

Summary
Background
Diadenosine 5′,5‴-P1,P4-tetraphosphate (Ap4A), a natural compound stored in platelet dense granules, inhibits ADP-induced platelet aggregation. Ap4A inhibits the platelet ADP receptors P2Y1 and P2Y12, is a partial agonist of P2Y12, and is a full agonist of the platelet ATP-gated ion channel P2X1. Modification of the Ap4A tetraphosphate backbone enhances inhibition of ADP-induced platelet aggregation. However, the effects of these Ap4A analogs on human platelet P2Y1, P2Y12 and P2X1 are unclear.
Objective
To determine the agonist and antagonist activities of diadenosine tetraphosphate analogs towards P2Y1, P2Y12, and P2X1.
Methods
We synthesized the following Ap4A analogs: P1,P4-dithiotetraphosphate; P2,P3-chloromethylenetetraphosphate; P1-thio-P2,P3-chloromethylenetetraphosphate; and P1,P4-dithio-P2,P3-chloromethylenetetraphosphate. We then measured the effects of these analogs on: (i) ADP-induced platelet aggregation; (ii) P2Y1-mediated changes in cytosolic Ca2+; (iii) P2Y12-mediated changes in vasodilator-stimulated phosphoprotein phosphorylation; and (iv) P2X1-mediated entry of extracellular Ca2+.
Results
Ap4A analogs with modifications in the phosphate backbone inhibited both P2Y1 and P2Y12, and showed no agonist activity towards these receptors. The dithio modification increased inhibition of P2Y1, P2Y12, and platelet aggregation, whereas the chloromethylene modification increased inhibition of P2Y12 and platelet aggregation, but decreased P2Y1 inhibition. Combining the dithio and chloromethylene modifications increased P2Y1 and P2Y12 inhibition. As compared with Ap4A, each modification decreased agonist activity towards P2X1, and the dual modification completely eliminated P2X1 agonist activity.
Conclusions
As compared with Ap4A, tetraphosphate backbone analogs of Ap4A have diminished activity towards P2X1 but inhibit both P2Y1 and P2Y12 and, with greater potency, inhibit ADP-induced platelet aggregation. Thus, diadenosine tetraphosphate analogs with dual receptor selectivity may have potential as antiplatelet drugs.
Keywords: diadenosine, platelet, receptor
Introduction
Combined antiplatelet therapy with aspirin and an inhibitor of the platelet ADP receptor P2Y12 significantly reduces the risk of ischemic events in patients with acute coronary syndromes and those undergoing percutaneous coronary intervention [1]. However, ischemic events still occur, and these agents do not inhibit platelet activation mediated through other receptors, thereby raising the question of whether inhibition of other pathways of platelet activation would be clinically beneficial.
Platelets possess three purinergic receptors: P2Y12, a Gi-linked ADP receptor that mediates the propagation of stable platelet aggregation and is the target of the Food and Drug Administration (FDA)-approved antiplatelet agents clopidogrel, prasugrel, and ticagrelor; P2Y1, a Gq-linked ADP receptor whose activation results in Ca2+ mobilization from intracellular pools, platelet shape change, and rapidly reversible platelet aggregation; and P2X1, an ATP-stimulated ligand-gated ion channel whose activation results in entry of extracellular Ca2+ and platelet shape change [2].
Diadenosine 5′,5‴-P1,P4-tetraphosphate (Ap4A), a natural compound stored in platelet dense granules, is released along with ADP, ATP and other diadenosine polyphosphates (Ap3–7A) upon platelet activation [3]. Ap4A inhibits ADP-induced platelet activation [4] but, until recently, its specific activity towards P2X1, P2Y1 and P2Y12 was unclear. We have demonstrated that Ap4A is an antagonist of platelet P2Y1 and P2Y12, a partial agonist of P2Y12, and an agonist of P2X1 [5]. Other previous studies also showed that the antiplatelet effect of Ap4A is enhanced by modification of the polyphosphate chain, yielding derivatives that resist degradation in plasma and may be clinically useful antithrombotic agents [4,6]. However, the relative antagonist potencies of these analogs against P2Y1 and P2Y12 have not been characterized, and their effects on platelet P2X1 have not been studied. Platelet P2X1 is involved in clot formation in high shear force conditions, such as arterial stenosis [7], and unmodified diadenosine polyphosphates are agonists of P2X1 expressed on a variety of human and rat cell types [8,9]. If Ap4A analogs are also P2X1 agonists, their potential as therapeutic antiplatelet agents may be limited. Therefore, the goal of the present study was to investigate the antiplatelet potency of Ap4A modified tetraphosphate backbone analogs, and their structure–activity relationships with regard to signaling through P2Y1, P2Y12, and P2X1.
Methods
Chemicals and reagents
Ap4A and its P1,P4-dithio derivative (subsequently referred to in this article as compound 1), P2,P3-chloromethylene derivative (subsequently referred to as compound 2) and P1,P4-dithio-P2,P3-chloromethylene derivative (subsequently referred to as compound 4) were synthesized as previously described [10]. The unsymmetrical P1-thio-P2,P3-chloromethylene derivative (subsequently referred to as compound 3) was prepared by extension of a method for the synthesis of unsymmetrical bis-nucleoside tetraphosphates [11]. The purity of compounds 1–4 was > 95%, as determined by reverse-phase HPLC, LCMS, and proton and phosphorus NMR (data not shown). The structures of Ap4A and its analogs are shown in Fig. 1. MRS2179, probenecid, adenosine 5′-(β,γ-methylene)triphosphate (β,γ-CH2-ATP), EGTA and apyrase (grade VII) were from Sigma-Aldrich (St Louis, MO, USA). D-Phenylalanyl-L-prolyl-L-arginine chloromethyl ketone (PPACK) was from Calbiochem (EMD Biosciences, La Jolla, CA, USA). FLUO-4 was from Invitrogen (Carlsbad, CA, USA). ADP was from Bio/Data (Horsham, PA, USA). CD41–phycoerythrin–Cy5 was from Beckman Coulter (Fullerton, CA, USA).
Fig. 1.
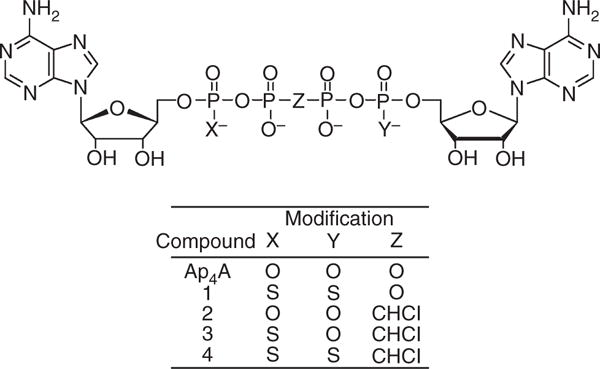
Chemical structure of diadenosine 5′,5‴-P1,P4-tetraphosphate (Ap4A) and its analogs. Compounds: 1, diadenosine 5′,5″-P1,P4-dithiotetraphosphate; 2, diadenosine 5′,5‴-P2,P3-chloromethylenetetraphosphate; 3, diadenosine-5′,5‴-P1-thio-P2,P3-chloromethylenetetraphosphate; 4, diadenosine-5′,5‴-P1,P4-dithio-P2,P3-chloromethylenetetraphosphate.
Blood collection and sample preparation
After Instutional Review Board-approved written informed consent had been obtained, blood was collected into tubes containing 3.2% sodium citrate from healthy aspirin-free (7 days) and non-steroidal anti-inflammatory drug-free (3 days) volunteers. Anticoagulated whole blood was used in vasodilator-stimulated phosphoprotein (VASP) phosphoryla tion and cytosolic Ca2+ assays. Platelet-rich plasma (PRP) and platelet-poor plasma for platelet aggregation assays were prepared as previously described [5]. For assays of platelet P2X1 function, blood was drawn into tubes containing PPACK (0.3 mM final concentration), apyrase (1.8 μM final concentration), and PRP prepared as previously described [5].
ADP-induced platelet shape change and platelet aggregation
ADP-stimulated (3 μM) platelet shape change in PRP with 10 mM EDTA was evaluated as previously described [5]. Light transmission platelet aggregation in response to ADP (3 μM) was measured with the 96-well microplate reader method, as previously described [5].
P2Y12-mediated VASP phosphorylation assay
P2Y12-mediated changes in VASP phosphorylation were measured by flow cytometry with a kit from BioCytex (Marseilles, France).
P2Y1-mediated cytosolic Ca2+ increase
The ADP-dependent, P2Y1-mediated increase in platelet cytosolic Ca2+ was measured by detection of changes in FLUO-4 fluorescence, as previously described [5]. The cytosolic Ca2+ increase was calculated as the ratio of the maximal post-stimulation FLUO-4 fluorescence to the baseline FLUO-4 fluorescence. The percentage inhibition of the ADP-induced Ca2+ increase caused by the addition of Ap4A analogs was calculated relative to ADP (3 μM) plus vehicle (Hepes–saline).
To test Ap4A analogs for their potential P2Y1 agonist properties, a high concentration of each compound (100 μM for compound 1, 250 μM for compound 2, 200 μM for compound 3, and 80 μM for compound 4), was added as a stimulant to whole blood incubated with FLUO-4 in the absence of additional ADP.
P2X1-mediated entry of extracellular Ca2+
Measurement of P2X1-mediated entry of extracellular Ca2+ based on changes in FLUO-4 fluorescence was performed as previously described [5]. The non-hydrolyzable ATP analog β,γ-CH2-ATP (20 μM) was used as a positive control. To confirm that any increases in intracellular Ca2+ observed were unrelated to P2Y1 activation, experiments were repeated with 100 μM MRS2179, a selective P2Y1 inhibitor, in the ambient buffer. The ability of high concentrations of Ap4A analogs to antagonize P2X1 activation by 20 μM β,γ-CH2-ATP was also tested.
Statistical analysis
The activation or inhibition parameters were analyzed with GRAPHPAD PRISM software, version 4.00 for Windows (Graph-Pad Software, San Diego, CA, USA). All data are expressed as mean ± standard error of the mean. Student’s t-test was used to determine statistical significance when two groups of data were compared. One-way ANOVA and Bonferroni’s multiple comparison test were used when three or more groups of data were compared.
Results
Inhibition of ADP-induced platelet shape change and platelet aggregation
All four Ap4A analogs inhibited ADP-stimulated (3 μM) platelet shape change (data not shown) and both primary and secondary platelet aggregation (Fig. 2). Figure 2A shows representative aggregation tracings for compound 4. Figure 2B shows the concentration-dependent inhibition of 3 μM ADP-induced aggregation of human platelets. Corresponding IC50s are shown in Table 1. As previously reported [4,6], both the P1,P4-dithio modification (compound 1) and the P2,P3-chloromethylene modification (compound 2) enhanced the inhibition of platelet aggregation: approximately two-fold for compound 1, and approximately six-fold for compound 2. The effect of these modifications was additive; that is, the greatest inhibition was observed with the dually modified P1,P4-dithio-P2,P3-chloromethylene derivative (compound 4), which was an approximately 60-fold more potent inhibitor than Ap4A. The previously unreported non-symmetrical P1-thio-P2,P3-chloromethylene Ap4A analog (compound 3) showed an inhibitory potency intermediate between those of compound 2 and compound 4.
Fig. 2.
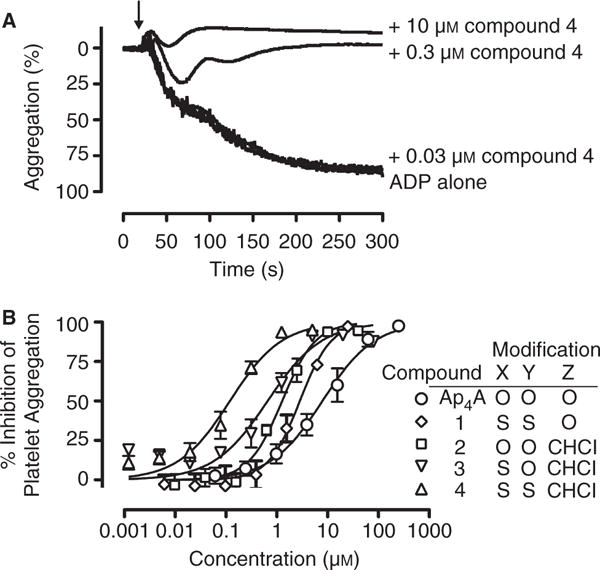
Inhibition of ADP-induced platelet aggregation by diadenosine 5′,5‴-P1,P4-tetraphosphate (Ap4A) analogs. (A) Representative turbidometric aggregation tracings for platelet-rich plasma stimulated with ADP alone (final concentration 3 μM) and in combination with 0.03, 0.3 or 10 μM compound 4. (B) Percentage inhibition of maximal platelet aggregation. The results shown are mean ± standard error of the mean. Data are from three independent experiments. Compounds: 1, diadenosine 5′,5‴-P1,P4-dithiotetraphosphate; 2, diadenosine 5′,5‴-P2,P3-chloromethylenetetraphosphate; 3, diadenosine-5′,5‴-P1-thio-P2,P3-chloromethylenetetraphosphate; 4, diadenosine-5′,5‴-P1,P4-dithio-P2,P3-chloromethylenetetraphosphate.
Table 1.
Inhibition of platelet aggregation and P2 receptor selectivity by diadenosine 5′,5‴-P1,P4-tetraphosphate (Ap4A) and its analogs
| Platelet aggregation
|
P2Y12 VASP
|
P2Y1 Ca2+ flux
|
|
|---|---|---|---|
| IC50 (μM) (95% CI) |
IC50 (μM) (95% CI) |
IC50 (μM) (95% CI) |
|
| Ap4A | 7.9 (5.5–11.4) | > 250 | 32.5 (22.1–47.8) |
| Compound 1 | 2.8 (2.0–4.0) | 88.6 (59.8–131.3) | 2.9 (2.0–4.2) |
| Compound 2 | 1.3 (1.0–1.7) | 19.3 (10.2–36.5) | 111.5 (51.8–239.9) |
| Compound 3 | 0.66 (0.43–1.01) | 23.9 (12.8–44.3) | 57.2 (15.5–211.5) |
| Compound 4 | 0.12 (0.08–0.17) | 6.4 (3.7–11.4) | 10.2 (6.7–15.7) |
CI, confidence interval; VASP, vasodilator-stimulated phosphoprotein. Compounds: 1, diadenosine 5′,5‴-P1,P4-dithiotetraphosphate; 2, diadenosine 5′,5‴-P2,P3-chloromethylenetetraphosphate; 3, diadenosine 5′,5‴-P1-thio-P2,P3-chloromethylenetetraphosphate; 4, diadenosine 5′,5‴-P1,P4-dithio-P2,P3-chloromethylenetetraphosphate.
Agonist and antagonist effects of Ap4A derivatives on the P2Y12-mediated decrease in VASP phosphorylation
VASP phosphorylation at Ser239, measured by flow cytometry with a specific mAb, was elevated in prostaglandin E1-treated platelets (Fig. 3, solid gray bars) and, as expected, was reduced by addition of the P2Y12 agonist ADP (Fig. 3, open bars). We previously reported that Ap4A is a partial P2Y12 agonist, and also produces a dose-dependent reduction in VASP phosphorylation [5]. In contrast, none of the four Ap4A analogs reduced VASP phosphorylation (Fig. 3A–D, striped bars), indicating that, at the concentrations tested, the compounds are not P2Y12 agonists. Compounds 1–4 each dose-dependently antagonized the ADP-induced reduction of VASP phosphorylation (Fig. 3A–D, hash-marked open bars). The concentrations of compounds 1–4 required for 50% inhibition of ADP-induced reduction of VASP phosphorylation were ~ 15–50-fold greater than that needed to inhibit ADP-induced platelet aggregation (Table 1). Nevertheless, the Ap4A and compound 1–4 IC50s for the ADP-induced reduction of VASP phosphorylation correlated with the IC50s for ADP-induced platelet aggregation (Pearson r2 = 0.98, P = 0.005).
Fig. 3.
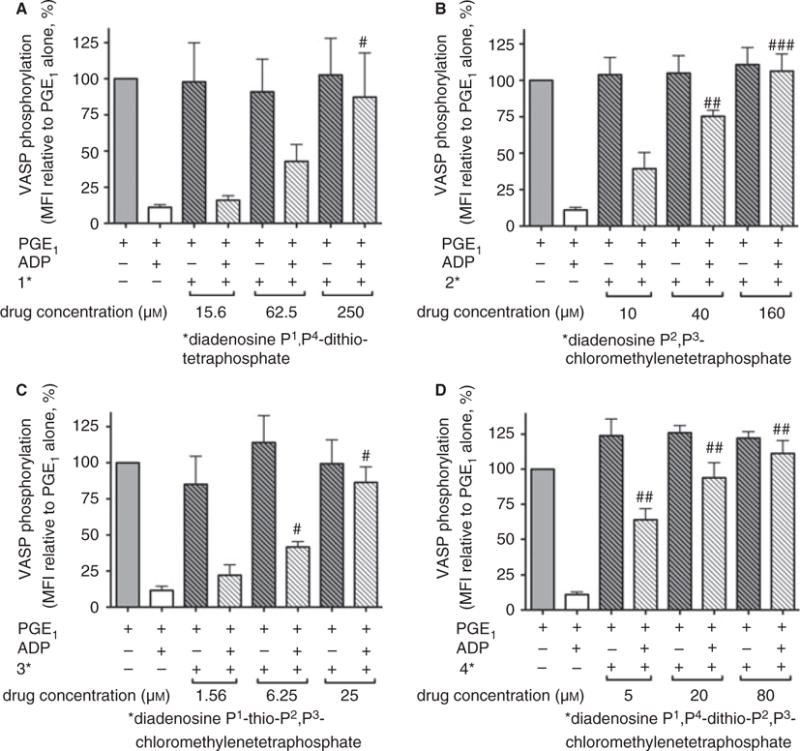
Inhibition of ADP-induced, P2Y12-mediated decrease in vasodilator-stimulated phosphoprotein (VASP) phosphorylation by diadenosine 5′,5‴-P1,P4-tetraphosphate (Ap4A) analogs. Prostaglandin E1 (PGE1)-stimulated VASP phosphorylation and its attenuation by ADP in the presence and absence of Ap4A analogs were measured by flow cytometry. (A) Compound 1. (B) Compound 2. (C) Compound 3. (D) Compound 4. The results shown are mean ± standard error of the mean. Data are from three independent experiments (P < 0.05, ##P < 0.01, and ### P < 0.001, as compared with PGE1 plus ADP). MFI, mean fluorescence intensity. Compounds: 1, diadenosine 5′,5‴-P1,P4-dithiotetraphosphate; 2, diadenosine 5′,5‴-P2,P3-chloromethylenetetraphosphate; 3, diadenosine-5′,5‴-P1-thio-P2,P3-chloromethylenetetraphosphate; 4, diadenosine-5′,5‴-P1,P4-dithio-P2,P3-chloromethylenetetraphosphate.
Agonist and antagonist effects of Ap4A derivatives on P2Y1-mediated cytosolic Ca2+ increase
Addition of 100 μM compound 1, 250 μM compound 2, 200 μM compound 3 or 80 μM compound 4 to FLUO-4-loaded platelets in the absence of ADP did not result in increased fluorescence (data not shown), indicating that, at these concentrations, these analogs lack P2Y1 agonist activity.
The dose-dependent inhibition of the ADP-induced P2Y1-mediated cytosolic Ca2+ increase by the four Ap4A analogs is shown in Fig. 4, and the corresponding IC50s are shown in Table 1. Relative to Ap4A, compound 1 had ~ 10-fold increased P2Y1 antagonistic potency, but compound 2 and compound 3 had < 50% of the potency of Ap4A. Compound 4 had a P2Y1 antagonist effect that was approximately three-fold greater than that of Ap4A, but less than that of compound 1. The concentrations of compounds 2–4 required for 50% inhibition of the ADP-stimulated Ca2+ increase were 85–150-fold greater than that needed for inhibition of ADP-induced platelet aggregation (Table 1). In contrast, compound 1 inhibited ADP-stimulated Ca2+ flux and ADP-stimulated platelet aggregation with nearly identical IC50s (Table 1). The Ap4A and compound 1–4 IC50s for ADP-stimulated Ca2+ flux and for ADP-stimulated platelet aggregation were not significantly correlated (P = 0.77).
Fig. 4.
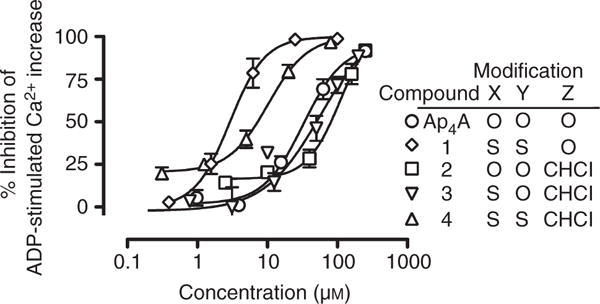
Inhibition of ADP-induced, P2Y1-mediated platelet Ca2+ increase by diadenosine 5′,5‴-P1,P4-tetraphosphate (Ap4A) analogs. The increase in platelet cytosolic Ca2+ in response to 3 μM ADP, with and without addition of Ap4A analogs, was measured by whole blood flow cytometry with the Ca2+ indicator FLUO-4. The percentage inhibition was calculated relative to ADP + vehicle (0% inhibition) and vehicle alone (100% inhibition). The results shown are mean ± standard error of the mean. Data are from three or four independent experiments. Compounds: 1, diadenosine 5′,5‴-P1,P4-dithiotetraphosphate; 2, diadenosine 5′,5‴-P2,P3-chloromethylenetetraphosphate; 3, diadenosine-5′,5‴-P1-thio-P2,P3-chloromethylenetetraphosphate; 4, diadenosine-5′,5‴-P1,P4-dithio-P2,P3-chloromethylenetetraphosphate.
Agonist effects of Ap4A derivatives on P2X1-mediated cytosolic Ca2+ influx
We previously reported that Ap4A is a potent (maximal response at 1 μM) agonist of P2X1 on human platelets, as shown by the influx of extracellular Ca2+ [5]. Figure 5 shows the increase in FLUO-4 fluorescence caused by entry of extracellular Ca2+ with the Ap4A derivatives. Relative to Ap4A, compound 1-stimulated Ca2+ entry was reduced (maximal extracellular Ca2+ influx at 10 μM), and compound 2-stimulated Ca2+ entry was strongly reduced (sub-maximal extracellular Ca2+ influx at 200 μM), and the effect of compound 3 was intermediate between those of compound 1 and compound 2, inducing strong Ca2+ influx at 100 μM (Fig. 5). Compound 4 did not induce influx of extracellular Ca2+ at concentrations up to 200 μM. To determine whether the observed Ap4A derivative-stimulated increase in cytosolic Ca2+ was attributable to P2Y1-mediated release of Ca2+ from intracellular stores, rather than P2X1-mediated influx of extracellular Ca2+, we evaluated FLUO-4 changes stimulated by compound 1 in the presence of the specific P2Y1 inhibitor MRS2179 (100 μM) and in samples where extracellular Ca2+ was chelated by EGTA (1 mM). MRS2179 did not alter the compound 1-stimulated increase in cytosolic Ca2+, whereas EGTA at 1 mM completely eliminated changes in cytosolic Ca2+ at all concentrations of compound 1 (data not shown).
Fig. 5.
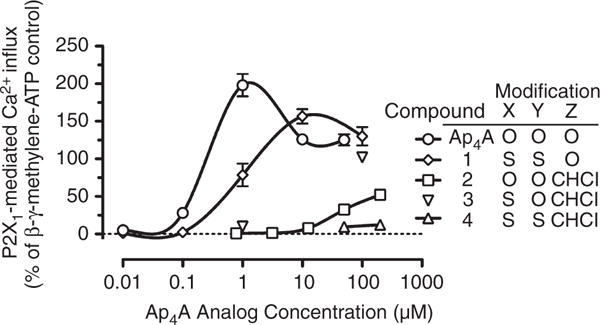
Agonist effects of diadenosine 5′,5‴-P1,P4-tetraphosphate (Ap4A) analogs on P2X1-mediated entry of extracellular Ca2+. Increasing concentrations of Ap4A analogs were added to FLUO-4-loaded platelets, and the change in FLUO-4 fluorescence was measured by flow cytometry. The results shown are mean ± standard error of the mean normalized to the response to adenosine 5′-(β,γ-methylene)triphosphate (β,γ-CH2-ATP) (20 μM) alone; compound 3 was evaluated at only 1 and 100 μM. The results for Ap4A and compounds 1, 2 and 4 are from four to eight independent experiments. Compounds: 1, diadenosine 5′,5‴-P1,P4-dithiotetraphosphate; 2, diadenosine 5′,5‴-P2,P3-chloromethylenetetraphosphate; 3, diadenosine-5′,5‴-P1-thio-P2,P3-chloromethylenetetraphosphate; 4, diadenosine-5′,5‴-P1,P4-dithio-P2,P3-chloromethylenetetraphosphate.
Maximal P2X1-mediated Ca2+ influx occurred at 10 μM compound 1; a higher concentration (100 μM compound 1) not only failed to generate a greater response, but caused significantly lower Ca2+ influx (Fig. 5). A similar biphasic dose response has been observed with the non-hydrolyzable ATP analog β,γ-CH2-ATP (data not shown) and with Ap4A [5].
None of the four Ap4A analogs, at any concentration tested, reduced the FLUO-4 fluorescence increase caused by 20 μM β,γ-CH2-ATP, indicating that these compounds are not platelet P2X1 antagonists (data not shown).
Discussion
The main findings of this study are as follows. (i) All four Ap4A analogs inhibit both platelet P2Y1 and P2Y12 function, but have no agonist activity towards either receptor. Thus, inhibition of both receptors contributes to the previously reported inhibition of ADP-induced platelet aggregation by compounds 1, 2, and 4 [4,6], and to the currently reported inhibition of platelet aggregation by compound 3. (ii) Dithio modification at P1 and P4 increased the inhibition of P2Y1, P2Y12, and platelet aggregation, and the P2,P3-chloromethylene modification increased the inhibition of P2Y12 and platelet aggregation, but decreased the inhibition of P2Y1. The simultaneous effect of both modifications was increased inhibition of both P2Y1 and P2Y12. (iii) Inhibition of platelet aggregation by Ap4A and compounds 1–4 correlates with their inhibition of the P2Y12-mediated VASP phosphorylation decrease, but not with their inhibition of P2Y1-mediated Ca2+ flux. However, the concentrations of compounds 2–4 required to inhibit platelet aggregation are many times lower than that required to inhibit either P2Y1 or P2Y12 activity, suggesting a synergistic effect, as previously suggested by the combined use of individual selective antagonists of P2Y1 and P2Y12 [12,13]. (iv) The P2X1 agonist property indigenous to Ap4A was diminished by the polyphosphate chain modifications, and completely abolished in the dithiochloromethylene derivative, compound 4, the most potent inhibitor of platelet aggregation. In sum, the potency and novel dual receptor antagonism mechanism of Ap4A analogs suggest that they have potential as antiplatelet drugs.
Platelet purinergic receptors are important targets for the development of antiplatelet agents. Thus far, only platelet P2Y12 inhibitors have been successfully applied in clinical practice [14]. P2Y1-selective antagonists have been developed and proposed as antiplatelet agents, but none has advanced to clinical development [15–22]. Likewise, Ap4A analogs with a modified polyphosphate chain have been proposed as potential antiplatelet drugs [4,6,23], but their development has not been pursued. Furthermore, the specific mechanism(s) for their enhanced potency and the structure–activity relationship with respect to platelet purinergic P2Y1 and P2Y12, and the effect on platelet P2X1, have not previously been described. Our recent study [5] showed that Ap4A can inhibit both platelet P2Y1 and P2Y12, but that the IC50 for inhibition of each of these receptors was much greater than the IC50 for inhibition of ADP-stimulated platelet aggregation, suggesting a possible synergistic effect of such dual inhibition.
Structure–activity relationships of Ap4A derivatives with regard to human platelet P2Y1 and P2Y12
The present study demonstrates that, like Ap4A, all four Ap4A derivatives possess antagonist activities towards both P2Y1 and P2Y12 receptors. However, the thio modification and chloromethylene modification have distinctly different effects on the ability of the corresponding Ap4A analogs to inhibit platelet P2Y1 and P2Y12. The dithio modification (compound 1) strongly increased (~ 10-fold relative to Ap4A) the inhibitory effect on P2Y1, whereas the chloromethylene modification (compound 2) decreased it (approximately three-fold relative to Ap4A). In agreement with the relative sizes of these two opposing effects, the dually modified compound 4 was apprximately three-fold stronger as an inhibitor of P2Y1 than Ap4A. Interestingly, the monothio derivative (compound 3) was not significantly different from compound 2, which has no thio substitution, in its P2Y1 inhibitory properties, suggesting that both P1 and P4 need to be thio-modified for the inhibition-enhancing effect of this thio modification to take place, and that perhaps both P1 and P4 are involved in a direct interaction with P2Y1.
In the case of P2Y12, and similar to the effect on P2Y1, the dithio modification increased (more than three-fold relative to Ap4A) the inhibitory properties of the Ap4A scaffold. However, in contrast to the negative effect that the chloromethylene modification has on the inhibition of P2Y1, the chloromethylene modification strongly increased the inhibition of P2Y12 (> 10-fold relative to Ap4A). Addition of a single thio group in the presence of the chloromethylene modification (i.e. compound 3) did not further enhance the inhibition of P2Y12, whereas the dithio modification did (approximately three-fold, compound 4 relative to compound 2). These results suggests that, as for P2Y1, both P1 and P4 need to be thio-modified for the P2Y12 inhibition-enhancing effect, and that perhaps both P1 and P4 are involved in a direct interaction with P2Y12.
The finding that Ap4A and its analogs antagonize both platelet P2Y1 and P2Y12 is remarkable when contrasted with distinctively different interactions of ATP and its analogs with these receptors. ATP is an inhibitor, albeit a weak one, of P2Y12 [24], and its modification led to the development of the highly active and selective P2Y12 inhibitor cangrelor (ARC69931MX) [25]. On the other hand, ATP is an agonist of P2Y1 [26], and ATP analogs have been developed as highly potent P2Y1 agonists. For example, 2-methylthio-ATP activates human P2Y1 (hP2Y1) in transfected HEK cells with an EC50 of 1 nM [27].
Structure–activity relationships of Ap4A derivatives with regard to human platelet P2X1
The emerging role of P2X1 in the activation of platelets has been documented [7]. Platelet P2X1 is activated by ATP, and is believed to be involved in platelet activation under high shear stress conditions, e.g. in partially occluded blood vessels. In addition, platelet P2X1 synergizes with other platelet purinergic receptors to enhance downstream signal transduction, such as the Ca2+ increase mediated by the P2Y1 pathway and the cAMP decrease mediated by the P2Y12 pathway. Ap4A is an agonist of rat and human P2X receptors from various tissues [28–30]. In platelets, Sage et al. [31] showed that Ap4A may induce a rise in cytosolic Ca2+, and suggested that this was mediated by P2X1. We have previously shown [5] that Ap4A increases platelet cytosolic Ca2+ levels by P2X1-mediated extracellular Ca2+ influx, and that this effect has an unusual dose-dependence, with a maximum at 1 μM Ap4A. In the present study, we have found that the P1,P4-thio and the P2,P3-chloromethylene modifications reduce the P2X1 agonist properties of the Ap4A scaffold. The dose–response curve of compound 1 was similar to that of Ap4A (Fig. 5), with a maximum effect ~ 150% of that of 20 μM β,γ-CH2-ATP, but with an approximately one order of magnitude rightward shift. The agonist properties were even more significantly reduced by the P2,P3-chloromethylene modifications, with compound 2 achieving only 50% of the 20 μM β,γ-CH2-ATP response at the highest studied concentration (200 μM). The effect of these modifications is additive, because compound 4, even at 200 μM, appears to be devoid of P2X1 agonist effect (Fig. 5). Elimination of P2X1 agonist activity is important in the development of antiplatelet agents, not only to avoid P2X1-mediated platelet activation, but also to avoid activation of P2X1 on cells in other tissues [32].
Like Ap4A, most analogs in the present study have IC50s for inhibition of ADP-induced platelet aggregation that are much lower than their IC50s for antagonism of P2Y1 or P2Y12 (Table 1), thus reaffirming the possibility of synergism in the simultaneous inhibition of P2Y1 and P2Y12. Both P2Y1 and P2Y12 are necessary for full-scale ADP-induced platelet aggregation [33,34]. Other derivatives of ADP, coenzyme A and acetyl-coenzyme A, also inhibit ADP-induced platelet aggregation [35], and inhibit both P2Y1 and P2Y12, albeit with micromolar activities [36], leading to the proposal that agents that act at both of these receptors may provide more protection against the effects of ADP than agents acting at only one of these receptors [36]. Synergism from inhibition of both platelet ADP receptors was previously suggested by the combined use of antagonists with individual selectivity for either P2Y1 or P2Y12: MRS2179 or A3P5P for P2Y1, and AR-C69931MX for P2Y12 [12,13]. This effect may be explained by the complex interplay between platelet ADP receptors [37,38]. However, in the case of Ap4A and its analogs, we cannot exclude the possibility that diadenosine polyphosphate compounds as a class inhibit platelet aggregation through an additional separate, as yet unknown, mechanism.
Conclusions
Ap4A analogs with modifications in the phosphate backbone inhibit both P2Y1 and P2Y12, and show no agonist activity towards these receptors. The dithio modifications at P1 and P4 increase the inhibitory activity towards P2Y1, P2Y12, and platelet aggregation, and the P2,P3-chloromethylene modification increases P2Y12 and platelet aggregation inhibition, but decreases P2Y1 inhibition. The simultaneous effect of both modifications is an increase in P2Y1 and P2Y12 inhibition. Both modifications decrease the agonist activity of Ap4A towards P2X1, and the dual modification completely eliminates P2X1 agonist activity.
Current FDA-approved (ticlopidine, clopidogrel, prasugrel, and ticagrelor) or experimental (cangrelor and elinogrel) ADP receptor antagonist antiplatelet therapy is solely directed against P2Y12. The present results identify derivatives of Ap4A as prototypical members of a class of antiplatelet agents that inhibit platelet function by targeting both P2Y1 and P2Y12, with an apparently synergistic effect on inhibition of platelet aggregation. Previous studies [4,6] and the present results suggest that such compounds would cause a rapid onset of platelet inhibition and, although the modifications allow them to resist degradation in plasma [4,6], they would probably be rapidly cleared from the bloodstream. These properties make Ap4A analogs potentially appropriate for clinical situations such as urgent percutaneous coronary intervention (where rapid platelet inhibition would be beneficial) and the need for emergency surgery, such as coronary artery bypass grafting (where rapid reversibility of platelet inhibition would be beneficial). Thus, Ap4A derivatives represent a unique class of antiplatelet agents with potential clinical benefits.
Acknowledgments
The authors gratefully acknowledge M. Yanachkova and L. Montville for their expert technical assistance.
This work was supported in part by SBIR grants HL081992 and HL088828 (to I. B. Yanachkov) from the National Heart, Lung and Blood Institute. A. L. Frelinger and A. D. Michelson have been investigators on research grants to Boston Children’s Hospital from GLSynthesis and Eli Lilly. A. D. Michelson has been a member of the Data Monitoring Committee of a clinical trial sponsored by Lilly. I. B. Yanachkov and G. E. Wright are employees of GLSynthesis, Inc. E. J. Dix has been a consultant to GLSynthesis, Inc.
Footnotes
Disclosure of conflict of interests
The other authors state that they have no conflict of interest.
References
- 1.Kushner FG, Hand M, Smith SC, Jr, King SB, 3rd, Anderson JL, Antman EM, Bailey SR, Bates ER, Blankenship JC, Casey DE, Jr, Green LA, Hochman JS, Jacobs AK, Krumholz HM, Morrison DA, Ornato JP, Pearle DL, Peterson ED, Sloan MA, Whitlow PL, et al. 2009 Focused Updates: ACC/AHA Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction (updating the 2004 Guideline and 2007 Focused Update) and ACC/AHA/SCAI Guidelines on Percutaneous Coronary Intervention (updating the 2005 Guideline and 2007 Focused Update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2009;120:2271–306. doi: 10.1161/CIRCULATIONAHA.109.192663. [DOI] [PubMed] [Google Scholar]
- 2.Cattaneo M. The platelet P2 receptors. In: Michelson AD, editor. Platelets. 2nd. San Diego, CA: Elsevier/Academic Press; 2007. pp. 201–20. [Google Scholar]
- 3.Jankowski J, Jankowski V, Laufer U, van der Giet M, Henning L, Tepel M, Zidek W, Schluter H. Identification and quantification of diadenosine polyphosphate concentrations in human plasma. Arterioscler Thromb Vasc Biol. 2003;23:1231–8. doi: 10.1161/01.ATV.0000075913.00428.FD. [DOI] [PubMed] [Google Scholar]
- 4.Zamecnik PC, Kim B, Gao MJ, Taylor G, Blackburn GM. Analogues of diadenosine 5′,5‴-P1,P4-tetraphosphate (Ap4A) as potential anti-platelet-aggregation agents. Proc Natl Acad Sci USA. 1992;89:2370–3. doi: 10.1073/pnas.89.6.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang H, Yanachkov IB, Michelson AD, Li Y, Barnard MR, Wright GE, Frelinger AL., 3rd Agonist and antagonist effects of diadenosine tetraphosphate, a platelet dense granule constituent, on platelet P2Y1, P2Y12 and P2X1 receptors. Thromb Res. 2010;125:159–65. doi: 10.1016/j.thromres.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan SW, Gallo SJ, Kim BK, Guo MJ, Blackburn GM, Zamecnik PC. P1,P4-dithio-P2,P3-monochloromethylene diadenosine 5′,5‴-P1,P4-tetraphosphate: a novel antiplatelet agent. Proc Natl Acad Sci USA. 1997;94:4034–9. doi: 10.1073/pnas.94.8.4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahaut-Smith MP, Tolhurst G, Evans RJ. Emerging roles for P2X1 receptors in platelet activation. Platelets. 2004;15:131–44. doi: 10.1080/09537100410001682788. [DOI] [PubMed] [Google Scholar]
- 8.Westfall TD, McIntyre CA, Obeid S, Bowes J, Kennedy C, Sneddon P. The interaction of diadenosine polyphosphates with P2X-receptors in the guinea-pig isolated vas deferens. Br J Pharmacol. 1997;121:57–62. doi: 10.1038/sj.bjp.0701099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewis CJ, Gitterman DP, Schluter H, Evans RJ. Effects of diadenosine polyphosphates (Ap(n)As) and adenosine polyphospho guanosines (Ap(n)Gs) on rat mesenteric artery P2X receptor ion channels. Br J Pharmacol. 2000;129:124–30. doi: 10.1038/sj.bjp.0702993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yanachkov IB, Dix EJ, Yanachkova MI, Wright GE. P1,P2-diimidazolyl derivatives of pyrophosphate and bis-phosphonates – synthesis, properties, and use in preparation of dinucleoside tetraphosphates and analogs. Org Biomol Chem. 2011;9:730–8. doi: 10.1039/c0ob00542h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han Q, Gaffney BL, Jones RA. One-flask synthesis of dinucleoside tetra- and pentaphosphates. Org Lett. 2006;8:2075–7. doi: 10.1021/ol060491d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nylander S, Mattsson C, Ramstrom S, Lindahl TL. Synergistic action between inhibition of P2Y12/P2Y1 and P2Y12/thrombin in ADP- and thrombin-induced human platelet activation. Br J Pharmacol. 2004;142:1325–31. doi: 10.1038/sj.bjp.0705885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turner NA, Moake JL, McIntire LV. Blockade of adenosine diphosphate receptors P2Y(12) and P2Y(1) is required to inhibit platelet aggregation in whole blood under flow. Blood. 2001;98:3340–5. doi: 10.1182/blood.v98.12.3340. [DOI] [PubMed] [Google Scholar]
- 14.Michelson AD. P2Y12 antagonism: promises and challenges. Arterioscler Thromb Vasc Biol. 2008;28:s33–8. doi: 10.1161/ATVBAHA.107.160689. [DOI] [PubMed] [Google Scholar]
- 15.Boyer JL, Romero-Avila T, Schachter JB, Harden TK. Identification of competitive antagonists of the P2Y1-receptor. Mol Pharmacol. 1996;50:1323–9. [PubMed] [Google Scholar]
- 16.Boyer J, Adams M, Ravi RG, Jacobson KA, Harden TK. 2-Chloro N6-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate is a selective high affinity P2Y1 receptor antagonist. Br J Pharmacol. 2002;135:2004–10. doi: 10.1038/sj.bjp.0704673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nandanan E, Jang SY, Moro S, Kim HO, Siddiqui MA, Russ P, Marquez VE, Busson R, Herdewijn P, Harden TK, Boyer JL, Jacobson KA. Synthesis, biological activity, and molecular modeling of ribose-modified deoxyadenosine bisphosphate analogues as P2Y1 receptor ligands. J Med Chem. 2000;43:829–42. doi: 10.1021/jm990249v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mathieu R, Baurand A, Schmitt M, Gachet C, Bourguignon JJ. Synthesis and biological activity of 2-alkylated deoxyadenosine bisphosphate derivatives as P2Y1 receptor antagonists. Bioorg Med Chem. 2004;12:1769–79. doi: 10.1016/j.bmc.2003.12.041. [DOI] [PubMed] [Google Scholar]
- 19.Xu B, Stephens A, Kirschenheuter G, Greslin AF, Cheng X, Sennelo J, Cattaneo M, Zighetti ML, Chen A, Kim SA, Kim HS, Bischofberger N, Cook G, Jacobson KA. Acyclic analogues of adenosine bisphosphates as P2Y receptor antagonists: phosphate substitution leads to multiple pathways of inhibition of platelet aggregation. J Med Chem. 2002;45:5694–709. doi: 10.1021/jm020173u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raboisson P, Baurand A, Cazenave JP, Gachet CRM, Speiss B, Bourgignon JJ. Novel antagonists acting at the P2Y(1) purinergic receptor: synthesis and conformational analysis using potentiometric and nuclear magnetic resonance titration techniques. J Med Chem. 2002;45:962–72. doi: 10.1021/jm0104062. [DOI] [PubMed] [Google Scholar]
- 21.Kim HS, Barak D, Harden TK, Boyer JL, Jacobson KA. Acyclic and cyclopropyl analogues of adenosine bisphosphate antagonists of the P2Y1 receptor: structure activity relationships and receptor docking. J Med Chem. 2001;44:3092–108. doi: 10.1021/jm010082h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HS, Ohno M, Xu B. 2-Substitution of adenine nucleotide analogues containing a bicyclo[3.1. ]hexane ring system locked in a Northern conformation: enhanced potency as P2Y1 receptor antagonists J Med Chem. 2003;46:4974–87. doi: 10.1021/jm030127+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Douglass JG, Patel RI, Yerxa BR, Shaver SR, Watson PS, Bednarski K, Plourde R, Redick CC, Brubaker K, Jones AC, Boyer JL. Lipophilic modifications to dinucleoside polyphosphates and nucleotides that confer antagonist properties at the platelet P2Y12 receptor. J Med Chem. 2008;51:1007–25. doi: 10.1021/jm701348d. [DOI] [PubMed] [Google Scholar]
- 24.Kauffenstein G, Hechler B, Cazenave JP, Gachet C. Adenine triphosphate nucleotides are antagonists at the P2Y receptor. J Thromb Haemost. 2004;2:1980–8. doi: 10.1111/j.1538-7836.2004.00926.x. [DOI] [PubMed] [Google Scholar]
- 25.Cattaneo M. ADP receptor antagonists. In: Michelson AD, editor. Platelets. 2nd. San Diego, CA: Elsevier/Academic Press; 2007. pp. 1127–44. [Google Scholar]
- 26.Palmer RK, Boyer JL, Schachter JB, Nicholas RA, Harden TK. Agonist action of adenosine triphosphates at the human P2Y1 receptor. Mol Pharmacol. 1998;54:1118–23. [PubMed] [Google Scholar]
- 27.Major DT, Nahum V, Wang Y, Reiser G, Fischer B. Molecular recognition in purinergic receptors. 2. Diastereoselectivity of the h-P2Y1-receptor. J Med Chem. 2004;47:4405–16. doi: 10.1021/jm049771u. [DOI] [PubMed] [Google Scholar]
- 28.Hoyle CHV, Hilderman RH, Pintor JJ, Schlüter H, King BF. Diadenosine polyphosphates as extracellular signal molecules. Drug Dev Res. 2001;52:260–73. [Google Scholar]
- 29.Delicado EG, Miras-Portugal MT, Carrasquero LM, Leon D, Perez-Sen R, Gualix J. Dinucleoside polyphosphates and their interaction with other nucleotide signaling pathways. Pflugers Arch. 2006;452:563–72. doi: 10.1007/s00424-006-0066-5. [DOI] [PubMed] [Google Scholar]
- 30.Pintor J, Diaz-Hernandez M, Gualix J, Gomez-Villafuertes R, Hernando F, Miras-Portugal MT. Diadenosine polyphosphate receptors from rat and guinea-pig brain to human nervous system. Pharmacol Ther. 2000;87:103–15. doi: 10.1016/s0163-7258(00)00049-8. [DOI] [PubMed] [Google Scholar]
- 31.Sage SO, MacKenzie AB, Jenner S, Mahaut-Smith MP. Purinoceptor-evoked calcium signalling in human platelets. Prostaglandins Leukot Essent Fatty Acids. 1997;57:435–8. doi: 10.1016/s0952-3278(97)90424-5. [DOI] [PubMed] [Google Scholar]
- 32.Longhurst PA, Schwegel T, Folander K, Swanson R. The human P2X1 receptor: molecular cloning, tissue distribution, and localization to chromosome 17. Biochim Biophys Acta. 1996;1308:185–8. doi: 10.1016/0167-4781(96)00112-1. [DOI] [PubMed] [Google Scholar]
- 33.Jin J, Kunapuli SP. Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Proc Natl Acad Sci USA. 1998;95:8070–4. doi: 10.1073/pnas.95.14.8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cattaneo M, Gachet C. ADP receptors and clinical bleeding disorders. Arterioscler Thromb Vasc Biol. 1999;19:2281–5. doi: 10.1161/01.atv.19.10.2281. [DOI] [PubMed] [Google Scholar]
- 35.Lin CY, Lubin B, Smith S. Inhibition of platelet aggregation by acyl-CoA thioesters. Biochim Biophys Acta. 1976;428:45–55. doi: 10.1016/0304-4165(76)90107-0. [DOI] [PubMed] [Google Scholar]
- 36.Manolopoulos P, Glenn JR, Fox SC, May JA, Dovlatova NL, Tang SW, Thomas NR, Ralevic V, Heptinstall S. Acyl derivatives of coenzyme A inhibit platelet function via antagonism at P2Y1 and P2Y12 receptors: a new finding that may influence the design of anti-thrombotic agents. Platelets. 2008;19:134–45. doi: 10.1080/09537100701708498. [DOI] [PubMed] [Google Scholar]
- 37.Tolhurst G, Vial C, Leon C, Gachet C, Evans RJ, Mahaut-Smith MP. Interplay between P2Y(1), P2Y(12), and P2X(1) receptors in the activation of megakaryocyte cation influx currents by ADP: evidence that the primary megakaryocyte represents a fully functional model of platelet P2 receptor signaling. Blood. 2005;106:1644–51. doi: 10.1182/blood-2005-02-0725. [DOI] [PubMed] [Google Scholar]
- 38.Hardy AR, Jones ML, Mundell SJ, Poole AW. Reciprocal cross-talk between P2Y1 and P2Y12 receptors at the level of calcium signaling in human platelets. Blood. 2004;104:1745–52. doi: 10.1182/blood-2004-02-0534. [DOI] [PubMed] [Google Scholar]