytes within the sinusoids, as well as in cholangiocytes. In the fNASH diseased tissue, GSTπ staining was prominent within scattered hepatocytes across the lobule as well as within cholangiocytes within and adjacent to fibrotic tissue (arrows). A similar phenotype was evident within nfNASH tissue (arrows). Ductular reactions occur in many different acute as well as chronic liver diseases and are associated with fibrosis38,39. To further delineate the ductular reaction in the end-stage NASH samples obtained in this study, cytokeratin 7 staining was performed. As shown in Panels G–I, in both fNASH and nfNASH, cytokeratin positive cells were present within and adjacent to fibrotic tissue demonstrating that these cells were part of the ductal response during injury and are cholangiocytes.
Figure 5. Immunohistochemical analysis of Trx1 and GSTπ in fNASH and nfNASH.

Top row panels (A–C) Trx1 staining, middle row (D–F) GSTπ, bottom row (G–I) cytokeratin 7. Arrows indicate cholangiocytes that are part of the ductal reaction. Figures are representative of hepatic tissue isolated from three normal, four nfNASH and four fNASH patients respectively.
We recently reported the use of BH derivatization followed by global LC-MS/MS analysis to identify carbonylated proteins in liver lysates isolated from ethanol-fed mice as well as tissue procured from end-stage human alcoholics16,18. In the present study, BH derivatization was performed on whole cell extracts prepared from fNASH and nfNASH tissue. Following avidin purification, carbonylated proteins were digested with trypsin and the resulting peptides were examined by LC-MS/MS. As shown in Table S1, a total of 778 carbonylated proteins, collectively, were identified in any sample.
To visualize proteins that were unique to each condition, a Venn diagram is presented in Figure 6A and Table S2. Of the 778 identified proteins, 194 were common to both normal and NASH tissue, 124 unique to tissue prepared from healthy individuals, 55 proteins were carbonylated in both fNASH and nfNASH and not healthy individuals, 124 proteins were unique to samples from patients with fatty NASH and 178 unique to nfNASH. To identify cellular pathways preferentially impacted by carbonylation in human NASH, proteins identified as carbonylated were subjected to bioinformatic analysis using the Panther Pathways database40,41. From Figure 6B and Tables S3, increased carbonylation of proteins regulating cholesterol and Huntington’s disease related pathways was evident in nfNASH (Table S3 red font). Examining fNASH, increased carbonylation was evident in proteins regulating Rho cytoskeletal pathways, nicotinic acetylcholine receptor signaling, and chemokine/cytokine inflammatory pathways (Table S3 blue font). Surprisingly, Normal tissue exhibited significant enrichment of carbonylated proteins in pathways regulating fructose/galactose metabolism, de novo purine synthesis and pyruvate metabolism (Table S3 green font). A similar enrichment was not detected in any of the NASH groups.
Figure 6. Bioinformatic analysis of carbonylated proteins in end-stage NASH.

A. Proteins identified in Supplemental Table S1 were analyzed using the VENN data analysis software at the Bioinformatics and Evolutionary Genomics (http://bioinformatics.psb.ugent.be/webtools/Venn/). B. For enrichment analysis, four groups of proteins were identified. Proteins were functionally annotated and pathways were examined for enrichment using EnrichR. Pathways that were nominally significant (p<0.01) in at least one of the 4 protein lists are included in the graphic. The colors of the heatmap range from white (unadjusted p-value>0.01) to bright red based on the log base 10 transformation of the unadjusted p-value. A p-value of 1 was used when the pathway was not represented by any proteins in the list. PANTHER pathways (rows) are ordered based on hierarchical clustering using the Euclidean distance and a binary indicator of significance (p<0.01 vs. p>=0.01).
An MS-MS approach was used in combination with the Mascot database to find individual peptides that are post-translationally modified by reactive aldehydes. Importantly, in addition to using the masses for modification by MDA, 4-HNE, 4-ONE, HHE and acrolein, Michael and Schiff base adduction of Lys, Cys and His residues by α/β unsaturated aldehydes crotonaldehyde, pentenaldehyde, hexenaldehyde, heptenaldehyde, octenaldehyde and nonenaldehyde was performed in the peptide searches (Table S4)42,43. From these searches, adducted peptides from 3 proteins were identified. As shown in in the MS-MS spectra (Figure S2), the cytoskeletal protein vimentin (Amino acid sequence: 223KVESLQEEIAFLK*K236 nonenaldehyde adduct, Mascot ion score 53.8), endoplasmin (ENPL/Grp94/Hsp90B1, Amino acid sequence: 703EDEDDK*TVLDLAVVLFETATLR724 heptenaldehyde adduct, Mascot ion score 32.4) and serum albumin (Amino acid sequence: 361C*CAAADPHECYAK373 4-ONE adduct, Mascot ion score 53.7) were identified in hepatic tissue isolated from patients with end stage nfNASH. In fNASH tissue wce, 2 different sites of carbonylation were identified in serum albumin (Amino acid sequence: 361C*CAAADPHECYAK373, 361CCAAADPH*ECYAK373 4-ONE adducts, Mascot ion scores 37.0, 26.2) in 6 different samples.
From bioinformatic data as well as the peptides identified, proteins regulating E.R. Stress are significant targets of carbonylation in end stage human NASH. To determine if overall expression of E.R. Stress responses is impacted in end stage fNASH or nfNASH a Western blot was used followed by quantification of expression using GAPDH normalization and densitometry. As shown in Figure 7 and Table 3, no significant changes in expression were evident when examining Grp78, ENPL/Grp94/Hsp90b, and Chop (GADD153). Expression of Hsp70 was significantly decreased in both fNASH and nfNASH tissue. We have previously shown that in end stage ALD, vimentin expression is significantly increased16. An examination of vimentin expression in the current samples also demonstrated a significant elevation of vimentin expression reflective of severe fibrosis. These data suggest that there may be selective mechanisms regulating E.R. stress and protein folding/heat shock responses in end stage NASH.
Figure 7. Expression of E.R. stress response proteins in end-stage NASH.

Overall expression of Grp94, Grp78, Hsp70, CHOP/GADD153, Vimentin and GAPDH in 10µg of whole cell extracts prepared from normal, fNASH and nfNASH hepatic tissue.
Table 3. Impact of fNASH and nfNASH on expression of enzymes regulating protein folding.
Quantitative analysis of Western blots presented in Figure 7.
| Normal fNASH | Protein folding Heat shock Response | ||||
| Grp94 | Hsp70 | Grp78 | Chop | Vimentin | |
|
| |||||
| 100±24.82 | 100±4.75 | 100±7.01 | 100±49.08 | 100±45.30 | |
|
| |||||
| 70.36±20.65 | 39.39±4.63a | 85.59±6.93 | 212.07±72.07 | 2696.27±588.96a | |
|
| |||||
| Normal nfNASH | Grp94 | Hsp70 | Grp78 | Chop | Vimentin |
|
| |||||
| 100±18.25 | 100±9.12 | 100±6.33 | 100±18.32 | 100±54.64 | |
|
| |||||
| 77.05±28.02 | 54.06±4.48a | 91.11±7.42 | 31.61±6.43a | 5528.46±3834.77a | |
Each band was quantified using NIH ImageJ and normalized against total GADPH expression. N=6 normal, 6 fNASH, 6 nfNASH, data are means +/− SEM,
= statistically significant change when compared to expression in normal tissue.
We have previously reported that Lys235 on vimentin is adducted in end stage alcoholic liver disease and that adduction perturbed protein structure16. To further explore the impact of adduction of heptenealdehyde on ENPL/Grp94 Lys708 (2o1v24) and 4-ONE on albumin Cys361, His367 (4k2c44), a similar approach was utilized. As shown in Figure 8A, the location of Lys708 on ENPL/Grp94 is in the periphery of the protein away from the ATP binding site. A closer examination revealed that Lys708 (purple) possesses electrostatic interactions with Asp712 (orange, Figure 8B). Examining serum albumin, both Cys361, His368 are located on the alpha helices that are connected by a loop on the periphery each protein subunit (Figure 8C, 8D).
Figure 8. Global molecular modeling of site of carbonylation on Grp94/endoplasmin Lys708 and serum albumin Cys361, His368.

A. Ribbon diagram from X-Ray crystallographic structure of endoplasmin (2o1v24) demonstrating location of Lys708 4-ONE adduct found on endoplasmin (Backbone-red, Lys708 CPK, ATP purple). B. Ribbon diagram demonstrating locations of electrostatic interactions between Lys708 (purple) and Asp712 (orange) residues (stick). C. Ribbon diagram from X-Ray crystallographic structure of serum albumin (4k2c44) demonstrating locations of Cys361, His368 4-ONE adducts found on serum albumin (Backbone-red, Cys361, His368 residues (CPK). D. Ribbon diagram demonstrating locations of electrostatic interactions between Cys361, His368 residues (CPK).
Discussion
Nonalcoholic liver disease is a multifactorial chronic inflammatory disease. A key contributor to the pathogenesis of NASH is enhanced hepatocellular oxidative stress resulting in the production of reactive species via inflammation as xanthine and NADPH oxidases which in turn induce lipid peroxidation of unsaturated fatty acids producing reactive aldehydes. In human NASH as well as murine models, antibodies against reactive aldehydes are increased2,3,29. As important, these antibodies further increased as the stage of disease progressed into fibrosis and cirrhosis. Supporting their contribution to pathogenesis, in obese NASH patients undergoing bariatric surgery, levels of lipid peroxidation and expression of Cyp2E1 decrease following surgery correlated with weight loss12. Previous studies from our laboratory have identified numerous proteins that are directly impacted by protein carbonylation in alcoholic liver disease in rodents as well as humans17,28,45–48. Although lipid peroxidation and the production of reactive aldehydes is widely accepted as a contributor to the pathogenesis of NASH, to date, global proteomic analysis of protein carbonylation has not been examined in human NASH. In the present report, we extend data concerning the pathology of NASH by using global proteomic approaches to examine lipid peroxidation in human end-stage fatty and nonfatty NASH livers that were procured during liver transplantation.
Our data clearly demonstrate increased lipid peroxidation is panlobular in end-stage NASH regardless of fatty or nonfatty pathology with staining increasing in areas surrounding fibrotic tissue. Although the overall MELD scores of the fNASH patients were higher when compared to nfNASH, carbonylation staining was not significantly different suggesting that at least at the end stages of disease protein carbonylation may not contribute to overall disease severity as calculated by MELD. Both groups however, possessed as direct contrast with lipid peroxidation in early stages of NASH which is primarily evident in the centri-lobular region49. In that report, the investigators only examined MDA-lysine adduction and did not examine other reactive aldehyde adducts. Comparing histology of the reported early stage study with end stage disease, we conclude that lipid peroxidation plays a continuing role in hepatocellular damage that progresses from primarily zone 3 to panlobular predominance as disease progresses. Increases in lipid peroxidation are evident in human NASH and correlate strongly with a reduction in GSH50. Although we do not demonstrate changes in GSH which would require fresh tissue for accurate measurements, the decrease in GST activity could be attributed to a reduction in GSH.
In normal hepatic tissue, both catalase and SOD2 expression is primarily localized in the centri-lobular region. From our histology, increased SOD2 and the lack of elevated catalase expression is evident in the periportal region of both fNASH and nfNASH. In some areas, expression is increased in hepatocytes surrounding fibrotic tissue but in other hepatocytes, expression is elevated in cells that are several layers removed from fibrotic tissue. From these data it can be predicted that the resulting decrease in catalase expression would contribute to elevated cellular concentrations of hydrogen peroxide which would contribute to oxidative damage. It may be that elevated SOD2 expression is directly due to cells attempting to mitigate increased levels of reactive intermediates. Furthermore from colocalization studies, expression of SOD2 and catalase are not necessarily in the same cell indicating that that oxidative stress is not consistent throughout areas of diseased tissue and that there may be differential responses. This is also supported by differences in 4-HNE and acrolein staining which also demonstrate areas of non-colocalization. Concurrently, elevated acrolein staining correlates with the increased expression of GSTπ. GSTπ has been reported to metabolize acrolein and in a cell culture model, transfection of catalytically inactive GSTπ resulted in enhanced production of lipid peroxidation products as well51,52. In addition to effects on lipid peroxidation, polymorphisms in GSTπ are risk factors for NAFLD and decreased GSTπ expression are associated with carcinogenesis53. The patients examined in this study did not possess hepatocellular carcinoma and but did exhibit elevated GSTπ expression. Furthermore, from the immunohistochemistry, GSTπ upregulation is especially notable in cholangiocytes that are part of the ductal response. In the biliary disorder primary biliary cholangitis (PBC), GSTπ expression is decreased suggesting that GSTπ modulation is disease dependent. GSTπ has been reported to contribute to detoxification of reactive aldehydes in other hepatic diseases which overall suggests that the upregulation of GSTπ in end stage NASH samples is an attempt to mitigate damage that is occurring due to lipid peroxidation/protein carbonylation52.
Enhancing these finding is the finding that the thioredoxin system is impacted in both NASH groups. Thioredoxin has previously been shown to be elevated in the serum of NASH patients with levels increasing as disease progresses33,34. This also correlated with mRNA data from human NAFLD patients54. In this report, Trx1 expression is only significantly elevated in fNASH. This is interesting due to the MELD scores of the fNASH patients was significantly higher than the MELD scores in nfNASH which further links Trx1 and disease severity. Elevated Trx1 could be a marker of disease progression or could be a marker of fNASH but not nfNASH.
Using LC-MS/MS approaches we have identified 778 proteins that are carbonylated in hepatic tissue. This is significantly less than what we have previously identified in human alcoholics16. We have previously demonstrated that the use of individual replicates significantly enhances identification of carbonylated proteins and hypothesize that this may be a contributing factor for the discrepancies18,47,55. Comparing the proteins identified in this study with our previous human end stage ALD study, 260 proteins were not identified in the ALD study (Figure S3A). Of these 103 were exclusive to fNASH, 124 exclusive to nfNASH and 33 proteins were identified in f/nfNASH16. Bioinformatic analysis of this comparison revealed a commonality of proteins implicated in Huntington’s disease and in Rho cytoskeletal regulation (Figure S3B, Table S4 Yellow highlight). This suggests that despite distinct origins in disease pathology that these pathways may both contribute to dysfunction in end-stage liver disease. Further comparison of NASH and end stage ALD samples, GSTπ, Trx1 and SOD2 are elevated during the ductular response to injury as is shown in Figure 5 and colocalize with carbonylated proteins (Figure S4). This suggests that upregulation of GSTπ and Trx1 in cholangiocytes and oxidative stress may be a general phenomenon of the ductular reaction during end stage liver disease. It should be noted however, that given the sample sizes, significantly larger studies will be necessary to further identify differences in carbonylation in patients that have been diagnosed with fatty NASH and non-fatty NASH. Concurrently, additional mass spectrometric and histological studies will be required to determine differences in carbonylation that occur in other hepatic diseases such as hepatitis C, primary sclerosing cholangitis and primary biliary cholangitis.
Increased carbonylation of vimentin was prominent in our previous report using samples isolated from ALD patients16. In this report, carbonylated vimentin was identified in all NASH samples examined but none of the normal samples. The fact that we have also identified the identical modification of vimentin in our NASH samples is not surprising. Carbonylation of vimentin may represent a common pathway that contributes to fibrosis. Vimentin is also known to contribute to pathogenesis of NASH56. In murine models of NASH vimentin expression is increased and also is strongly correlated with serum albumin expression. During fibrotic conditions, vimentin expression is elevated and is indicative of liver undergoing repair and fibrosis. In cell culture models, vimentin significantly increases in cirrhotic hepatocytes as they undergo hepatocyte epithelial mesenchymal transition57. In hepatic stellate cells, vimentin expression increases as fibrosis forms58. Increased vimentin expression is associated with increased tissue remodeling and fibrosis in methionine-choline-deficient models of NASH and correlates with increased hepatocellular death59,60. The finding of adducted serum albumin is intriguing. In a recent report using the methionine choline deficient model of NASH in mice, prior immunization using serum albumin adducted by malondialdehyde potentiated hepatocellular inflammation and cytokine levels2. As important, increased fibrosis was evident in immunized animals when compared to controls. Further stressing the importance of oxidation of albumin in end stage liver disease. In human alcoholics with severe hepatitis, hyperoxidized serum albumin contributes to neutrophil activation and enhanced oxidative stress61. Our data combined with these previous reports support the proposition that adduction of serum proteins by lipid aldehydes and the initiation of both innate and adaptive immune response contribute to human NASH progression mimicking evidence in animal models.
The endoplasmic reticulum (E.R.) is an important hepatocellular mediator of protein synthesis, folding, post-translational modification and trafficking. In diseases such as ALD, exposure of hepatocytes to elevated oxidative stress will often induce E.R. stress characterized by disruption of normal protein folding and induction of E.R. Stress response proteins. We have previously shown that in models of alcoholic liver disease, carbonylation of E.R. stress chaperone proteins that assist in protein folding such as Glucose Response Protein 78 (Grp78) and Hsp70 disrupts their function contributing to the unfolded protein response and impacting hepatocellular metabolism62–64. Disruption of E.R. responses is closely linked to oxidative stress and is a known contributor of NASH pathogenesis65. In NASH, E.R. stress decreases following bariatric surgery and as does lipid peroxidation as shown in a separate study12,66. From the crystal structure along with our data demonstrating no significant changes in Grp94 expression, the impact of carbonylation of Grp94 Lys708 is unclear. It should be noted that there was considerable variability in Grp94 expression on both NASH groups. Furthermore, although the adducted peptide for Grp94 was only identified in a single patient, carbonylation of Grp94 was present in all fNASH and nfNASH samples (Data not shown). If Grp94 function is impaired in NASH patients, we anticipate that it would contribute to elevated E.R. stress enhancing pathogenesis. Adduction would disrupt electrostatic interactions with the adjacent Asp712 but both of these residues are far removed from the ATP binding site. Although we did not find carbonylated peptides for other E.R. stress chaperone proteins, many including Grp78, Hsp72, protein disulfide isomerases and peptidyl prolyl isomerases were identified in our global screen. Hsp70 expression however is significantly decreased in both NASH patient groups. Hsp70 has been shown to decrease as NAFLD progresses to NASH in human patients and has been demonstrated to inhibit inflammatory signals by its ability to associate with IκB kinase gamma (IKKγ)67,68. We demonstrate increased inflammation in the form of MPO positive neutrophils in our human NASH samples which colocalized with increased protein carbonylation. We hypothesize that increased carbonylation combined with decreased expression of Hsp70 may be contributing to elevated inflammation enhancing carbonylation. This further supports the contribution of carbonylation on dysregulation of protein folding and E.R. stress in human endstage NASH.
In summary, this study is the first to examine and identify hepatic proteins that are post-translationally modified by reactive aldehydes in liver tissue isolated from patients diagnosed with end-stage NASH. Furthermore, although they are phenotypically similar with respect to carbonylation as determined by histology, fatty NASH and non-fatty NASH exhibit significant differences in the pathways targeted by reactive aldehydes. As important, inflammation, E.R. Stress and oxidative stress all are contributing to hepatocellular dysfunction. This report combined with previous reports also provides additional evidence that oxidative stress is an ongoing active process in NASH continuing from simple steatosis during early stages30 throughout the course of the disease and suggests that the use of adjuvant therapeutics that target oxidative stress may still have some benefit in end stage liver disease.
Supplementary Material
Supplemental Figure S1. Quantification of picrosirius red stained sections exposed to polarized light. Quantitative analysis of PSR staining (N=4 normal, 7 fNASH, 7 nfNASH, data are means +/− SEM *p<0.05, **p<0.01).
Supplemental Figure S2. MS/MS analysis of carbonylated peptides identified in NASH. A. Nonenaldehyde (2-nonenal) modified vimentin peptide containing Lys235 adduct, Mascot score 53.8. MS/MS analysis was performed and the resulting b/y ion fragmentation confirmed peptide identity (KVESLQEEIAFLK*K). B. Heptenaldehyde modified endoplasmin (ENPL/Grp94/Hsp90B1), peptide containing Lys708 adduct, Mascot score 32.4. MS/MS analysis was performed and the resulting b/y ion fragmentation confirmed peptide identity (703EDEDDK*TVLDLAVVLFETATLR724). C–E. 4-ONE modified serum albumin peptides containing Cys361 adduct isolated from human nfNASH, Mascot ion scores 32.8, 53.7, 22.6. MS/MS analysis was performed and the resulting b/y ion fragmentation confirmed peptide identity (361C*CAAADPHECYAK373). F,G. 4-ONE modified serum albumin peptides containing Cys361 adduct isolated from human fNASH, Mascot ion scores 31.8, 37.0. MS/MS analysis was performed and the resulting b/y ion fragmentation confirmed peptide identity (361C*CAAADPHECYAK373). H. 4-ONE modified serum albumin peptides containing His368 adduct isolated from human fNASH, Mascot ion score 24.8. MS/MS analysis was performed and the resulting b/y ion fragmentation confirmed peptide identity (31CCAAADPH*ECYAK373).
Supplemental Figure S3. Bioinformatic analysis of carbonylated proteins in end-stage NASH compared to end stage ALD. A. Proteins identified in Supplemental Table S1 and in REF 16 were analyzed using the VENN data analysis software at the Bioinformatics and Evolutionary Genomics (http://bioinformatics.psb.ugent.be/webtools/Venn/). B. For enrichment analysis, five groups of proteins were identified. Proteins were functionally annotated and pathways were examined for enrichment using EnrichR. Pathways that were nominally significant (p<0.01) in at least one of the 5 protein lists are included in the graphic. The colors of the heatmap range from white (unadjusted p-value>0.01) to bright red based on the log base 10 transformation of the unadjusted p-value. A p-value of 1 was used when the pathway was not represented by any proteins in the list. PANTHER pathways (rows) are ordered based on hierarchical clustering using the Euclidean distance and a binary indicator of significance (p<0.01 vs. p>=0.01).
{kind=link}
Supplemental Figure S4. Immunohistochemical analysis of Cytokeratin 7, 4-HNE, Trx1 and GSTπ in fNASH and nfNASH. Tissue sections isolated from human patients diagnosed with end-stage ALD REF 16 were probed for cytokeratin 7, 4-HNE, Trx1, GSTπ and SOD2 (Green=SOD2, Red=Catalase, Blue= Hoechst 33342 nuclear staining). Arrows represent cells that are part of the ductal reaction that stain positive for elevated markers of oxidative stress. Figures are representative of hepatic tissue isolated from four end stage ALD patients.
{kind=link}
Supplemental Table S1. Table Summary of carbonylated proteins identified in whole cell extract prepared from hepatic tissue isolated from normal and end stage fNASH and nfNASH human patients.
Supplemental Table S2. Proteins identified by VENN analysis in Normal tissue, fNASH and nfNASH.
Supplemental Table S3. Protein lists obtained from PANTHER Bioinformatics obtained from carbonylated proteins. Red text indicates increased in nfNASH, blue text indicates increased in fNASH, green text indicates increased in Normal tissue and yellow highlights indicates increased in ALD.
Supplemental Table S4. Carbonylated peptides identified in human fNASH and nfNASH.
Supplemental Table S5. Proteins identified by VENN analysis in ALD, fNASH and nfNASH.
Highlights.
We demonstrate increased production of reactive aldehydes in human fatty and nonfatty NASH
Increased reactive aldehydes corresponds to elevated expression of antioxidant proteins within the ductular reaction.
Using LC-MS/MS we identify 357 carbonylated proteins unique to human NASH
Pathway analysis revealed different pathways are preferentially targeted when comparing fatty NASH and nonfatty NASH.
Using MS/MS, sites of adduction were identified on peptides isolated from vimentin, endoplasmin and serum albumin
Acknowledgments
Financial Support and Acknowledgements: This research was supported by the following grants from the National Institutes of Health; F32 AA018613-03 CTS, R37AA009300-22 DRP. The authors wish to acknowledge Joe Gomez and the Skaggs School of Pharmacy and Pharmaceutical Sciences Mass Spectrometry Core Facility for assistance in analysis of normal and end-stage NASH carbonylation. The authors also wish to thank E. Erin Smith, HTL(ASCP)CMQIHC of the University of Colorado Denver Cancer Center Research Histology Core for assistance in preparing histology slides. The UCDCCRHC is supported in part by NIH/NCRR Colorado CTSI Grant Number UL1 RR025780 and the University of Colorado Cancer Center Grant (P30 CA046934).
Abbreviations
- ALD
alcoholic liver disease
- ALT
alanine aminotransferase
- CID
collision-induced dissociation
- Cyp2E1
Cytochrome P4502E1
- 4-HNE
4-hydroxy-2-nonenal
- 4-ONE
4-oxononenal
- MDA
malondialdehyde
- NAFLD
Nonalcoholic fatty liver disease
- NASH
Nonalcoholic Steatohepatitis
- ROS
Reactive oxidative species
- SOD2
mitochondrial superoxide dismutase
- Trx1
thioredoxin
- TrxR1
Thioredoxin reductase
- TxNIP
Thioredoxin-interacting protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors have no conflict of interest to report.
References
- 1.Valle A, Catalan V, Rodriguez A, Rotellar F, Valenti V, Silva C, Salvador J, Fruhbeck G, Gomez-Ambrosi J, Roca P, Oliver J. Identification of liver proteins altered by type 2 diabetes mellitus in obese subjects. Liver Int. 2012;32:951–61. doi: 10.1111/j.1478-3231.2012.02765.x. [DOI] [PubMed] [Google Scholar]
- 2.Sutti S, Jindal A, Locatelli I, Vacchiano M, Gigliotti L, Bozzola C, Albano E. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology. 2014;59:886–97. doi: 10.1002/hep.26749. [DOI] [PubMed] [Google Scholar]
- 3.Nobili V, Parola M, Alisi A, Marra F, Piemonte F, Mombello C, Sutti S, Povero D, Maina V, Novo E, Albano E. Oxidative stress parameters in paediatric non-alcoholic fatty liver disease. Int J Mol Med. 2010;26:471–6. doi: 10.3892/ijmm_00000487. [DOI] [PubMed] [Google Scholar]
- 4.Basaranoglu M, Basaranoglu G, Senturk H. From fatty liver to fibrosis: a tale of "second hit". World J Gastroenterol. 2013;19:1158–65. doi: 10.3748/wjg.v19.i8.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Enomoto N, Takei Y, Yamashina S, Ikejima K, Kitamura T, Sato N. Anti-inflammatory strategies in alcoholic steatohepatitis. Journal of gastroenterology and hepatology. 2007;22(Suppl 1):S59–61. doi: 10.1111/j.1440-1746.2006.04652.x. [DOI] [PubMed] [Google Scholar]
- 6.Albano E. Alcohol, oxidative stress and free radical damage. The Proceedings of the Nutrition Society. 2006;65:278–90. doi: 10.1079/pns2006496. [DOI] [PubMed] [Google Scholar]
- 7.Osna NA, Carter WG, Ganesan M, Kirpich IA, McClain CJ, Petersen DR, Shearn CT, Tomasi ML, Kharbanda KK. Aberrant post-translational protein modifications in the pathogenesis of alcohol-induced liver injury. World J Gastroenterol. 2016;22:6192–6200. doi: 10.3748/wjg.v22.i27.6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aubert J, Begriche K, Knockaert L, Robin MA, Fromenty B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: mechanisms and pathophysiological role. Clinics and research in hepatology and gastroenterology. 2011;35:630–7. doi: 10.1016/j.clinre.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 9.Leung TM, Nieto N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. Journal of hepatology. 2013;58:395–8. doi: 10.1016/j.jhep.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 10.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 11.Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med. 2012;52:59–69. doi: 10.1016/j.freeradbiomed.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 12.Bell LN, Temm CJ, Saxena R, Vuppalanchi R, Schauer P, Rabinovitz M, Krasinskas A, Chalasani N, Mattar SG. Bariatric surgery-induced weight loss reduces hepatic lipid peroxidation levels and affects hepatic cytochrome P-450 protein content. Ann Surg. 2010;251:1041–8. doi: 10.1097/SLA.0b013e3181dbb572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Videla LA, Rodrigo R, Orellana M, Fernandez V, Tapia G, Quinones L, Varela N, Contreras J, Lazarte R, Csendes A, Rojas J, Maluenda F, Burdiles P, Diaz JC, Smok G, Thielemann L, Poniachik J. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin Sci (Lond) 2004;106:261–8. doi: 10.1042/CS20030285. [DOI] [PubMed] [Google Scholar]
- 14.Shimomura Y, Takaki A, Wada N, Yasunaka T, Ikeda F, Maruyama T, Tamaki N, Uchida D, Onishi H, Kuwaki K, Nakamura S, Nouso K, Miyake Y, Koike K, Tomofuji T, Morita M, Yamamoto K, Okada H. The Serum Oxidative/Anti-oxidative Stress Balance Becomes Dysregulated in Patients with Non-alcoholic Steatohepatitis Associated with Hepatocellular Carcinoma. Intern Med. 2017;56:243–251. doi: 10.2169/internalmedicine.56.7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Casoinic F, Sampelean D, Buzoianu AD, Hancu N, Baston D. Serum Levels of Oxidative Stress Markers in Patients with Type 2 Diabetes Mellitus and Non-alcoholic Steatohepatitis. Rom J Intern Med. 2016;54:228–236. doi: 10.1515/rjim-2016-0035. [DOI] [PubMed] [Google Scholar]
- 16.Shearn CT, Orlicky DJ, Saba LM, Shearn AH, Petersen DR. Increased hepatocellular protein carbonylation in human end-stage alcoholic cirrhosis. Free Radic Biol Med. 2015 doi: 10.1016/j.freeradbiomed.2015.10.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shearn CT, Smathers RL, Backos DS, Reigan P, Orlicky DJ, Petersen DR. Increased carbonylation of the lipid phosphatase PTEN contributes to Akt2 activation in a murine model of early alcohol-induced steatosis. Free Radic Biol Med. 2013;65:680–92. doi: 10.1016/j.freeradbiomed.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shearn CT, Fritz KS, Shearn AH, Saba LM, Mercer KE, Engi B, Galligan JJ, Zimniak P, Orlicky DJ, Ronis MJ, Petersen DR. Deletion of GSTA4-4 results in increased mitochondrial post-translational modification of proteins by reactive aldehydes following chronic ethanol consumption in mice. Redox Biol. 2015;7:68–77. doi: 10.1016/j.redox.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shearn CT, Backos DS, Orlicky DJ, Smathers-McCullough RL, Petersen DR. Identification of 5' AMP-activated kinase as a target of reactive aldehydes during chronic ingestion of high concentrations of ethanol. J Biol Chem. 2014;289:15449–62. doi: 10.1074/jbc.M113.543942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shearn CT, Fritz KS, Reigan P, Petersen DR. Modification of Akt2 by 4-hydroxynonenal inhibits insulin-dependent Akt signaling in HepG2 cells. Biochemistry. 2011;50:3984–96. doi: 10.1021/bi200029w. [DOI] [PubMed] [Google Scholar]
- 21.Shearn CT, Smathers RL, Jiang H, Orlicky DJ, Maclean KN, Petersen DR. Increased dietary fat contributes to dysregulation of the LKB1/AMPK pathway and increased damage in a mouse model of early-stage ethanol-mediated steatosis. J Nutr Biochem. 2013;24:1436–45. doi: 10.1016/j.jnutbio.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45:D183–D189. doi: 10.1093/nar/gkw1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma'ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–7. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol Cell. 2007;28:41–56. doi: 10.1016/j.molcel.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D, Gamblin SJ. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472:230–3. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chernyatina AA, Nicolet S, Aebi U, Herrmann H, Strelkov SV. Atomic structure of the vimentin central alpha-helical domain and its implications for intermediate filament assembly. Proc Natl Acad Sci U S A. 2012;109:13620–5. doi: 10.1073/pnas.1206836109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamath PS, Kim WR Advanced Liver Disease Study G. The model for end-stage liver disease (MELD) Hepatology. 2007;45:797–805. doi: 10.1002/hep.21563. [DOI] [PubMed] [Google Scholar]
- 28.Shearn CT, Backos DS, Orlicky DJ, Smathers-McCullough RL, Petersen DR. Identification of 5' AMP activated kinase as a target of reactive aldehydes during chronic ingestion of high concentrations of ethanol. J Biol Chem. 2014 doi: 10.1074/jbc.M113.543942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Albano E, Mottaran E, Vidali M, Reale E, Saksena S, Occhino G, Burt AD, Day CP. Immune response towards lipid peroxidation products as a predictor of progression of non-alcoholic fatty liver disease to advanced fibrosis. Gut. 2005;54:987–93. doi: 10.1136/gut.2004.057968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shearn CT, Mercer KE, Orlicky DJ, Hennings L, Smathers-McCullough RL, Stiles BL, Ronis MJ, Petersen DR. Short Term Feeding of a High Fat Diet Exerts an Additive Effect on Hepatocellular Damage and Steatosis in Liver-Specific PTEN Knockout Mice. PLoS One. 2014;9:e96553. doi: 10.1371/journal.pone.0096553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sreekumar R, Rosado B, Rasmussen D, Charlton M. Hepatic gene expression in histologically progressive nonalcoholic steatohepatitis. Hepatology. 2003;38:244–51. doi: 10.1053/jhep.2003.50290. [DOI] [PubMed] [Google Scholar]
- 32.Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295:C849–68. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sumida Y, Nakashima T, Yoh T, Furutani M, Hirohama A, Kakisaka Y, Nakajima Y, Ishikawa H, Mitsuyoshi H, Okanoue T, Kashima K, Nakamura H, Yodoi J. Serum thioredoxin levels as a predictor of steatohepatitis in patients with nonalcoholic fatty liver disease. J Hepatol. 2003;38:32–8. doi: 10.1016/s0168-8278(02)00331-8. [DOI] [PubMed] [Google Scholar]
- 34.Nakashima T, Sumida Y, Furutani M, Hirohama A, Okita M, Mitsuyoshi H, Itoh Y, Okanoue T. Elevation of serum thioredoxin levels in patients with nonalcoholic steatohepatitis. Hepatol Res. 2005;33:135–7. doi: 10.1016/j.hepres.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 35.He K, Zhu X, Liu Y, Miao C, Wang T, Li P, Zhao L, Chen Y, Gong J, Cai C, Li J, Li S, Ruan XZ, Gong J. Inhibition of NLRP3 inflammasome by thioredoxin-interacting protein in mouse Kupffer cells as a regulatory mechanism for non-alcoholic fatty liver disease development. Oncotarget. 2017;8:37657–37672. doi: 10.18632/oncotarget.17489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Noike T, Miwa S, Soeda J, Kobayashi A, Miyagawa S. Increased expression of thioredoxin-1, vascular endothelial growth factor, and redox factor-1 is associated with poor prognosis in patients with liver metastasis from colorectal cancer. Hum Pathol. 2008;39:201–8. doi: 10.1016/j.humpath.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 37.Nakamura H, Masutani H, Tagaya Y, Yamauchi A, Inamoto T, Nanbu Y, Fujii S, Ozawa K, Yodoi J. Expression and growth-promoting effect of adult T-cell leukemia-derived factor. A human thioredoxin homologue in hepatocellular carcinoma. Cancer. 1992;69:2091–7. doi: 10.1002/1097-0142(19920415)69:8<2091::aid-cncr2820690814>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 38.Gouw AS, Clouston AD, Theise ND. Ductular reactions in human liver: diversity at the interface. Hepatology. 2011;54:1853–63. doi: 10.1002/hep.24613. [DOI] [PubMed] [Google Scholar]
- 39.Gadd VL, Skoien R, Powell EE, Fagan KJ, Winterford C, Horsfall L, Irvine K, Clouston AD. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology. 2014;59:1393–405. doi: 10.1002/hep.26937. [DOI] [PubMed] [Google Scholar]
- 40.Saba LM, Flink SC, Vanderlinden LA, Israel Y, Tampier L, Colombo G, Kiianmaa K, Bell RL, Printz MP, Flodman P, Koob G, Richardson HN, Lombardo J, Hoffman PL, Tabakoff B. The sequenced rat brain transcriptome - its use in identifying networks predisposing alcohol consumption. FEBS J. 2015 doi: 10.1111/febs.13358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ichihashi K, Osawa T, Toyokuni S, Uchida K. Endogenous formation of protein adducts with carcinogenic aldehydes: implications for oxidative stress. J Biol Chem. 2001;276:23903–13. doi: 10.1074/jbc.M101947200. [DOI] [PubMed] [Google Scholar]
- 43.Ferre N, Girona J, Cabre M, Joven J, LaVille A, Masana L, Paternain JL, Camps J. Hepatic production of apolar aldehydes in rats with carbon tetrachloride-induced cirrhosis. Mol Cell Biochem. 1999;198:57–60. doi: 10.1023/a:1006998028528. [DOI] [PubMed] [Google Scholar]
- 44.Huang X, Begley M, Morgenstern KA, Gu Y, Rose P, Zhao H, Zhu X. Crystal structure of an inactive Akt2 kinase domain. Structure. 2003;11:21–30. doi: 10.1016/s0969-2126(02)00937-1. [DOI] [PubMed] [Google Scholar]
- 45.Sampey BP, Stewart BJ, Petersen DR. Ethanol-induced modulation of hepatocellular extracellular signal-regulated kinase-1/2 activity via 4-hydroxynonenal. J Biol Chem. 2007;282:1925–37. doi: 10.1074/jbc.M610602200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roede JR, Carbone DL, Doorn JA, Kirichenko OV, Reigan P, Petersen DR. In vitro and in silico characterization of peroxiredoxin 6 modified by 4-hydroxynonenal and 4-oxononenal. Chem Res Toxicol. 2008;21:2289–99. doi: 10.1021/tx800244u. [DOI] [PubMed] [Google Scholar]
- 47.Smathers RL, Galligan JJ, Stewart BJ, Petersen DR. Overview of lipid peroxidation products and hepatic protein modification in alcoholic liver disease. Chem Biol Interact. 2011;192:107–12. doi: 10.1016/j.cbi.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fritz KS, Galligan JJ, Smathers RL, Roede JR, Shearn CT, Reigan P, Petersen DR. 4-Hydroxynonenal inhibits SIRT3 via thiol-specific modification. Chem Res Toxicol. 2011;24:651–62. doi: 10.1021/tx100355a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.MacDonald GA, Bridle KR, Ward PJ, Walker NI, Houglum K, George DK, Smith JL, Powell LW, Crawford DH, Ramm GA. Lipid peroxidation in hepatic steatosis in humans is associated with hepatic fibrosis and occurs predominately in acinar zone 3. J Gastroenterol Hepatol. 2001;16:599–606. doi: 10.1046/j.1440-1746.2001.02445.x. [DOI] [PubMed] [Google Scholar]
- 50.Malaguarnera L, Madeddu R, Palio E, Arena N, Malaguarnera M. Heme oxygenase-1 levels and oxidative stress-related parameters in non-alcoholic fatty liver disease patients. J Hepatol. 2005;42:585–91. doi: 10.1016/j.jhep.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 51.Manevich Y, Hutchens S, Tew KD, Townsend DM. Allelic variants of glutathione S-transferase P1-1 differentially mediate the peroxidase function of peroxiredoxin VI and alter membrane lipid peroxidation. Free Radic Biol Med. 2013;54:62–70. doi: 10.1016/j.freeradbiomed.2012.10.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen WY, Zhang J, Ghare S, Barve S, McClain C, Joshi-Barve S. Acrolein Is a Pathogenic Mediator of Alcoholic Liver Disease and the Scavenger Hydralazine Is Protective in Mice. Cell Mol Gastroenterol Hepatol. 2016;2:685–700. doi: 10.1016/j.jcmgh.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hashemi M, Eskandari-Nasab E, Fazaeli A, Bahari A, Hashemzehi NA, Shafieipour S, Taheri M, Moazeni-Roodi A, Zakeri Z, Bakhshipour A, Ghavami S. Association of genetic polymorphisms of glutathione-S-transferase genes (GSTT1, GSTM1, and GSTP1) and susceptibility to nonalcoholic fatty liver disease in Zahedan, Southeast Iran. DNA Cell Biol. 2012;31:672–7. doi: 10.1089/dna.2011.1343. [DOI] [PubMed] [Google Scholar]
- 54.Mitsuyoshi H, Yasui K, Harano Y, Endo M, Tsuji K, Minami M, Itoh Y, Okanoue T, Yoshikawa T. Analysis of hepatic genes involved in the metabolism of fatty acids and iron in nonalcoholic fatty liver disease. Hepatol Res. 2009;39:366–73. doi: 10.1111/j.1872-034X.2008.00464.x. [DOI] [PubMed] [Google Scholar]
- 55.Carbone DL, Doorn JA, Kiebler Z, Ickes BR, Petersen DR. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J Pharmacol Exp Ther. 2005;315:8–15. doi: 10.1124/jpet.105.088088. [DOI] [PubMed] [Google Scholar]
- 56.Wang S, Song K, Srivastava R, Dong C, Go GW, Li N, Iwakiri Y, Mani A. Nonalcoholic fatty liver disease induced by noncanonical Wnt and its rescue by Wnt3a. FASEB J. 2015;29:3436–45. doi: 10.1096/fj.15-271171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nitta T, Kim JS, Mohuczy D, Behrns KE. Murine cirrhosis induces hepatocyte epithelial mesenchymal transition and alterations in survival signaling pathways. Hepatology. 2008;48:909–19. doi: 10.1002/hep.22397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, Pradere JP, Schwabe RF. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. doi: 10.1038/ncomms3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee SJ, Yoo JD, Choi SY, Kwon OS. The expression and secretion of vimentin in the progression of non-alcoholic steatohepatitis. BMB Rep. 2014;47:457–62. doi: 10.5483/BMBRep.2014.47.8.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Csak T, Bala S, Lippai D, Satishchandran A, Catalano D, Kodys K, Szabo G. microRNA-122 regulates hypoxia-inducible factor-1 and vimentin in hepatocytes and correlates with fibrosis in diet-induced steatohepatitis. Liver Int. 2015;35:532–41. doi: 10.1111/liv.12633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Das S, Maras JS, Hussain MS, Sharma S, David P, Sukriti S, Shasthry SM, Maiwall R, Trehanpati N, Singh TP, Sarin SK. Hyperoxidized albumin modulates neutrophils to induce oxidative stress and inflammation in severe alcoholic hepatitis. Hepatology. 2017;65:631–646. doi: 10.1002/hep.28897. [DOI] [PubMed] [Google Scholar]
- 62.Galligan JJ, Fritz KS, Backos DS, Shearn CT, Smathers RL, Jiang H, MacLean KN, Reigan PR, Petersen DR. Oxidative stress-mediated aldehyde adduction of GRP78 in a mouse model of alcoholic liver disease: functional independence of ATPase activity and chaperone function. Free Radic Biol Med. 2014;73:411–20. doi: 10.1016/j.freeradbiomed.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Galligan JJ, Smathers RL, Shearn CT, Fritz KS, Backos DS, Jiang H, Franklin CC, Orlicky DJ, Maclean KN, Petersen DR. Oxidative Stress and the ER Stress Response in a Murine Model for Early-Stage Alcoholic Liver Disease. J Toxicol. 2012;2012:207594. doi: 10.1155/2012/207594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carbone DL, Doorn JA, Kiebler Z, Sampey BP, Petersen DR. Inhibition of Hsp72-mediated protein refolding by 4-hydroxy-2-nonenal. Chem Res Toxicol. 2004;17:1459–67. doi: 10.1021/tx049838g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ashraf NU, Sheikh TA. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease. Free Radic Res. 2015;49:1405–18. doi: 10.3109/10715762.2015.1078461. [DOI] [PubMed] [Google Scholar]
- 66.Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS, Klein S. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009;58:693–700. doi: 10.2337/db08-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Di Naso FC, Porto RR, Fillmann HS, Maggioni L, Padoin AV, Ramos RJ, Mottin CC, Bittencourt A, Marroni NA, de Bittencourt PI., Jr Obesity depresses the anti-inflammatory HSP70 pathway, contributing to NAFLD progression. Obesity (Silver Spring) 2015;23:120–9. doi: 10.1002/oby.20919. [DOI] [PubMed] [Google Scholar]
- 68.Chen HW, Kuo HT, Wang SJ, Lu TS, Yang RC. In vivo heat shock protein assembles with septic liver NF-kappaB/I-kappaB complex regulating NF-kappaB activity. Shock. 2005;24:232–8. doi: 10.1097/01.shk.0000174020.87439.f2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. Quantification of picrosirius red stained sections exposed to polarized light. Quantitative analysis of PSR staining (N=4 normal, 7 fNASH, 7 nfNASH, data are means +/− SEM *p<0.05, **p<0.01).
Supplemental Figure S2. MS/MS analysis of carbonylated peptides identified in NASH. A. Nonenaldehyde (2-nonenal) modified vimentin peptide containing Lys235 adduct, Mascot score 53.8. MS/MS analysis was performed and the resulting b/y ion fragmentation confirmed peptide identity (KVESLQEEIAFLK*K). B. Heptenaldehyde modified endoplasmin (ENPL/Grp94/Hsp90B1), peptide containing Lys708 adduct, Mascot score 32.4. MS/MS analysis was performed and the resulting b/y ion fragmentation confirmed peptide identity (703EDEDDK*TVLDLAVVLFETATLR724). C–E. 4-ONE modified serum albumin peptides containing Cys361 adduct isolated from human nfNASH, Mascot ion scores 32.8, 53.7, 22.6. MS/MS analysis was performed and the resulting b/y ion fragmentation confirmed peptide identity (361C*CAAADPHECYAK373). F,G. 4-ONE modified serum albumin peptides containing Cys361 adduct isolated from human fNASH, Mascot ion scores 31.8, 37.0. MS/MS analysis was performed and the resulting b/y ion fragmentation confirmed peptide identity (361C*CAAADPHECYAK373). H. 4-ONE modified serum albumin peptides containing His368 adduct isolated from human fNASH, Mascot ion score 24.8. MS/MS analysis was performed and the resulting b/y ion fragmentation confirmed peptide identity (31CCAAADPH*ECYAK373).
Supplemental Figure S3. Bioinformatic analysis of carbonylated proteins in end-stage NASH compared to end stage ALD. A. Proteins identified in Supplemental Table S1 and in REF 16 were analyzed using the VENN data analysis software at the Bioinformatics and Evolutionary Genomics (http://bioinformatics.psb.ugent.be/webtools/Venn/). B. For enrichment analysis, five groups of proteins were identified. Proteins were functionally annotated and pathways were examined for enrichment using EnrichR. Pathways that were nominally significant (p<0.01) in at least one of the 5 protein lists are included in the graphic. The colors of the heatmap range from white (unadjusted p-value>0.01) to bright red based on the log base 10 transformation of the unadjusted p-value. A p-value of 1 was used when the pathway was not represented by any proteins in the list. PANTHER pathways (rows) are ordered based on hierarchical clustering using the Euclidean distance and a binary indicator of significance (p<0.01 vs. p>=0.01).
Supplemental Figure S4. Immunohistochemical analysis of Cytokeratin 7, 4-HNE, Trx1 and GSTπ in fNASH and nfNASH. Tissue sections isolated from human patients diagnosed with end-stage ALD REF 16 were probed for cytokeratin 7, 4-HNE, Trx1, GSTπ and SOD2 (Green=SOD2, Red=Catalase, Blue= Hoechst 33342 nuclear staining). Arrows represent cells that are part of the ductal reaction that stain positive for elevated markers of oxidative stress. Figures are representative of hepatic tissue isolated from four end stage ALD patients.
Supplemental Table S1. Table Summary of carbonylated proteins identified in whole cell extract prepared from hepatic tissue isolated from normal and end stage fNASH and nfNASH human patients.
Supplemental Table S2. Proteins identified by VENN analysis in Normal tissue, fNASH and nfNASH.
Supplemental Table S3. Protein lists obtained from PANTHER Bioinformatics obtained from carbonylated proteins. Red text indicates increased in nfNASH, blue text indicates increased in fNASH, green text indicates increased in Normal tissue and yellow highlights indicates increased in ALD.
Supplemental Table S4. Carbonylated peptides identified in human fNASH and nfNASH.
Supplemental Table S5. Proteins identified by VENN analysis in ALD, fNASH and nfNASH.