

Abstract
Many regulated epigenetic elements and base lesions found in genomic DNA can both directly impact gene expression and play a role in disease processes. However, due to their non-canonical nature, they are challenging to assess with conventional technologies. Here, we present a new approach for the targeted detection of diverse modified bases in DNA. We first use enzymatic components of the DNA base excision repair pathway to install an individual affinity label at each location of a selected modified base with high yield. We then probe the resulting material with a solid-state nanopore assay capable of discriminating labeled DNA from unlabeled. The technique features exceptional modularity via selection of targeting enzymes, which we establish through the detection of four DNA base elements: uracil, 8-oxoguanine, T:G mismatch, and the methyladenine analog 1,N6-ethenoadenine. Our results demonstrate the potential for quantitative nanopore assessment of a broad range of base modifications.
Keywords: nanopore, base lesion, epigenetics, detection, cancer
A variety of non-canonical bases are prevalent in genomic DNA and play crucial roles in cell functions that include gene expression and suppression1, transposon expression2,3, stem cell differentiation4, and chromosomal inactivation5. For example, the abundance and position of epigenetic modifications are tightly regulated and errors in this regulation have been linked to a wide range of diseases6 including cancer. In addition, DNA base damage elements generated both endogenously and exogenously are a major source of point mutations if not correctly repaired by cellular processes. The locations of these elements can be random or could be linked to sequence accessibility in chromatin structures. While the impact of modified DNA bases is clear, their detection can be challenging. For example, direct sequencing approaches typically lack the ability to identify non-canonical bases that may be present in DNA7, with some extension to the epigenetic modification methylcytosine, specifically8. Conventional technologies like mass spectrometry and high-performance liquid chromatography are burdensome, expensive, and destructive to the DNA, and in some cases can induce additional lesions9, leading to misrepresentation of density. In addition, immunological methods like the enzyme-linked immunosorbent assay (ELISA) rely on antibodies that can suffer from cross-reactivity10. One approach that addresses some of these concerns has been pioneered recently by Song, et al11 in which a single, high-affinity tag was attached enzymatically to 5-hydroxymethylcytosine bases, permitting downstream analysis, enrichment, or sequencing. While the process has been adapted to access some additional elements of the demethylation pathway12–14, only a limited suite of modifications are suitable for such tagging. As the ability to probe a wide variety of base modifications would be of significant value, we set out to develop a new, modular strategy employing solid-state (SS-) nanopore technology that would be permissive for a host of different DNA modifications.
SS-nanopores15,16 have been widely studied as a means to assess biological molecules like DNA17,18, RNA19,20, and proteins21–23 using the principle of resistive pulse sensing. The platform consists of an insulating thin-film membrane that contains a nanometer-scale pore, positioned in an electrolyte solution. Application of an electrical bias across the membrane generates an electric field through the pore, and as charged molecules are threaded electrophoretically one-by-one, they temporarily occlude the aperture and interrupt the measured ionic current. These brief electrical disruptions are designated as “events”, and their properties have been used to study molecular attributes24, probe intermolecular interactions25,26, and determine analyte concentration27,28. Historically, a significant limitation of this measurement approach has been a lack of selectivity: all molecules of like-charge will translocate and contribute to the overall signal, thus requiring differentiation ex post facto via often subtle differences in event characteristics. We have developed a SS-nanopore assay that enables nearly binary detection and quantification of DNA featuring a single biotin affinity tag29. Briefly, when target DNA fragments (below ~250 bp) or a key chaperone protein (monovalent streptavidin30, MS) are introduced individually to a SS-nanopore of appropriate diameter, their rapid translocations prevent events from being resolved by conventional electronics (Fig. 1a, left and center). However, when the two molecules bind, the larger nucleoprotein complex interacts with the walls of the nanopore during passage, slowing its translocation to a resolvable speed and yielding events (Fig. 1a, right). Recently, we expanded this basic approach to assess hydroxymethylcytosine epigenetic modifications28 by employing an established method for specific biotin labeling of the base11, enabling direct assessment of a base modification with physiological relevance. However, the scope of possible targets for the labeling approach was limited intrinsically by enzymatic recognition. Here, we enhance our SS-nanopore measurement scheme significantly by integrating it with an alternative, modular labeling technique that enables the targeted detection of diverse base modifications.
Figure 1.
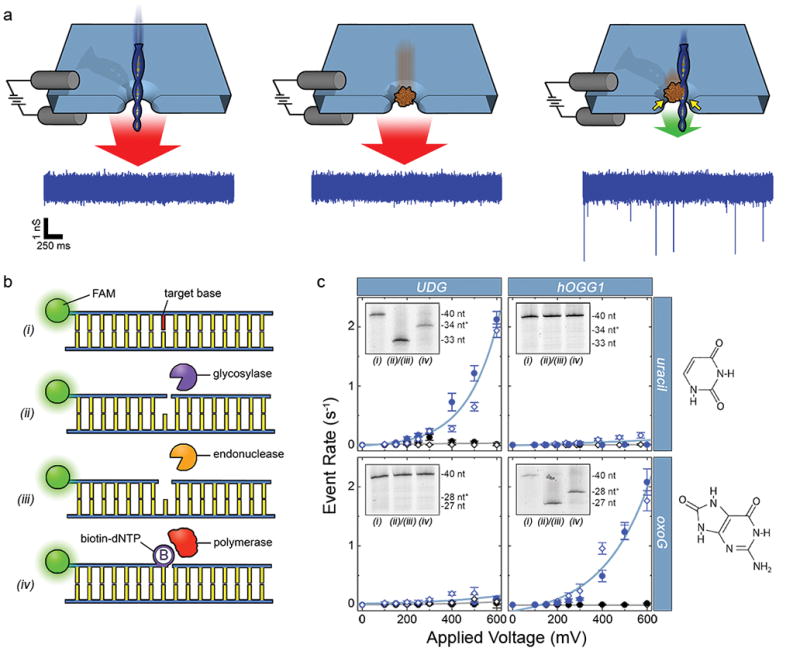
(a) Depiction of the selective SS-nanopore assay. Individual passage of a short DNA (left) or a chaperone protein (MS, center) yields no events due to the high translocation speed (red arrows); a DNA-protein complex (right) interacts with the pore walls (yellow arrows), resulting in slower translocation speed (green arrow) and resolvable events. Sample conductance traces at bottom were measured at 300 mV using 75 bp DNA (500 nM) with a synthetic biotin. (b) Schematic representation of the general labeling approach. (i) A duplex DNA molecule featuring a target base element (red). (ii) A glycosylase recognizes and excises the base element (diagram shows activity of a bifunctional glycosylase that nicks the phosphate backbone 3′ to the excision). (iii) An AP endonuclease cuts the backbone 5′ to the excision. (iv) A gap-filling polymerase incorporates a single biotinylated nucleotide at the modification position. (c) SS-nanopore analyses of 250 nM DNA oligonucleotides featuring either a single uracil (at nucleotide position 34, top) or a single oxoG (at nucleotide position 28, bottom). Data points indicate measurements on treated DNA with (blue) and without (black) MS. Filled circles and open diamonds are independent measurements on different SS-nanopore devices and all lines are exponential fits to the data. Dramatic increases in event rate are measured for DNA-MS when a glycosylase specific for the target base is used (blue data, upper left and lower right). Almost no effect is observed for mismatched glycosylase (blue data, upper right and lower left). Insets: denaturing gel analyses of the same DNA constructs (steps numbered as in (b)). Lane 1: annealed oligonucleotide; lane 2: following glycosylase/endonuclease treatment; lane 3: following T4(exo-) fill-in. * indicates DNA length plus biotin tag. Right: molecular structures of the target bases.
Similar to a recent report by Riedl, et al.31, our methodology exploits the enzymatic machinery of the DNA base excision repair (BER) pathway, which identifies and restores base lesions in vivo. In our approach (Fig. 1b), a modified base element was first excised from the DNA using a DNA glycosylase, which removed the target base from the phosphate backbone, leaving an abasic (AP) site. If the glycosylase was bifunctional (i.e. had AP lyase activity), the phosphodiester bond was also cleaved 3′ to the modification, leaving a single strand nick. Ensuing steps were not affected by this activity. Next, an AP endonuclease was used to cleave the phosphodiester bond 5′ to the abasic site and remove the exposed 3′ phosphate, leaving a hydroxyl group that was amenable to the final step: treatment with a gap-filling polymerase to incorporate a biotin-conjugated nucleotide into the DNA structure. For this, we used a mutant polymerase lacking 3′-5′ exonuclease activity (T4(exo-)) and provided it with only of the cognate biotin-dNTP, resulting in the insertion of a single affinity label at the precise location of the modified base. We have found the absence of exonuclease activity to be particularly important as processive cleavage of nucleotides from the modification site can result in prevention or misincorporation of the biotinylated nucleotide label. We note that this methodology ultimately resulted in a nick 3′ to the inserted biotin-dNTP. While it should be possible to repair this nick through ligation, we did not include such a step because of the potential for reduced product yield. The presence of the nick did not negatively impact subsequent measurements.
Crucially, this general approach could be used to target a variety of distinct modified bases through variation of two central components: the DNA glycosylase, selected for recognition of a particular lesion, and the biotin-conjugated nucleotide, selected to match the canonical identity of the target modified base (or in the case of a mismatch target, the appropriate nucleotide for Watson-Crick base-pairing with the opposite strand). As an initial demonstration of this modularity, we first showed selective detection of uracil and oxoguanine (oxoG) bases. Uracils arise in DNA upon deamination of cytosine, resulting in a mutagenic U:G mismatch, or upon misincorporation of dUTP, resulting in a genotoxic U:A pair32. Meanwhile, oxoG is the major oxidative base damage associated with reactive oxygen species (ROS) due to the low redox potential of guanine and has known mutagenic potential via transversion during DNA replication33. For these measurements, we used synthetic 40 bp double-strand (ds-) DNA oligonucleotides, with one strand containing the target modified base at a known position and a fluorescent FAM label at the 5′ end. We utilized endonuclease IV (EndoIV) to prime the excised gap for T4(exo-) incorporation of a biotin-dNTP.
Denaturing gel analysis (Fig. 1c insets) of each sequential step for the two bases using an appropriate glycosylase/nucleotide combination showed excision of the modified base and incorporation of the biotin-dNTP. Labeling of uracil was achieved using a combination of uracil DNA glycosylase (UDG) and biotin-dUTP while oxoG labeling employed human oxoG DNA glycosylase (hOGG1) and biotin-dGTP. Notably, we made use of a “one-pot” treatment for each of these targets (see Materials and Methods) that minimized material loss and enabled high product yields of ~92% and ~83%, respectively. In addition, UDG is a monofunctional glycosylase while hOGG1 is bifunctional, showing that the approach was not affected significantly by either absence or presence of AP lyase activity in the glycosylase. Identical treatments of each base modification with non-target components showed no detectable labeling, highlighting process selectivity that was facilitated by the low cross-recognition of each glycosylase (Supplementary Fig. S1–2).
SS-nanopore analyses of the same labeling products demonstrated clear specificity in the resulting electrical signal as well. For the appropriate combinations of base modification and enzymes, we observed exponential voltage-dependent event rates (Fig. 1c), characteristic of the assay28,34. Provided with the same total DNA concentrations (250 nM), the nearly identical event rate trends for both cases further indicated not only the similarity of the yields for the two labeling protocols, but also the reproducibility of the assay. In contrast, mismatched components yielded negligible event rates that were indistinguishable from negative controls across the entire investigated range of applied voltage (Fig. 1c, black). These results suggested that non-specific labeling of DNA was insignificant, including at the 3′ ends of the molecule (Supplementary Fig. S3), and confirmed intrinsic discrimination for an intended base element.
While these data clearly demonstrated a flexible approach that could in principle be extended to a broad range of base targets35, glycosylases can also have additional activities that could interfere with the labeling procedure as described. For example, thymine DNA glycosylase (TDG) is a major component of the cytosine demethylation process, recognizing T:G mismatches36 among other elements37, but it also recruits additional enzymes like histone acetyltransferases38. Because of this latter role, TDG has a high affinity for the AP site resulting from base excision, making it difficult to detach for subsequent labeling steps (Fig. 2a). To address this, we sought to promote enzyme disengagement through the incorporation into the protocol of an additional endonuclease, AP endonuclease 1 (APE1). The extensive dsDNA binding surface of APE1 and the prominent kinking it induces in the DNA helix39 have been suggested as means to promote displacement of glycosylases more efficiently than EndoIV40. However, the improved activity of APE1 comes at the expense of 3′-5′ exonuclease activity not found in the other enzyme, especially under key buffer conditions41. To partially mitigate this effect, we used the APE1 D308A mutant42, which features reduced 3′-5′ exonuclease activity. This inclusion improved yield significantly over wild type APE1 (Supplementary Fig. S4), but the remaining nucleotide digestion activity still necessitated a supplementary purification step prior to polymerase gap-filling to limit decomposition of the DNA. While this increased the number of steps and decreased overall product yield somewhat, the resulting material showed successful incorporation of biotin-labeled nucleotides on gel (Fig. 2b) at a high yield (~73%), as well as selective detection by our SS-nanopore assay (Fig. 2c). Indeed, we recovered the same exponential trend in measured SS-nanopore event rate and the same selectivity over a negative control as found for uracil and oxoG. The event rate dependence was slightly higher for TDG labeling than for previous examples, which could be due to minor residual enzyme binding or small differences in pore attributes (diameter, shape, etc.).
Figure 2.
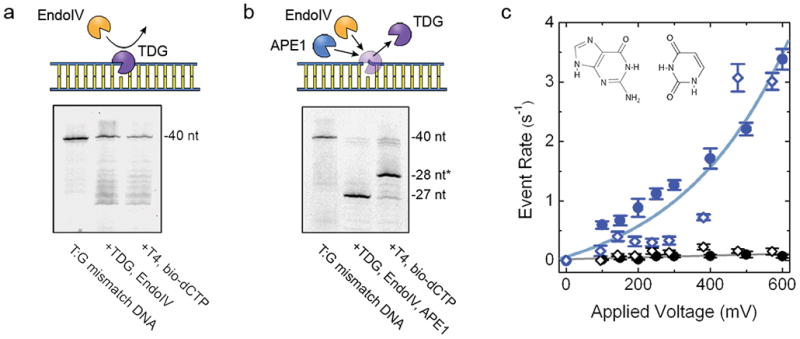
(a) Top: Schematic showing inaccessibility of AP site by EndoIV caused by TDG binding. Bottom: denaturing gel of labeling steps for a T:G mismatch oligonucleotide using EndoIV only. No significant labeling is observed. (b) Top: schematic showing release of TDG by APE1, leaving DNA accessible by EndoIV. Bottom: denaturing gel of labeling steps for a T:G mismatch oligonucleotide using both EndoIV and APE1 (D308A mutant), indicating recovery of high yield labeling. (c) SS-nanopore analysis of 250 nM labeled construct from (b) both with (blue) and without (black) MS. Filled circles and open diamonds are independent measurements on different SS-nanopore devices and lines are exponential fits to the data. Inset: molecular structure of the T:G mismatch base element.
Notably, this alternative method could be used to incorporate other glycosylases with similar behavior as well. As an example, we utilized human alkyladenine DNA glycosylase (hAAG), which excises alkylated bases from DNA, but has also been observed to bind tightly to its DNA template43. The major target of hAAG is the important epigenetic element methyladenine44, but this base is known to be unstable for in vitro measurements. Consequently, we instead used for our demonstration a synthetic oligonucleotide featuring the methyladenine analog 1,N6-ethenoadenine, and employed hAAG and biotin-dATP for labeling. Subsequent analyses of the product again indicated efficient (~74%) labeling on gel (Fig. 3a) and a selective event rate increase in SS-nanopore measurements (Fig. 3b). We noted additional spread in the data at low voltages (<400 mV) specific to the labeled 1,N6-ethenoadenine DNA, which we suggest may have been due to structural irregularities associated with the modified base itself (see Supplementary Fig. S5). We also observed a lower maximum rate at 600 mV, which we attributed to the smaller length28 of this DNA as compared to the other constructs described in this report. However, the selective rate difference is easily resolved, demonstrating the broad modularity of both the labeling scheme and the measurement approach.
Figure 3.
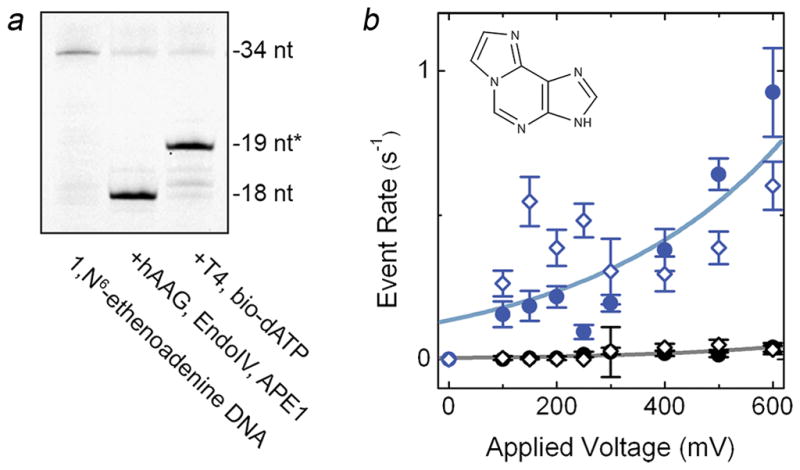
(a) Denaturing gel of labeling steps for a 1,N6-ethenoadenine oligonucleotide using EndoIV and APE1 (D308A mutant), showing high yield biotin labeling. (b) SS-nanopore analysis of 250 nM labeled construct from (a) both with (blue) and without (black) MS. Filled circles and open diamonds are independent measurements on different SS-nanopore devices and lines are exponential fits to the data. Inset: molecular structure of 1,N6-ethenoadenine.
In conclusion, we have shown that a variety of single-base modifications can be assessed with a selective SS-nanopore assay. This was achieved by incorporating an efficient and targeted affinity-labeling technique that exploited the physiological activities of enzymes involved in the BER pathway to install a single biotin tag at the precise location of a given base element. We first showed selective recognition of uracil and oxoG bases with the glycoslyases UDG and hOGG1, respectively. Next, we sought to utilize other glycosylases by integrating a mixture of endonucleases designed to promote enzyme release and limit DNA digestion. While the alternative procedure entailed some loss of material due to increased exonuclease activity, it enabled the use of glycosylases that are specifically challenging to incorporate in the labeling approach due to strong AP binding capacity. As a demonstration, we showed that this approach could be used for the study of T:G mismatch bases with TDG and the methyladenine analog 1,N6-ethenoadenine with hAAG. Therefore, with the flexible protocols established here, nearly any glycosylase could be integrated, facilitating the labeling and analysis of a broad range of bases that they target, including the widely studied methylcytosine45. The central limiting factor for this capacity is in the specificities of the glycosylases themselves, since many have recognition for multiple elements. However, the affinity for specific targets can vary wildly, offering a potential pathway to high selectivity and we expect that the use of point mutations in the glycosylases may also be able to tailor their specificity and enable high certainty in recognition.
These validatory measurements were performed using concentrations of 250 nM, corresponding to ~65 ng of DNA per run in our current protocols. Even taking into account sample loss during the labeling procedure and the general low physiological abundance of modified bases, amounts of genomic DNA capable of supporting this assessment could in principle be obtained from μL volumes of whole blood using commercial kits (e.g. QIAamp DNA Mini Blood Mini Kit). We also note that our approach has been verified34 to resolve concentrations down to at least 10 nM without adjustment, making pertinent, clinically-derived materials still more easily attainable. Coupled with the quantitative nature of the technique28 and its viability among a background of non-target components29,34, our results establish a highly selective and translational SS-nanopore assay. The physiologically-relevant base modifications that it targets may have important impacts on biology and disease, but are challenging to probe through conventional means. Furthermore, the modular labeling approach itself could also be employed independently in applications like affinity enrichment and genomic analyses11–13, and is amenable to the integration of any label that can be incorporated by polymerase activity, including fluorescent tags.
SS-Nanopore measurements
Fabricated silicon chips (4×4 mm), each supporting a 10–20 μm thin film silicon nitride window (20 nm thickness) were obtained commercially (Norcada, Inc., Alberta, Canada). A single SS-nanopore (diameter 7.5–9.0 nm, as determined from resistance measurement27) was produced in each membrane using a helium ion milling technique described elsewhere46. Prior to measurement, a chip was rinsed with deionized water and ethanol, dried under filtered air flow, and then exposed to air plasma (30 W) for 2 min on each side before being placed into a custom Ultem 1000 flow cell that enabled introduction of measurement buffer (1 M NaCl, 10 mM PBS buffer) to independent reservoirs on each side of the device. Ionic current measurements were performed with a patch clamp amplifier (Axopatch 200B) through Ag/AgCl electrodes and used to verify pore diameter. After introducing biomolecules in measurement buffer to the cathode chamber, current was recorded at a bandwidth of 200 kHz with a 100 kHz four-pole Bessel filter. Analysis was performed with custom software and an additional low-pass filter of 25 kHz. The event threshold for analysis was set at 4.5 standard deviations above the RMS noise level and only events with durations between 12.5 μs and 2.5 ms were considered. Each rate measurement was determined by considering at least 3.5 min of uninterrupted trace recording, broken into segments of 3.2 s. The standard deviation between segments was taken as the measurement error.
Gel Electrophoresis
The denaturing gel was prepared by mixing thoroughly 70 mL of 23% gel matrix (22% acrylamide, 1% bis-acrylamide, 7 M urea in 1X tris/borate/EDTA (3:1:1) (TBE) buffer), 240 μL of 25% ammonium persulfate, and 42 μL tetramethylethylenediamine. The gel mixture was cast and allowed to polymerize for 30 minutes before running samples with dye in 1X TBE (3:1:1) at 55 W for 90 minutes. Yields were approximated by measuring product band intensity relative to intermediates in the final lane using ImageJ analysis software47. For electromobility shift assays (EMSA) gels (see Supplementary Fig. S3), 3.5% agarose gels were prepared in 1X TBE buffer with GelRed nucleic acid stain (Phenix Research Products, Candler, NC). Gel images were acquired using a Gel Doc™ system (Bio-Rad, Hercules, CA).
APE1 D308A protein expression
APE1 D308A plasmid (provided by the Demple Lab, Stony Brook University) was transformed into BL21*(DE3) cells and grown in 1 L LB broth at 37°C. After bacterial cell cultures reached OD600=0.6, expression was induced with 0.5 mM isopropyl β-D-thiogalactopyranoside (IPTG). The cultures were then incubated for another 90 minutes before being harvested by centrifugation, resuspended in 50 mM HEPES-KOH (pH 7.5), 100 mM KCl, 1 mM EDTA, 0.1 mM DTT, and 10% (v/v) glycerol, and lysed by two passes through an EmulsiFlex-C5 (Avestin, Ottawa, Canada). The lysate was cleared by centrifugation at 20,000×g for 20 minutes,loaded onto a 15 mL SP Sepharose column (GE Healthcare, Pittsburgh, PA), and eluted with a linear gradient of 100–750 mM KCl. Elutions were analyzed by SDS-PAGE and fractions containing the protein were pooled and dialyzed overnight at 4°C against APE1 storage buffer (50 mM HEPES-KOH (pH 7.5), 200 mM KCl, 1 mM EDTA, 0.1 mM DTT, 10% (v/v) glycerol) and concentrated using 10 kDa MWCO centrifugal spin filter columns (EMD Millipore, Billerica, MA). Final protein concentration was determined with the Bio-Rad Protein Assay (Bio-Rad) and aliquots were stored at −20°C prior to use.
TDG protein expression from E. coli
We followed a protocol adapted by Liu, et al48 from earlier work49 with minor modifications. An expression plasmid for human TDG based on pET28 was transformed into BL21(DE3) cells and grown in 1 L LB broth at 37°C. Once the cultures reached OD600=0.6, they were gradually cooled to 16°C, induced with 0.25 mM IPTG and grown overnight. Cells were harvested by centrifugation, resuspended in 20 mL TDG lysis buffer (50 mM sodium phosphate, pH 8.0, 300 mM NaCl, 25 mM imidazole) with protease inhibitors, and lysed by two passes through an EmulsiFlex-C5. The lysate was cleared by centrifugation at 20,000×g for 20 minutes, loaded onto a 1 mL column of HisPur cobalt resin (Fisher Scientific) equilibrated with TDG lysis buffer, and bound by two applications of the lysate to the column under gravity flow. The column was washed with 20 mL of TDG lysis buffer and subsequently eluted in 1 mL aliquots by a linear gradient of 100–500 mM imidazole. Elutions were analyzed by SDS-PAGE and fractions containing the protein were pooled and dialyzed overnight at 4°C against TDG storage buffer (20 mM HEPES, pH 7.5, 100 mM NaCl, 1 mM DTT, 0.5 mM EDTA, 1% v/v glycerol). Dialyzed proteins were concentrated using 10 kDa MWCO centrifugal spin filter columns. Final TDG concentration was determined with the Bio-Rad Protein Assay and aliquots were stored at −80°C prior to use.
Uracil labeling
A custom 40 nt oligonucleotide with a 5′ FAM (sequence: TCA CGA CTA GTG TTA ACA TGT GCA CCT GCA GAA UGA GAA T) was annealed to a complementary sequence by mixing both at an equimolar ratio, incubating in deionized water at 95°C for 10 minutes, and cooling to room temperature over 1 hour. To cap the 3′ ends of the DNA, a 100 μL aliquot was prepared containing 385 pmol duplex DNA, 30 nmol 2′,3′-dideoxyadenosine 5′-triphosphate (ddATP) (GE Healthcare), 500 U Terminal Transferase (New England Biolabs, Ipswich, MA), and 25 mmol CoCl2 (New England Biolabs) and incubated in 1X Terminal Transferase Reaction Buffer (New England Biolabs) at 37°C for 1.5 hrs. The resulting material was purified with the QIAquick PCR purification kit (Qiagen, Valencia, CA) to allow for buffer exchange. To excise uracil, a 30 μL aliquot was prepared containing 100 pmol of capped duplex DNA, 20 U E. coli UDG (New England Biolabs), 40 U EndoIV (New England Biolabs), 3 μg bovine serum albumin (BSA, New England Biolabs), and incubated in 1X NEB2 buffer (New England Biolabs) at 37°C for 1 hr. Next, 1.5 nmol of biotinylated dUTP (Perkin Elmer, Waltham, MA) and 0.12 U T4(exo-) (Lucigen, Middleton, WI) were added to a final volume of 40 μL in 1X NEB2 buffer and the mixture was incubated at 37°C for 30 minutes. Finally, the mixture was subjected to purification by QIAquick PCR purification kit to remove proteins and excess nucleotides.
OxoG labeling
A custom 40 nt oligonucleotide with a 5′ FAM (sequence: TCA CGA CTA GTG TTA ACA TGT GCA CCT GoCA GAA TGA GAA T, where Go is oxoG) was annealed to a complementary sequence by mixing both at an equimolar ratio, incubating in deionized water at 95°C for 10 minutes, and cooling to room temperature over 1 hour. To excise oxoG, a 30 μL aliquot was prepared containing 100 pmol of duplex, 6.5 U hOGG1 (New England Biolabs), 40 U EndoIV, 3 μg BSA, and incubated in 1X NEB2 buffer at 37°C for 1 hr. Next, 1.5 nmol of biotinylated dGTP (Perkin Elmer) and 0.12 U T4(exo-) were added to a final volume of 40 μL in 1X NEB2 buffer and the mixture was incubated at 37°C for 30 minutes. Finally, the mixture was subjected to QIAquick PCR purification kit purification to remove proteins and excess nucleotides.
T:G mismatch labeling
A custom 40 nt oligonucleotide with a 5′ FAM (sequence: TCA CGA CTA GTG TTA ACA TGT CGA CCT TGA GAA TGA GAA T) was annealed to a complementary sequence (except with a guanine opposite the indicated thymine) by mixing both at an equimolar ratio, incubating in deionized water at 95°C for 10 minutes, and cooling to room temperature over 1 hour. To excise target thymine, a 30 μL aliquot was prepared containing 100 pmol of duplex, 7.5 mg human TDG49, 40 fg APE1 (D308A mutant42), 3 μg BSA, and incubated in 1X HEMN.1 buffer (20 mM HEPES (pH 7.3), 100 mM NaCl, 2.5 mM MgCl2, 0.2 mM EDTA) at 37°C for 1 hr. The mixture was purified with a QIAquick PCR purification kit. Then, 40 U EndoIV and 3 μg BSA were added to a total volume of 30 μL in 1X NEB2 buffer and incubated at 37°C for 30 min. 1.5 nmol biotinylated dCTP (Perkin Elmer, Waltham, MA) and 0.12 U T4(exo-) were added to a final volume of 40 μL in 1X NEB2 buffer and the mixture was further incubated at 37°C for 30 minutes. Finally, the mixture was subjected to a second purification to remove proteins and excess nucleotides.
1,N6-ethenoadenine labeling
A custom 34 nt oligonucleotide with a 5′ FAM (sequence: CAG TTG AGG ATC CCC ATA AeTG CGG CTG TTT TCT G, where Ae is 1,N6-ethenoadenine) was annealed to a complementary sequence by mixing both at an equimolar ratio, incubating in deionized water at 95°C for 10 minutes, and cooling to room temperature over 1 hour. To cap the 3′ ends of the DNA, a 100 μL aliquot containing 385 pmol duplex DNA, 30 nmol ddATP, 500 U of Terminal Transferase, and 25 mmol CoCl2 was incubated in 1X Terminal Transferase reaction buffer at 37°C for 1.5 hrs. The resulting material was purified with a QIAquick PCR purification kit to allow for buffer exchange. To excise target 1,N6-ethenoadenine, a 80 μL aliquot was prepared containing 100 pmol of duplex, 425 U hAAG (New England Biolabs), 200 fg APE1 D308A mutant, 8 μg BSA, and 1X Thermopol buffer (New England Biolabs) and incubated at 37°C for 1 hr. The mixture was purified with a QIAquick PCR purification kit. Then, 40 U EndoIV and 3 μg BSA were added to a total volume of 30 μL in 1X NEB2 buffer and incubated at 37°C for 30 min. 1.5 nmol biotinylated dATP (Perkin Elmer, Waltham, MA) and 0.12 U T4(exo-) were added to a final volume of 40 μL in 1X NEB2 buffer and the mixture was further incubated at 37°C for 30 minutes. Finally, the mixture was subjected to a second QIAquick purification to remove proteins and excess nucleotides.
Supplementary Material
Acknowledgments
We gratefully acknowledge Susan Wallace (University of Vermont) for helpful discussions about DNA damage repair, the Howarth Lab (Oxford University) for supplying MS proteins, Bruce Demple (Stony Brook University) for supplying APE1 D308A expression vector, and the Wake Forest Comprehensive Cancer Center’s Crystallography and Computational Biology shared resource (through NCI Support Grant P30CA012197) for assistance with protein expression. F.W. was supported by a pre-doctoral fellowship through the Wake Forest University Structural and Computational Biophysics training program (T32GM095440). The authors also acknowledge research funding provided by NIH grant R21CA193067 and by a pilot award from the Wake Forest Comprehensive Cancer Center.
Footnotes
Author Contributions
F.W. developed the labeling protocols, expressed proteins, performed SS-nanopore measurements, analyzed data, and contributed to manuscript preparation. O.K.Z. contributed to protein expression, experimental design and data analysis. B.S. performed labeling procedures and SS-nanopore measurements. T.H. and D.P. facilitated protein expression. S.H. and F.W.P. contributed to labeling protocol design and gel analysis. R.M.K. made contributions to protein expression and experimental design. E.W.T. made material contributions to the project. A.R.H. designed and oversaw the project, analyzed data, and wrote the manuscript. All authors reviewed the manuscript.
Competing Interests
A.R.H declares the following financial interest. He is listed as inventor on patents covering the presented labeling approach and the SS-nanopore assay it enhances. The other authors declare no competing interests.
Supporting Information is available free of charge on the ACS Publications website at DOI:XXX. Gel analyses exploring cross-recognition of glycosylases, gels showing the effect of terminal transferase capping on inhibiting off-target labeling, gel analysis of APE1 WT and D308A mutant exonuclease activity, and additional SS-nanopore data for labeled 1,N6-ethenoadenine DNA are shown in Supplementary Figures S1–S7.
References
- 1.Jaenisch R, Bird A. Nat Genet. 2003;33:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 2.Slotkin RK, Martienssen R. Nat Rev Genet. 2007;8:272–285. doi: 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- 3.Zhang G, Huang H, Liu D, Cheng Y, Liu X, Zhang W, Yin R, Zhang D, Zhang P, Liu J, Li C, Liu B, Luo Y, Zhu Y, Zhang N, He S, He C, Wang H, Chen D. Cell. 2015;161:893–906. doi: 10.1016/j.cell.2015.04.018. [DOI] [PubMed] [Google Scholar]
- 4.Reik W, Dean W, Walter J. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 5.Okamoto I, Otte AP, Allis CD, Reinberg D, Heard E. Science. 2004;303:644–649. doi: 10.1126/science.1092727. [DOI] [PubMed] [Google Scholar]
- 6.Egger G, Liang GN, Aparicio A, Jones PA. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 7.Korlach J, Turner SW. Curr Opin Struct Biol. 2012;22:251–261. doi: 10.1016/j.sbi.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Grunau C, Clark SJ, Rosenthal A. Nucleic Acids Res. 2001;29:e65–e72. doi: 10.1093/nar/29.13.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cadet J, Douki T, Ravanat JL. Environ Health Perspect. 1997;105:1034–1039. doi: 10.1289/ehp.105-1470384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker M. Nature. 2015;521:274–276. doi: 10.1038/521274a. [DOI] [PubMed] [Google Scholar]
- 11.Song CX, Clark TA, Lu XY, Kislyuk A, Dai Q, Turner SW, He C, Korlach J. Nat Methods. 2012;9:75–U188. doi: 10.1038/nmeth.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song CX, Szulwach KE, Dai Q, Fu Y, Mao SQ, Lin L, Street C, Li Y, Poidevin M, Wu H, Gao J, Liu P, Li L, Xu GL, Jin P, He C. Cell. 2013;153:678–691. doi: 10.1016/j.cell.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L, Szulwach KE, Hon GC, Song CX, Park B, Yu M, Lu XY, Dai Q, Wang X, Street CR, Tan HP, Min JH, Ren B, Jin P, He C. Nat Commun. 2013;4:1517. doi: 10.1038/ncomms2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu X, Han D, Zhao BS, Song CX, Zhang LS, Doré LC, He C. Cell Res. 2015;25:386–389. doi: 10.1038/cr.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dekker C. Nat Nanotechnol. 2007;2:209–215. doi: 10.1038/nnano.2007.27. [DOI] [PubMed] [Google Scholar]
- 16.Wanunu M. Phys Life Rev. 2012;9:125–158. doi: 10.1016/j.plrev.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li JL, Gershow M, Stein D, Brandin E, Golovchenko JA. Nat Mater. 2003;2:611–615. doi: 10.1038/nmat965. [DOI] [PubMed] [Google Scholar]
- 18.Storm AJ, Chen JH, Zandbergen HW, Dekker C. Phys Rev E. 2005;71:051903. doi: 10.1103/PhysRevE.71.051903. [DOI] [PubMed] [Google Scholar]
- 19.Skinner GM, van den Hout M, Broekmans O, Dekker C, Dekker NH. Nano Lett. 2009;9:2953–2960. doi: 10.1021/nl901370w. [DOI] [PubMed] [Google Scholar]
- 20.van den Hout M, Skinner GM, Klijnhout S, Krudde V, Dekker NH. Small. 2011;7:2217–2224. doi: 10.1002/smll.201100265. [DOI] [PubMed] [Google Scholar]
- 21.Firnkes M, Pedone D, Knezevic J, Doeblinger M, Rant U. Nano Lett. 2010;10:2162–2167. doi: 10.1021/nl100861c. [DOI] [PubMed] [Google Scholar]
- 22.Plesa C, Kowalczyk SW, Zinsmeester R, Grosberg AY, Rabin Y, Dekker C. Nano Lett. 2013;13:658–663. doi: 10.1021/nl3042678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Larkin J, Henley RY, Muthukumar M, Rosenstein JK, Wanunu M. Biophys J. 2014;106:696–704. doi: 10.1016/j.bpj.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Storm AJ, Storm C, Chen JH, Zandbergen H, Joanny JF, Dekker C. Nano Lett. 2005;5:1193–1197. doi: 10.1021/nl048030d. [DOI] [PubMed] [Google Scholar]
- 25.Smeets RMM, Kowalczyk SW, Hall AR, Dekker NH, Dekker C. Nano Lett. 2009;9:3089–3095. doi: 10.1021/nl803189k. [DOI] [PubMed] [Google Scholar]
- 26.Marshall MM, Ruzicka J, Zahid OK, Henrich VC, Taylor EW, Hall AR. Langmuir. 2015;31:4582–4588. doi: 10.1021/acs.langmuir.5b00457. [DOI] [PubMed] [Google Scholar]
- 27.Wanunu M, Dadosh T, Ray V, Jin J, McReynolds L, Drndic M. Nat Nanotechnol. 2010;5:807–814. doi: 10.1038/nnano.2010.202. [DOI] [PubMed] [Google Scholar]
- 28.Zahid OK, Zhao BS, He C, Hall AR. Sci Rep. 2016;6:29565. doi: 10.1038/srep29565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlsen AT, Zahid OK, Ruzicka JA, Taylor EW, Hall AR. Nano Lett. 2014;14:5488–5492. doi: 10.1021/nl501340d. [DOI] [PubMed] [Google Scholar]
- 30.Howarth M, Chinnapen DJF, Gerrow K, Dorrestein PC, Grandy MR, Kelleher NL, El-Husseini A, Ting AY. Nat Methods. 2006;3:267–273. doi: 10.1038/NMETHXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riedl J, Ding Y, Fleming AM, Burrows CJ. Nat Commun. 2015;6:8807. doi: 10.1038/ncomms9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kavli B, Otterlei M, Slupphaug G, Krokan HE. DNA Repair. 2007;6:505–516. doi: 10.1016/j.dnarep.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 33.Grollman AP, Moriya M. Trends Genet. 1993;9:246–249. doi: 10.1016/0168-9525(93)90089-z. [DOI] [PubMed] [Google Scholar]
- 34.Zahid OK, Wang F, Ruzicka JA, Taylor EW, Hall AR. Nano Lett. 2016;16:2033–2039. doi: 10.1021/acs.nanolett.6b00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacobs AL, Schär P. Chromosoma. 2012;121:1–20. doi: 10.1007/s00412-011-0347-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiebauer K, Jiricny J. Proc Natl Acad Sci U S A. 1990;87:5842–5845. doi: 10.1073/pnas.87.15.5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maiti A, Drohat AC. J Biol Chem. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tini M, Benecke A, Evans RM, Chambon P. Mol Cell. 2002;9:265–277. doi: 10.1016/s1097-2765(02)00453-7. [DOI] [PubMed] [Google Scholar]
- 39.Mol CD, Izumi T, Mitra S, Tainer JA. Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 40.Waters TR, Gallinari P, Jiricny J, Swann PF. J Biol Chem. 1999;274:67–74. doi: 10.1074/jbc.274.1.67. [DOI] [PubMed] [Google Scholar]
- 41.Chou KM, Cheng YC. J Biol Chem. 2003;278:18289–18296. doi: 10.1074/jbc.M212143200. [DOI] [PubMed] [Google Scholar]
- 42.Masuda Y, Bennett RAO, Demple B. J Biol Chem. 1998;273:30360–30365. doi: 10.1074/jbc.273.46.30360. [DOI] [PubMed] [Google Scholar]
- 43.Abner CW, Lau AY, Ellenberger T, Bloom LB. J Biol Chem. 2001;276:13379–13387. doi: 10.1074/jbc.M010641200. [DOI] [PubMed] [Google Scholar]
- 44.Wyatt MD, Allan JM, Lau AY, Ellenberger TE, Samson LD. BioEssays News Rev Mol Cell Dev Biol. 1999;21:668–676. doi: 10.1002/(SICI)1521-1878(199908)21:8<668::AID-BIES6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 45.Morales-Ruiz T, Ortega-Galisteo AP, Ponferrada-Marín MI, Martínez-Macías MI, Ariza RR, Roldán-Arjona T. Proc Natl Acad Sci. 2006;103:6853–6858. doi: 10.1073/pnas.0601109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang J, Ferranti DC, Stern LA, Sanford CA, Huang J, Ren Z, Qin LC, Hall AR. Nanotechnology. 2011;22:285310. doi: 10.1088/0957-4484/22/28/285310. [DOI] [PubMed] [Google Scholar]
- 47.Collins TJ. BioTechniques. 2007;43:25–30. doi: 10.2144/000112517. [DOI] [PubMed] [Google Scholar]
- 48.Liu MY, Torabifard H, Crawford DJ, DeNizio JE, Cao XJ, Garcia BA, Cisneros GA, Kohli RM. Nat Chem Biol. 2017;13:181–187. doi: 10.1038/nchembio.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morgan MT, Bennett MT, Drohat AC. J Biol Chem. 2007;282:27578. doi: 10.1074/jbc.M704253200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.