


Abstract
Cell-surface engineering strategies that permit long-lived display of well-defined, functionally active molecules are highly attractive for eliciting desired cellular responses and for understanding biological processes. Current methodologies for the exogenous introduction of synthetic biomolecules often result in short-lived presentations, or require genetic manipulation to facilitate membrane attachment. Herein, we report a cell-surface engineering strategy that is based on the use of a CMP-Neu5Ac derivative that is modified at C-5 by a bifunctional entity composed of a complex synthetic heparan sulfate (HS) oligosaccharide and biotin. It is shown that recombinant ST6GAL1 can readily transfer the modified sialic acid to N-glycans of glycoprotein acceptors of living cells resulting in long-lived display. The HS oligosaccharide is functionally active, can restore protein binding, and allows activation of cell signaling events of HS-deficient cells. The cell-surface engineering methodology can easily be adapted to any cell type and is highly amenable to a wide range of complex biomolecules.
Graphical abstract
INTRODUCTION
Cell-surface engineering provides attractive opportunities to modulate cellular functions such as cell differentiation and proliferation, cell–cell adhesion and communication, and immune detection.1–6 It provides a powerful means to elucidate molecular mechanisms by which specific biomolecules perform their functions and may find application in cell therapy by targeting therapeutic cells to specific tissues,2,7–9 endowing cells with new functions such as recruiting/restoring protein binding or directing cell signaling or stem cell differentiation,10–13 and altering immune cell responses toward cancerous cells.14,15
Genetic approaches such as gene deletion, knockdown or overexpression enables the manipulation of proteins of interest in a controlled and inducible manner.16–19 Engineering of other cell-surface biomolecules, such as glycans of glycoproteins and glycolipids, is much more challenging because their structures are not precisely encoded in the genome.4,6,20 For example, cell-surface glycans can be modulated by disruption of specific glycosyltransferases; however, this will influence the assembly of a variety of different glycans and therefore is not amenable to examine the involvement of a single compound on biological activity.
Emergent technologies to engineer cell surfaces by exogenous introduction of chemically defined structures are attractive for the controlled display of a wide variety of compounds. Peptides, proteins, and glycans have been attached to hydrophobic lipids to facilitate cell-surface display.12–15,21–23 These presentations are often short-lived due to rapid endocytosis. In part, this problem can be addressed by using lipids, such as cholesterylamine, that can recycle back to the cell surface after internalization.15 Continual recycling may, however, lead to remodeling of the displayed compound, which in turn may influence biological responsiveness. HaloTag protein technology offers an alternative means to display ligands on the surface of cells and results in relatively long residency times.10 This approach requires genetic manipulations to introduce the HaloTag protein and therefore cannot easily be adapted to primary cells. A foreign protein may also elicit immune responses complicating in vivo applications. Thus, there is an unmet need for cell-surface engineering methodologies that can be applied to many cell types including primary cells, have minimal impact on cell viability, and allow long-lived display of an exogenously provided ligand.
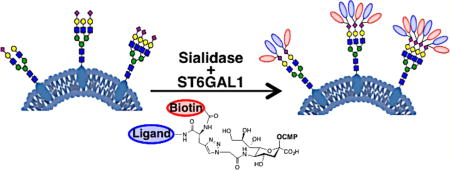
We report here a methodology that makes it possible to attach highly complex bioactive compounds to cell-surface glycoproteins of living cells. It uses an exogenously administered CMP-Neu5Ac derivative that is modified at C-5 with a bifunctional entity composed of a complex carbohydrate and biotin (e.g., 1, Figure 1). Recombinant ST6GAL1,24,25 which is selective for N-glycans,26,27 can transfer this donor to appropriate glycoprotein acceptors. As a proof of principle, cells deficient in heparan sulfate (HS) biosynthesis were modified with a synthetic HS oligosaccharide, which resulted in long-lived display and gain of function by endowing the cells with an ability to recruit FGF2, leading to the phosphorylation of the transcription factor ERK1/2 and the induction of cell proliferation.
Figure 1.

Cell-surface engineering using an exogenously administered bifunctional CMP-Neu5Ac derivative and recombinant ST6GAL1.
RESULTS AND DISCUSSION
Chemical Synthesis
It is known that sialyltransferases can tolerate simple modifications such as azide or biotin at C-5 of CMP-sialic acid (CMP-Neu5Ac) and such derivatives can readily be transferred to N-acetyllactosamine (LacNAc) acceptors.26–31 Almost all cell-surface proteins are modified by O-linked or N-linked glycans that commonly are extended or terminated by LacNAc moieties that can serve as acceptor substrates for sialyltransferases.32–35 A recent crystal structure of the sialyltransferase ST6GAL1 combined with computational substrate docking of ST6GAL1 complexed with CMP-Neu5Ac and LacNAc indicates that the C-5 acetamido moiety of Neu5Ac is solvent exposed.25 Thus, we were compelled to explore whether CMP-Neu5Ac derivatives such as 1, which is modified with a highly complex bifunctional entity composed of a biotin moiety and a synthetic HS-oligosaccharide with two carboxylate and six sulfate groups, can be synthesized and transferred to glycoprotein acceptors of living cells resulting in long-lived and functional display.
A convergent strategy was developed for the preparation of compounds such as 1 by a copper-catalyzed alkyne–azide cycloaddition (CuAAC)36 of CMP-Neu5Ac (5) having an azide at C-5 and an alkyne-functionalized biomolecule such as 4 (Scheme 1). The attraction of such a strategy is that it is modular, allowing for the convenient preparation of a range of donors modified by different entities for structure activity relationship studies. Furthermore, CMP-Neu5Ac is modified at a late stage in the synthesis under mild conditions, which is desirable due to its chemical lability.30,37
Scheme 1.

Synthesis and Enzymatic Transfer Using ST6GAL1 of HS-Tetrasaccharide-Modified CMP-Neu5Ac 1
To demonstrate the feasibility of this synthetic strategy, we employed the complex, highly charged HS-tetrasaccharide 2. This tetrasaccharide is composed of two disaccharide units containing 2-O-sulfo-iduronic acid and N-sulfo-6-O-sulfo-glucosamine, and it was prepared by a modular synthetic approach.38,39 The amine of the anomeric linker of 2 was reacted with NHS-activated propargyl glycine in the presence of DIPEA to give 3. Removal of the Fmoc protecting group of 3 with Et3N provided an amine that was biotinylated with NHS-activated biotin furnishing 4. A CuAAC of 4 with 5 in the presence of copper sulfate, sodium ascorbate, and TBTA afforded the bifunctional CMP-Neu5Ac derivative 1 in good overall yield after purification by size exclusion chromatography over P2 Biogel. The successful preparation of 1 demonstrates that highly complex compounds having chemically sensitive functional groups such as N- and O-sulfates can be conjugated to a labile sugar-nucleotide such as CMP-Neu5Ac for use in glycosyltransferase reactions.
With the bifunctional CMP-Neu5Ac derivative 1 in hand, we explored the substrate tolerance with the sialyltransferase ST6GAL1. Interestingly, ST6GAL1 could efficiently transfer 1 to LacNAc acceptor 6 providing α-2,6-sialyl-LacNAc derivative 7, bearing the biotin moiety and HS-tetrasaccharide on the C5-position of the sialoside (Scheme 1). Furthermore, the C5-modified sialoside of 7 was resistant to treatment with a sialidase from Clostridium perfringens (C. perfringens) (Scheme S2, Supporting Information (SI)). This observation is important because while LacNAc moieties are highly abundant on cell-surface glycoproteins they can be terminated with native sialosides, and thus concurrent treatment of cells with a neuraminidase and ST6GAL1 creates additional acceptors for cell-surface glycosylation with 1.
Cell-Surface Labeling
Next, attention was focused on the use of compound 1 for the cell-surface labeling of Ext1−/− mouse lung endothelial cells (Ext1−/− cells). These cells have a mutation in the Ext1 gene and lack the enzyme exostosin 1, and thus cannot biosynthesize HS. Ext1−/− cells were incubated with 1 and ST6GAL1 in the presence or absence of C. perfringens neuraminidase, and subsequently stained with avidin-AlexaFluor488 (AF488). After staining, the fluorescence intensity of cell lysates was measured, and 1 was also detected on the surface of live Ext1−/− cells by flow cytometry. Robust avidin staining was observed upon treatment with 1, ST6GAL1 and neuraminidase, whereas untreated control Ext1−/− and CMP-Neu5Ac treated cells resulted in no avidin binding (Figure 2a,b). The transfer efficiency of 1 is both time-, temperature- and dose-dependent with optimal conditions being 100 µM of 1 for 2 h at 37 °C (Figure 2c, Figure S1a). At 100 µM a high degree of labeling is achieved, however the extent of labeling can be controlled by reducing the concentration of the modified CMP-Neu5Ac 1 (Figure S1). Concurrent neuraminidase treatment greatly enhanced the display of 1 (Figure 2a, Figure S1b) highlighting the importance of creating additional LacNAc acceptor sites during enzymatic labeling. Finally, imaging by fluorescence microscopy confirmed the introduction of 1 onto the Ext1−/− cell surface (Figure 2d).
Figure 2.

Cell-surface display of 1 on Ext1−/− cells. (a) Fluorescence intensity of cell lysates after treatment with ST6GAL1 and 1 at the indicated concentration, with or without neuraminidase (Neu), followed by avidin-AF488 staining. (b) Detection of 1 on live Ext1−/− cell surfaces by flow cytometry. Cells were stained with avidin-AF488 and co-stained with PI to exclude non-viable cells. (c) Fluorescence intensity of cell lysates after treatment with 100 µM of 1 or CMP-Neu5Ac, ST6GAL1, and Neu for the indicated time and temperature, followed by avidin-AF488 staining. (d) Fluorescence microscopy imaging of Ext1−/− cells and Ext1−/− cells displaying 1. Cell surfaces were stained with avidin-AF488, and nuclei were co-stained with Hoescht 33342.
Ext1−/− cells labeled with 1 and cultured for 24 h exhibited robust avidin binding by flow cytometry (Figure 3a). Remarkably, after culturing for 72 h, 1 could still be detected on the cell-surface indicating the new cell-surface engineering strategy can provide persistent display of a desired ligand. This observation was confirmed by detecting proteins modified with 1 by Western blotting using an anti-biotin antibody (Figure 3b). A strong biotin signal was detected immediately after treatment with 1, neuraminidase and ST6GAL1 (t = 0 h). It was observed that after culturing for 24 or 72 h, certain bands had disappeared whereas other glycoproteins retained an oligosaccharide modified by a biotin tag. This highlights that a sub-population of glycoproteins turnover slowly allowing long-lasting display of a synthetic compound introduced by enzymatic transfer of a modified CMP-Neu5Ac with ST6GAL1. Persistent and stable display of synthetic ligands is crucial for cell-surface engineering techniques to probe biological processes that occur on longer times scales.10,15
Figure 3.

Persistent display of 1 on Ext1−/− cells. (a) Detection of 1 on live Ext1−/− cell surfaces over time by flow cytometry. Cells were stained with avidin-AF488 and co-stained with PI to exclude nonviable cells. (b) Western blot analysis using an anti-biotin antibody of cell lysates after treatment with 1, neu. and ST6GAL1, and subsequent culturing in medium for the indicated time.
To confirm the selectivity of ST6GAL1 for N-glycans of cell-surface glycoproteins,40,41 cell lysates of labeled Ext1−/− cells were treated with peptide-N-glycosidase F (PNGase F),42 which is an amidase that cleaves between the asparagine residue and innermost GlcNAc moiety of N-glycoproteins. This treatment completely abolished biotin staining by Western blot (Figure S1b), confirming that only N-linked glycans were modified. We also compared the exogenous enzymatic labeling methodology to a two-step metabolic labeling protocol in which Ext1−/− cell were cultured for 24 h in the presence peracylated azide-modified N-acetylmannosamine (Ac4ManNAz) to display azido-containing sialosides at the surface of the cells.43,44 The resulting cells were subjected to a strain-promoted alkyne–azide cycloaddition (SPAAC) and were examined for avidin binding by flow cytometry (Figure S2) and cell lysates were analyzed by Western blotting with an anti-biotin antibody (Figure S1a). Metabolic labeling showed much weaker labeling compared to exogenous enzymatic labeling employing compound 1, ST6GAL1 and neuraminidase. The two step metabolic labeling protocol also showed residual staining when cell lysates were treated with PNGase F, indicating that in this case N- and O-linked glycans were labeled (Figure S1b).
Having established the persistent display of 1 on the cell-surface 72 h after labeling on certain glycoproteins, we enriched labeled glycoproteins by immuno-precipitation using an anti-biotin antibody and conducted LC-MS/MS proteomic analysis to examine alteration in protein abundance immediately after (T = 0 h) and 72 h after labeling. Proteins were identified at a 1% false-discovery rate at the protein level, and those identified in the negative controls were excluded from the final protein list. A summary of the top 20 proteins identified at T = 0 sorted by total spectral counts is presented in Table 1. Interestingly, four of these top 20 proteins were still detected 72 h after labeling, including integrin β-1, integrin β-3, cadherin-13, and 4F2 cell-surface antigen heavy chain. The identified proteins have multiple N-glycosylation sites, and glycoproteins such as Integrin β-1 and 4F2 cell-surface antigen heavy chain, which were detected 72 h after labeling, have been reported to have half-lives greater than 24 h.45,46 Other proteins such as basal cell adhesion molecule and CD109, which were not detected 72 h after labeling, have been reported to have half-lives less than 10 h.45 The data indicate that, as expected, certain glycoproteins have extended half-lives and more importantly, that they can retain the introduced HS-tetrasaccharide on the cell surface for an extended period of time.
Table 1.
Top 20 Proteins Identified after Immunoprecipitation and Proteomic Analysis Immediately after Labeling (0 h) and Proteins That Were Detected after Culturing Labeled Cells for 72 h (Indicated by +)
| time cultured post- labeling |
||||
|---|---|---|---|---|
|
|
||||
| UniProt ID | protein name | protein weight (kDa) |
0 h | 72 h |
| P09055 | integrin β-1 | 88.2 | + | + |
| Q61739 | integrin α-6 | 122.1 | + | − |
| A0A087WRT4 | FAT atypical cadherin 1 | 507.2 | + | − |
| A0A1L1SQU7 | uncharacterized protein | 511.9 | + | − |
| F2Z4A3 | FAT atypical cadherin 1 | 505.9 | + | − |
| Q61738 | integrin α-7 | 129.2 | + | − |
| G3X9Q1 | integrin α-7 | 124.5 | + | − |
| Q9R069 | basal cell adhesion molecule | 67.6 | + | − |
| O54890 | integrin β-3 | 86.7 | + | + |
| Q8R422 | CD109 antigen | 161.5 | + | − |
| B2RXS4 | plexin-B2 | 206.1 | + | − |
| P10852 | 4F2 cell-surface antigen heavy chain | 58.3 | + | + |
| Q61490 | CD166 antigen | 65.0 | + | − |
| E9Q3Q6 | CD166 antigen | 63.6 | + | − |
| E9Q4G8 | CD166 antigen | 61.8 | + | − |
| P15116 | cadherin-2 | 99.7 | + | − |
| D3YYT0 | cadherin-2 | 93.8 | + | − |
| P97333 | neuropilin-1 | 102.9 | + | − |
| Q9WTR5 | cadherin-13 | 78.1 | + | + |
| P15306 | thrombomodulin | 61.8 | + | − |
Next, we explored whether the introduced HS oligosaccharide can restore cellular function by recruiting a protein such as FGF2, which can then activate various cell signaling events. In this respect, HS functions as a co-receptor to facilitate FGF2 binding to its protein receptor, FGFR. A ternary complex of FGFR-FGF2-HS initiates signal transduction leading to ERK1/2 phosphorylation that promotes cell proliferation.47–49 By displaying 1 on Ext1−/− cells, a significant increase in FGF2 binding was observed by flow cytometry using an anti-FGF2 antibody (Figure 4a). The recruited FGF2 is biologically active and Ext1−/− cells displaying 1 and stimulated with FGF2 exhibited a 1.9-fold increase in phospho-ERK1/2 compared to control cells (Figure 4b,c). Cell proliferation rates of cells displaying 1 were also explored using real-time cell analysis (RTCA). This non-invasive, electrical impedance assay facilitates label-free quantification of cell growth rates by continuous monitoring of the magnitude of impedance, which is dependent upon the number of adhered cells over time.50,51 We observed a significant increase in the growth rate of Ext1−/− cells displaying 1 compared to control cells (doubling time of 26 vs 40 h, Figures 4d and S3).
Figure 4.

Display of 1 on Ext1−/− cells using ST6GAL1 modulates FGF2 binding and cell growth rate. (a) FGF2 binding by flow cytometry. (b) Representative Western blots of phospho-ERK1/2 and total ERK1/2 levels of Ext1−/− cells and Ext1−/− cells displaying 1 stimulated with FGF2. (c) Relative phospho-ERK1/2 levels with respect to total ERK1/2 levels 30 min after stimulation with FGF2. (d) Cell growth rate of Ext1−/− control cells and Ext1−/− cells displaying 1 for 48 h. *P < 0.01.
CONCLUSIONS
Exogenous glycans have previously been displayed on the surface of cells through a lipid anchor or HaloTag.10,12–15 We have demonstrated that well-defined glycans can be presented in the context of glycoproteins through enzymatic labeling using a glycosyltransferase and a glycosyl donor modified by a complex and biologically active entity such as a heparan sulfate (HS) oligosaccharide. It was found that ST6GAL1 is not limited to only transfer CMP-Neu5Ac analogues modified at C-5 by simple entities such as azide or biotin,26–31 but can also transfer highly complex and biologically relevant entities such as HS oligosaccharides. A key feature of the approach is a synthetic route for the preparation of rather labile CMP-Neu5Ac analogues by CuAAC of CMP-Neu5Ac having an azide at C-5 and an alkyne-functionalized biomolecule. After enzymatic display of the modified CMP-Neu5Ac, a subset of the labeled glycoproteins persist on the cell surface, resulting in long-lasting display of the well-defined oligosaccharide. Presenting exogenously delivered biomolecules in the context of glycoproteins is particularly relevant for those that are naturally displayed in this fashion. In addition, this approach does not require genetic engineering, making it easily adaptable to a wide range of cell types including primary cells. It can also be applied to cells that have defects in the biosynthesis of specific biomolecules such as HS, and we have shown through a gain of function study, that biological properties of individual molecules can be examined.
Cell-surface engineering is particularly attractive to study the biology of individual HS oligosaccharides.10,12 These linear polysaccharides are heavily O- and N-sulfated and reside on the cell surface and extracellular matrix of virtually all mammalian tissue types where they interact with numerous signaling proteins, growth factors, and ECM components.47–49 Examination of biological properties of individual HS oligosaccharides is very challenging, and by combining chemical or enzymatic synthesis with cell-surface display technologies this deficiency can be addressed.
A remarkable finding of this study is that ST6GAL1 can transfer CMP-Neu5Ac analogues that are modified by highly complex entities such as sulfated oligosaccharides, and thus it is expected that the methodology can be extended to a wide range of complex biomolecules. Displaying biologically active molecules on cell surfaces will provide opportunities to manipulate cellular physiology and phenotypic outcomes and may find application for cellular therapies.
EXPERIMENTAL SECTION
Methods
Methods, associated references, and additional information and data are available in the Supporting Information.
Materials
Recombinant β-galactoside α-2,6-sialyltransferase 1 (ST6GAL1) was generously provided by Dr. Kelley W. Moremen (University of Georgia).24,25 Calf intestinal alkaline phosphatase (CIAP) was purchased from Sigma. Clostridium perfringens neuraminidase was purchased from New England BioLabs. HS-tetrasaccharide 2 and CMP-Neu5Ac 5 were prepared as previously described.28,38,39 CMP-Neu5Ac (Carbosynth LLC) and compound 1 were stored as dry solids at −20 °C and were dissolved in culture medium without FBS just before use. Avidin-AlexaFluor-488 conjugate, propidium iodide (PI) and Hoescht 33342 were purchased from ThermoFisher Scientific; mouse monoclonal anti-biotin antibody conjugated to peroxidase (HRP) was from Jackson ImmunoResearch Laboratories; mouse monoclonal antibody (mAb) to β-actin conjugated to HRP, anti-FGF2 antibody, goat anti-rabbit IgG conjugated to HRP, and goat anti-rabbit IgG conjugated to AlexaFluor-488 were purchased from Abcam; recombinant human FGF2 basic was purchased from R&D Systems; p44/42 MAPK (ERK1/2, rabbit mAb) and phospho-p44/42 MAPK (phospho-ERK, rabbit mAb) were purchased from Cell Signaling Technology Inc.; Dulbecco’s phosphate-buffered saline (DPBS) with Ca2+/Mg2+, DPBS without Ca2+/Mg2+, and Dulbecco’s modified eagle’s medium (DMEM) high glucose were purchased from ATCC. Trypsin EDTA (1×) and penicillin/streptomycin (P/S, 100×) were purchased from Corning Life Sciences. E-plate VIEW 16 PET was purchased from ACEA Biosciences, Inc.
Cell Culture
Ext1−/− mouse lung endothelial cells were generously provided by Dr. Lianchun Wang (University of Georgia). Ext1−/− cells were cultured on tissue culture plates in DMEM supplemented with 1× P/S and 10% fetal bovine serum (FBS). Cells were maintained in a humid 5% CO2 atmosphere at 37 °C and were passaged using 1× trypsin-EDTA after reaching ~80% confluency.
Enzymatic Cell-Surface Display of 1 or CMP-Neu5Ac
Ext1−/− cells were plated in 12-well plates (200 000 cells/well) and were grown to 80% confluency. Cells were washed with culture medium without FBS and incubated in a mixture of 300 µL culture medium without FBS containing 100 µM of 1, 10 µg/mL ST6GAL1, 50 U/mL C. perfringens neuraminidase, 10 U/mL CIAP and 0.1% BSA for 2 h at 37 °C. Untreated control experiments were treated in a mixture of 300 µL culture medium without FBS containing 10 µg/mL ST6GAL1, 10 U/mL CIAP and 0.1% BSA for 2 h at 37 °C. For control experiments with CMP-Neu5Ac, Ext1−/− cells were washed with culture medium without FBS and then pretreated for 1 h at 37 °C with 50 U/mL C. perfringens neuraminidase in 300 µL culture medium without FBS. Following neuraminidase pretreatment, cells were incubated in a mixture of 300 µL culture medium without FBS containing 100 µM of CMP-Neu5Ac, 10 µg/mL ST6GAL1, 10 U/mL CIAP and 0.1% BSA for 2 h at 37 °C. The labeled cells were washed with 1% FBS/DPBS then treated as indicated.
Detection of Cell-Surface Display of 1 by Fluorescence Intensity
After enzymatic cell-surface display of 1, cells were incubated with avidin-AlexaFluor-488 (2.5 µg/mL) in 1% FBS/DPBS for 15 min at 4 °C in the dark. Cells were washed with cold DPBS and lysed in 0.5% Triton X-100 in DPBS lysis buffer. Lysates were analyzed for fluorescence intensity (λem = 520/λex = 485) using a microplate reader (BMG Labtech). Data points were collected in triplicate and are representative of three separate experiments (n = 9). Cell lysates were assayed for total protein by using the bicinchoninic acid assay (BCA assay, Pierce Biotechnology) and fluorescence intensity was expressed as fluorescence (AU) per µg total protein.
Enzymatic Cell-Surface Display of 1 and Detection by Fluorescence Microscopy
Ext1−/− cells were seeded at a density of 25 000 cells/coverslip (22 mm) and were grown to 60% confluency then labeled as described above with a total volume of 100 µL. After staining with avidin-AlexaFluor-488 (2.5 µg/mL) in 1% FBS/DPBS for 20 min at 4 °C in the dark, cells were washed with 1% FBS/DPBS and fixed with formaldehyde (3.7% in PBS) at room temperature for 15 min, then washed and permeabilized for 10 min at room temperature using 0.1% Triton X-100 in PBS. After washing the nucleus was labeled Hoescht 33342 (1 µg/mL), and the cells were washed and mounted with Prolong Gold (ThermoFisher) before being imaged by fluorescent microscopy using a 20× air objective. Data was collected in triplicates (n = 3).
Flow Cytometry Analysis
For the detection of 1 using avidin, following enzymatic cell-surface display cells were stained with avidin-AlexaFluor-488 (2.5 µg/mL) in 1% FBS/DPBS for 20 min at 4 °C in the dark. The cells were washed with DPBS without Ca/Mg, then detached using 150 µL of trypsin-EDTA for 2 min at 37 °C. The cells were suspended in 1% FBS/DPBS, centrifuged gently (500 rpm for 3 min), and resuspended in 500 µL of 1% FBS/DPBS and transferred to polystyrene tubes for flow cytometric analysis (Beckman Coulter HyperCyAn, CTEGD Cytometry Center, University of Georgia). Cell viability was determined by adding PI to cell suspensions 5 min prior to analysis. The live population of cells was gated based on forward and side scatter emission, and exclusion of PI positive cells on the FL3 (613/20 BP filter) emission channel. Avidin-AlexaFluor-488 binding was determined by fluorescence intensity on the FL1(530/30 BP filter) emission channel. Data points were collected in duplicates and are representative of three separate experiments (n = 6).
For analysis of FGF-2 binding, following enzymatic cell-surface display cells were incubated with human FGF-2 basic (2.5 µg/mL) in 1% FBS/DPBS for 20 min at 4 °C. Cells were washed, then incubated with anti-FGF-2 antibody (2.5 µg/mL) for 20 min at 4 °C. Following washing, cells were incubated with goat anti-rabbit IgG conjugated with AlexaFluor-488 (2.5 µg/mL) for 20 min at 4 °C in the dark. Cells were washed with DPBS without Ca/Mg and were detached, resuspended and analyzed as described above. Data points were collected in duplicates and are representative of three separate experiments (n = 6).
Western Blot Analyses
Following enzymatic cell-surface display of 1, cells were washed with cold DPBS and lysed with RIPA lysis buffer with 1× protease inhibitor cocktail. The cell lysates were clarified by centrifugation at 22000g for 15 min and the total protein content of the clear supernatants was assessed using the BCA assay. The samples (~5 µg of protein) were resolved on a 4–15% SDS-PAGE gel (Bio-Rad) and transferred to a PVDF membrane. Next, the membrane was blocked in blocking buffer (5% nonfat dry milk in PBST (PBS containing 0.1% Tween-20 and 0.1% Triton X-100)) for 2 h at room temperature. The blocked membrane was incubated for 1 h at room temperature with an anti-biotin antibody conjugated to HRP (1:75 000) in blocking buffer and washed with PBST (4 × 10 min). Final detection of HRP activity was performed using ECL Plus chemiluminescent substrate (Amersham), exposure to film (Kodak) and development using a digital X-ray imaging machine (Kodak). Next, the blot was stripped and reprobed for loading control (β-actin) using an anti-β-actin antibody (1:10 000) for 1 h at room temperature. Data were collected in triplicates (n = 3).
Supplementary Material
Acknowledgments
This project was supported by the National Institute of General Medical Sciences (NIGMS; P01GM107012 and P41GM-103390) from the U.S. National Institutes of Health. We thank Dr. Kelley W. Moremen (University of Georgia) for providing recombinant ST6GAL1, and Dr. Lianchun Wang (University of Georgia) for providing mouse Ext1−/− endothelial cells.
Footnotes
ASSOCIATED CONTENT
- Figures S1–S3, chemical synthesis, biological assays, and NMR spectra (PDF)
The authors declare no competing financial interest.
References
- 1.Armstrong JP, Perriman AW. Exp. Biol. Med. 2016;241:1098–1106. doi: 10.1177/1535370216650291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stephan MT, Irvine DJ. Nano Today. 2011;6:309–325. doi: 10.1016/j.nantod.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Custódio CA, Mano JF. ChemNanoMat. 2016;2:376–384. doi: 10.1002/cnma.201600047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffin ME, Hsieh-Wilson LC. Cell Chem. Biol. 2016;23:108–121. doi: 10.1016/j.chembiol.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hudak JE, Bertozzi CR. Chem. Biol. 2014;21:16–37. doi: 10.1016/j.chembiol.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nischan N, Kohler JJ. Glycobiology. 2016;26:789–796. doi: 10.1093/glycob/cww045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarkar D, Vemula PK, Zhao W, Gupta A, Karnik R, Karp JM. Biomaterials. 2010;31:5266–5274. doi: 10.1016/j.biomaterials.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sackstein R. Ann. Biomed. Eng. 2012;40:766–776. doi: 10.1007/s10439-011-0461-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stephan MT, Moon JJ, Um SH, Bershteyn A, Irvine DJ. Nat. Med. 2010;16:1035–1041. doi: 10.1038/nm.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pulsipher A, Griffin ME, Stone SE, Hsieh-Wilson LC. Angew. Chem., Int. Ed. 2015;54:1466–1470. doi: 10.1002/anie.201409258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srivastava G, Kaur KJ, Hindsgaul O, Palcic MM. J. Biol. Chem. 1992;267:22356–22361. [PubMed] [Google Scholar]
- 12.Huang ML, Smith RAA, Trieger GW, Godula K. J. Am. Chem. Soc. 2014;136:10565–10568. doi: 10.1021/ja505012a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pulsipher A, Griffin ME, Stone SE, Brown JM, Hsieh-Wilson LC. J. Am. Chem. Soc. 2014;136:6794–6797. doi: 10.1021/ja5005174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hudak JE, Canham SM, Bertozzi CR. Nat. Chem. Biol. 2014;10:69–75. doi: 10.1038/nchembio.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woods EC, Yee NA, Shen J, Bertozzi CR. Angew. Chem., Int. Ed. 2015;54:15782–15788. doi: 10.1002/anie.201508783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Medof ME, Nagarajan S, Tykocinski ML. FASEB J. 1996;10:574–86. doi: 10.1096/fasebj.10.5.8621057. [DOI] [PubMed] [Google Scholar]
- 17.Rieder E, Berinstein A, Baxt B, Kang A, Mason PW. Proc. Natl. Acad. Sci. U. S. A. 1996;93:10428–10433. doi: 10.1073/pnas.93.19.10428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muenchmeier N, Boecker S, Bankel L, Hinz L, Rieth N, Lapa C, Mendler AN, Noessner E, Mocikat R, Nelson PJ. PLoS One. 2013;8:e72749. doi: 10.1371/journal.pone.0072749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stachel G, Trenkwalder T, Götz F, El Aouni C, Muenchmeier N, Pfosser A, Nussbaum C, Sperandio M, Hatzopoulos AK, Hinkel R, Nelson PJ, Kupatt C. Stem Cells. 2013;31:1795–1805. doi: 10.1002/stem.1439. [DOI] [PubMed] [Google Scholar]
- 20.Saxon E, Bertozzi CR. Annu. Rev. Cell Dev. Biol. 2001;17:1–23. doi: 10.1146/annurev.cellbio.17.1.1. [DOI] [PubMed] [Google Scholar]
- 21.Ko IK, Kean TJ, Dennis JE. Biomaterials. 2009;30:3702–3710. doi: 10.1016/j.biomaterials.2009.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuda M, Hatanaka W, Takeo M, Kim CW, Niidome T, Yamamoto T, Kishimura A, Mori T, Katayama Y. Bioconjugate Chem. 2014;25:2134–2143. doi: 10.1021/bc500465j. [DOI] [PubMed] [Google Scholar]
- 23.Rabuka D, Forstner MB, Groves JT, Bertozzi CR. J. Am. Chem. Soc. 2008;130:5947–5953. doi: 10.1021/ja710644g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barb AW, Meng L, Gao Z, Johnson RW, Moremen KW, Prestegard JH. Biochemistry. 2012;51:4618–4626. doi: 10.1021/bi300319q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meng L, Forouhar F, Thieker D, Gao Z, Ramiah A, Moniz H, Xiang Y, Seetharaman J, Milaninia S, Su M, Bridger R, Veillon L, Azadi P, Kornhaber G, Wells L, Montelione GT, Woods RJ, Tong L, Moremen KW. J. Biol. Chem. 2013;288:34680–34698. doi: 10.1074/jbc.M113.519041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mbua NE, Li X, Flanagan-Steet HR, Meng L, Aoki K, Moremen KW, Wolfert MA, Steet R, Boons G-J. Angew. Chem., Int. Ed. 2013;52:13012–13015. doi: 10.1002/anie.201307095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu S-H, Zhao P, Sun T, Gao Z, Moremen KW, Boons G-J, Wells L, Steet R. J. Biol. Chem. 2016;291:3982–3989. doi: 10.1074/jbc.M115.700369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun T, Yu S-H, Zhao P, Meng L, Moremen KW, Wells L, Steet R, Boons G-J. J. Am. Chem. Soc. 2016;138:11575–11582. doi: 10.1021/jacs.6b04049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu C-C, Withers SG. Adv. Synth. Catal. 2015;357:1633–1654. [Google Scholar]
- 30.Chen X, Varki A. ACS Chem. Biol. 2010;5:163–176. doi: 10.1021/cb900266r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeFrees S, Wang Z-G, Xing R, Scott AE, Wang J, Zopf D, Gouty DL, Sjoberg ER, Panneerselvam K, Brinkman-Van der Linden ECM, Bayer RJ, Tarp MA, Clausen H. Glycobiology. 2006;16:833–843. doi: 10.1093/glycob/cwl004. [DOI] [PubMed] [Google Scholar]
- 32.Ohtsubo K, Marth JD. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 33.Moremen KW, Tiemeyer M, Nairn AV. Nat. Rev. Mol. Cell Biol. 2012;13:448–462. doi: 10.1038/nrm3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palcic MM. Curr. Opin. Chem. Biol. 2011;15:226–233. doi: 10.1016/j.cbpa.2010.11.022. [DOI] [PubMed] [Google Scholar]
- 35.Schmaltz RM, Hanson SR, Wong C-H. Chem. Rev. 2011;111:4259–4307. doi: 10.1021/cr200113w. [DOI] [PubMed] [Google Scholar]
- 36.Meldal M, Tornøe CW. Chem. Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 37.Yu H, Yu H, Karpel R, Chen X. Bioorg. Med. Chem. 2004;12:6427–6435. doi: 10.1016/j.bmc.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 38.Arungundram S, Al-Mafraji K, Asong J, Leach FE, Amster IJ, Venot A, Turnbull JE, Boons G-J. J. Am. Chem. Soc. 2009;131:17394–17405. doi: 10.1021/ja907358k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zong C, Venot A, Dhamale O, Boons G-J. Org. Lett. 2013;15:342–345. doi: 10.1021/ol303270v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dall’Olio F. Glycoconjugate J. 2000;17:669–676. doi: 10.1023/a:1011077000164. [DOI] [PubMed] [Google Scholar]
- 41.Zhuo Y, Bellis SL. J. Biol. Chem. 2011;286:5935–5941. doi: 10.1074/jbc.R110.191429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maley F, Trimble RB, Tarentino AL, Plummer TH. Anal. Biochem. 1989;180:195–204. doi: 10.1016/0003-2697(89)90115-2. [DOI] [PubMed] [Google Scholar]
- 43.Dube DH, Bertozzi CR. Curr. Opin. Chem. Biol. 2003;7:616–625. doi: 10.1016/j.cbpa.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 44.Du J, Meledeo MA, Wang Z, Khanna HS, Paruchuri VDP, Yarema KJ. Glycobiology. 2009;19:1382–1401. doi: 10.1093/glycob/cwp115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao H, Wu R. Chem. Sci. 2017;8:268–277. doi: 10.1039/c6sc01814a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu J, He X, Qi Y, Tian X, Monkley SJ, Critchley DR, Corbett SA, Lowry SF, Graham AM, Li S. Mol. Cell. Biol. 2011;31:3366–3377. doi: 10.1128/MCB.01403-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell. 1991;64:841–848. doi: 10.1016/0092-8674(91)90512-w. [DOI] [PubMed] [Google Scholar]
- 48.Ornitz DM, Itoh N. Wiley Interdiscip. Rev.: Dev. Biol. 2015;4:215–266. doi: 10.1002/wdev.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu D, Esko JD. Annu. Rev. Biochem. 2014;83:129–157. doi: 10.1146/annurev-biochem-060713-035314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dowling CM, Herranz Ors C, Kiely PA. Biosci. Rep. 2014;34:415–427. doi: 10.1042/BSR20140031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roshan Moniri M, Young A, Reinheimer K, Rayat J, Dai L-J, Warnock GL. Cytotechnology. 2015;67:379–386. doi: 10.1007/s10616-014-9692-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.