

Abstract
Stimulation of Na+/H+ exchanger isoform 1 (NHE1) in astrocytes causes ionic dysregulation under ischemic conditions. In this study, we created a Nhe1flox/flox (Nhe1f/f) mouse line with exon 5 of Nhe1 flanked with two loxP sites and selective ablation of Nhe1 in astrocytes was achieved by crossing Nhe1f/f mice with Gfap-CreERT2 Cre-recombinase mice. Gfap-CreERT2+/-;Nhe1f/f mice at postnatal day 60-90 were treated with either corn oil or tamoxifen (Tam, 75mg/kg/day, i.p.) for 5 days. After 30-day post injection, mice underwent transient middle cerebral artery occlusion (tMCAO) to induce ischemic stroke. Compared to the oil-vehicle group (control), Tam-treated Gfap-CreERT2+/-;Nhe1f/f (Nhe1 KO) mice developed significantly smaller ischemic infarction, less edema, and less neurological function deficits at 1-5 days after tMCAO. Immunocytochemical analysis revealed less astrocytic proliferation, less cellular hypertrophy, and less peri-lesion gliosis in Nhe1 KO mouse brains. Selective deletion of Nhe1 in astrocytes also reduced cerebral micro-vessel damage and blood-brain barrier (BBB) injury in ischemic brains. The BBB micro-vessels of the control brains show swollen endothelial cells, opened tight junctions, increased expression of pro-inflammatory protease MMP-9 and significant loss of tight junction protein occludin. In contrast, the Nhe1 KO mice exhibited reduced BBB breakdown and normal tight junction structure, with increased expression of occludin and reduced MMP-9. Most importantly, deletion of astrocytic Nhe1 gene significantly increased regional cerebral blood flow in the ischemic hemisphere at 24 hours post-MCAO. Taken together, our study provides the first line of evidence for a causative role of astrocytic NHE1 protein in reactive astrogliosis and ischemic neurovascular damage.
Keywords: astrocyte end-feet, blood-brain barrier, cerebral edema, gliosis, MMP, neurovascular unit
Introduction
Astrocytes are the ubiquitous glial cells that play many critical roles in supporting normal brain function, including maintenance of ionic and osmotic homeostasis, regulation of metabolism of major neurotransmitters, remodeling extracellular space, and providing inflammatory and antioxidant defense in the brain (Sofroniew et al., 2010; Verkhratsky et al., 2015). In addition, astrocytes interact with neurons, microglia, endothelial cells and pericytes in regulating neurovascular functions and blood-brain barrier (BBB) integrity (Alvarez et al., 2013). Thus, this functional versatility of astrocytes makes them critical regulators of brain microenvironment (Kimelberg et al., 2010; Schitine et al., 2015).
Astrocytes respond to brain injury by a hallmark process of reactive astrogliosis, characterized by a hypertrophic phenotype and increased expression of the intermediate filaments protein, glial fibrillary acidic protein (GFAP) (Sofroniew, 2009; Zamanian et al., 2012; Hol et al., 2015). Accumulating evidence indicates that astrogliosis could be involved in exacerbating injury and/or tissue repair depending on the brain injury time and context (Colombo et al., 2016). Upon ischemic stroke, reactive astrocytes are known to amplify injury via the release of neurotoxic levels of reactive oxygen species, exacerbate inflammation via cytokine production, compromise BBB function via VEGF production, and aggravate cytotoxic edema through stimulation of AQP4 (Swanson et al., 2004; Brambilla et al., 2005; Argaw et al., 2009; Zador et al., 2009; Cengiz et al., 2014). Concomitantly, reactive astrocytes are also involved in tissue repair via secretion of anti-inflammatory factors, uptake of excess glutamate and isolation of undamaged tissue (Li et al., 2008; Kim et al., 2010; Wanner et al., 2013). The complex aspects of astrogliosis function drive profound interest in developing a better understanding of the signals regulating reactive astrogliosis and, in particular, in searching for the molecular targets that promote tissue functional recovery, which is comprehensively reviewed by Liu et al (Liu et al., 2017). Previous studies indicate that deletion or inactivation of astrocyte-specific TGFβ signaling in mice exacerbates motor function deficits and glial scar formation without altering overall disease severity after ischemic stroke (Cekanaviciute et al., 2014). Selective deletion of Notch 1 signaling in astrocytes was shown to reduce astrocyte proliferation in the ischemic peri-infarct region at 24 h of reperfusion, but the functional outcomes were not evaluated (Shimada et al., 2011). Selective deletion of STAT3 in reactive astrocytes also results in increased white matter damage, neuronal death and slower motor recovery after spinal cord injury (Okada et al., 2006; Herrmann et al., 2008). On the other hand, conditional knockout of p38 MAPK in GFAP-Cre/loxp mice prevented astrocytic hypertrophy and GFAP upregulation and enhanced motor functional recovery after ischemic stroke (Roy Choudhury et al., 2014). These findings suggest that multiple but distinct signaling cascades are involved in regulating reactive gliosis and have differential impacts on neurological damage and recovery of neurological function after brain injury.
In this study, we investigated roles of astrocytic Na+/H+ exchanger isoform 1 (NHE1) in reactive astrocyte formation and in neurological damage after ischemic stroke. NHE1 protein represents a major pathway for H+ extrusion in exchange for Na+ in astrocytes (Kirischuk et al., 2012; Rose et al., 2013). Our previous studies show that oxygen-glucose deprivation of cortical astrocytes leads to sustained NHE1 activation, resulting in Na+ overload and swelling (Kintner et al., 2004; Luo et al., 2005). Genetic ablation of Nhe1 or pharmacological inhibition with its potent inhibitor HOE 642 protects against astrocyte damage (Yao et al., 1999; Kintner et al., 2004; Luo et al., 2005). These findings demonstrate the importance of astrocytic NHE1 in ischemic astrocyte injury. However, since NHE1 is expressed in all cell types, using either the global Nhe1 knockout or pharmacological inhibition of NHE1 does not allow a detailed analysis of the specific roles of astrocytic NHE1 protein in astrocyte dysfunction after ischemic stroke. In this study, we created a Nhe1f/f mouse line and produced Gfap-CreERT2+/-;Nhe1f/f littermates by crossing Nhe1f/f with an inducible Gfap-Cre transgenic mouse line Gfap-CreERT2+/-. Selective deletion of astrocytic Nhe1 in Gfap-CreERT2+/-; Nhe1f/f mice led to reduced stroke volume, inhibition of reactive astrocyte formation and improved neurological function after ischemic stroke. Our study shows for the first time that deletion of Nhe1 gene specifically in astrocytes has a beneficial effect in ischemic stroke.
Materials and Methods
Materials
5-Bromo-2′-Deoxyuridine (BrdU), Evans blue dye, tamoxifen, rabbit anti-laminin antibody, and 2,3,5-triphenyl-tetrazolium chloride (TTC) were from Sigma-Aldrich (St. Louis, MO). Osmium Tetroxide and Uranyl Acetate were from Electron Microscopy Sciences (Hatfield, PA). Propylene oxide, Polybed 812 epoxy resin were from Polysciences (Warrington, PA). Toluidine Blue and Potassium Ferricyanide was from Fischer chemicals. Rabbit anti-Aquaporin 4 (AQP4) antibody was from Milipore (Billerica, MA). Mouse anti-GFAP antibodies were from Cell Signaling Technology (Danvers, MA). Sheep anti-BrdU antibody and mouse monoclonal antibody to S100β were from Abcam (Cambridge, MA). Anti-MMP-9 and anti NHE1 andibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal antibodies to Occludin, and Claudin antibody were from Thermoscientific Life Technologies Corporation (Grand Island, NY). Donkey anti-goat Alexa Fluor® 488-conjugated IgG, goat anti-rat Alexa Fluor® 488-conjugated IgG, donkey anti-rabbit Alexa Fluor® 546-cojugated IgG, goat anti-rabbit Alexa Fluor® 546-cojugated IgG, and TO-PRO®-3 iodide were from Invitrogen (Carlsbad, CA).
Mice and genotyping
All studies were in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals from the U.S. Department of Health and Human Services and were approved by the University of Pittsburgh Medical Center Institutional Animal Care and Use Committee.
Generation of Nhe1f/f mice
Design and construction of the Nhe1 targeting vector and subsequent steps to generate heterozygous Nhe1flox/+ mice were performed by the Gene Targeted Mouse Service Core at the University of Cincinnati. To engineer the targeting vector for the Nhe1-loxP locus, two loxP motifs were introduced into a 0.9-kb fragment of Nhe1 gene that flank exon 5. This targeting construct (Nhe1-loxP) also contained a neomycin-resistance gene flanked by two FRT sites located next to the 5′-LoxP site (Figure 1 A). The floxed region and the homologus arms, 2.6 kb and 3.4 kb, were PCR amplified from mouse genomic DNA and cloned into the vector. Subsequent homologous recombination was performed in embryonic stem (ES) cells derived from a 129S6/SvEv Tac strain (Taconic, Hudson, NY). Correctly targeted ES cells, were verified by southern blotting (Figure 1 B). Chimeric mice were generated and bred with Black Swiss (Taconic) females and germline transmission was identified by PCR analysis. Mice carrying the targeted allele were then bred with a mouse expressing FLP recombinase in order to remove the Neo cassette via recombination between the two FRT sites flanking the Neo gene. PCR genotyping analysis of Nhe1f/f mice was carried out using genomic DNA from tail biopsies and the following primers: Forward primer: 5′-CTGCTGCATTCTCTATCTTACTC-3′, P3 in Fig. 1C; Reverse primer: 5′-GTTTCGAAGTGTAGGCTGTGAG-3′, P4 in Fig. 1C. The reaction mix was composed of 10 μl GC 20× buffer (New Englang Biolabs), 8.6 μl H2O, 1 μl DNA, using the following amplification protocol (94°C 2 min; 94°C 30 s; 55°C 30 s; 72°C 1 min; 30 cycles; 72°C 3 min). Amplification product sizes were as follows: wild type (280 bp), Nhe1f/+ (280 bp and 406 bp), Nhe1f/f, 406 bp. In parallel, forward primer 5′- GTCAATCAGTATATGAAGTGACG-3′, P1 in Fig. 1C and reverse primer 5′- GAACTGCTCGA ATATGATAAC-3′, P5 in Fig 1C primers were used to amplify a 150-base pair product from the mutant to confirm the genotype.
Figure 1. Creation of Nhe1f/f conditional knockout mice.
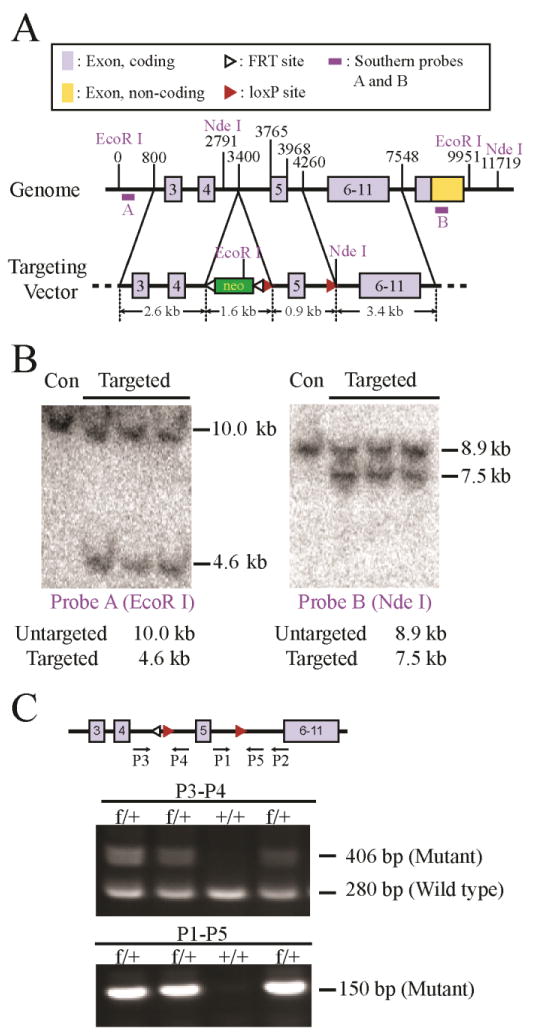
A. Nhe1 conditional knockout strategy and position of southern blots probes. A targeting construct was made with two loxP sites flanking exon 5 with a PGKneo cassette flanked by two FRT sites located in intron 4. The construct was transfected into ES cells and the clones with appropriate homologous recombination were identified by Southern blot analysis using probes A and B, which were from regions 5′ and 3′, respectively, to the fragments used to prepare the construct.
B. Genomic DNA from three targeted clones and untargeted ES cells (Con) was digested with EcoR1 or Nde I and the products were detected by Southern blot analysis using probes A and B, respectively. C. Diagram of the targeted allele after deletion of the PGKneo cassette and PCR genotyping of Nhe1f/+ and Nhe1+/+ animals using P1 – P5 PCR Primers. Primers P3 and P4 amplify a 406-bp product in the floxed allele, and a 280-bp product in the wild type gene. Primers P1and P5 (which includes sequences from the LoxP site) amplifies a 150-bp product in the floxed allele only.
Generation of Gfap-CreERT2+/-;Nhe1f/f mice
The Gfap-CreERT2+/- transgenic mouse line (C57BL/6 genetic background) was generated as described previously (Ganat et al., 2006) and purchased from Jackson Laboratory (Stock # 012849). The mice express a Cre recombinase–estrogen receptor type 2 fusion protein (CreERT2) in radial glia, astrocytes, and neuro stem cells under control of the Gfap2 upstream fragment of the human GFAP (hGFAP) promoter (Ganat et al., 2006). Genotyping was done by PCR using primers to the Cre gene (5′- GCA ACG AGT GAT GAG GTT CGC AAG -3′, forward; 5′-TCC GCC GCA TAA CCA GTG AAA CAG -3′, reverse) to generate a band of 307 bp (Ganat et al., 2006).
Nhe1f/f mice were cross bred with Gfap-CreERT2+/- mice to generate F1 Gfap-CreERT2+/-;Nhe1f/+ mice. The F1 mice were subsequently mated with Nhe1f/f mice to generate Gfap-CreERT2+/-;Nhe1f/f mice. Mice were genotyped by standard PCR reaction protocols. All the exeriments were carried out on Gfap-CreERT2+/-;Nhe1f/f mice. Astrocyte specific knockout of Nhe1 was induced by a 5 day i.p. injection of tamoxifen (Tam) in corn oil at a dosage of 75 mg/kg (i.p.) starting at postnatal day 60 to 90 (P60-90). Mice injected with corn oil only served as controls. Scientists were blinded to the allocation of the mice to the groups. Litters were caged in mixed groups after i.p. injection with either corn oil or Tam and also upon surgery. Animals for experiments were gender and age matched. A total of 80 mice used for the study consisted of 8-12 week old males (n=56; weight: 20±0.5 g) and females (n=40, weight: 25 ± 0.2 g). For analyses of infarct volume measurement 12 mice in total were used (male oil: n=3; female oil: n=2; male tam: n=5; female tam: n=2). Behavioral studies and immunocytochemical analyses were performed on the same cohort of animals and brain samples were extracted at different experimental times. Mice were excluded from the study if they died prior to 24 h reperfusion (n=6).
Middle cerebral artery occlusion and reperfusion
Transient focal cerebral ischemia was induced by intraluminal occlusion of left middle cerebral artery (MCA), as previously described (Begum et al., 2015). Under 1.5% isoflurane, the left common carotid artery (CCA) was exposed via a midline pre-tracheal incision and the external carotid artery (ECA) and the CCA were ligated. To occlude the MCA, a rubber silicon-coated monofilament suture (Filament size 6-0, diameter 0.09-0.11 mm, length 20 mm; diameter with coating 0.23 ± 0.02 mm; coating length 5 mm) was inserted into the ECA and advanced along the internal carotid artery 18-20 mm from the bifurcation of the carotid artery. For reperfusion, the suture was gently withdrawn 60 min after ischemia. Tympanic membrane and rectal temperature probes were inserted and cranial and body temperatures were maintained at 36.5±0.5 °C throughout the experiment by a heating blanket.
Regional cerebral blood flow (rCBF) measurements
Cerebral blood flow was monitored using a two-dimensional laser speckle contrast analysis system (PeriCam PSI High Resolution with PIMSoft; Perimed, Järfälla, Sweden). Mice were anesthetized with isofluorane and maintained at physiological body temperature as described above. The skull of the animal was secured in a stereotactic frame (David Kopf Instruments, Tujunga, CA). A midline incision was made in the scalp and the skull surface cleaned with sterile normal saline. A charged-coupled device camera was placed 10 cm above the skull using a Pericam PSI System and blood perfusion images were taken at 5 min prior to MCAO, every 15-min interval during MCAO or during 0-90 min reperfusion, and at 24 h reperfusion. Raw speckle images were taken in a 1.6 cm × 1.4 cm field (at 19 frames/second). 57 frames averaging, with the resolution of 0.02 mm3 consecutive images at each time point per animal were averaged for analysis using equal-sized, oval-shaped regions of interest (ROI) covering the frontal and parietal bone plates of the ipsilateral (IL) and contralateral (CL) hemispheres of the Con and Nhe1 KO mice. Only animals with at least a 50% decrease in the ipsilateral blood perfusion following MCAO were included in the study. Cerebral blood perfusion is expressed in arbitrary units (Perfusion Units).
Measurement of infarct volume
Infarct volume was determined by 2,3,5-triphenyl-tetrazolium chloride (TTC) staining at 48 h reperfusion, as described before (Begum et al., 2015). Infarction volume was quantified using Imaje J software as described previously (Swanson et al., 1990). The extent of hemispheric swelling was calculated using the equation:volume of ipsilateral hemisphere−volume of contralateral hemisphere)/volume of contralateral hemisphere.
Evaluation of Evans blue dye extravasation
Changes in the BBB permeability were assessed by the leakage of Evans blue (EB) into the brain parenchyma following intravenous injection of EB dye (2% in PBS, 10 μl/g body weight at 22 h reperfusion). EB (961 Da dye) strongly binds to the albumin fraction of proteins to form a high molecular weight complex (68.5 kDa) and EB complex does not permeate the intact BBB unless the BBB is compromised after stroke (Belayev et al., 1996). Following 2 h injection, mice were transcardially perfused with PBS to clear the blood and any remaining EB in the vascular system. The brain was quickly removed, fixed in 4% PFA in 0.1 M PBS for 24 h and then cryoprotected in 30% sucrose in 0.1 M PBS overnight. Coronal brain sections (40 μm) were mounted onto glass slides and coverslipped with Vectashield mounting medium. The fluorescence signals of EB (excited at 635 nm, emitted at 665 and 735 nm) were obtained with a confocal microscope (Leica DMIRE2) using 20× ojective. The confocal images were analyzed using ImageJ software (NIH). The corresponding EB signal was background subtracted and the intensity of EB was quantified. Three sections per brain were imaged and 3-4 images per section was used for analysis.
Cerebral blood vessel isolation
Cerebral blood vessels were isolated from mouse brains as described previously (Boulay et al., 2015). Briefly, 48 h after MCAO mice were anesthetized with 3% isfluorane and decapitated and placed in HBSS containing 1M HEPES. The choroid plexus was dissected out from the lateral ventricles and the CL and IL cortices were separated. The brain tissue was then chopped into small pieces and gently homogenized with a automatized Dounce homogenizer (20 strokes at 400 rpm) in HBSS, followed by centrifugation at 2000 g for 10 min at 4 °C. Pellets were resuspended in HBSS with 18% dextran (MW 70 000) and centrifuged at 4,400 g for 15 min at 4 °C. Pelleted blood vessels were collected on top of a nylon mesh (20 μm) and washed extensively with HBSS containing 1% BSA. The vessels were resuspended in ice cold HBBS with 1% BSA and centrifuged at 2000 g for 5 min at 4 °C. Isolated vessels were processed immediately for immunostaning as described previously (Boulay et al., 2015). Vessels were incubated with primary antibodies for occludin (1:100) overnight at 4 °C followed by goat anti-mouse Alexa 488–conjugated secondary antibodies for 1 h at room temperatutre (RT). Images were captured using Leica DMIRE2 inverted confocal laser scanning microscope.
Immunofluorescencent staining and image analysis
Mice were anesthetized and transcardially perfused with 4% PFA in 0.1 M PBS. The brains were postfixed (4% PFA, 4° C, 24 h) and equilibrated in 30% sucrose at 4°C. Coronal brains sections (25 μm, at -0.38 mm bregma) were washed with PBS and incubated with blocking solution (10% NGS, 0.5% TX-100 in 0.1 M PBS) for 1 h at room temperature. Reactive astrocytes were identified by incubating sections with mouse anti- GFAP (1:200) or rabbit anti-GFAP (1:200), and mouse anti-S100 β (1:200), antibodies. BBB tight junction damage was identified by mouse anti-Occludin (1:100). Astrocyte end feet were labeled by rabbit anti-Aquaporin-4, and rabbit anti-Laminin (1:200) antibodies. Mouse anti-GLUT1 (1:100) antisera was used for labeling the endothelial cells, and mouse anti-MMP-9 (1:100), antisera was used for labeling the endothelial cells and perivascular astrocyte end feet. All the primary antibodies were diluted in the blocking solution and incubated with brain sections over night at 4°C. On the following day, the sections were washed with PBS and then incubated with secondary goat anti-mouse Alexa 488–conjugated IgG and goat anti-rabbit Alexa Flour 546–conjugated IgG; or donkey anti-rabbit Alexa Fluor 488–conjugated IgG and donkey anti-mouse Alexa Fluor 546–conjugated IgG. Nuclear stain TO-PRO-3 iodide (1:1000 in blocking solution) was used to stain the nucleus. For negative controls, brain sections were stained with the secondary antibody only.
For cellular morphological and quantitative analysis of astroglyosis, Bitplane Imaris software was used (Version 7.4.2, Bitplane, Zurich, Switzerland). Briefly, images were imported and subjected to surface reconstruction. Surface reconstruction parameters were set to appropriately label all astroglia and their processes and the voxels within one stack, were rendered into three dimensional objects and the volume was analysed. The volume of the obtained objects was expressed as summated soma volume of the ROI. Next, we used the FilamentTracer module (Imaris) to quantify morphological changes of astrocyte processes using the following endpoints: mean process length and the summarized process volume.
For the quantification of microvessels, skeleton analysis of AQP4-labeled vessels was performed as previously described (Morrison et al., 2013). Briefly, 3-4 confocal images in each ROI were despeckled to reduce background noise. The resulting image was converted into a binary image and then skeletonized using ImageJ software. The AnalyzeSkeleton plugin (http://imagejdocu.tudor.lu/) was applied to all skeletonized images to quantify the number of vessels, branches and vessel length. Data were expressed as the summed number of vessels, vessel length and the summed branches per frame. For the quantification of immunofluorescent signals, 3-6 fields of view per section were acquired from the perilesional IL and corresponding CL cortex. Immunoreactivity was quantified with the ImageJ software by measuring the corrected total cell fluorescence or mean gray values. The results were expressed in arbitrary units.
BrdU immunostaining analysis
To label newly synthesized DNA, mice received injections of 5-bromo-2-deoxyuridine (BrdU, 50 mg/kg in saline, i.p.) with an initial dose at day 3 after MCAO and daily for two consecutive days. Mice were killed 2 h after the last BrdU injection at day 5 after MCAO. For BrdU immunostaining, the brain sections mounted on slides were pretreated with 2 N HCL for 30 min at 37° C followed by 3 washes in 0.1 M PBS and then incubated with polyclonal sheep anti-BrdU (1:50) and mouse anti-GFAP (1:200). Slides were then rinsed with 0.1 M PBS and incubated with Alexa 546 donkey anti-sheep (1:200) and Alexa 488 donkey anti-mouse (1:200) for 2 h RT, washed with 0.1 M PBS, mounted, and coverslipped with mounting medium. Fluorescent images were captured under 20× or 40× lens using a Leica DMIRE2 inverted confocal laser scanning microscope. Identical digital imaging acquisition parameters were used throughout the study. Gliogenesis in the peri-infarct region was evaluated by counting the number of GFAP+/BrdU+ cells using Image J software. The total number of cells in the region of interest was determined by the DRQ5 nuclear staining. The number of positively stained cells in the ROI was normalized by the total number of cells and data were expressed as an average of the number of positive cells/total cells ×100.
Transmission electron microscopy
Structural integrity of cerebral microvessels was analyzed in the CL and IL peri-infarct areas in the cortex as desribed previously (Kimbrough et al., 2015) with modifications. Briefly, mice were transcardially perfused with PBS followed by fixation with 4% PFA for 24 h. After fixation, the brain was removed and sectioned into 1 mm thick slices and post fixed in 2.5% glutaraldehyde in PBS. Under a dissection microscope, tissue punches were taken to capture the IL and CL peri-infarct areas in the cortex. Tissues were washed three times in PBS then, post-fixed in 1% Osmium Tetroxide with 1% potassium ferricyanide for 1 hour. Following three additional PBS washes, the pellet was dehydrated through a graded series of 30-100% ethanol, 100% propylene oxide and then infiltrated in 1:1 mixture of propylene oxide: Polybed 812 epoxy resin for 1 hr. After several changes of 100% resin over 24 hrs, pellet was embedded in a final change of resin, cured at 37°C overnight, followed by additional hardening at 65°C for two more days. Ultrathin (70 nm) sections were collected on 200 mesh copper grids, stained with 2% uranyl acetate in 50% methanol for 10 minutes, followed by 1% lead citrate for 7 min. Sections were imaged using a JEOL JEM 1011 transmission electron microscope (Peabody, MA) at 80 kV fitted with a side mount AMT 2k digital camera (Advanced Microscopy Techniques, Danvers, MA).
Neurological function tests
Neurological functional deficits in mice were screened in a blinded manner with the following tests: neurological score, corner test, adhesive removal test, and rotarod test, all considered reliable for identifying and quantifying sensorimotor deficits and postural asymmetries (Zhang et al., 2002; Schaar et al., 2010).
Neurological score
A neurological deficit grading system was used to evaluate neurological deficit at 1 - 5 days after MCAO as described previously (Zhang et al., 2002) according to the following scale: 0 = no observable deficit; 1 = forelimb flexion; 2 = forelimb flexion and decreased resistance to lateral push; 3 = forelimb flexion, decreased resistance to lateral push and unilateral circling; and 4 = forelimb flexion and impaired or absent ambulation.
Adhesive removal test
The adhesive removal test, the most effective assesment of sensorimotor function impairments after ischemic stroke (Bouet et al., 2007; Bouet et al., 2009), was used to measure somatosensory deficits as described previously (Zhao et al., 2016). Two pieces of adhesive tape (4 mm × 3 mm) were attached to the forepaws in an alternating sequence and with equal pressure by the experimenter before each trial. The mouse was placed in a transparent plastic box and the times to contact and to remove each adhesive tape were collected with a maximum of 120 s. Experimenters were blind with regard to the mice groups. Pre-operative training was carried twice per day for 3 days. Animals were tested on day 1, 2, 3, 4 and 5 after MCAO. Two parameters were monitored for each paw: contact time (point that the mouse reacts to the presence of adhesive strips) and adhesive removal point.
Rotarod test
The rotarod test was used to assess motor coordination and balance alterations. The apparatus consisted of a black striated rod separated in four compartments (Model 755, IITC Life Science Inc., Woodland Hills, CA). Animals were first habituated to a stationary rod for 2 min, then placed on a rotating drum accelerating from 4 to 40 rpm over a 5 min period. The time the animal stayed on the drum was recorded. Three trials were performed with 15-min interval rest periods. Then, the mice were placed on the rotating drum at 40rpm/s and observed over 60s. The time at which the animal fell off the drum was recorded. Animals were tested on day 1, 2, 3, 4 and 5 after MCAO.
Statistical analysis
All statistical tests were performed using GraphPad Prisim 6.0. or SigmaPlot 11. The data are reported with mean values ± standard error or deviation (as indicated). Statistical significance was determined by Student t test or one-way analysis of variance (ANOVA) in case of multiple comparisions. Differences between groups were tested using Student-Newman-Keuls post-hoc test. Behavioral data was analyzed using nonparametric Kruskal-Wallis test followed by the Mann-Whitney U test with Bonferroni correctios. Values of p < 0.05 were considered statistically significant.
Results
Generation of the Nhe1f/f allele
To generate the Nhe1 conditional knockout mouse line (Nhe1f/f), we performed homologous recombination in embryonic stem (ES) cells in order to introduce loxP sites that flank exon 5. A targeting construct with two loxP sites flanking exon 5 with a PGKneo cassette flanked by two FRT sites was engineered (Figure 1 A). Out of 396 drug resistant ES cell clones, 11 were found to be homologously recombined at the target site, as determined by PCR analysis. Seven of them were further analyzed and confirmed by Southern blot analysis (Figure 1 B) using probes from genomic DNA that was 5′ or 3′ to the region used to prepare the construct (probes A or B, respectively, in Figure 1A). The targeted allele displayed a 4.6 kb band after EcoRI digestion and a 7.5 kb band after NdeI digestion, whereas the wild-type allele displayed a 10.0 kb band after EcoRI digestion and an 8.9 kb band after NdeI digestion (Figure 1 B). Three clones, with highest degrees of euploidy, were chosen for C57BL/6 host blastocyst injection to generate chimeric mice. Chimeras from one clone (K2F10) were germline competent and produced offspring derived from the ES cells as determined by PCR analysis of tail biopsies. After germline transmission, additional breeding with a mouse expressing FLP recombinase was performed to remove the Neo cassette and leave LoxP sites flanking exon 5 (Figure 1 C). Nhe1f/+ mice lacking the Neo cassette were mated with C57BL/6 wild-type mice for nine generations before being considered on a pure C57BL/6 background, and subsequently intercrossed to obtain homozygous Nhe1f/f mice. All the genotypes were confirmed by PCR analysis (Figure 1 C). Nhe1f/f mice were born at a normal Mendelian ratio and were viable without displaying any obvious defect as compared to wild-type littermates. Adult Nhe1f/f females generated normal-sized litters and showed normal maternal care behaviour.
Selective deletion of Nhe1 in Gfap-CreER+/-;Nhe1f/f mice reduces infarction and improves neurological functions after ischemic stroke
Generation of Gfap-Cre ER+/-;Nhe1f/f mice was made by crossing Gfap-CreER+/-mice with Nhe1f/f mice (Figure 2 A). Astrocyte specific deletion of Nhe1 in Gfap-CreER+/-;Nhe1f/f mice was induced by administration of Tam (75 mg/kg, i.p.) daily for five consecutive days to achieve maximum Cre-mediated recombination as described before (Madisen et al., 2010). Corn oil-treated Gfap-CreER+/-;Nhe1f/f mice served as controls (Figure 2 A). 30-day post injection waiting period was used for tamoxifen clearance. Transient MCAO was induced in both male and female mice and biochemical and neurological functional evaluations were conducted at day 1-5 post-MCAO (Figure 2 B). Kaplan-Meier survival curves of the control and Nhe1 KO mice following ischemic stroke have been included in Supporting Information Figure 2. Mortality data analyzed using the Mantel-Cox log-rank test revealed no significant differences in the survival rates between the two groups (p = 0.589).
Figure 2. Deletion of Nhe1 in Gfap-CreER+/-;Nhe1 f/f mice decreases ischemic infarct, swelling, and improves neurological function.
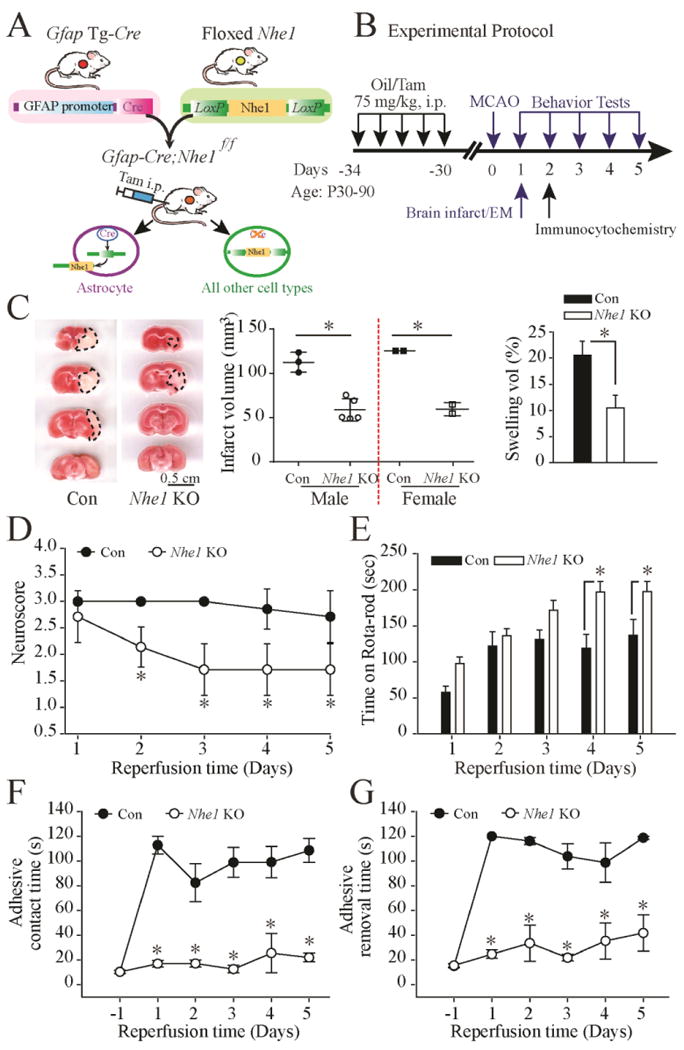
A. Breeding scheme for generation of astrocyte-specific Nhe1 knockout mice. Nhe1f/f mice were crossed with Gfap-CreER+/- transgenic mice to generate Gfap-CreER+/-;Nhe1f/f mice.
B. Experimental protocol. Gfap-CreER+/-;Nhe1f/f mice at postnatal days P60-90 were treated with either corn oil or Tam in corn oil (75mg/kg/day, ip) for 5 days. After 30 days, ischemic stroke was induced by transient middle cerebral artery occlusion (MCAO) at P90-120. Biochemical or neurological assays were conducted at 1-5 days post-stroke.
C. Representative TTC-stained coronal sections of control (oil-) and Nhe1 KO (Tam-treated) brains at 48 h reperfusion. Infarct volume and % of hemispheric swelling were shown. Data are mean ± SD. n = 5-7 (Oil: 3 male, 2 female; Tam: 5 male, 2 female). *p < 0.05 vs. oil.
D. Neurological scores. Data are mean ± SD. n = 7 (Con: 1 male; 6 female; Nhe1 KO: 5 male, 2 female). * p < 0.05 vs. oil.
E. Rotarod test with accelerating speed. Data are mean ± SEM. n = 9-11 (Con: 2 male, 7 female; Nhe1 KO: 6 male, 5 female). * p < 0.05 vs. oil.
F. Contact time of adhesive test. Data are mean ± SEM. n = 7 (Con: 4 male, 3 female; Nhe1 KO: 4 male, 3 female). * p < 0.05 vs. oil.
G. Removal time of adhesive test. Data are mean ± SEM. n = 7 (the same cohort). * p < 0.05 vs. Con.
TTC staining of brain sections at 1, 3, 5 and 7 mm from the frontal pole revealed the extent of brain damage at 48 h reperfusion (Figure 2 C). The control mice exhibited large infarcts (males =112.4 ± 6.4 mm3; females = 125 ± 0.1 mm3) and hemispheric swelling (male = 24 ± 1.5%; female = 15 ± 1.7 %). There were no significant differences between male and female mice (p > 0.05). In contrast, astrocytic Nhe1 KO mice exhibited reduced infarct volume (male = 58.7 ± 5.6 mm3; female = 59 ± 5.3 mm3) and smaller swelling volume (male =11 ± 3.3%; female = 7.4 ± 1.4 %, p < 0.05). In the arrays of neurological function tests, the control mice illustrated classical neurological deficit development at 1-5 days after MCAO, including poor neurological scores (Figure 2 D), a significant deficit in locomotor coordination in the accelerated rotarod test (Figure 2 E) with no signs of recovery at day 5 reperfusion. In contrast, the Nhe1 KO mice exhibited significantly better neurological scores as early as day 1 of reperfusion and showed significant improvement in motor functions at day 4-5 post injury (Figure 2 D, E). Figure 2 F and G show that the oil-treated mice exhibited significantly prolonged adhesive contact time and removal time at all the time points. No improvement was detected until day 5 post-stroke. In contrast, Nhe1 KO mice showed almost no deficits in the adhesive contact and removal tests and significantly better neurological function, compared to the oil-treated mice (p < 0.05). Taken together, these findings clearly illustrate that deletion of Nhe1 gene in GFAP+ astrocytes not only reduces ischemic stroke-mediated infarct but also improves their neurological function.
Supporting Information Figure 1 illustrates the efficacy of Tam-induced specific knockout of Nhe1 on NHE1 protein expression in ischemic brains. Using immunofluorescence staining assay, at day 3 post-stroke, the control mouse (Con) brain expressed abundant NHE1 protein in both GFAP+ astrocytes and neurons in the Ipsilateral hemispheres (Supporting Information Figure 1 A). In contrast, NHE1 protein expression was absent in GFAP+ astrocytes (arrow-heads) but remained unchanged in neurons (arrows) in the Tam-treated Nhe1 KO mouse (Supporting Information Figure 1 B). Low basal levels of NHE1 protein expression was detected in non-ischemic brains (data not shown).
Deletion of Nhe1 in Gfap-CreER+/-;Nhe1f/f mice reduces gliogenesis and reactive astrocyte formation after ischemic stroke
Next, we evaluated whether NHE1 activity plays a role in reactive astrocyte formation after transient ischemic stroke. At day 2 after MCAO, little expression of GFAP was detected in the CL uninjured brains (data not shown). However, astrocytes with elevated GFAP expression in the processes and soma formed an astroglial barrier around the peri-lesion area in the control mice (Figure 3 A). In contrast, the Nhe1 KO mice exhibited significantly reduced GFAP staining intensity (Figure 3 A). We further evaluated whether the reduced numbers of GFAP+ astrocytes represent less astrocyte proliferation. We injected mice with the mitotic marker BrdU at day 3 after MCAO to determine proliferation when astrocytes are activated. As shown in Figure 3 A (Lower panel), the control mice showed significantly higher Brdu+/GFAP+ cells in the ischemic peri-lesion area than the Nhe1 KO brains. These data suggest that deletion of astrocytic Nhe1 also inhibits reactive astrocyte proliferation after ischemic stroke.
Figure 3. Deletion of Nhe1 in Gfap-CreER+/-;Nhe1f/f mice inhibits reactive gliosis and astrocytic BrdU labeling.
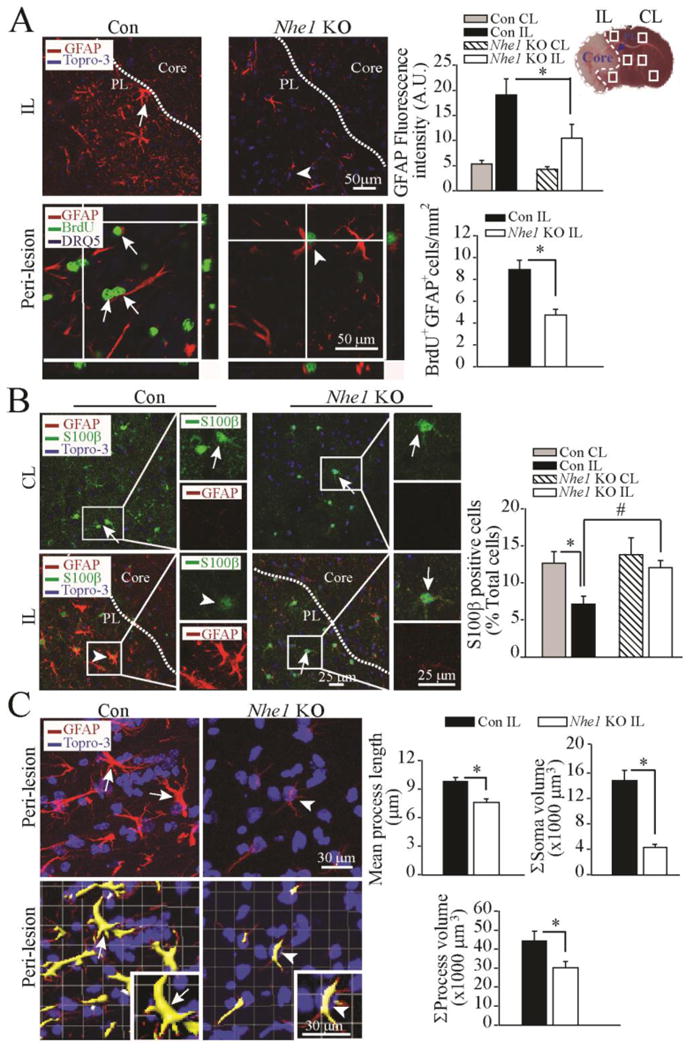
A. Representative images of GFAP+ astrocytes were shown in the peri-infarct zones of the Con or Nhe1 KO mice. A TTC-stained brain section at 48 h after MCAO was marked for the ischemic core, and sample collection areas (white box) in the peri-lesion tissue (PL, dotted lines) and contralateral control. Arrows: Increased GFAP expression or BrdU labeling. Arrowhead: Reduced GFAP expression or BrdU labeling. Data are mean ± SEM, n = 3-4. * p < 0.05 vs. Con.
B. Representative images of GFAP, S100β, and To-pro-3 staining in the CL and IL cortex regions of the Con and Nhe1 KO mice. Arrows: Preserved S100 β expression. Arrowheads: loss of S100 β in GFAP+ astrocytes. Data are ± SEM, n = 3. *p < 0.05 vs. CL; # p < 0.05 vs. Con.
C. Representative immunofluorescent Z-stack images of brain sections showing changes of GFAP+ reactive astrocyte morphology in the Con and Nhe1 KO mice. Lower panel shows cropped view of 3D rendering. Arrows: increased soma area of GFAP+ cells. Arrowheads: Reduced soma area of GFAP+ cells. Summary data of changes of GFAP+ cell processes, mean process length, and soma volume were shown. Data are mean ± SEM, n = 6. * p < 0.05 vs. Con.
Moreover, selective knockout of astrocytic Nhe1 in GFAP+ astrocytes preserves S100β expression. Figure 3 B shows that in the CL regions of both the control and Nhe1 KO brains, S100β+ cells spread throughout the brain parenchyma (arrows). Astrocytes express S100β in the cell bodies and thin processes. After 48 h of reperfusion, loss of S100β and increased GFAP expression (arrow head) were seen in the peri-lesion areas of the control mice. In contrast, the Nhe1 KO ischemic brains exhibited preserved S100β expression and lack of GFAP+ astrogliosis (arrows).
We further quantified changes of reactive astrocyte morphology in the control and Nhe1 KO ischemic brains. Representative confocal Z stacks of GFAP-stained images in the peri-lesion area were taken and analysed (Figure 3 C). GFAP+ astocytes in the control brains had highly ramified morphology with increase in the astrocyte soma volume (15 ± 0.2 μm3), mean process length (10 ± 0.3 μm), and process volume (44 ± 0.1 μm3). In contrast, the Nhe1 KO ischemic brains showed significantly less ramified morphology with smaller cell body volume (4 ± 0.1 μm3), and reduced process length (7 ± 0.4 μm) and process volume (28.3 ± 0.2 μm3) (Figure 3 C). Taken together these results strongly suggest that NHE1 protein is involved in the regulation of reactive astrocyte proliferation and astrocyte function in ischemic brains.
Deletion of Nhe1 in Gfap-CreER+/-;Nhe1f/f mice reduces cerebral vessel damage and preserves BBB integrity after ischemic stroke
We then studied the impact of deleting astrocytic Nhe1 gene on cerebral vessel damage and BBB integrity in ischemic brains. First, we examined expression of laminin, a key extracellular matrix protein associated with inflammation and the BBB damage. Neurovascular units were concurrently stained with GFAP and laminin. The vessel walls show increased laminin expression with aggregated (or disrupted) pattern in the control mice at 48 h after MCAO, indicating extracellular matrix remodeling (arrow, Figure 4 A). Quantification of fluorescence intensity signals revealed a significant increase of laminin in the control brains. Interestingly, the Nhe1 KO mice illustrated uniform distribution of laminin in the vessel walls and abundant expression of laminin in the astrocytic endfeet, reflecting an intact basement membrane (arrowhead, Figure 4 A).
Figure 4. Deletion of Nhe1 in Gfap-CreER+/-;Nhe1f/f mice promotes less cerebral vessel damage and improved BBB morphology after ischemic stroke.
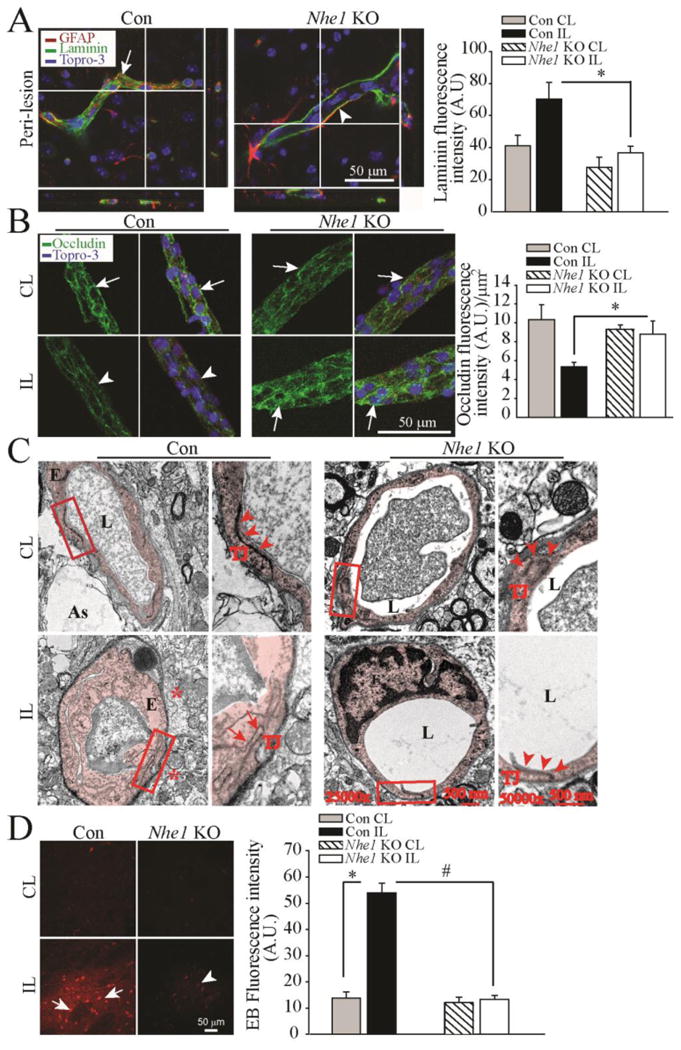
A. Representative confocal Z-stack images of laminin-stained microvessels in the Con and Nhe1 KO ischemic brains after 48 h reperfusion. Arrow: accumulation of degraded laminin. Arrowheads: less laminin accumulation. Summary data of laminin fluorescence intensity are mean ± SEM, n=4. *p < 0.05 vs Con.
B. Representative confocal images of tight junction protein occludin stained vessels isolated from Con and Nhe1 KO ischemic brains at 48 h reperfusion. Arrows: Preserved occludin expression. Arrow heads: Reduced occludin expression. Data are mean ± SEM, n=4 *p < 0.05 vs Con
C. Transmission electron microscopy images of cerebral vessels in the peri-lesion areas from the Con and Nhe1 KO mouse brains at 24 h reperfusion. Extensive perivascular damage, swollen endothelial cells and opened tight junctions were shown. Arrow: opened tight junctions. Arrowhead: intact tight junctions. As: Astrocyte endfeet; L: capillary lumen; E: endothelial cell; TJ: tight junctions. Box: to be enlarged.
D. Changes of the BBB permeability in the ischemic brain parenchyma at 48 h reperfusion. EB extravasation was detected in the IL hemispheres of the Con (arrow) but not in the Nhe1 KO mice (arrowhead). Data are mean ± SEM, n=3. *p < 0.05 vs CL. #p < 0.05 vs Con.
We further evaluated expression of endothelial tight junction proteins in vessels isolated from the control and Nhe1 KO brains (Figure 4 B). The abundant endothelial tight junction protein occludin was detected especially at the junctions between endothelial cells of the healthy CL microvessels (Figure 4 B, arrow). However, overall occludin expression was significantly reduced and its continuous distribution pattern was disrupted (Figure 4 B, arrowhead) in the IL peri-lesion areas of control brains. Interestingly, occludin staining in Nhe1 KO brains showed concentrated distribution at the borders of endothelial cells without reduction (Figure 4 B, arrow), suggesting that tight junctions were intact. These findings were further supported by the electron microscopic analysis of BBB structures in control and Nhe1 KO brains. Under TEM, compared to the normal tight junction structure and basement membrane in the healthy CL hemisphere microvessels, the control brain showed swollen endothelial cells and opened tight junctions (Figure 4 C, arrows), as well as extensive damage to perivascular astrocytes. Swollen astrocyte endfeet were visible in areas with diffuse cytoplasm in contact with the basal lamina (Figure 4 C *). In contrast, Nhe1 KO brains displayed a nearly normal cerebral microvascular structure with smooth inner surface and closely connected endothelial cells via tight junction (Figure 4 C, arrow heads). No swollen astrocytic endfeet structures were observed surrounding the microvessels in the Nhe1 KO mice. These data suggest that selective deletion of astrocytic Nhe1 in the Gfap-CreER+/-;Nhe1f/f mice preserves perivascular astrocyte function and microvessel tight junction structure.
We then determined whether Nhe1 KO mice show improved BBB integrity by testing its permeability to high molecular weight protein albumin extravasation in the brain parenchyma. As shown in Figure 4 D, EB leakage was minimal at 24 h reperfusion in both CL hemisphere tissues of the oil- and Tam-treated mice. However, EB leakage was markedly increased in the IL cortex of the control brains. In contrast, no significant leakage of EB was detected in Nhe1 KO brains (Figure 4 D). These findings strongly suggest that deletion of Nhe1 in GFAP+ astrocytes not only reduces microvessel damage but also improves BBB integrity after ischemic stroke.
Deletion of Nhe1 in Gfap-CreER+/-;Nhe1f/f mice abrogates MMP-9 expression in the perivascular regions
Proteases such as MMP-9 plays an important role in proteolytic disruption of the BBB and in cerebrovascular damage. We therefore assessed the expression of MMP-9 in control and Nhe1 KO mice at 48 h after MCAO. Extensive MMP-9 immunoreactivity was detected in the perivascular regions in the IL hemispheres of control mice (Figure 5 A, arrow), but not in the CL hemisphere (data not shown). Most MMP-9 immunoreactive signals was located at the astrocyte endfeet and co-localized with AQP4 in the endothelial cells bordering the vessels (Figure 5 A, arrows). In contrast, no MMP-9-positive signals were detected in the IL regions of Nhe1 KO mouse brains (Figure 5 A, arrow heads). These findings strongly suggest that deletion of Nhe1 in GFAP+ astrocytes ameliorates MMP-9 secretion/activation upon ischemic stroke.
Figure 5. Selective deletion of Nhe1 in Gfap-CreER+/-;Nhe1f/f mice inhibits MMP9 expression in cerebral vessels.
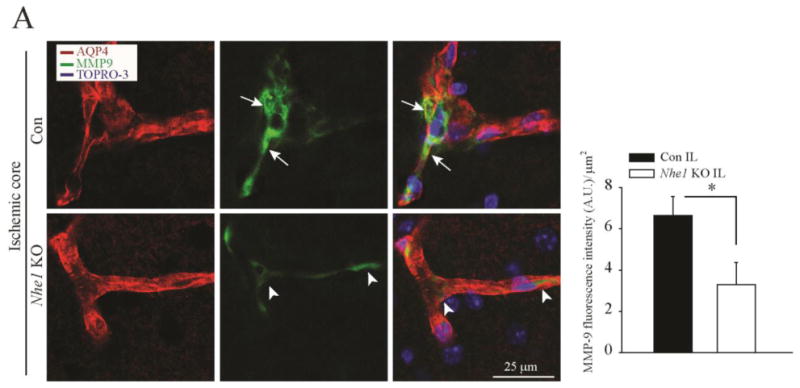
A. Representative confocal images of MMP-9 and AQP4 expression in the Con and Nhe1 KO ischemic brains at 48 h reperfusion. Increased perivascular MMP9 expression (green) is associated with AQP4 signals (red) only in the Con but not in Nhe1 KO mice. Arrows: Increased perivascular MMP-9 expression. Arrowheads: Absence of perivascular MMP9 expression. Summary data of MMP-9 fluorescence intensity are mean ± SEM, n=5. *p < 0.05 vs Con.
Deletion of Nhe1 in Gfap-CreER+/-;Nhe1f/f mice exhibit reduced perivascular damage and increased micro vessel density after ischemic stroke
We employed aquaporin 4 (AQP4) immunocytochemical analyses to study the extent of micro vessel damage upon ischemia in control and Nhe1 KO mice. Figure 6 A shows that the IL cortex exhibited an increase in the number of perivascular reactive GFAP+ astrocytes, which are closely associated with AQP4-stained microvessels at the peri-lesion areas (arrow, white box). Strikingly, elevated GFAP+ expression and hypertrophic AQP4+ astrocytic end-foot processes were co-localized in the dilated blood vessels of the control brains (arrow, Figure 6 A). In contrast, the Nhe1 KO brains appeared to show less expression of AQP4 and less co-localization of AQP4 and GFAP immunosignals in the peri-vascular astrocyte processes, but there are no statistically significant differences. (Supporting Information Figure 3).
Figure 6. Deletion of Nhe1 in Gfap-CreER+/-;Nhe1f/f mice reduced perivascular damage and preserved microvessel density after ischemic stroke.
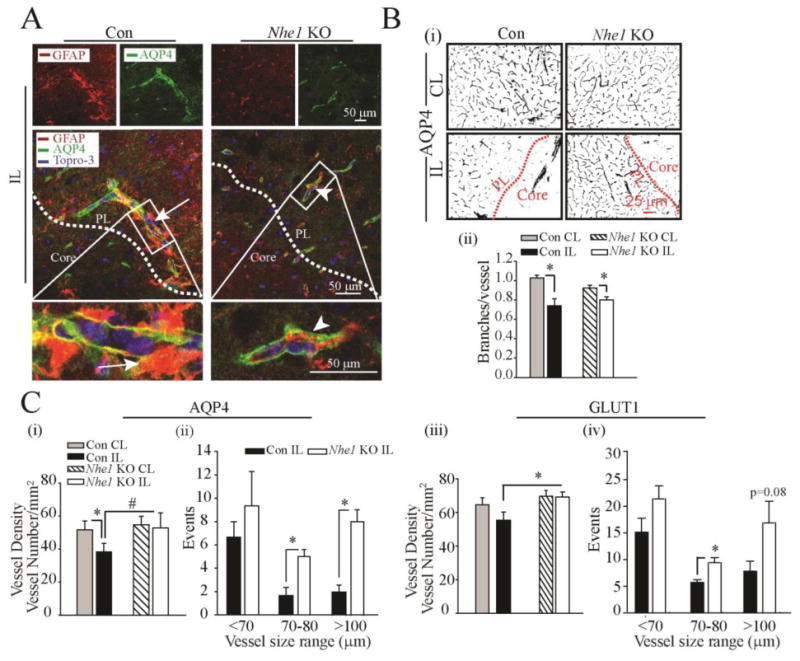
A. Representative confocal Z-stack images of AQP4-stained microvessels in the Con and Nhe1 KO ischemic brains at 48 h reperfusion. Increased numbers of perivascular reactive GFAP+ astrocytes (red) were associated with AQP4 stained microvessels (green), more clearly shown in the Con than in the KO mice. Arrows: increased perivascular reactive astrogliosis. Arrowheads: reduced perivascular reactive astrogliosis. White box: enlarged in the lower panel.
B. Representative binary images of AQP4-labeled microvessels show different cerebral microvessel density in the peri-lesion regions of the Con and Nhe1 KO mice at 48 h reperfusion (i). Summary data of branches/vessel (ii). C. Summary data of vessel density and size distribution of AQP4-stained vessels are shown in (i) and (ii). Summary data of GLUT1 vessel density (iii), and size distribution of GLUT1-stained vessels (iv) were shown. Data are mean ± SEM, n=3-5. *p < 0.05 vs CL; #p < 0.05 vs Con.
In evaluation of the peri-vascular AQP4-labeled vessels, we observed differences in the change of microvessel density and branching between control and Nhe1 KO ischemic brains. At 2 days after ischemic stroke, AQP4-labeled microvessels were decreased or nearly abolished in the ischemic core (Figure 6 B (i)). Vascular density reductions also occurred in the ischemic peri-lesion areas. However, a significant loss of microvasculature density in the ischemic peri-lesion areas was detected only in the control but not in the Nhe1 KO mice (Figure 6 C, (i)). Interestingly, deletion of astrocytic Nhe1 profoundly preserved larger microvessels (> 100 μm in length) with less effect on small vessels (< 70 μm in length, Figure 6 C (ii)). We also performed the quantification of microvessels by immunostaining for endothelial cell specific marker glucose transporter 1 (GLUT1). As shown in the Figure 6 C (iii, iv) and Supporting Information Figure 4, the Nhe1 KO mice illustrated significant higher density of larger microvessels in the ischemic peri-lesion areas at 48 h of reperfusion, compared to that in control mouse brains. This is consistent with the findings using the analysis of the AQP4-stained microvessels. These results suggest that deletion of Nhe1 gene in GFAP+ astrocytes preserved the density of microvessels in the ischemic brains.
Astrocytic Nhe1 KO mice exhibit improved regional collateral blood flow (rCBF) after ischemic stroke
Using a PeriCam PSI System imager with a speckle laser, we monitored changes of cortical blood flow in real time by measuring the movement of red blood cells in the cortical vessels, which provides information about changes of cerebral blood perfusion over time. Baseline measurements were recorded over a 5-min period prior to subjecting the animals to the MCAO. Immediately following vessel occlusion or upon reperfusion, rCBF was recorded at 5-min intervals. Figure 7 A shows representative images of cerebral perfusion in the control and Nhe1 KO mice. A significant decrease of rCBF from the baseline within the IL hemisphere was detected in both control (88.1 ± 11.4 vs. 246.9 ± 24.5, p < 0.0001) and Nhe1 KO mice (92.3 ± 8.4 vs. 232.0 ± 18.8, p < 0.0001) at 60 min post-MCAO (Figure 7 B). Interestingly, a significantly higher perfusion was observed at 24 h of reperfusion in Nhe1 KO mice than in control mice (220.0 ± 10.4 vs. 174.6 ± 7.0, p < 0.01). These findings further suggest that Nhe1 KO mice exhibited higher cerebral perfusion, which could result from improved microvessel density and improved collateral flow.
Figure 7. Selective deletion of GFAP+ astrocytic Nhe1 improves rCBF in ischemic brains.
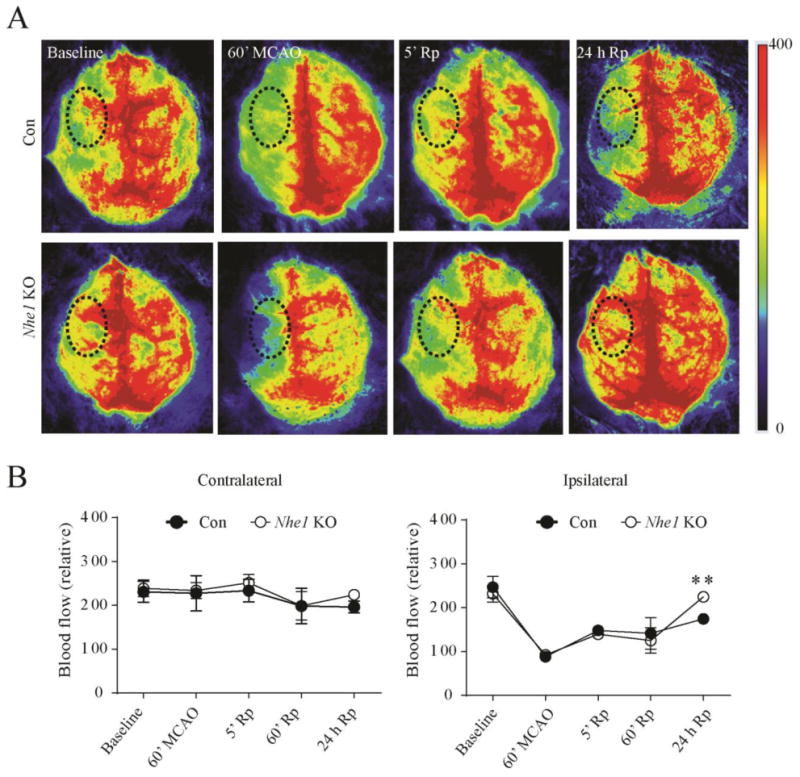
A. Representative images of Laser Speckle analysis of blood perfusion in the Con and Nhe1 KO mice. Baseline and occlusion images show areas of red as high blood perfusion and areas of blue as low blood perfusion. Graph indicates quantification of blood perfusion in MCA regions of interest (black dotted circle) in the CL and IL hemispheres. B. A significantly higher improvement in the rCBF was observed in the IL hemisphere of Nhe1 KO brains at 24 h of reperfusion. n = 3-7. ** p < 0.01 compared to baseline measurements.
Changes of pHi and gene expression in Nhe1 KO astrocytes
To further understand the impact of NHE1 in astrocyte pHi regulation, we measured changes of pHi in astrocytes in acutely isolated cortical slices from the Con and Nhe1 KO mice. Astrocytes in the slice loaded with a pH indicator SNARF-5F were identified by their selective uptake of SR101 dye (Supporting Information Figure 5 A). The basal pHi of astrocytes in the presence of HEPES-buffered solution (pH 7.4, CO2/HCO3-free) was calculated as 7.8 ± 0.2 in the Con brains and 7.9 ± 0.2 in the Nhe1 KO brains (Supporting Information Figure 5 A-D). These values are in agreement with previously reported alkaline basal pHi values (7.5 - 7.7) in the brain slices (Lin et al., 1996). Upon 2 min exposure to chemical ischemia (NaN3, 2-deoxyglucose), reduction in pHi was detected in both Con astrocytes (0.4 ± 0.03 pH units) and Nhe1 KO astrocytes (0.5 ± 0.1 pH units), but they were not significantly different (Supporting Information Figure 5 D). The average pHi remains reduced in the Con and Nhe1 KO astrocytes (0.3 ± 0.1 pH units) at 5-minute post ischemia. The lack of excess acidosis in the Nhe1 KO astrocytes may in part result from compensatory mechanisms from other acid extrusion pathways. A recent study shows that sodium bicarbonate cotransporter NBCe1 in cortical astrocytes can robustly regulate pHi when NHE1 activity is blocked by its inhibitor EIPA (Theparambil et al., 2014). Even when using HEPES-buffered solution (pH 7.4, CO2/HCO3-free), NBCe1 in astrocytes remains active with sufficient residual interstitial HCO3 levels (Brookes et al., 1994; Theparambil et al., 2014). On the other hand, it may require longer ischemic challenge to determine the impact of deleting astrocyte Nhe1 on astrocyte pHi since we detected NHE1-dependent pHi recovery at 15 min re-oxygenation following 2 h OGD in astrocyte cultures (Kintner et al., 2004). Taken together, additional studies are needed to investigate impact of deleting astrocyte Nhe1 in pHi regulation following ischemia and possible activation of other pH regulatory mechanisms including NBCe1, and the voltage-gated proton channel Hv1, both of which mRNAs were detected in astrocytes in our study.
Discussion
NHE1 activation is required for astrogliosis after ischemic stroke
The hallmarks of reactive astrogliosis after ischemic injury include a pronounced increase in expression of GFAP and astrocyte hypertrophy that occurs over the first few days, along with scar formation by ten days (Ding, 2014). Activation of NHE1 protein may be essential for forming reactive astrocytes. A positive correlation between NHE1 activation and reactive astrocyte formation has been observed in hippocampal astrocytes after in vitro ischemia (Cengiz et al., 2014). Upregulation of NHE1 protein in reactive astrocytes is also detected in ischemic brains (Shi et al., 2011). In the present study, establishing the Gfap-CreER+/-;Nhe1f/f mice allowed us to decipher the direct role of astrocytic NHE1 in reactive astrocyte formation and in neurological damage after ischemic stroke. We demonstrated that the astrocytic Nhe1 KO mice were healthy and did not show any discernible phenotypes. After acute ischemic stroke, Nhe1 KO mice exhibited significantly reduced reactive astrogliosis, attenuated hypertrophy and reduced BrdU+ labeling in GFAP+ cells in the peri-ischemic region of ischemic brains. These findings suggest that NHE1 protein is involved in astrocyte proliferation.
NHE1-mediated change in pHi and/or intracellular Na+ concentration is important for the regulation of cell proliferation (Nakamura et al., 2008). Intracellular alkalinization-mediated by NHE1 activation is required for efficient progression from S to G2/M phase (Putney et al., 2003). Studies have also shown that H+ ion translocation by NHE1 is necessary for controlling both the activity and expression of various M phase regulators, including cyclin B1 and the cyclin-dependent kinases (Cdks), in promoting mitotic activity (Putney et al., 2003). In our current study, we conducted initial RNAseq analysis of GFAP+ astrocytes and found key cell proliferation genes, such as Cdk7, Cdk12, Cdk18 and Cdkn1a, were significantly elevated in the control brains at 24 h post-MCAO (Supporting Information Figure 6). In contrast, the Nhe1 KO astrocytes expressed significantly lower amounts of these key Cdk genes (Supporting Information Figure 6). CDK7 is an important component of activated CDK complex that regulates cell cycle progression (Wu et al., 2011; Wang et al., 2016). Administration of CDK inhibitors reduces the aberrant cell cycle activation and ischemia-induced reactive astrogliosis (Byrnes et al., 2007; Zhu et al., 2007; Hilton et al., 2008). Thus, our findings suggest that NHE1 protein affects a check point that controls the key cell cycle activator gene expression in GFAP+ astrocytes. Therefore, selective deletion of Nhe1 in GFAP+ astrocytes inhibits astrogliosis in part by decreasing cell cycle gene transcription and astrocyte proliferation after ischemic stroke.
NHE1 activation in astrocyte dysfunction after ischemic stroke
Our study illustrated that deletion of Nhe1 in astrocytes is associated with reduced reactive astrogliosis and less ischemic brain damage. We speculate that may result from reducing astrocyte death and preserving normal astrocyte function in ischemic brains. Astrocytes regulate uptake and release of many signaling molecules including growth factors, cytokines, neurotransmitters, and ATP (Sofroniew, 2009). Hypoxia, extracellular acidosis, and reduction of extracellular Na+ concentration (shifts of cellular Na+ gradient) lead to astrocyte death (Bondarenko et al., 2001b; a). The underlying mechanisms involve toxic intracellular Na+ overload mediated by activation of NHE1 protein and reverse mode operation of the Na+/Ca2+ exchanger (Rose et al., 1998; Bondarenko et al., 2001a). Inhibition of NHE1 protein with HOE642, blocking the Na+/Ca2+ exchanger with KB-R7943, or removal of extracellular Na+ all prevented hypoxia-induced astrocyte death (Bondarenko et al., 2001a; Kintner et al., 2004). Therefore, the neuroprotective effects seen in the current study by deletion of Nhe1 in astrocytes are likely due to reduced intracellular Na+ overload, decreased astrocyte death, and restoration of normal astrocyte functions. This view is further supported by our S100β data. Brain injury triggers astrocytes to release S100β protein into the extracellular space (Morrison et al., 2016). Elevated S100β levels have been detected in CSF and serum samples after ischemic stroke and traumatic head injuries, which corelates with severity of brain damage (Vos et al., 2010; Sun et al., 2013). In our study, we found that S100β protein is preserved in the GFAP+ cell soma of Nhe1 KO brains and, in contrast, extensive loss of S100β from GFAP+ cells was detected in control mouse brains. Recent findings show that preventing astrocyte scar formation, attenuating scar forming astrocytes or ablating chronic astrocytic scars blocks CNS axon regeneration, and exacerbates neurological functional deficits in spinal cord injury, brain injury or experimental autoimmune encephalomyelistis models (Anderson et al., 2016). Our study suggests that NHE1 activity is an important contributing factor for astrocyte dysfunction, and targeting NHE1 presents a better strategy in maintaining normal astrocytic homeostasis and function.
Roles of NHE1 in peri-vascular astrocyte endfeet and AQP4 expression
Astrocyte endfeet together with pericytes and basal lamina play an important role in the regulation of rCBF and maintenance of BBB integrity (Haydon et al., 2006; Takano et al., 2006; Attwell et al., 2010; Liebner et al., 2011). The endfeet of astrocytes are densely packed with AQP4 and allow astrocytes to regulate the transport of water and other substances between the intravascular space and brain parenchyma (Iliff et al., 2012; Iliff et al., 2013; Yang et al., 2013). AQP4 also plays a major role in ischemic edema after ischemia (Manley et al., 2000; Papadopoulos et al., 2004a; Papadopoulos et al., 2004b; Ribeiro Mde et al., 2006; Fu et al., 2007). Increased AQP4 expression has been demonstrated to coincide with perivascular astrocyte swelling (Manley et al., 2000). In a mouse model of transient cerebral ischemia, AQP4 expression was upregulated in the perivascular endfeet at 1 h post occlusion and correlated with cerebral swelling in the core and border of the lesion (Ribeiro Mde et al., 2006). AQP4 expression in cultured astrocytes is enhanced by lactic acidosis after ischemia (Morishima et al., 2008). Although disruption of AQP4 anchoring proteins or deletion of AQP4 from perivascular astrocytes in mice leads to a reduction in cerebral edema after ischemic stroke (Manley et al., 2000; Amiry-Moghaddam et al., 2003; Zeynalov et al., 2008; Zeng et al., 2010), deletion of AQP4 also increases vasogenic edema upon ischemic stroke (Saadoun et al., 2010; Papadopoulos et al., 2013). These findings suggest that AQP4-mediated cellular damage in ischemic stroke is complicated and associated with other biochemical changes.
In our study, an increased number of perivascular astrocytes with intense AQP4 immunopositive signals were detected in the endfeet structures of the control brains at 48 h post-MCAO (Figure 6 A). Astrocytic Nhe1 KO mice show reduced perivascular reactive astrocytes without changing the overall brain expression of AQP4 (Supporting Information Figure 2). Most importantly, a significant reduction in hemispheric swelling volumes was detected in Nhe1 KO mice (Figure 2 C). This indicates that astrocytic NHE1 protein plays an important role in progression of edema. The coupled operation of NHE1 and bicarbonate antiporters, and the H+ monocarboxylate symporters causes accumulation of cellular Na+, Cl- and water and astrocyte swelling (Staub et al., 1990; Yao et al., 2001). We have previously shown that pharmacological inhibition of NHE1 in astrocytes reduces the rise in Na+ and swelling after in vitro ischemia (Kintner et al., 2004). Therefore, deletion of astrocytic Nhe1 may decrease ischemic edema by several mechanisms, including reducing astrocytic inflammation, AQP4 expression, and astrocyte swelling.
Astrocytic NHE1 protein in microvessel damage and BBB leakage
Endothelial tight junctions (TJs), the basal lamina and perivascular astrocytes together play a crucial role in the formation and maintenance of the BBB (Ronaldson et al., 2012). TJ transmembrane proteins, such as occludin and claudin-5 or Zo-1, play a key role in the functional preservation of the BBB (Bazzoni et al., 2000; Hawkins et al., 2005). Since astrocyte endfeet cover over 95% of the brain capillary surface, the swelling of astrocyte endfeet causes disassembly of the tight junction network and decreased expression of TJ proteins such as occludin and claudin-5 (Willis et al., 2004; Shin et al., 2015). In the current study, we observed extensive EB extravasation into brain parenchyma in the IL hemispheres of the control mouse brains, indicating vascular leakage. This is accompanied with opened TJ structures and reduced expression of occludin in the microvessels of the control mouse brains at 48 h post-MCAO. In contrast, intact TJ structures and stable occludin expression was seen in the Nhe1 KO mouse brains. Disrupting occludin alone could alter functions of the TJs and vascular leakage (Tavelin et al., 2003; Bauer et al., 2010; Liu et al., 2012). We did not detect any changes in the expression of claudin-5 protein in the two treatment groups (Data not shown). Therefore, deleting astrocytic Nhe1 may preserve BBB integrity in part by maintaining occludin expression and TJ structures in ischemic brains.
Activated astrocytes secrete chemokines (MCP-1, Rantes), pro- and anti-inflammatory cytokines (IL-1α, IL-1β, TNFα, interferon-γ) and growth factors (TGF-β), which are known to directly damage the cerebral endothelium and disrupt the BBB, (Lau et al., 2001; Lee et al., 2007; Doyle et al., 2010; Vangilder et al., 2011; Ronaldson et al., 2012). Increased NHE1 activity in astrocytes aggravates pro-inflammatory stimulation (Cengiz et al., 2014). Increased Ca2+ influx activates many signaling pathways and molecular events, especially calpain proteases and MMPs, which are widely regarded as hallmarks of the breakdown of the BBB (Gu et al., 2002; Zhao et al., 2006; Cao et al., 2007). NHE1 activation in astrocytes causes intracellular Ca2+ overload via reverse activation of the Na+/Ca2+ exchanger (Kintner et al., 2007). During the acute phase of post-ischemic stroke, increased NHE1 activity may contribute to high influx of Ca2+ at the astrocyte endfeet and damage to the BBB. Thus, the better-preserved TJ structure and BBB function in Nhe1 KO mouse brains may result from decreased MMP activation at the BBB.
Association between NHE1 and MMP9 in ischemic brains
In acute ischemia, activation of MMPs (especially, MMP-9) causes neurovascular matrix degradation, BBB disruption leading to edema, and increased inflammation (Planas et al., 2001; Horstmann et al., 2003; Montaner et al., 2003; Yang et al., 2007; Rosell et al., 2008; Dejonckheere et al., 2011). Activated astrocytes are the most important source of MMP-9 during brain injury (Zhang et al., 2004; Tejima et al., 2007). Increased MMP-9 is known to cause cleavage of basement membrane laminin and degradation of the TJ protein occludin (Liu et al., 2012). We found increased perivascular expression of MMP-9 protein and degradation of microvessel laminin in the control brains, but deletion of astrocytic Nhe1 abolished upregulation of MMP-9 expression in the perivascular astrocyte endfeet. Activation of MMP-9 depends on various factors including circulating inflammatory cytokines, endogenous tPA, and oxygen free radicals (Gasche et al., 2001; Gu et al., 2002; Amantea et al., 2014; Turner et al., 2016). In addition, acidic extracellular pH (pHe) is also known to trigger MMP-9 expression via the NFkB pathway since the NFkB and AP-1 binding sites of the MMP-9 promoter are sensitive to acidic pHe (Kato et al., 2005). Focal cerebral ischemia causes a rapid fall in pHe from 7.2 to below 6.6 (Nedergaard et al., 1991) and increased NHE1 activity accelerates H+ efflux in astrocytes (Kintner et al., 2007). Based on these observations, deletion of astrocytic Nhe1 may reduce the perivascular expression and function of MMP-9 via multiple mechanisms in the Nhe1 KO brains.
Astrocytic NHE1 protein in regulating cerebral vascular function after ischemic stroke
Vascular remodeling after stroke is a critical determinant of stroke outcome (Gertz et al., 2006; Gertz et al., 2012). Angiogenesis through new blood vessel formation results in improved collateral circulation and has an impact on the long-term recovery of stroke patients (Slevin et al., 2006; Font et al., 2010). Our results demonstrate that deletion of Nhe1 in the GfapCreER/+;Nhe1f/f brains leads to improved rCBF and higher microvessel density (especially larger microvessels) in the ischemic peri lesion area. We speculate that deletion of astrocytic Nhe1 may preserve vascular function by reducing ischemia-induced vascular damage at the BBB. In addition, the Nhe1 KO mouse brain may exhibit altered expression and secretion of angiogenic factors from astrocytes that stimulate angiogenesis and long term vascular remodeling. Additional studies are required to investigate these cellular and molecular mechanisms and identify the specific factors involved. Taken together, our findings demonstrate that selective deletion of the astrocytic Nhe1 gene reduces cerebral microvessel damage and improves rCBF after acute ischemic stroke in mice.
Conclusions
In summary, previous studies have shown that pharmacological inhibition of NHE1 protein with its inhibitors has neuroprotective effects in experimental stroke models (Luo et al., 2005; Shi et al., 2011; O'Donnell et al., 2013). Here, we show for the first time that astrocytic NHE1 protein plays a critical role in reactive astrogliosis and neurovascular damage after ischemic stroke. By using inducible, astrocyte specific Gfap-CreER+/-;Nhe1f/f conditional knockout mice, we demonstrated that selective deletion of astrocytic Nhe1 gene in astrocytes reduced stroke volume, inhibited reactive astrocyte formation, prevented BBB damage, and improved rCBF and neurological function (as summarized in Supporting Information Figure 7). Our results indicate that selective deletion of Nhe1 in reactive astrocytes enables them to maintain astrocytic cellular homeostasis and physiological functions in ischemic brains. Additional studies are warranted for a more complete understanding of the underlying mechanisms regarding specific signaling molecules and pathways. Our new findings add incentives for developing more potent and selective NHE1 inhibitors for preventing astrocyte damage, inhibiting overstimulation of reactive astrogliosis, and maintaining normal astrocyte function in post-ischemic stroke.
Supplementary Material
Main Points.
-
➢
Stimulation of astrocytic NHE1 protein at the BBB is detrimental in ischemic brains.
-
➢
Deletion of astrocytic Nhe1 gene inhibits astrogliosis, reduces stroke volume, prevents BBB damage, and improves rCBF and neurological function after stroke.
Acknowledgments
Grant sponsors: NIH R01 NS048216 (D.S); HL130363 and DK097391 (P.H); 1S10OD021540-01 (SC.W).
References
- Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia. 2013;61:1939–1958. doi: 10.1002/glia.22575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amantea D, Certo M, Russo R, Bagetta G, Corasaniti MT, Tassorelli C. Early reperfusion injury is associated to MMP2 and IL-1beta elevation in cortical neurons of rats subjected to middle cerebral artery occlusion. Neuroscience. 2014;277:755–763. doi: 10.1016/j.neuroscience.2014.07.064. [DOI] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Otsuka T, Hurn PD, Traystman RJ, Haug FM, Froehner SC, Adams ME, Neely JD, Agre P, Ottersen OP, Bhardwaj A. An alpha-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc Natl Acad Sci U S A. 2003;100:2106–2111. doi: 10.1073/pnas.0437946100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MA, Burda JE, Ren Y, Ao Y, O'Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, Sofroniew MV. Astrocyte scar formation aids central nervous system axon regeneration. Nature. 2016;532:195–200. doi: 10.1038/nature17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci U S A. 2009;106:1977–1982. doi: 10.1073/pnas.0808698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer AT, Burgers HF, Rabie T, Marti HH. Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J Cereb Blood Flow Metab. 2010;30:837–848. doi: 10.1038/jcbfm.2009.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzoni G, Martinez-Estrada OM, Orsenigo F, Cordenonsi M, Citi S, Dejana E. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J Biol Chem. 2000;275:20520–20526. doi: 10.1074/jbc.M905251199. [DOI] [PubMed] [Google Scholar]
- Begum G, Yuan H, Kahle KT, Li L, Wang S, Shi Y, Shmukler BE, Yang SS, Lin SH, Alper SL, Sun D. Inhibition of WNK3 Kinase Signaling Reduces Brain Damage and Accelerates Neurological Recovery After Stroke. Stroke. 2015;46:1956–1965. doi: 10.1161/STROKEAHA.115.008939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belayev L, Busto R, Zhao W, Ginsberg MD. Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 1996;739:88–96. doi: 10.1016/s0006-8993(96)00815-3. [DOI] [PubMed] [Google Scholar]
- Bondarenko A, Chesler M. Calcium dependence of rapid astrocyte death induced by transient hypoxia, acidosis, and extracellular ion shifts. Glia. 2001a;34:143–149. doi: 10.1002/glia.1049. [DOI] [PubMed] [Google Scholar]
- Bondarenko A, Chesler M. Rapid astrocyte death induced by transient hypoxia, acidosis, and extracellular ion shifts. Glia. 2001b;34:134–142. doi: 10.1002/glia.1048. [DOI] [PubMed] [Google Scholar]
- Bouet V, Boulouard M, Toutain J, Divoux D, Bernaudin M, Schumann-Bard P, Freret T. The adhesive removal test: a sensitive method to assess sensorimotor deficits in mice. Nat Protoc. 2009;4:1560–1564. doi: 10.1038/nprot.2009.125. [DOI] [PubMed] [Google Scholar]
- Bouet V, Freret T, Toutain J, Divoux D, Boulouard M, Schumann-Bard P. Sensorimotor and cognitive deficits after transient middle cerebral artery occlusion in the mouse. Exp Neurol. 2007;203:555–567. doi: 10.1016/j.expneurol.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Boulay AC, Saubamea B, Decleves X, Cohen-Salmon M. Purification of Mouse Brain Vessels. J Vis Exp. 2015:e53208. doi: 10.3791/53208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes N, Turner RJ. K(+)-induced alkalinization in mouse cerebral astrocytes mediated by reversal of electrogenic Na(+)-HCO3- cotransport. Am J Physiol. 1994;267:C1633–1640. doi: 10.1152/ajpcell.1994.267.6.C1633. [DOI] [PubMed] [Google Scholar]
- Byrnes KR, Stoica BA, Fricke S, Di Giovanni S, Faden AI. Cell cycle activation contributes to post-mitotic cell death and secondary damage after spinal cord injury. Brain. 2007;130:2977–2992. doi: 10.1093/brain/awm179. [DOI] [PubMed] [Google Scholar]
- Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, Clark RS, Graham SH, Chen J. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J Neurosci. 2007;27:9278–9293. doi: 10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cekanaviciute E, Fathali N, Doyle KP, Williams AM, Han J, Buckwalter MS. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia. 2014;62:1227–1240. doi: 10.1002/glia.22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cengiz P, Kintner DB, Chanana V, Yuan H, Akture E, Kendigelen P, Begum G, Fidan E, Uluc K, Ferrazzano P, Sun D. Sustained Na+/H+ exchanger activation promotes gliotransmitter release from reactive hippocampal astrocytes following oxygen-glucose deprivation. PLoS One. 2014;9:e84294. doi: 10.1371/journal.pone.0084294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo E, Farina C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016;37:608–620. doi: 10.1016/j.it.2016.06.006. [DOI] [PubMed] [Google Scholar]
- Dejonckheere E, Vandenbroucke RE, Libert C. Matrix metalloproteinases as drug targets in ischemia/reperfusion injury. Drug Discov Today. 2011;16:762–778. doi: 10.1016/j.drudis.2011.06.009. [DOI] [PubMed] [Google Scholar]
- Ding S. Dynamic reactive astrocytes after focal ischemia. Neural Regen Res. 2014;9:2048–2052. doi: 10.4103/1673-5374.147929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle KP, Cekanaviciute E, Mamer LE, Buckwalter MS. TGFbeta signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J Neuroinflammation. 2010;7:62. doi: 10.1186/1742-2094-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Font MA, Arboix A, Krupinski J. Angiogenesis, neurogenesis and neuroplasticity in ischemic stroke. Curr Cardiol Rev. 2010;6:238–244. doi: 10.2174/157340310791658802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Li Q, Feng Z, Mu D. The roles of aquaporin-4 in brain edema following neonatal hypoxia ischemia and reoxygenation in a cultured rat astrocyte model. Glia. 2007;55:935–941. doi: 10.1002/glia.20515. [DOI] [PubMed] [Google Scholar]
- Ganat YM, Silbereis J, Cave C, Ngu H, Anderson GM, Ohkubo Y, Ment LR, Vaccarino FM. Early postnatal astroglial cells produce multilineage precursors and neural stem cells in vivo. Journal of Neuroscience. 2006;26:8609–8621. doi: 10.1523/JNEUROSCI.2532-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasche Y, Copin JC, Sugawara T, Fujimura M, Chan PH. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:1393–1400. doi: 10.1097/00004647-200112000-00003. [DOI] [PubMed] [Google Scholar]
- Gertz K, Kronenberg G, Kalin RE, Baldinger T, Werner C, Balkaya M, Eom GD, Hellmann-Regen J, Krober J, Miller KR, Lindauer U, Laufs U, Dirnagl U, Heppner FL, Endres M. Essential role of interleukin-6 in post-stroke angiogenesis. Brain. 2012;135:1964–1980. doi: 10.1093/brain/aws075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz K, Priller J, Kronenberg G, Fink KB, Winter B, Schrock H, Ji S, Milosevic M, Harms C, Bohm M, Dirnagl U, Laufs U, Endres M. Physical activity improves long-term stroke outcome via endothelial nitric oxide synthase-dependent augmentation of neovascularization and cerebral blood flow. Circ Res. 2006;99:1132–1140. doi: 10.1161/01.RES.0000250175.14861.77. [DOI] [PubMed] [Google Scholar]
- Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton GD, Stoica BA, Byrnes KR, Faden AI. Roscovitine reduces neuronal loss, glial activation, and neurologic deficits after brain trauma. J Cereb Blood Flow Metab. 2008;28:1845–1859. doi: 10.1038/jcbfm.2008.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hol EM, Pekny M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr Opin Cell Biol. 2015;32:121–130. doi: 10.1016/j.ceb.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Horstmann S, Kalb P, Koziol J, Gardner H, Wagner S. Profiles of matrix metalloproteinases, their inhibitors, and laminin in stroke patients: influence of different therapies. Stroke. 2003;34:2165–2170. doi: 10.1161/01.STR.0000088062.86084.F2. [DOI] [PubMed] [Google Scholar]
- Iliff JJ, Nedergaard M. A link between glial Ca2+ signaling and hypoxia in aging? J Cereb Blood Flow Metab. 2013;33:170. doi: 10.1038/jcbfm.2012.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Lambert CA, Colige AC, Mineur P, Noel A, Frankenne F, Foidart JM, Baba M, Hata R, Miyazaki K, Tsukuda M. Acidic extracellular pH induces matrix metalloproteinase-9 expression in mouse metastatic melanoma cells through the phospholipase D-mitogen-activated protein kinase signaling. J Biol Chem. 2005;280:10938–10944. doi: 10.1074/jbc.M411313200. [DOI] [PubMed] [Google Scholar]
- Kim JH, Min KJ, Seol W, Jou I, Joe EH. Astrocytes in injury states rapidly produce anti-inflammatory factors and attenuate microglial inflammatory responses. J Neurochem. 2010;115:1161–1171. doi: 10.1111/j.1471-4159.2010.07004.x. [DOI] [PubMed] [Google Scholar]
- Kimbrough IF, Robel S, Roberson ED, Sontheimer H. Vascular amyloidosis impairs the gliovascular unit in a mouse model of Alzheimer's disease. Brain. 2015;138:3716–3733. doi: 10.1093/brain/awv327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelberg HK, Nedergaard M. Functions of astrocytes and their potential as therapeutic targets. Neurotherapeutics. 2010;7:338–353. doi: 10.1016/j.nurt.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kintner DB, Su G, Lenart B, Ballard AJ, Meyer JW, Ng LL, Shull GE, Sun D. Increased tolerance to oxygen and glucose deprivation in astrocytes from Na(+)/H(+) exchanger isoform 1 null mice. Am J Physiol Cell Physiol. 2004;287:C12–21. doi: 10.1152/ajpcell.00560.2003. [DOI] [PubMed] [Google Scholar]
- Kintner DB, Wang Y, Sun D. Role of membrane ion transport proteins in cerebral ischemic damage. Front Biosci. 2007;12:762–770. doi: 10.2741/2099. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Parpura V, Verkhratsky A. Sodium dynamics: another key to astroglial excitability? Trends Neurosci. 2012;35:497–506. doi: 10.1016/j.tins.2012.04.003. [DOI] [PubMed] [Google Scholar]
- Lau LT, Yu AC. Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J Neurotrauma. 2001;18:351–359. doi: 10.1089/08977150151071035. [DOI] [PubMed] [Google Scholar]
- Lee SR, Guo SZ, Scannevin RH, Magliaro BC, Rhodes KJ, Wang X, Lo EH. Induction of matrix metalloproteinase, cytokines and chemokines in rat cortical astrocytes exposed to plasminogen activators. Neurosci Lett. 2007;417:1–5. doi: 10.1016/j.neulet.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Li L, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, Nodin C, Stahlberg A, Aprico K, Larsson K, Yabe T, Moons L, Fotheringham A, Davies I, Carmeliet P, Schwartz JP, Pekna M, Kubista M, Blomstrand F, Maragakis N, Nilsson M, Pekny M. Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab. 2008;28:468–481. doi: 10.1038/sj.jcbfm.9600546. [DOI] [PubMed] [Google Scholar]
- Liebner S, Czupalla CJ, Wolburg H. Current concepts of blood-brain barrier development. Int J Dev Biol. 2011;55:467–476. doi: 10.1387/ijdb.103224sl. [DOI] [PubMed] [Google Scholar]
- Lin CW, Kalaria RN, Kroon SN, Bae JY, Sayre LM, LaManna JC. The amiloride-sensitive Na+/H+ exchange antiporter and control of intracellular pH in hippocampal brain slices. Brain Res. 1996;731:108–113. doi: 10.1016/0006-8993(96)00512-4. [DOI] [PubMed] [Google Scholar]
- Liu B, Teschemacher AG, Kasparov S. Astroglia as a cellular target for neuroprotection and treatment of neuro-psychiatric disorders. Glia. 2017;65:1205–1226. doi: 10.1002/glia.23136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Jin X, Liu KJ, Liu W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci. 2012;32:3044–3057. doi: 10.1523/JNEUROSCI.6409-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Chen H, Kintner DB, Shull GE, Sun D. Decreased neuronal death in Na+/H+ exchanger isoform 1-null mice after in vitro and in vivo ischemia. J Neurosci. 2005;25:11256–11268. doi: 10.1523/JNEUROSCI.3271-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- Montaner J, Molina CA, Monasterio J, Abilleira S, Arenillas JF, Ribo M, Quintana M, Alvarez-Sabin J. Matrix metalloproteinase-9 pretreatment level predicts intracranial hemorrhagic complications after thrombolysis in human stroke. Circulation. 2003;107:598–603. doi: 10.1161/01.cir.0000046451.38849.90. [DOI] [PubMed] [Google Scholar]
- Morishima T, Aoyama M, Iida Y, Yamamoto N, Hirate H, Arima H, Fujita Y, Sasano H, Tsuda T, Katsuya H, Asai K, Sobue K. Lactic acid increases aquaporin 4 expression on the cell membrane of cultured rat astrocytes. Neurosci Res. 2008;61:18–26. doi: 10.1016/j.neures.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Morrison HW, Filosa JA. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation. 2013;10:4. doi: 10.1186/1742-2094-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison HW, Filosa JA. Sex differences in astrocyte and microglia responses immediately following middle cerebral artery occlusion in adult mice. Neuroscience. 2016;339:85–99. doi: 10.1016/j.neuroscience.2016.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Kamouchi M, Kitazono T, Kuroda J, Matsuo R, Hagiwara N, Ishikawa E, Ooboshi H, Ibayashi S, Iida M. Role of NHE1 in calcium signaling and cell proliferation in human CNS pericytes. Am J Physiol Heart Circ Physiol. 2008;294:H1700–1707. doi: 10.1152/ajpheart.01203.2007. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Kraig RP, Tanabe J, Pulsinelli WA. Dynamics of interstitial and intracellular pH in evolving brain infarct. Am J Physiol. 1991;260:R581–588. doi: 10.1152/ajpregu.1991.260.3.R581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Manley GT, Krishna S, Verkman AS. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004a;18:1291–1293. doi: 10.1096/fj.04-1723fje. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Saadoun S, Binder DK, Manley GT, Krishna S, Verkman AS. Molecular mechanisms of brain tumor edema. Neuroscience. 2004b;129:1011–1020. doi: 10.1016/j.neuroscience.2004.05.044. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. Aquaporin water channels in the nervous system. Nat Rev Neurosci. 2013;14:265–277. doi: 10.1038/nrn3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas AM, Sole S, Justicia C. Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischemia. Neurobiol Dis. 2001;8:834–846. doi: 10.1006/nbdi.2001.0435. [DOI] [PubMed] [Google Scholar]
- Putney LK, Barber DL. Na-H exchange-dependent increase in intracellular pH times G2/M entry and transition. J Biol Chem. 2003;278:44645–44649. doi: 10.1074/jbc.M308099200. [DOI] [PubMed] [Google Scholar]
- Ribeiro Mde C, Hirt L, Bogousslavsky J, Regli L, Badaut J. Time course of aquaporin expression after transient focal cerebral ischemia in mice. J Neurosci Res. 2006;83:1231–1240. doi: 10.1002/jnr.20819. [DOI] [PubMed] [Google Scholar]
- Ronaldson PT, Davis TP. Blood-brain barrier integrity and glial support: mechanisms that can be targeted for novel therapeutic approaches in stroke. Curr Pharm Des. 2012;18:3624–3644. doi: 10.2174/138161212802002625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Karus C. Two sides of the same coin: sodium homeostasis and signaling in astrocytes under physiological and pathophysiological conditions. Glia. 2013;61:1191–1205. doi: 10.1002/glia.22492. [DOI] [PubMed] [Google Scholar]
- Rose CR, Waxman SG, Ransom BR. Effects of glucose deprivation, chemical hypoxia, and simulated ischemia on Na+ homeostasis in rat spinal cord astrocytes. J Neurosci. 1998;18:3554–3562. doi: 10.1523/JNEUROSCI.18-10-03554.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosell A, Lo EH. Multiphasic roles for matrix metalloproteinases after stroke. Curr Opin Pharmacol. 2008;8:82–89. doi: 10.1016/j.coph.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Roy Choudhury G, Ryou MG, Poteet E, Wen Y, He R, Sun F, Yuan F, Jin K, Yang SH. Involvement of p38 MAPK in reactive astrogliosis induced by ischemic stroke. Brain Res. 2014;1551:45–58. doi: 10.1016/j.brainres.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadoun S, Papadopoulos MC. Aquaporin-4 in brain and spinal cord oedema. Neuroscience. 2010;168:1036–1046. doi: 10.1016/j.neuroscience.2009.08.019. [DOI] [PubMed] [Google Scholar]
- Schaar KL, Brenneman MM, Savitz SI. Functional assessments in the rodent stroke model. Exp Transl Stroke Med. 2010;2:13. doi: 10.1186/2040-7378-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schitine C, Nogaroli L, Costa MR, Hedin-Pereira C. Astrocyte heterogeneity in the brain: from development to disease. Front Cell Neurosci. 2015;9:76. doi: 10.3389/fncel.2015.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Chanana V, Watters JJ, Ferrazzano P, Sun D. Role of sodium/hydrogen exchanger isoform 1 in microglial activation and proinflammatory responses in ischemic brains. J Neurochem. 2011;119:124–135. doi: 10.1111/j.1471-4159.2011.07403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada IS, Borders A, Aronshtam A, Spees JL. Proliferating reactive astrocytes are regulated by Notch-1 in the peri-infarct area after stroke. Stroke. 2011;42:3231–3237. doi: 10.1161/STROKEAHA.111.623280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JA, Kim YA, Jeong SI, Lee KE, Kim HS, Park EM. Extracellular signal-regulated kinase1/2-dependent changes in tight junctions after ischemic preconditioning contributes to tolerance induction after ischemic stroke. Brain Struct Funct. 2015;220:13–26. doi: 10.1007/s00429-013-0632-5. [DOI] [PubMed] [Google Scholar]
- Slevin M, Kumar P, Gaffney J, Kumar S, Krupinski J. Can angiogenesis be exploited to improve stroke outcome? Mechanisms and therapeutic potential. Clin Sci (Lond) 2006;111:171–183. doi: 10.1042/CS20060049. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staub F, Baethmann A, Peters J, Weigt H, Kempski O. Effects of lactacidosis on glial cell volume and viability. J Cereb Blood Flow Metab. 1990;10:866–876. doi: 10.1038/jcbfm.1990.143. [DOI] [PubMed] [Google Scholar]
- Sun BD, Liu HM, Nie SN. S100B protein in serum is elevated after global cerebral ischemic injury. World J Emerg Med. 2013;4:165–168. doi: 10.5847/wjem.j.issn.1920-8642.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Ying W, Kauppinen TM. Astrocyte influences on ischemic neuronal death. Curr Mol Med. 2004;4:193–205. doi: 10.2174/1566524043479185. [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- Tavelin S, Hashimoto K, Malkinson J, Lazorova L, Toth I, Artursson P. A new principle for tight junction modulation based on occludin peptides. Mol Pharmacol. 2003;64:1530–1540. doi: 10.1124/mol.64.6.1530. [DOI] [PubMed] [Google Scholar]
- Tejima E, Zhao BQ, Tsuji K, Rosell A, van Leyen K, Gonzalez RG, Montaner J, Wang X, Lo EH. Astrocytic induction of matrix metalloproteinase-9 and edema in brain hemorrhage. J Cereb Blood Flow Metab. 2007;27:460–468. doi: 10.1038/sj.jcbfm.9600354. [DOI] [PubMed] [Google Scholar]
- Theparambil SM, Ruminot I, Schneider HP, Shull GE, Deitmer JW. The electrogenic sodium bicarbonate cotransporter NBCe1 is a high-affinity bicarbonate carrier in cortical astrocytes. J Neurosci. 2014;34:1148–1157. doi: 10.1523/JNEUROSCI.2377-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RJ, Sharp FR. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front Cell Neurosci. 2016;10:56. doi: 10.3389/fncel.2016.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangilder RL, Rosen CL, Barr TL, Huber JD. Targeting the neurovascular unit for treatment of neurological disorders. Pharmacol Ther. 2011;130:239–247. doi: 10.1016/j.pharmthera.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Nedergaard M, Hertz L. Why are astrocytes important? Neurochem Res. 2015;40:389–401. doi: 10.1007/s11064-014-1403-2. [DOI] [PubMed] [Google Scholar]
- Vos PE, Jacobs B, Andriessen TM, Lamers KJ, Borm GF, Beems T, Edwards M, Rosmalen CF, Vissers JL. GFAP and S100B are biomarkers of traumatic brain injury: an observational cohort study. Neurology. 2010;75:1786–1793. doi: 10.1212/WNL.0b013e3181fd62d2. [DOI] [PubMed] [Google Scholar]
- Wang Q, Li M, Zhang X, Huang H, Huang J, Ke J, Ding H, Xiao J, Shan X, Liu Q, Bao B, Yang L. Upregulation of CDK7 in gastric cancer cell promotes tumor cell proliferation and predicts poor prognosis. Exp Mol Pathol. 2016;100:514–521. doi: 10.1016/j.yexmp.2016.05.001. [DOI] [PubMed] [Google Scholar]
- Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, Ao Y, Sofroniew MV. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci. 2013;33:12870–12886. doi: 10.1523/JNEUROSCI.2121-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis CL, Leach L, Clarke GJ, Nolan CC, Ray DE. Reversible disruption of tight junction complexes in the rat blood-brain barrier, following transitory focal astrocyte loss. Glia. 2004;48:1–13. doi: 10.1002/glia.20049. [DOI] [PubMed] [Google Scholar]
- Wu J, Stoica BA, Faden AI. Cell cycle activation and spinal cord injury. Neurotherapeutics. 2011;8:221–228. doi: 10.1007/s13311-011-0028-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Li MX, Luo Y, Chen T, Liu J, Fang P, Jiang B, Hu ZL, Jin Y, Chen JG, Wang F. Chronic ceftriaxone treatment rescues hippocampal memory deficit in AQP4 knockout mice via activation of GLT-1. Neuropharmacology. 2013;75:213–222. doi: 10.1016/j.neuropharm.2013.08.009. [DOI] [PubMed] [Google Scholar]
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- Yao H, Gu XQ, Douglas RM, Haddad GG. Role of Na(+)/H(+) exchanger during O(2) deprivation in mouse CA1 neurons. Am J Physiol Cell Physiol. 2001;281:C1205–1210. doi: 10.1152/ajpcell.2001.281.4.C1205. [DOI] [PubMed] [Google Scholar]
- Yao H, Ma E, Gu XQ, Haddad GG. Intracellular pH regulation of CA1 neurons in Na(+)/H(+) isoform 1 mutant mice. J Clin Invest. 1999;104:637–645. doi: 10.1172/JCI6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zador Z, Stiver S, Wang V, Manley GT. Role of aquaporin-4 in cerebral edema and stroke. Handb Exp Pharmacol. 2009:159–170. doi: 10.1007/978-3-540-79885-9_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng HK, Wang QS, Deng YY, Fang M, Chen CB, Fu YH, Jiang WQ, Jiang X. Hypertonic saline ameliorates cerebral edema through downregulation of aquaporin-4 expression in the astrocytes. Neuroscience. 2010;166:878–885. doi: 10.1016/j.neuroscience.2009.12.076. [DOI] [PubMed] [Google Scholar]
- Zeynalov E, Chen CH, Froehner SC, Adams ME, Ottersen OP, Amiry-Moghaddam M, Bhardwaj A. The perivascular pool of aquaporin-4 mediates the effect of osmotherapy in postischemic cerebral edema. Crit Care Med. 2008;36:2634–2640. doi: 10.1097/CCM.0b013e3181847853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Schallert T, Zhang ZG, Jiang Q, Arniego P, Li Q, Lu M, Chopp M. A test for detecting long-term sensorimotor dysfunction in the mouse after focal cerebral ischemia. J Neurosci Methods. 2002;117:207–214. doi: 10.1016/s0165-0270(02)00114-0. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chintala SK. Influence of interleukin-1 beta induction and mitogen-activated protein kinase phosphorylation on optic nerve ligation-induced matrix metalloproteinase-9 activation in the retina. Exp Eye Res. 2004;78:849–860. doi: 10.1016/j.exer.2003.10.018. [DOI] [PubMed] [Google Scholar]
- Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ, Wang X, Lo EH. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;12:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]
- Zhao H, Nepomuceno R, Gao X, Foley LM, Wang S, Begum G, Zhu W, Pigott VM, Falgoust LM, Kahle KT, Yang SS, Lin SH, Alper SL, Hitchens TK, Hu S, Zhang Z, Sun D. Deletion of the WNK3-SPAK kinase complex in mice improves radiographic and clinical outcomes in malignant cerebral edema after ischemic stroke. J Cereb Blood Flow Metab. 2016 doi: 10.1177/0271678X16631561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Zhang Q, Yu Z, Zhang L, Tian D, Zhu S, Bu B, Xie M, Wang W. Inhibiting cell cycle progression reduces reactive astrogliosis initiated by scratch injury in vitro and by cerebral ischemia in vivo. Glia. 2007;55:546–558. doi: 10.1002/glia.20476. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.