

Abstract
NFKB2 encodes the p100/p52 protein, a critical mediator of the canonical and noncanonical NFkB signaling pathways. Here we report the comprehensive immune evaluation of a child with a novel NFKB2 mutation and provide evidence that aberrant NFKB2 signaling not only causes humoral immune deficiency, but also interferes with the TCR-mediated proliferation of T cells. These observations expand the known phenotype associated with NFKB2 mutations.
Keywords: NFKB2, CVID, Common Variable Immunodeficiency, CID, Combined Immune Deficiency, TCR Signaling, Memory T cells, autoimmune alopecia, TNFRSF13B, TACI
INTRODUCTION
NFκB2 (nuclear factor of kappa light polypeptide gene enhancer in B-cells 2) is a central molecule in the noncanonical NFκB pathway and exists in two forms — the intact, full-length p100 protein, which can inhibit canonical NFκB signaling, and the cleaved p52 fragment, which translocates to the nucleus to drive targeted gene transcription (1). In the quiescent state, the p100 molecule forms inactive cytoplasmic heterodimers with 3 of the 5 known NFκB transcription factors (RelA, RelB, and c-Rel), inhibiting their nuclear translocation. In T-cells, this p100-mediated mechanism appears to negatively regulate TCR signaling after canonical pathway activation, preventing excessive T-cell activation (2). Following activation of the noncanonical pathway, NIK (NFκB-inducing kinase) phosphorylates IKKα, which subsequently activates p100 by phosphorylating two conserved serine residues (Ser866, Ser870) that prime the full-length inhibitory protein for ubiquitination (at Lys855) and proteasomal cleavage to generate the active p52 fragment (3–5). Multiple mouse models have shown that disrupted Nfkb2 signaling adversely affects both T & B cell function (3, 6, 7). Recently, two novel human mutations in NFKB2 were described in patients with humoral immune deficiency, but no T cell functional evaluations were reported(5). Herein we report the comprehensive immune evaluation of a child with a novel NFKB2 mutation and provide evidence that aberrant NFκB2 signaling not only causes humoral immune deficiency, but also interferes with the T-cell receptor (TCR)–mediated proliferation of T cells.
CASE
A 2.5-year-old, male hypogammaglobulinemia patient first came to clinical attention when he was approximately 18 months of age after the insidious development of alopecia universalis, trachonychia, and recurrent sinopulmonary infections. Following immune evaluation, the patient was discovered to have diminished levels of all antibody isotypes, poor responses to vaccination, and an increased CD4:CD8 ratio but otherwise normal lymphocyte subpopulations (Table I). B-cell analysis revealed a reduced fraction of age-adjusted class-switched memory B-cells. Serum BAFF (B–cell activating factor) levels were elevated (4156 pg/mL, Ref: 241–1748 pg/mL), but B cell BAFFR (BAFF receptor) and TACI (transmembrane activator and CAML interactor) surface expression were normal by flow cytometry (Table 1). Given the patient’s presumed autoimmune-mediated alopecia, he was screened for anti-thyroid antibodies and anti-nuclear antibodies (ANA), which were not present. The patient’s T-cell compartment showed a reduced fraction of CD8+/CD45RO+ memory cells and CD4+ effector memory cells (Tem, CD45RO+/CCR7Neg/dim)(Table 1). No significant T cell oligoclonality was noted by TCR variable chain beta analysis (Table 1). Initial T-cell function assays showed a reassuring response to mitogens, IL-2 stimulation (normal STAT5 phosphorylation), and normal surface expression of CD40L and ICOS following stimulation (Table 1). Surprisingly, serial antigen stimulation assays with Tetanus (post booster vaccination) and Candida antigens failed to stimulate the patient’s T-cells to proliferate (Table 1). An expanded mitogens panel (anti-CD3 antibody ± anti-CD28 antibody or IL-2) confirmed a pronounced defect in TCR-driven proliferation in total CD3+ cells which was only partially rescued by CD28 or IL-2 co-stimulation (Table 1). Shortly after being diagnosed with hypogammaglobulinemia, the patient was started on IVIG for recurrent sinopulmonary infection. He was subsequently transitioned to subcutaneous replacement therapy, which has been well tolerated.
Table 1.
Clinical Laboratory Studies
| Patient – 2.5 yoa | Father – 37 yoa | |
|---|---|---|
| Lymphocyte Subpopulations | ||
| Absolute lymphocyte count (lymphocytes/μl) | 5840† (3000–9500) | 1270 (1000–4800) |
| CD3+ | 5251 (1400–8000) | 916 (700–2100) |
| CD4+ | 4210 (900–5500) | 533 (300–1400) |
| CD8+ | 967 (400–2300) | 344 (200–900) |
| CD4/CD8 Ratio | 4.5* (0.9–3.7) | 1.5 (1.0–3.6) |
| CD16+/CD56+ | 673 (100–1400) | 175 (90–600) |
| CD19+ | 1336 (600–3100) | 219 (100–500) |
| Naïve, mature B cells (CD19+/CD27−/IgD+/IgM+) % of CD19+ |
80% (72–87%) | 54% (33–76%) |
| Marginal zone memory B cells (CD19+/CD27+/IgD+/IgM+) % of CD19+ |
16% (7–20%) | 22% (8–34%) |
| Class-switched memory B cells (CD19+/CD27+/IgM−/IgD−) % of CD19+/% of CD27+ |
0.7%/4%* (0.7–7.2%)/(6–30%) |
13.6%/33% (4.7–22.1%)/(19–53%) |
| CD19+/BAFFR+ % of CD19+ |
92.4% (>90.2%) | ND |
| CD19+/TACI+ (CD267) % of CD19+ |
22.8% (>3.4%) | ND |
| CD4+ Naïve T cells (CD45RA+/CD27+) (CD45RA+/RO−) % of CD3+CD4+ |
85.7% (73.6–91.7%) 83% (46–83%) |
ND |
| CD4+ Total Memory T cell (CD45RA−/CD27+) (CD45RA−/RO+) % of CD3+CD4+ |
14.6% (13.9–32.3%) 5% (4–36%) |
ND |
| CD4+ Memory Tcm cells (CD45RO+/CCR7+) % of CD4+ memory cells |
21.1% (20.7–46.0%) | ND |
| CD4+ Memory Tem cells (CD45RO+/CCR7−) % of CD4+ memory cells |
16.8%* (30.5–61.7%) | ND |
| CD8+ Naïve T cells (CD45RA+/CD27+) (CD45RA+/RO−) % of CD3+CD8+ |
94.5% (77.7–98.4%) 93% (41–100%) |
ND |
| CD8+ Total Memory T cell (CD45RA−/CD27+) (CD45RA−/RO+) % of CD3+CD8+ |
5.3%* (7.2–23.9%) 2% (0–40%) |
ND |
| CD8+ Memory Tcm cells (CD45RO+/CCR7+) % of CD8+ memory cells |
8.4% (0.0–29.7%) | ND |
| CD4+ Memory Tem cells (CD45RO+/CCR7−) % of CD4+ memory cells |
20.6% (19.7–59.1%) | ND |
| TCR-variable β chain: 24 marker panel β-2 β-9 β-11 β-12 β-13.6 β-20 β-21.3 % of CD3+ |
All markers within normal range, except those minor deviations listed below 9.53%* (4.16–8.91%) 1.01%* (2.62–4.15%) 3.62%* (0.81–1.5%) 2.28%* (1.08–1.93%) 2.83%* (1.29–2.5%) 0.52%* (1.19–4.6%) 2.66%* (1.69–2.56%) |
ND |
| Immunoglobulins | ||
| IgG (mg/dL) | 173* (400–1250) | 1160 (600–1500) |
| IgG1 | 134* (290–850) | ND |
| IgG2 | 28* (45–260) | ND |
| IgG3 | 9* (15–113) | ND |
| IgG4 | 21 (≤79) | ND |
| IgM (mg/dL) | 12.1* (41–164) | 77 (60–263) |
| IgA (mg/dL) | <6* (14–105) | 111 (68–378) |
| IgE (IU/mL) | 3 (2–97) | 19 (2–214) |
| Vaccine Response | ||
| Tetanus IgG (IU/mL) | 0.1* (>0.1) | 27.1 (>0.1) |
| Diphtheria IgG | 0* (>0.1) | 3.1 (>0.1) |
| Pneumococcal IgG (mcg/mL) |
≥1.5 in 2 of 14 serotypes (post Prevnar-13 series) |
≥1.5 in 8 of 14 serotypes (pre-vaccination) |
| T-Cell Function | ||
| Mitogens (PHA, ConA, PWM) | All Normal (>135190 cpm, >73522 cpm, >26677 cpm, respectively) | ND |
| pSTAT5 Tyrosine Phosphorylation, CD4+ T cells | 40% (10–47%) | ND |
| Antigen stimulation: Tetanus | 243*, 1 month post-DTaPˆ booster. 416 * 12 months post-booster. (≥4761 cpm) |
ND |
| Antigen stimulation: Candida | 786* (≥15289 cpm) | ND |
| TCR stimulation: PBMCs -Anti-CD3 Ab alone -Anti-CD3 Ab+anti-CD28 Ab - Anti-CD3 Ab+IL-2 % CD3+ proliferating. |
3.8%* (≥20.3%) 20.6%* (≥44.6%) 30.6%* (≥46.2%) |
ND |
| Endocrinology | ||
| Cortisol # (mcg/dL) | 19 (3–21) | ND |
| ACTH # (pg/mL) | 17 (4–46) | ND |
| Anti-thyroid antibodies (IU/mL) | Thyroglobulin Ab, <10 (10–114) Thyroid Peroxidase Ab, <5 (5–33) |
ND |
| Genetics: Exome Sequencing, Select Variants | ||
| Patient | Parents (Father/Mother) | |
| NFKB2 | c.2598-2599insT (p.A867Cfs*19), heterozygous | WT/WT |
| TNFRSF13B (aka TACI, CD267) | c.706G>T (p.E236*) rs201021960, heterozygous | c.706G>T (p.E236*) rs201021960, heterozygous/WT |
Values in parentheses indicate clinically validated, age matched control ranges/values.
Average value during a 1-year period
Indicates abnormal value
Abbreviations: yoa, years of age; moa, months of age; Tcm, Central Memory T cell; Tem, Effector Memory T cell; PHA, phytohemagglutinin; ConA, concanavalin A; PWM, pokeweed mitogen; ND, not done; cpm, count per minute; PBMC, peripheral blood mononuclear cells; ACTH, Adrenocorticotropic hormone; Ab, antibody; WT, wild-type.
DTaP = Diphtheria, Tetanus, acellular Pertussis vaccine, 4th dose at 18 moa
= Assay drawn at 8 am.
After being appropriately consented, the family underwent parent-child trio exome sequencing using the NimbleGen V3 capture array followed by sequencing on the Illumina HiSeq 2500 system (100 bp pair-end reads). After filtering for sequence quality, the resulting reads were mapped onto the human reference genome (Genome Reference Consortium, GRCh37). Subsequent bioinformatic filtering, based on multiple models of inheritance, frequency, variant type, in silico predictions, and mutation database searches, highlighted two candidate variants (Table 1,Fig. 1A), which were confirmed by Sanger sequencing. Notably, a de novo, patient-specific T-nucleotide insertion was observed at c.2598 of NFKB2, creating a frameshift, heterozygous mutation (p.A867Cfs*19) that deletes 1 of 2 regulatory phosphorylation sites (Fig. 1B). Western blot analysis confirmed the presence of a truncated NFκB2 protein in patient-derived lysate from fresh, unstimulated PBMCs (Fig. 1C). To clarify the molecular weight and characteristics of the mutant NFKB2 protein, HEK293 cells were co-transfected with recombinant NFKB2 (WT or mutantA867C), with or without NIK (Fig. 1C, far right). The mutant NFKB protein was truncated and was not processed into the active p52 fragment. Given our patient’s severely blunted responses to both Tetanus and Candida antigens but normal responses to mitogens, we hypothesized that p100-inhibited TCR signaling may be contributing to his phenotype. We isolated CD4+ memory T cells from the patient and two unaffected adult controls, stimulated the cells with anti-CD3 antibody or human IL-2 for 120 hours and quantified proliferation. Patient-derived memory T cells showed significantly reduced TCR-mediated and normal IL-2–mediated proliferation, compared to normal controls (Fig. 1D).
Figure 1. Experimental Results.
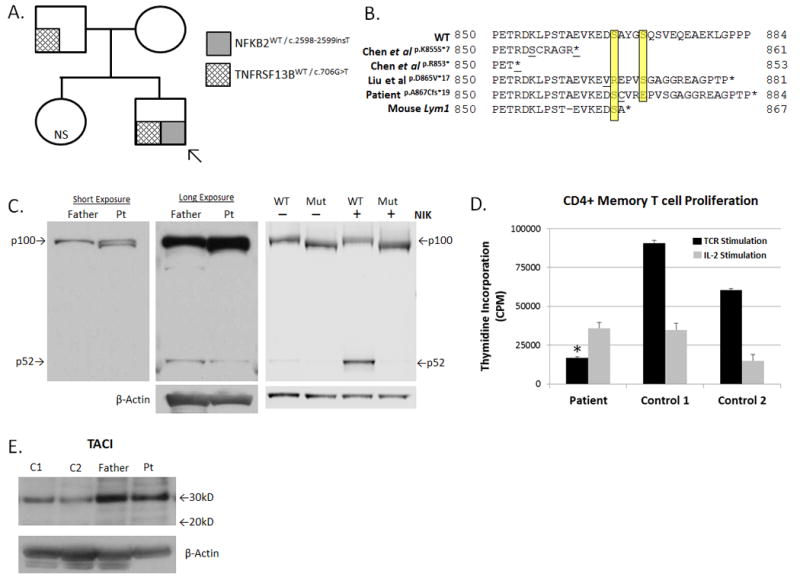
(A). Pedigree and Mutational Analysis. Arrow indicates proband (“patient”).
(B). Predicted NFκB2 Protein Sequence Alignment. Yellow highlighting denotes Ser866 and Ser870. Underlined amino acids indicate sites of the first divergent codon.
(C). NFκB2 Protein Analysis: (Left & Middle panels) Fresh PBMC-derived protein lysates from the patient (pt) and his father were probed with an anti-NFKB2 antibody (Cell Signaling #4882). Note “double” p100 band and a weak p52 band in the patient lane. (Right Panel): Lysates from NFKB2-transfected HEK293 cells, with or without NIK co-transfection, followed by NFKB2 western blot. In all panels, protein loading was normalized by BCA quantification and β-actin immunoblot.
(D). TCR-Mediated Proliferation: Negatively selected, untouched CD4+ memory T cells were isolated from fresh PBMCs (Miltenyi Biotec). Cells were stimulated with an anti-CD3 antibody (1μg/mL, Biolegend, Clone OKT3, black bars) or human IL-2 (2000 IU/mL, gray bars) for 120 hrs, and proliferation was quantified by tritiated thymidine incorporation performed in triplicate. Data are given as mean ±SEM. *p < 0.05 for patient versus controls (individually and mean).
(E). TACI Protein Analysis: Fresh PBMC-derived protein lysates from the patient, his father, and two unrelated controls (C1, C2) were probed with an anti-TACI antibody (Sigma, PRS2395). The wild-type TACI protein yields a band of ~32 kD, while the p.E236* truncated TACI protein is predicted to migrate at ~26 kD. No truncated TACI protein bands are observed. Protein loading was normalized by BCA quantification & β-actin immunoblot.
Abbreviations: NS, not sequenced; MW, molecular weight; CPM, counts per minute.
Whole-exome sequencing also revealed a second, potentially relevant familial mutation in TNFRSF13B (also known as TACI or CD267), a gene which encodes a BAFF/APRIL receptor and is associated with common variable immunodeficiency (CVID). The patient inherited this variant from his father (Fig. 1A). This heterozygous, nonsense mutation (c.706G>T, p.E236*) is predicted to truncate TACI’s terminal intracytoplasmic region by 57 amino acids. Western blot analysis of lysate from fresh, unstimulated PBMCs from the patient and father revealed normal full-length TACI (32 kD, Fig. 1E). No truncated band (predicted size ~26 kD) was observed, suggesting that the mutant transcript may undergo nonsense-mediated decay. To detect possible clinical effects of the TACI mutation, the patient’s father also underwent immune screening, which showed no evidence of immune deficiency (Table 1). This TACI variant is reported in the dbSNP database (rs201021960) and likely is a benign heterozygous polymorphism, however, we cannot exclude a role for this variant as a disease modifier in certain individuals. The patient’s elevated serum BAFF level may be secondary to impaired ligand feedback inhibition in BAFF-secreting cells, however, little is known about the role of TACI or NFKB2 in regulating serum BAFF levels.
DISCUSSION
NFKB2 functions at the interface of the canonical and noncanonical NFkB pathways and multiple in vitro and in vivo studies have shown that changes to the p100/p52 ratio has profound effects on the observed immune phenotype (3, 6–8). While the Nfkb2 knockout (KO) mice have abnormal splenic architecture, no gross hypogammagloblinemia occurs in these mice (6). Nfkb2 KO mice actually have elevated IgM and IgG2a and reduced IgG3 and IgA relative to wild-type (WT) littermates, whereas the halpoinsufficient mice have normal immunoglobulin levels. In contrast to the null mouse, the Nfkb2Lym1 mutant mouse harbors a unique Nfkb2-truncating mutation that blocks proteolytic generation of p52 while preserving the inhibitor p100Lym1 protein (3)(Fig1B). The resulting Nfkb2Lym1/Lym1 phenotype is characterized by peripheral lymphocytosis with an expanded CD4+ fraction, panhypogammagloblinemia, diminished B-cell response to CD40L, disrupted splenic architecture, pulmonary and hepatic inflammatory infiltrates, and osteopetrosis. The heterozygous Nfkb2Lym1/+ phenotype is highly similar to that of Nfkb2Lym1/Lym1, with lymphocytosis, splenic architectural defects and impaired RelA nuclear translocation in activated B-cells (3). A related model, the Nikaly/aly mutant mouse, also has impaired noncanonical pathway activation which leads to increased cytoplasmic p100 levels. These mice have a complex T-cell phenotype where suppressive CD4+ memory T cells (CD44hi) blunt the proliferation of naïve CD4+ T cells (4). Collectively, these mouse models show that the abnormal accumulation of p100 disrupts noncanonical (i.e. CD40L-mediated) and canonical (i.e. TCR-mediated) NFkB signaling in murine lymphocytes.
Similar to these mouse models, our patient’s phenotype of combined immune deficiency (CID) with alopecia universalis and impaired T-cell antigen response is consistent with abnormal noncanonical and canonical signaling. While the detailed molecular mechanism is not yet fully resolved, plausible possibilities include a dominant negative effect from accumulated uncleavable mutant p100 in the lymphocytes or a haploinsufficiency effect from diminished p52 production. Our T cell functional testing expands upon the humoral immune deficiency phenotypes described in two recent reports detailing patients with similar mutations (Fig 1B)(5, 9). Of note, Liu et al showed diminished T follicular helper (Tfh) cells in one NFKB2 patient, and the authors mention, but do not show, normal to slightly reduced T cell mitogen responses in two patients. Unlike the previously reported human mutations, our patient’s mutation is more like the Nfkb2Lym1/+ mouse mutation in that it selectively disrupts just the Ser870 residue, thus suggesting that loss of the distal serine alone is sufficient to induce a CID-like phenotype in humans. Notably, our patient also differs from the previously reported cohort in that he does not yet suffer from central adrenal insufficiency (Table 1), but we are concerned that he may develop this defect in later life. He is undergoing regular endocrine evaluation to monitor for signs and symptoms of polyendocrine autoimmune disease. His case poses a clinical management dilemma regarding how best to suppress ongoing autoimmunity in patients with CID and also highlights the clinical utility of whole-exome sequencing technology in diagnosing rare and/or newly described diseases.
Acknowledgments
Special thanks to Joyce Collett, Rebecca Marsh, Jack Bleesing, and Lisa Filipovich of the Cincinnati Children’s Hospital Medical Center Diagnostic Immunology Laboratory for their technical support. The authors also thank Kimberly Risma, David Hildeman and Joseph Sherrill for their thoughtful scientific advice and Shawna Hottinger for her editorial assistance. This research was funded in part by the Cincinnati Children’s Hospital Research Foundation (Procter Scholarship awarded to AWL) and an NIH K12 (HD028827) Child Health Research Career Development Award (AWL).
References
- 1.Ruland J, Mak TW. Transducing signals from antigen receptors to nuclear factor kappaB. Immunological reviews. 2003;193:93–100. doi: 10.1034/j.1600-065x.2003.00049.x. [DOI] [PubMed] [Google Scholar]
- 2.Legarda-Addison D, Ting AT. Negative regulation of TCR signaling by NF-kappaB2/p100. Journal of immunology. 2007;178(12):7767–78. doi: 10.4049/jimmunol.178.12.7767. [DOI] [PubMed] [Google Scholar]
- 3.Tucker E, O’Donnell K, Fuchsberger M, Hilton AA, Metcalf D, Greig K, et al. A novel mutation in the Nfkb2 gene generates an NF-kappa B2 “super repressor”. Journal of immunology. 2007;179(11):7514–22. doi: 10.4049/jimmunol.179.11.7514. [DOI] [PubMed] [Google Scholar]
- 4.Ishimaru N, Kishimoto H, Hayashi Y, Sprent J. Regulation of naive T cell function by the NF-kappaB2 pathway. Nature immunology. 2006;7(7):763–72. doi: 10.1038/ni1351. [DOI] [PubMed] [Google Scholar]
- 5.Chen K, Coonrod EM, Kumanovics A, Franks ZF, Durtschi JD, Margraf RL, et al. Germline mutations in NFKB2 implicate the noncanonical NF-kappaB pathway in the pathogenesis of common variable immunodeficiency. American journal of human genetics. 2013;93(5):812–24. doi: 10.1016/j.ajhg.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caamano JH, Rizzo CA, Durham SK, Barton DS, Raventos-Suarez C, Snapper CM, et al. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. The Journal of experimental medicine. 1998;187(2):185–96. doi: 10.1084/jem.187.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rowe AM, Murray SE, Raue HP, Koguchi Y, Slifka MK, Parker DC. A cell-intrinsic requirement for NF-kappaB-inducing kinase in CD4 and CD8 T cell memory. Journal of immunology. 2013;191(7):3663–72. doi: 10.4049/jimmunol.1301328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giardino Torchia ML, Conze DB, Jankovic D, Ashwell JD. Balance between NF-kappaB p100 and p52 regulates T cell costimulation dependence. Journal of immunology. 2013;190(2):549–55. doi: 10.4049/jimmunol.1201697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, Hanson S, Gurugama P, Jones A, Clark B, Ibrahim MA. Novel NFKB2 Mutation in Early-Onset CVID. Journal of clinical immunology. 2014 doi: 10.1007/s10875-014-0064-x. [DOI] [PubMed] [Google Scholar]