

Summary
Mutations in presenilin-1 and -2 (PSENs), which encode components of the γ-secretase (GS) complex, cause familial Alzheimer’s disease (FAD). It is hypothesized that altered GS-mediated processing of the amyloid precursor protein (APP) to Aβ42 fragment, which is accumulated in disease brain, may be pathogenic. Here we describe an in vitro model system that enables the facile analysis of neuronal disease mechanisms in non-neuronal patient cells using CRISPR gene activation of endogenous disease-relevant genes. In FAD patient-derived fibroblast cultures, CRISPR activation of APP or BACE unmasked an occult processivity defect in downstream GS-mediated carboxypeptidase cleavage of APP, ultimately leading to higher Aβ42 levels. These data suggest that, selectively in neurons, relatively high levels of BACE1 activity lead to substrate pressure on FAD mutant GS complexes, thus promoting CNS Aβ42 accumulation. Our results introduce an additional platform for analysis of neurological disease.
Keywords: CRISPR/Cas9, Synergistic Activation Mediator, Alzheimer’s disease, amyloid precursor protein, β-amyloid, BACE1, Presenilin, fibroblast, induced neuronal cells, γ-secretase processivity
eTOC Blurb
Availability of facile cell-based models is a challenge in the study of neurodegenerative diseases. Using CRISPR activation Inoue et al. demonstrate that activation of APP and/or BACE1 genes unmasks a γ-secretase carboxypeptidase deficiency in patient fibroblasts, promoting Aβ42 accumulation.
Introduction
A challenge to the study of human neurodegenerative diseases, such as Alzheimer’s disease (AD), is the limited availability of facile cell-based disease models. Patient neurons are typically not easily available, particularly during the early phase of disease. Stem cell-based models such as human induced pluripotent stem cell (hIPSC)-derived neurons have been illuminating for some neurological diseases (Yagi et al., 2011; Israel et al., 2012; Koch et al., 2012; Kondo et al., 2013; Woodruff et al., 2013; Mahairaki et al., 2014; Muratore et al., 2014b; Moore et al., 2015; Oliveira et al., 2015), but require significant resources and effort, particularly for comparing large cohorts of patients and controls (Muratore et al., 2014a; Qiang et al., 2014). In some cases, patient skin fibroblasts, which are more easily accessed than CNS neurons, can reveal disease-relevant cellular mechanisms, such as with lysosomal storage disorders (Connolly, 1998; Parenti et al., 2015; Coutinho and Alves, 2016). However, fibroblasts may not be useful to model CNS disorders if key disease-relevant genes are not expressed sufficiently to unlock relevant disease mechanisms in the fibroblast system.
Molecular re-engineering of the CRISPR-Cas system has generated a broad precision mammalian genome regulatory toolbox, enabling gene knockout, insertion, activation, and suppression (Dominguez et al., 2016). Precision genome activation or suppression can be attained by introducing catalytically dead Cas9 (dCas9), together with transcriptional regulatory elements such as VP64 or KRAB, and sgRNAs targeting dCas9 to specific transcription sites (Dominguez et al., 2016). Recently, a powerful genome activation system termed Synergistic Activation Mediator (SAM) was developed, by incorporating an additional activation helper protein P65-HSF1 into the dCas9-VP64 and sgRNA complex (Konermann et al., 2015).
We focused our analysis here on a prototypical pathological process, familial AD (FAD) due to mutations in presenilin (PSEN)-1 or -2 genes, as the underlying genetic cause is partly established and molecular assays for PSEN-associated activities are well described (Levy-Lahad et al., 1995b; Levy-Lahad et al., 1995a; Rogaev et al., 1995; Sherrington et al., 1995; Li et al., 2014; De Strooper and Chavez Gutierrez, 2015). PSENs are essential components of the γ-secretase (GS) complex, which is a heteromultimeric complex required for intramembranous cleavage of substrates such as the βCTF/C99 fragment of amyloid precursor protein (APP) (De Strooper et al., 2012; Andrew et al., 2016; Selkoe and Hardy, 2016). Initial APP cleavage by BACE1 generates a secreted fragment, sAPPβ, and a 99 amino acid (aa) transmembrane stub, βCTF. The GS complex subsequently cleaves βCTF into a 48 or 49 aa peptide (Aβ48 or Aβ49), as well as the APP intracellular domain (AICD), by GS endopeptidase activity. Finally, GS carboxypeptidase activity is thought to further trim Aβ48 by steps of 3 or 4 aa, to Aβ42 or Aβ38 (termed the Aβ48 product line), whereas Aβ49 is processed to Aβ43 or Aβ40 (termed the Aβ49 product line) (Figure 1A). Thus, the ratio of the major amyloidogenic peptide, Aβ42, to the major non-amyloidogenic peptide, Aβ40, may be determined by alterations in either endopeptidase or carboxypeptidase activity. FAD-associated mutations in PSEN genes have long been known to alter the processing of APP in the FAD brain, leading to an increased accumulation of Aβ42 (De Strooper et al., 2012; Tomita, 2014; Selkoe and Hardy, 2016). Altered APP processing has been described even in skin fibroblasts from FAD patients, despite the apparent absence of AD pathology in the skin (Scheuner et al., 1996; Connolly, 1998; Etcheberrigaray and Bhagavan, 1999; Kondo et al., 2013).
Figure 1. Altered APP processing in skin fibroblasts from FAD patients.
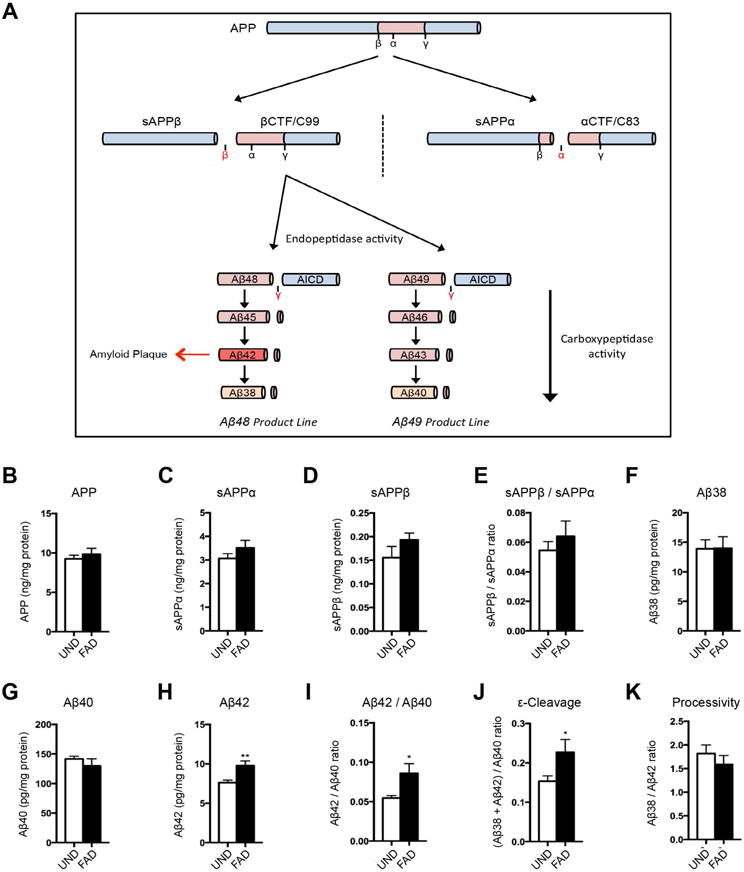
(A) Schematic of APP processing. (B–E) Levels of intracellular APP (B), extracellular sAPPα (C), and sAPPβ (D) in FAD or UND fibroblasts by ELISA, as well as the sAPPβ/sAPPα ratio (E). (F–I) The levels of extracellular Aβ38 (F), Aβ40 (G), and Aβ42 (H) by multiplex-ELISA. The Aβ42/Aβ40 ratio was significantly increased in FAD fibroblasts (I). (J–K) γ-secretase activities in fibroblasts. The endopeptidase product line preference was increased in FAD fibroblasts (J). The carboxypeptidase activity did not differ between UND and FAD fibroblasts (K).
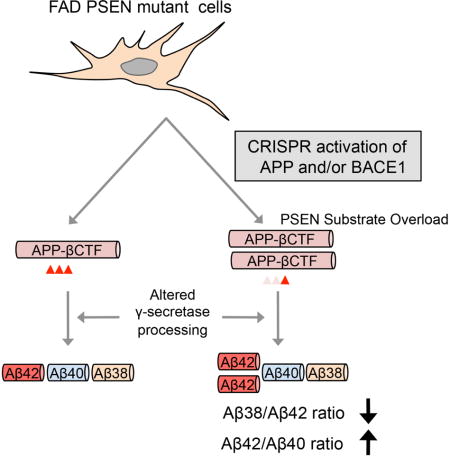
Here we sought to apply the SAM methodology with the intent of identifying genetic factors that typically limit the utility of fibroblasts in modeling neuropathological processes such as AD. One goal of this effort is to generate a simple, minimalist skin fibroblast-based model system that can recapitulate aspects of AD pathology. We show that SAM transcriptional activation of APP and/or BACE1 is sufficient to unmask an occult GS carboxypeptidase processivity defect in patient skin fibroblasts relative to fibroblasts from unaffected individuals. A secondary consequence of such a carboxypeptidase processivity defect is a further increase in the Aβ42/Aβ40 ratio. Thus, by inducing a small handful of genes in patient skin cells – particularly genes that are not typically expressed in these cultures, or harbor known disease mutations or variants – it may be possible to probe disease mechanisms in patient skin fibroblasts that would otherwise be inaccessible.
Results
Comparison of APP processing in primary human skin fibroblasts and induced neuronal cells
Analysis of fibroblast cultures from two cohorts of FAD patients harboring PSEN1 or PSEN2 mutations (FAD lines) or unaffected individuals without PSEN mutations (UND lines) (Table S1), demonstrated altered APP processing to Aβ (Figure 1B–I and S1A–H), in that the ratio of extracellular Aβ42/Aβ40 was increased in FAD cultures, consistent with prior reports (Scheuner et al., 1996; Kondo et al., 2013). The increased Aβ42/Aβ40 ratio resulted from the accumulation of Aβ42 and the reduction of Aβ40 levels in the FAD cultures (Figure 1G–I and S1F–H). Accumulation of APP, or of the soluble products of APP processing (sAPPα and sAPPβ), did not appear significantly altered (Figure 1B–E and S1A–D), suggesting that PSEN1 or PSEN2 mutations in FAD did not affect APP processing upstream of the γ-secretase (GS) cleavage.
Previous studies have provided evidence that FAD-associated PSEN mutations may impair two distinct GS activities: an endopeptidase activity that cleaves the C-terminal fragment of APP protein into Aβ and AICD fragments, and a carboxypeptidase activity that processes longer Aβ fragments into shorter ones (Figure 1A)(Chavez-Gutierrez et al., 2012). To address which GS activities are altered in skin fibroblasts from FAD patients, we estimated the endopeptidase product line preference in terms of the ratio of products from the Aβ48 product line (Aβ42+Aβ38): products of the Aβ49 product line (Aβ40), whereas we determined the carboxypeptidase activity as the ratio of Aβ38/Aβ42 (Moore et al., 2015). In FAD fibroblasts, we found that endopeptidase product line preference was significantly shifted, favoring the Aβ48 product line, whereas carboxypeptidase activity did not appear altered (Figure 1J–K and S1I–J). The latter finding contrasted with published studies using human patient brain tissues, cell-culture systems, or overexpression models, which suggested that most PSEN mutations affect GS carboxypeptidase activity more prominently (Quintero-Monzon et al., 2011; Chavez-Gutierrez et al., 2012; Fernandez et al., 2014; Szaruga et al., 2015). We therefore went on to establish a second model system: human induced neuronal cells (hiNs) generated by direct conversion of both UND and FAD fibroblasts. Using a cocktail of transcription factors, as previously described, we generated a population of cells with neuronal features (Figure S1L–M) (Vierbuchen et al., 2010; Pang et al., 2011). Analysis of endogenous APP processing in hiN cultures revealed evidence of alterations both in endopeptidase product line preference and carboxypeptidase activity (Figure 2A–F). Specifically, in addition to favoring the Aβ48 product line, FAD mutations were also associated with reduced processivity (Figure 2F). The reduced carboxypeptidase activity contributed to the altered Aβ42/Aβ40 ratio (Figure 2D). Similar to the fibroblast cultures, the levels of APP, sAPPα and sAPPβ in hiN were unchanged in the FAD lines compared to the UND lines (Figure S1N–Q). Further comparison of the levels of APP derived fragments in hiNs versus fibroblasts in both patient and control lines, demonstrated an overall increase of APP levels and processing in hiNs, substantiated by increased levels of all the different APP-derived fragments (Figure 2G and S1K). Interestingly, sAPPβ had the mostly prominent increase upon hiN conversion, consistent with a higher BACE1 activity and a relative increase of the BACE1-dependent amyloidogenic pathway, which is predicted to also increase accumulation of βCTF, the intracellular substrate of GS complex (Figure 1A).
Figure 2. APP processing in hiN cells converted from fibroblasts.
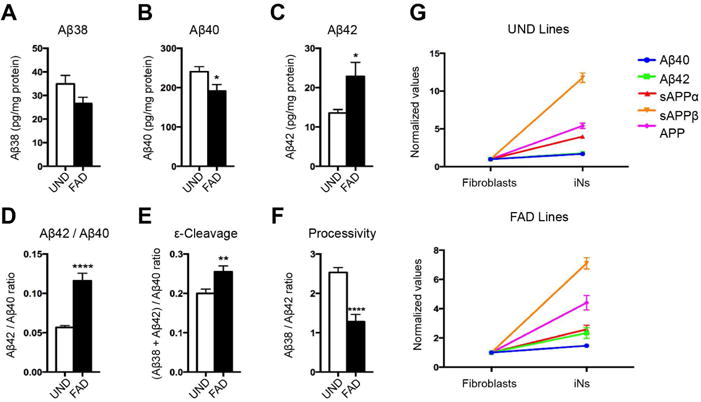
(A–F) Levels of extracellular Aβ38 (A), Aβ40 (B), and Aβ42 (C) quantified by multiplex-ELISA. Significant differences between UND and FAD hiNs in the Aβ42/Aβ40 ratio (D), ε-cleavage (E), and processivity (F). (G) Comparison of the levels of Aβ40, Aβ42, sAPPα, sAPPβ, and APP between fibroblasts and hiNs of UND and FAD lines.
Validation of sgRNAs for CRISPR-mediated activation of APP and BACE1
We next sought to test the hypothesis that skin fibroblasts may differ from CNS neurons, in terms of their GS processing of APP, as a consequence of a lower level of BACE1 activity in fibroblasts, and thus less GS substrate. We applied CRISPR/Cas9-mediated SAM technology to enhance the expression of endogenous APP or BACE1 (Figure 3A) (Konermann et al., 2015). Multiple candidate sgRNAs for either APP (18 sgRNAs candidates) or BACE1 (3 sgRNAs candidates), targeting sites across the proximal promoters (between −200 bp and the +1 transcription start site [TSS]) of the 2 genes (Table S2 and Figure 3B), were designed. These sgRNAs incorporated two MS2 RNA aptamers at the tetraloop and stemloop-2 to enable transcriptional activation.
Figure 3. SAM-mediated transcriptional activation of APP and BACE1.
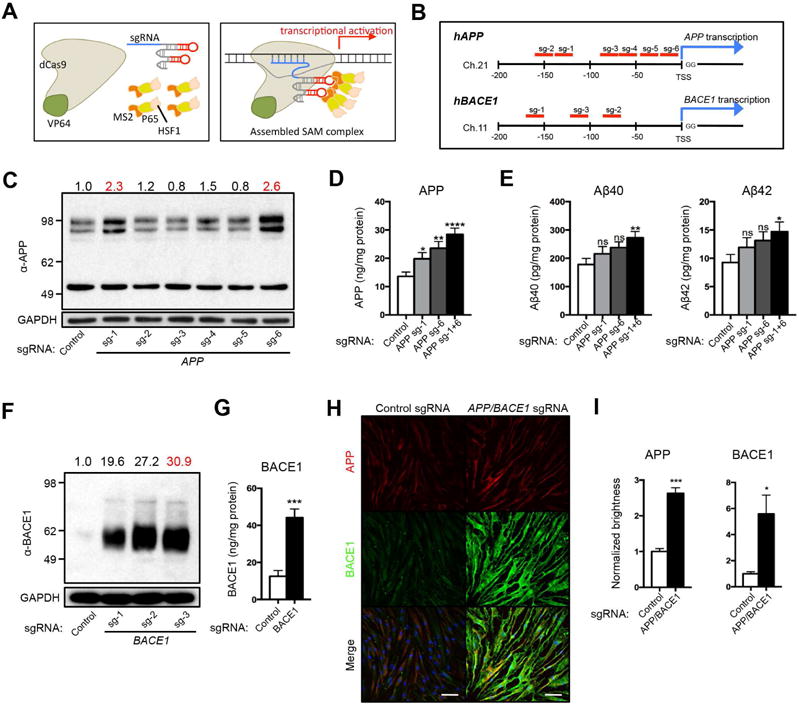
(A) Schematic of the SAM-mediated transcriptional activation. Three components -dCas9-VP64, MS2-P65-HSF1, and gene specific sgRNA (left) - are recruited near the TSS to activate transcription (right). (B) Design of individual sgRNAs for APP and BACE1. Candidate sgRNAs are shown in red. The sequences are shown in Table S2. (C) Screening of APP sgRNAs by Western Blotting. APP sgRNA-1 (sg-1) and sgRNA-6 (sg-6) elevated endogenous APP relative to control sgRNA (2.3- and 2.6-fold, respectively). (D) Additive elevation of endogenous APP by simultaneous treatment of APP sg-1 and sg-6 (sg-1+6). (E) Significant increases in the levels of Aβ40 and Aβ42 by simultaneous treatment of APP sg-1 and sg-6. (F) Screening of BACE1 sgRNAs by Western Blotting. BACE1 sgRNAs (sg-1 to -3) elevated endogenous BACE1 relative to control sgRNA (19.6- to 30.9-fold). (G) Elevation of endogenous BACE1 by BACE1 sg-3. (H) Immunofluorescent images of APP (red) and BACE1 (green) in fibroblasts with SAM activation of APP/BACE1 or control sgRNA. DNA counterstaining in blue. (I) Quantification of APP and BACE1 total brightness. n = 3 independent wells from HDFa. Bars, 100 μm.
Human fibroblasts were simultaneously infected with three lentiviral SAM components including a dCas9-VP64 fusion protein, a gene-specific sgRNA-MS2, and a MS2-P65-HSF1 helper protein to form the transcriptional activation complex. Initial qPCR screening narrowed down the APP sgRNA candidates to sgRNA-1 to -6 (Table S2). Western Blotting analysis showed that fibroblasts expressing APP sgRNA-1 or sgRNA-6 induced approximately 2.3 or 2.6-fold endogenous APP with proper molecular weight, relative to fibroblasts expressing control sgRNA, whereas the other sgRNAs tested showed no increase in protein levels (Figure 3C). qPCR analysis using two primer sets confirmed the significant increase of APP expression at the transcriptional level (Figure S2A). Interestingly, concomitant activation by APP sgRNA-1 and sgRNA-6 (sgRNA-1/6) further induced the APP protein, leading to significantly increased accumulation of Aβ40 and Aβ42 (Figure 3D–E), as determined by ELISA. Fibroblasts expressing BACE1 sgRNA-1 to -3 induced 19.6 to 30.9-fold endogenous BACE1 protein relative to fibroblasts expressing control sgRNA. BACE1 sgRNA-3 was the most efficient in the induction of endogenous BACE1 (Figure 3F). qPCR and ELISA analyses confirmed that BACE1 sgRNA-3 increased BACE1 at the transcriptional and protein levels (Figure 3G, S2B). Furthermore, significantly increased levels of APP and BACE1 were detected in fibroblasts expressing APP and BACE1 sgRNAs as quantified by immunocytochemical analyses (Figure 3H–I). Higher magnification images did not show evidence of altered protein localization or distribution (Figure S2C). Thus, we chose APP sgRNA-1/sgRNA-6 and BACE1 sgRNA-3 for subsequent activation studies.
These sgRNAs appeared highly specific, as confirmed by the lack of induction of a panel of 26 genes, quantified by qPCR analysis (Figure S2D–E). We also compared the SAM activation technology with typical lentiviral overexpression of APP and/or BACE1. Lentiviral overexpression of exogenous cDNA resulted in much higher levels of expression (Figure S3A), but appeared toxic to the fibroblasts leading to cell loss (data not shown). As previously described, very high levels of BACE1 led to a paradoxical suppression of Aβ production, due to the generation, by the excess BACE1 enzyme, of abnormal APP-derived intracellular cleavage products, that are not efficient substrates for GS (Figure S3B–E) (Zhou et al., 2011).
SAM activation of APP and/or BACE1 unmasks a deficit in PSEN-mediated Aβ processing in FAD fibroblasts
We next investigated whether SAM transcriptional activation of endogenous APP and/or BACE1 would elicit aspects of AD pathology in FAD fibroblasts. Transcriptional activation of APP, but not BACE1, significantly increased APP levels similarly in both UND and FAD fibroblasts (Figure 4A). Combined activation of APP and BACE1 diminished the APP sgRNA-mediated APP increase in either line (Figure 4A). sAPPα and sAPPβ fragments were also elevated with SAM activation, and combined activation of APP and BACE1 significantly increased the sAPPβ/sAPPα ratio (Figure S3F–H) and βCTF (Figure S3I), consistent with a relative increase of the amyloidogenic pathway (Figure 1A). These results suggested a further increase in Aβ fragments by combined activation of APP and BACE1. Transcriptional activation of either APP or BACE1 significantly increased the production of Aβ40, Aβ42 and Aβ38 in both UND and FAD lines, and the combined activation led to an additive increase as expected (Figure 4B, C, and S3J). Interestingly, the Aβ38 increase was strikingly suppressed in FAD lines relative to UND lines, especially in the combined activation group (Figure S3J). The suppressed induction of Aβ38 in FAD lines indicated a decrease in GS processivity ([Aβ38/Aβ42]), which trims longer Aβ peptides to produce shorter ones (Figure 4F). Endopeptidase activity (ε-cleavage) in FAD cultures displayed a significant shift, relative to UND cultures, towards the production of the Aβ48 product line relative to the Aβ49 product line, regardless of SAM activation (Figure 4E). Both the decreased carboxypeptidase activity and the shifted endopeptidase activity led to an increased ratio of Aβ42/Aβ40 in FAD lines, resulting in a higher relative accumulation of the longer and more aggregation-prone Aβ42 species (Figure 4D).
Figure 4. SAM activation of APP and/or BACE1 unmasks an occult g-secretase carboxypeptidase defect in FAD fibroblasts.
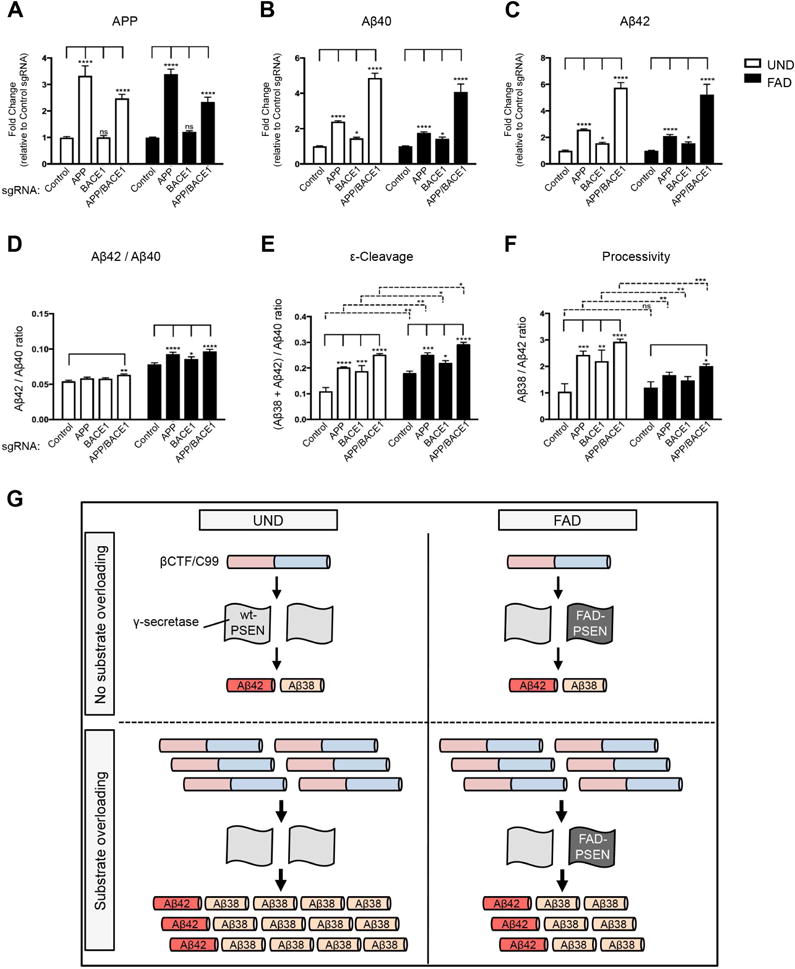
(A–F) SAM activation of APP, BACE1, or both in UND and FAD fibroblasts. Levels of APP (A), Aβ40 (B), and Aβ42 (C) in SAM-activated UND and FAD fibroblast cultures, as well as the Aβ42/Aβ40 ratio (D). The levels of ε-cleavage (E) and processivity (F) in SAM-activated UND and FAD fibroblast cultures. (G) Schematic of Aβ processivity defect in SAM-activated FAD fibroblasts. The Aβ38/Aβ42 ratio is similar in UND and FAD fibroblasts without substrate overloading. Induction of high substrate levels by SAM activation of APP and/or BACE1 unveils the PSEN-mediated γ-secretase processivity defect in FAD fibroblasts.
We sought to investigate whether the observed impairment in GS carboxypeptidase activity was specific to the Aβ48 product line and also quantified Aβ43 to determine processivity in the Aβ49 product line (Aβ40/Aβ43) (Nakaya et al., 2005; Takami et al., 2009; Saito et al., 2011). The levels of released Aβ43 in the fibroblast culture media were generally below the detection limit of our assay and only reliably measured in the presence of APP and BACE1 co-activation. Nonetheless, a similar trend was observed, with FAD cultures displaying apparently decreased carboxypeptidase activity in the Aβ49 product line as well (Figure S3K). Taken together, these data point to a model where transcriptional activation of endogenous APP or BACE1 expression, or both, increased GS substrate levels and thus unmasked an intrinsic defect in carboxypeptidase processivity in FAD fibroblasts harboring PSEN mutations. Thus, increased BACE1 activity in neurons, relative to skin fibroblasts, puts CNS neurons at higher risk for accumulation of the AD-associated Aβ42 peptide.
SAM activation of APP and BACE1 does not modify tau accumulation in fibroblasts
Previous studies have linked the accumulation of APP fragments such as AICD, βCTF or Aβ to alterations in the generation or phosphorylation of tau (p-tau) (Gotz et al., 2001; Lewis et al., 2001; Ryan and Pimplikar, 2005; Bolmont et al., 2007; Ghosal et al., 2009; Moore et al., 2015). We did not observe any significant changes in the levels of tau, p-tau (at Thr231 [T231]) or the ratio (p-tau per total tau) between APP and/or BACE1-activated and control cells. The presence of PSEN mutations in FAD cultures also did not affect the levels of those (Figure S4A–C). However, a limitation to the analysis of tau in fibroblasts is its very low expression level; total tau and p-tau levels were nearly undetectable in naïve UND and FAD fibroblasts, in contrast to hiNs. After fibroblast conversion to hiNs we observed a ~60× increase in the levels of total tau, although the ratio of p-tau over total tau remained conserved (Figure S4D–F). We therefore used SAM technology to induce endogenous expression of tau, alone or together with APP and BACE1. We designed sgRNAs for MAPT as previously described (Figure S4G) and chose MAPT sgRNA-3 as this sgRNA induced approximately 19.5-fold endogenous tau with different isoforms, relative to control sgRNA (Figure S4H). qPCR and ELISA analysis confirmed the elevations at the transcriptional and protein levels (Figure S4I–J).
We also determined the identity of the tau isoforms induced by MAPT sgRNA-3, as six isoforms exist in vivo in CNS, generated by alternative splicing at N-terminal (either 0N, 1N, or 2N repeats) and C-terminal (either 3R or 4R repeats) domains. Fibroblasts expressing control sgRNA expressed only 3R-type isoforms (0N3R, 1N3R and 2N3R). In contrast, fibroblasts expressing MAPT sgRNA-3 expressed these 3R isoforms as well as a subset of 4R isoforms (0N4R and 2N4R; Figure S4K and L). We next used MAPT sgRNA-3 to examine the pathological interaction between tau and APP fragments in FAD fibroblasts (Moore et al., 2015). SAM activation with MAPT sgRNA alone increased total tau as well as p-tau in both lines (Figure S4M and N). However, the ratio of p-tau was not significantly changed between UND and FAD lines by MAPT activation (Figure S4O). Additional activation of APP or APP/BACE1 in the context of MAPT activation did not alter the ratio of p-tau between UND and FAD lines (Figure S4M-O).
Finally, we examined the effect of tau induction on APP processing in fibroblasts. Again no interaction between the two pathways was observed and the levels of APP, Aβ40, Aβ42, or the Aβ42/Aβ40 ratio, in either UND or FAD fibroblasts remained unchanged in the context of MAPT activation (Figure S4P–S). Thus, these studies failed to reveal evidence for an effect of increased APP processing on tau levels or phosphorylation, or an effect of tau levels on APP processing. As APP processing has previously been implicated in tau modification (Gotz et al., 2001; Lewis et al., 2001; Ryan and Pimplikar, 2005; Bolmont et al., 2007; Ghosal et al., 2009; Moore et al., 2015), such a pathway may require other yet to be identified neuron-specific factors.
Discussion
The present study supports the utility of CRISPR-mediated transcriptional activation in modeling pathological processes in patient-derived skin fibroblasts, as disease-relevant genes or biological processes may not be present in naïve fibroblasts. FAD mutations in PSENs serve as prototypical examples here, as their activities can be easily quantified in different cell types. Our findings suggest that, at endogenous levels in human fibroblasts, GS carboxypeptidase activity, even in the context of PSEN mutations, is sufficiently robust in converting Aβ42 into Aβ38. We hypothesize that once GS carboxypeptidase activity is placed under substrate pressure, due to increased expression of APP or BACE1, such as seen in neuronal cultures, a carboxypeptidase activity deficit is revealed in the FAD fibroblast cultures, leading to a further shift in the Aβ42/Aβ40 ratio (Figure 4G). PSEN FAD-associated mutations have been reported to cause a premature release of longer, intermediary substrates/products during the sequential carboxypeptidase cleavage (Chavez-Gutierrez et al., 2012), and a recent publication showed that this is a consequence of impaired catalytic γ-Secretase activity (Szaruga et al., 2017). It is possible that increased concentrations of longer Aβ forms, in the context of PSEN mutations, drive competition at the GS catalytic site in a concentration-dependent manner, thus impairing GS processivity (Figure 4G), but this remains to be demonstrated.
How mutations in PSEN cause AD remains a contentious issue. Gain of function models suggest that FAD-mutated PSENs primarily increase Aβ42 through modified endopeptidase activity and/or a shift in product line preference (Hardy and Selkoe, 2002; Potter et al., 2013; Selkoe and Hardy, 2016), although the precise mechanism is unknown. A traditional loss of function hypothesis propose that overall loss of PSEN function, due to the lower endopeptidase activity, causes age-dependent neurodegeneration, and cognitive impairment, without increasing Aβ (Saura et al., 2004; Shen and Kelleher, 2007; Heilig et al., 2010; Wines-Samuelson et al., 2010; Watanabe et al., 2012; Heilig et al., 2013; Xia et al., 2015; Xia et al., 2016; Sun et al., 2017). Recently, several reports have proposed that PSEN mutations increase the Aβ42/Aβ40 due to a dominant reduction in GS carboxypeptidase activity (Quintero-Monzon et al., 2011; Chavez-Gutierrez et al., 2012; Fernandez et al., 2014; Szaruga et al., 2015; Szaruga et al., 2017). Data herein obtained from human fibroblasts suggest that both a neomorphic mutation shifting GS endopeptidase activity to the Aβ48 product line, and a loss of carboxypeptidase function that is unmasked under substrate pressure contribute to the increased Aβ42/Aβ40 ratio. Although we focus here on the processing of Aβ42:Aβ40 in part because of the amyloidogenic properties of Aβ42, which is thus implicated in disease pathology, it is also important to emphasize that the substrate pressure on mutant PSENs, in some contexts, may be more broadly damaging due to defective processing or cleavage of many other GS carboxypeptidase substrates, or the altered accumulation of other APP fragments.
Our simple fibroblast model system, with activation of endogenous APP and/or BACE1, has significant advantages over hIPSC-based neuronal models: first, this fibroblast model system is inherently isogenic, as select genes are precisely induced by CRISPR methodology; second, the fibroblast culture procedure for CRISPR activation is relatively short (approximately one week) and facile. In contrast, conventional protocols for neuronal differentiation from hIPSCs generally require extensive manipulations (Muratore et al., 2014a; Qiang et al., 2014). CRISPR-induced patient skin fibroblasts may enable faster genetic or drug screenings, and mechanistic studies of APP processing in the context of larger cohorts of sporadic AD patients. In future studies, it will be important to further characterize the kinetics of APP processing to fragments within distinct Aβ product lines, in these skin fibroblast-based in vitro models; as accumulation of endogenous Aβ fragments is modest in these models (relative to exogenous APP overexpression-based in vitro model systems), biochemical techniques such as mass spectrometry are technically challenging. Finally, it will also be of particular interest in the future to utilize this activated CRISPR skin fibroblast model system to probe the roles of additional genetic modifiers of sporadic AD risk, such as APOE isoforms.
Methods
hiN generation from human fibroblasts
Human dermal fibroblasts used in this study are listed in Table S1. Conversion of fibroblasts into neuronal cells was driven by forced expression of 4 genes according to a prior paper with minor modifications (Vierbuchen et al., 2010; Pang et al., 2011).
Construction of gene-specific sgRNAs
Individual gene-specific sgRNA vectors were constructed following the SAM target sgRNA cloning protocol (sam.genome-engineering.org) (Konermann et al., 2015). Candidate sgRNA oligonucleotides were designed using the ‘SAM Cas9 activator design tool’, and synthesized after the additions of 5′-CACCG to the sense oligonucleotide and 5′-AAAC and C-3′ to the antisense oligonucleotide (Integrated DNA Technologies). Synthesized 25 base sense and antisense oligonucleotides (100 μM each) were mixed with T4 polynucleotide kinase (#M0201, New England Biolab; 10 μM each oligonucleotide, 1× T4 ligase buffer, 0.5 units/μl T4 polynucleotide kinase) and annealed in a thermal cycler with the following conditions: 37°C for 30 min, 95°C for 5 min, and decreasing the temperature to 25°C at 5°C/min. The annealed oligonucleotides were ligated into the BsmBI digested site of the lenti sgRNA(MS2)_zeo backbone vector by T7 ligase (#L6020F, Enzymatics; 1× ligase buffer, 0.1 mg/ml BSA, 15 units/μl T7 ligase, 1 μg/μl vector) in a thermal cycler with the following conditions: 37°C for 5 min, 20°C for 5 min, and repeating for 15 cycles total. Ligated plasmid was transformed into Stbl3 competent cells (#C737303, Thermo Fisher Scientific), and plated onto a LB agar plate containing 100 μg/ml Ampicillin. Inserted sgRNA sequences were confirmed by Sanger sequencing using the 5′ sequencing primer (5′-GAG GGC CTA TTT CCC ATG ATT CCT TCA TAT-3′). The constructed sgRNAs are listed in Table S2.
SAM genome activation in fibroblasts
Human fibroblasts were plated onto a 0.1% gelatin (#ES006B, Millipore)-coated 24-well plate at the density of 0.7×104 cells/cm2 for 24 hours and then infected with concentrated lentiviral particles in D-MEM containing 10% FBS and 8 μg/ml polybrene (#H9268, Sigma-Aldrich). Virus containing each SAM component were equally applied into the fibroblast cultures (dCas9-VP64:MS2-P65-HSF1:sgRNA-MS2 = 1:1:1). For APP sgRNA lentivirus, equal volumes of APP sgRNA-1 and sgRNA-6 lentiviral particles were used. Virus-containing medium was removed after 20 hours and replaced with fresh medium. Infected fibroblasts were cultured without medium change after viral removal. For ELISA analyses except for Aβ43, cells were cultured in 250 μl of culture medium per well for 7 days. For ELISA analyses of Aβ43, cells were cultured in 200 μl of culture medium per well for 12 days to increase the concentration. For immunofluorescence analyses, fibroblasts were plated onto 0.1% gelatin-coated 24 Well ibiTreat μ-Plates (#82406, Ibidi).
ELISA, Western Blotting, qPCR, and immunfluorescence analyses
Levels of sAPPα, sAPPβ, Aβ38, Aβ40, Aβ42, and Aβ43 fragments were determined in the extracellular media of control (CTR) and FAD fibroblasts, while the concentrations of APP, APP-βCTF, BACE1, total tau and p-tau (T231) were determined in cell lysates. Previous to sample analysis by ELISA, cell culture medium was collected and centrifuged at 6,000 × g for 5 min to remove cell debris. Cell lysates were prepared by incubating the cells with RIPA buffer (#89900, Thermo Fisher Scientific) supplemented with protease inhibitor cocktail (#P8340, Sigma-Aldrich) and phosphatase inhibitor cocktail (#78420, Thermo Fisher Scientific) for 30 min on ice. Extracts were centrifuged at 20,000 × g at 4°C for 30 min and the supernatant was collected. Aβ40, Aβ42, Aβ38, sAPPα, sAPPβ, total tau and p-tau (T231) levels were quantified with multiplex ELISA kits from Meso Scale Discovery (#K15200E-2, #K15120E-2, and #K15121D-2 respectively), and plates were read on a MSD Sector Imager 2400. APP levels were measured using an APP human ELISA kit (#KHB0051, Thermo Fischer Scientific). APP-βCTF levels were measured using a human APP-βCTF Assay kit (#27776, Immuno-Biological Laboratories). BACE1 levels were measured using a BACE1 Assay kit (#27752, Immuno-Biological Laboratories). Aβ43 levels were measured using a Human Aβ (1–43) (FL) Assay kit (#27710, Immuno-Biological Laboratories). Plates were read in a Molecular Devices UVmax plate reader at 450 nm and concentrations were calculated using SoftMax® Pro 5 software. Measured concentrations were normalized to total protein concentration determined with a bicinchoninic acid assay (Pierce™ BCA Protein Assay Kit, #23225, Thermo Fisher Scientific). All datasets are shown in Table S4.
Statistical Analysis
Statistical analyses were performed using GraphPad PRISM 6.0 software. Values are expressed as mean ± SEM. Statistical differences were tested using unpaired t tests for two samples and a two-way ANOVA with Turkey’s post-hoc test for multiple samples. p-values of * p˂0.05, ** p˂0.01, *** p˂0.001, and **** p˂0.0001.
Supplementary Material
Highlights.
CRISPR activation of APP and/or BACE1 increases Aβ in skin fibroblasts
CRISPR activation unveils a γ-secretase processivity defect in FAD fibroblasts
γ-secretase processivity defects contribute to relatively higher levels of Aβ42
CRISPR activation allows for the selective study of CNS-disease genes in fibroblasts
Acknowledgments
We thank D. Trono, M. Wernig, R. Jaenisch, and F. Zhang for providing the plasmids. This work was supported by NIH grant 4R01AG042317-05 (K.I., L.M.A.O, and A.A.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
K.I. designed the studies and performed all experiments, analyzed data, and wrote the manuscript. L.M.A.O. designed the studies and carried out qPCR, immunocytochemisty and ELISA experiments, analyzed data, and wrote the manuscript. A.A. designed the studies, oversaw the data acquisition and analysis, and wrote the manuscript.
Competing Interests
A.A. is a co-founder and consultant of Alector LLC. The other authors declare no competing financial interest.
References
- Andrew RJ, Kellett KA, Thinakaran G, Hooper NM. A Greek Tragedy: The Growing Complexity of Alzheimer Amyloid Precursor Protein Proteolysis. The Journal of biological chemistry. 2016;291:19235–19244. doi: 10.1074/jbc.R116.746032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T, Clavaguera F, Meyer-Luehmann M, Herzig MC, Radde R, Staufenbiel M, Lewis J, Hutton M, Tolnay M, Jucker M. Induction of tau pathology by intracerebral infusion of amyloid-beta-containing brain extract and by amyloid-beta deposition in APP × Tau transgenic mice. The American journal of pathology. 2007;171:2012–2020. doi: 10.2353/ajpath.2007.070403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez-Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. The EMBO journal. 2012;31:2261–2274. doi: 10.1038/emboj.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly GP. Fibroblast models of neurological disorders: fluorescence measurement studies. Trends in pharmacological sciences. 1998;19:171–177. doi: 10.1016/s0165-6147(98)01202-4. [DOI] [PubMed] [Google Scholar]
- Coutinho MF, Alves S. From rare to common and back again: 60years of lysosomal dysfunction. Molecular genetics and metabolism. 2016;117:53–65. doi: 10.1016/j.ymgme.2015.08.008. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Chavez Gutierrez L. Learning by failing: ideas and concepts to tackle gamma-secretases in Alzheimer’s disease and beyond. Annual review of pharmacology and toxicology. 2015;55:419–437. doi: 10.1146/annurev-pharmtox-010814-124309. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Iwatsubo T, Wolfe MS. Presenilins and gamma-secretase: structure, function, and role in Alzheimer Disease. Cold Spring Harbor perspectives in medicine. 2012;2:a006304. doi: 10.1101/cshperspect.a006304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez AA, Lim WA, Qi LS. Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nature reviews Molecular cell biology. 2016;17:5–15. doi: 10.1038/nrm.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etcheberrigaray R, Bhagavan S. Ionic and signal transduction alterations in Alzheimer’s disease: relevance of studies on peripheral cells. Molecular neurobiology. 1999;20:93–109. doi: 10.1007/BF02742436. [DOI] [PubMed] [Google Scholar]
- Fernandez MA, Klutkowski JA, Freret T, Wolfe MS. Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid beta-peptides (Abeta) by gamma-secretase to increase 42-to-40-residue Abeta. The Journal of biological chemistry. 2014;289:31043–31052. doi: 10.1074/jbc.M114.581165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, Pimplikar SW. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:18367–18372. doi: 10.1073/pnas.0907652106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Heilig EA, Xia W, Shen J, Kelleher RJ., 3rd A presenilin-1 mutation identified in familial Alzheimer disease with cotton wool plaques causes a nearly complete loss of gamma-secretase activity. The Journal of biological chemistry. 2010;285:22350–22359. doi: 10.1074/jbc.M110.116962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig EA, Gutti U, Tai T, Shen J, Kelleher RJ., 3rd Trans-dominant negative effects of pathogenic PSEN1 mutations on gamma-secretase activity and Abeta production. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:11606–11617. doi: 10.1523/JNEUROSCI.0954-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, Goldstein LS. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch P, Tamboli IY, Mertens J, Wunderlich P, Ladewig J, Stuber K, Esselmann H, Wiltfang J, Brustle O, Walter J. Presenilin-1 L166P mutant human pluripotent stem cell-derived neurons exhibit partial loss of gamma-secretase activity in endogenous amyloid-beta generation. The American journal of pathology. 2012;180:2404–2416. doi: 10.1016/j.ajpath.2012.02.012. [DOI] [PubMed] [Google Scholar]
- Kondo T, et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell stem cell. 2013;12:487–496. doi: 10.1016/j.stem.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, Weber JL, Bird TD, Schellenberg GD. A familial Alzheimer’s disease locus on chromosome 1. Science. 1995a;269:970–973. doi: 10.1126/science.7638621. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995b;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- Li Y, Bohm C, Dodd R, Chen F, Qamar S, Schmitt-Ulms G, Fraser PE, St George-Hyslop PH. Structural biology of presenilin 1 complexes. Molecular neurodegeneration. 2014;9:59. doi: 10.1186/1750-1326-9-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahairaki V, Ryu J, Peters A, Chang Q, Li T, Park TS, Burridge PW, Talbot CC, Jr, Asnaghi L, Martin LJ, Zambidis ET, Koliatsos VE. Induced pluripotent stem cells from familial Alzheimer’s disease patients differentiate into mature neurons with amyloidogenic properties. Stem cells and development. 2014;23:2996–3010. doi: 10.1089/scd.2013.0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore S, Evans LD, Andersson T, Portelius E, Smith J, Dias TB, Saurat N, McGlade A, Kirwan P, Blennow K, Hardy J, Zetterberg H, Livesey FJ. APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell reports. 2015;11:689–696. doi: 10.1016/j.celrep.2015.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratore CR, Srikanth P, Callahan DG, Young-Pearse TL. Comparison and optimization of hiPSC forebrain cortical differentiation protocols. PloS one. 2014a;9:e105807. doi: 10.1371/journal.pone.0105807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratore CR, Rice HC, Srikanth P, Callahan DG, Shin T, Benjamin LN, Walsh DM, Selkoe DJ, Young-Pearse TL. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Human molecular genetics. 2014b;23:3523–3536. doi: 10.1093/hmg/ddu064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya Y, Yamane T, Shiraishi H, Wang HQ, Matsubara E, Sato T, Dolios G, Wang R, De Strooper B, Shoji M, Komano H, Yanagisawa K, Ihara Y, Fraser P, St George-Hyslop P, Nishimura M. Random mutagenesis of presenilin-1 identifies novel mutants exclusively generating long amyloid beta-peptides. The Journal of biological chemistry. 2005;280:19070–19077. doi: 10.1074/jbc.M501130200. [DOI] [PubMed] [Google Scholar]
- Oliveira LM, Falomir-Lockhart LJ, Botelho MG, Lin KH, Wales P, Koch JC, Gerhardt E, Taschenberger H, Outeiro TF, Lingor P, Schule B, Arndt-Jovin DJ, Jovin TM. Elevated alpha-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson’s patient-derived induced pluripotent stem cells. Cell death & disease. 2015;6:e1994. doi: 10.1038/cddis.2015.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, Citri A, Sebastiano V, Marro S, Sudhof TC, Wernig M. Induction of human neuronal cells by defined transcription factors. Nature. 2011;476:220–223. doi: 10.1038/nature10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annual review of medicine. 2015;66:471–486. doi: 10.1146/annurev-med-122313-085916. [DOI] [PubMed] [Google Scholar]
- Potter R, Patterson BW, Elbert DL, Ovod V, Kasten T, Sigurdson W, Mawuenyega K, Blazey T, Goate A, Chott R, Yarasheski KE, Holtzman DM, Morris JC, Benzinger TL, Bateman RJ. Increased in vivo amyloid-beta42 production, exchange, and loss in presenilin mutation carriers. Science translational medicine. 2013;5:189ra177. doi: 10.1126/scitranslmed.3005615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang L, Inoue K, Abeliovich A. Instant neurons: directed somatic cell reprogramming models of central nervous system disorders. Biological psychiatry. 2014;75:945–951. doi: 10.1016/j.biopsych.2013.10.027. [DOI] [PubMed] [Google Scholar]
- Quintero-Monzon O, Martin MM, Fernandez MA, Cappello CA, Krzysiak AJ, Osenkowski P, Wolfe MS. Dissociation between the processivity and total activity of gamma-secretase: implications for the mechanism of Alzheimer’s disease-causing presenilin mutations. Biochemistry. 2011;50:9023–9035. doi: 10.1021/bi2007146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- Ryan KA, Pimplikar SW. Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. The Journal of cell biology. 2005;171:327–335. doi: 10.1083/jcb.200505078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Suemoto T, Brouwers N, Sleegers K, Funamoto S, Mihira N, Matsuba Y, Yamada K, Nilsson P, Takano J, Nishimura M, Iwata N, Van Broeckhoven C, Ihara Y, Saido TC. Potent amyloidogenicity and pathogenicity of Abeta43. Nature neuroscience. 2011;14:1023–1032. doi: 10.1038/nn.2858. [DOI] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ, 3rd, Kandel ER, Duff K, Kirkwood A, Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Scheuner D, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nature medicine. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO molecular medicine. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Kelleher RJ., 3rd The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:403–409. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Sun L, Zhou R, Yang G, Shi Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:E476–E485. doi: 10.1073/pnas.1618657114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaruga M, Veugelen S, Benurwar M, Lismont S, Sepulveda-Falla D, Lleo A, Ryan NS, Lashley T, Fox NC, Murayama S, Gijsen H, De Strooper B, Chavez-Gutierrez L. Qualitative changes in human gamma-secretase underlie familial Alzheimer’s disease. The Journal of experimental medicine. 2015;212:2003–2013. doi: 10.1084/jem.20150892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaruga M, Munteanu B, Lismont S, Veugelen S, Horre K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, Gallardo R, Schymkowitz J, Rousseau F, Fox NC, Hopf C, De Strooper B, Chavez-Gutierrez L. Alzheimer’s-Causing Mutations Shift Abeta Length by Destabilizing gamma-Secretase-Abetan Interactions. Cell. 2017;170:443–456.e414. doi: 10.1016/j.cell.2017.07.004. [DOI] [PubMed] [Google Scholar]
- Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima-Kawashima M, Funamoto S, Ihara Y. gamma-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:13042–13052. doi: 10.1523/JNEUROSCI.2362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita T. Molecular mechanism of intramembrane proteolysis by gamma-secretase. Journal of biochemistry. 2014;156:195–201. doi: 10.1093/jb/mvu049. [DOI] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Xia D, Kanekiyo T, Kelleher RJ, 3rd, Shen J. Familial frontotemporal dementia-associated presenilin-1 c.548G>T mutation causes decreased mRNA expression and reduced presenilin function in knock-in mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:5085–5096. doi: 10.1523/JNEUROSCI.0317-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wines-Samuelson M, Schulte EC, Smith MJ, Aoki C, Liu X, Kelleher RJ, 3rd, Shen J. Characterization of age-dependent and progressive cortical neuronal degeneration in presenilin conditional mutant mice. PloS one. 2010;5:e10195. doi: 10.1371/journal.pone.0010195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff G, Young JE, Martinez FJ, Buen F, Gore A, Kinaga J, Li Z, Yuan SH, Zhang K, Goldstein LS. The presenilin-1 DeltaE9 mutation results in reduced gamma-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell reports. 2013;5:974–985. doi: 10.1016/j.celrep.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia D, Kelleher RJ, 3rd, Shen J. Loss of Abeta43 Production Caused by Presenilin-1 Mutations in the Knockin Mouse Brain. Neuron. 2016;90:417–422. doi: 10.1016/j.neuron.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia D, Watanabe H, Wu B, Lee SH, Li Y, Tsvetkov E, Bolshakov VY, Shen J, Kelleher RJ., 3rd Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron. 2015;85:967–981. doi: 10.1016/j.neuron.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T, Yamanaka S, Okano H, Suzuki N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Human molecular genetics. 2011;20:4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
- Zhou L, Brouwers N, Benilova I, Vandersteen A, Mercken M, Van Laere K, Van Damme P, Demedts D, Van Leuven F, Sleegers K, Broersen K, Van Broeckhoven C, Vandenberghe R, De Strooper B. Amyloid precursor protein mutation E682K at the alternative beta-secretase cleavage beta’-site increases Abeta generation. EMBO molecular medicine. 2011;3:291–302. doi: 10.1002/emmm.201100138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.