

SUMMARY
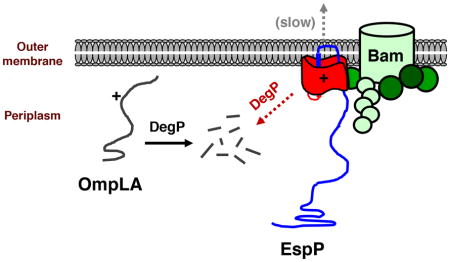
Almost all bacterial outer membrane proteins (OMPs) contain a β barrel domain that serves as a membrane anchor, but the assembly and quality control of these proteins are poorly understood. Here we show that the introduction of a single lipid-facing arginine residue near the middle of the β barrel of the E. coli OMPs OmpLA and EspP creates an energy barrier that impedes membrane insertion. Although several unintegrated OmpLA mutants remained insertion-competent, they were slowly degraded by the periplasmic protease DegP. Two EspP mutants were also gradually degraded by DegP, but were toxic because they first bound to the Bam complex, an essential heteroligomer that catalyzes the membrane insertion of OMPs. Interestingly, another EspP mutant likewise formed a prolonged, deleterious interaction with the Bam complex, but was protected from degradation and eventually inserted into the membrane in a native conformation. The different types of interactions between the EspP mutants and the Bam complex that we observed may correspond to distinct stages in OMP assembly. Our results show that sequences that significantly delay assembly are disfavored not only because unintegrated OMPs are subject to degradation, but also because OMPs that assemble slowly can form dominant-negative interactions with the Bam complex.
Keywords: β barrel, Bam complex, outer membrane, protein folding, protein quality control
ABBREVIATED SUMMARY
The introduction of lipid-facing arginine residues near the middle of the β barrel of bacterial outer membrane proteins hinders membrane insertion. An analysis of the mutant proteins shows that there is a strong selection against sequences that delay assembly because unintegrated outer membrane proteins are gradually degraded by DegP, even if they remain insertion-competent, and because outer membrane proteins that assemble slowly can form toxic interactions with the Bam complex.
INTRODUCTION
The vast majority of integral membrane proteins that reside in the outer membrane (OM) of Gram-negative bacteria and organelles of bacterial origin have a unique architecture. Most proteins that reside in the bacterial inner membrane (IM) and other biological membranes contain hydrophobic ~20 residue α-helical membrane spanning segments. In contrast, OM proteins (OMPs) are anchored to the membrane by amphipathic β sheets that fold into a closed cylindrical structure known as a “β barrel”. Whereas the presence of a single hydrophobic α-helix is sufficient to embed a protein in the IM, the stable integration of a protein into the OM requires the formation of a fully folded β barrel structure that exposes a hydrophobic exterior surface.
The process by which OMPs are assembled is still poorly understood. While IM proteins are integrated into the membrane co-translationally through the opening of a lateral gate in the Sec complex and the partitioning of α-helical segments into the lipid bilayer (White and von Heijne, 2008), OMPs are translocated across the IM through the Sec complex post-translationally. OMPs interact with molecular chaperones (e.g., Skp and SurA) in the periplasm that presumably maintain them in an assembly-competent conformation (Chen and Henning, 1996; Rouviere and Gross, 1996; Walton et al., 2009). Subsequently they interact with the β barrel assembly machine (Bam) complex, a hetero-oligomer that catalyzes their integration into the OM (Voulhoux et al., 2003; Wu et al., 2005; Hagan et al., 2010). In Escherichia coli, the Bam complex consists of BamA, an integral OMP that contains five periplasmic polypeptide transport associated (POTRA) domains, and four lipoproteins (BamB-E) that interact with the POTRA domains (Wu et al., 2005; Kim et al., 2007; Sklar et al., 2007). Only BamA and BamD are highly conserved and essential for viability (Wu et al., 2005; Malinverni et al., 2006; Webb et al., 2012). Although the mechanistic details of Bam complex-mediated OMP assembly are unknown, it has been proposed that OMPs partition into the lipid bilayer in a stepwise fashion through a lateral gate formed by an unstable region in the BamA β barrel (Noinaj et al., 2013; Noinaj et al., 2014). In an alternative model, it has also been proposed that the Bam complex promotes the integration of OMPs by destabilizing the lipid bilayer (Gessmann et al., 2014; Fleming, 2015; Plummer and Fleming, 2015). The crystal structure of the Bam complex show that the POTRA domains and lipoprotein subunits adopt a ring like architecture that might induce integration of OMPs via a rotational motion, but do not shed light on the exact mechanism of integration (Bakelar et al., 2016; Gu et al., 2016; Han et al., 2016). It should also be noted that while the IM is composed entirely of phospholipids, the outer leaflet of the OM consists of a unique glycolipid known as lipopolysaccharide (LPS) that might also play a role in assembly.
In addition to the OMP assembly machinery, bacteria produce periplasmic quality control systems that eliminate β barrel proteins that fail to insert into the OM. Many mislocalized OMPs are degraded by DegP, a protease that functions as a chaperone at low temperature (Misra et al., 1991; Spiess et al., 1999; CastilloKeller and Misra, 2003; Ge et al., 2014), but some OMPs are degraded by other proteases (Echenique-Rivera, et al., 2011; Weski and Ehrmann, 2012; Narita, et al., 2013). DegP is essential for viability in E. coli at high temperature and becomes essential at lower temperatures when the assembly of OMPs is impaired (Lipinska et al. 1989; Strauch et al., 1989; Charlson et al., 2006). It is widely believed that DegP performs a dual function by degrading severely misfolded OMPs that are aggregation prone and therefore potentially toxic while protecting proteins that are present in insertion competent conformations (Sawa et al., 2011; Lyu and Zhao, 2015). This view emerged partly from experiments that showed that the binding of misfolded proteins transforms inactive DegP hexamers into proteolytically active 12- and 24-mers and that folded OMPs co-purify with recombinant DegP (Krojer et al., 2008). While the results of structural studies are intriguing, the basis by which defective OMPs are recognized by DegP and other periplasmic quality control systems remains unclear. It has been proposed that quality control In the cytoplasm and the endoplasmic reticulum is a time-dependent stochastic process in which a protein will be degraded if it encounters a protease or is otherwise earmarked for destruction before reaching its native state (Wickner et al., 1999; Buchberger et al., 2010). This idea has been confirmed by mathematical modeling of the periplasmic quality control system (Costello et al., 2016)
Given that the accurate biogenesis of proteins depends on their amino acid sequences, the predominance of hydrophobic residues and the underrepresentation of ionizable residues in membrane-exposed regions of OMPs is striking (Ulmschneider and Sansom, 2001; Chamberlain and Bowie, 2004; Jackups and Liang, 2005; Jimenez-Morales and Liang, 2011; Slusky and Dunbrack, 2013). Several lines of evidence indicate that there is a position-dependent thermodynamic penalty for inserting an ionizable residue into a biological membrane. The penalty is greatest when the residue is located near the middle of the lipid bilayer (Moon and Fleming, 2011; Hessa et al., 2007). Experimentally determined hydrophobicity scales that measure the tendency of individual amino acid side chains to partition into a lipid bilayer, however, suggest that even the maximum thermodynamic penalty (~2–3 kcal mol−1) is rather modest (Wimley and White, 1996; Hessa et al., 2007; Moon and Fleming, 2011). These results suggest that there are kinetic as well as thermodynamic factors that prohibit the localization of ionizable residues at lipid-exposed positions.
To gain mechanistic insight into OMP assembly and quality control, we examined the consequences of introducing lipid-facing arginine mutations into two E. coli proteins, outer membrane phospholipase A (OmpLA) and EspP. Our mutagenesis scheme was based on the expectation that surface-exposed arginine residues would perturb the membrane insertion reaction per se but, unlike previously studied mutations (CastilloKeller and Misra, 2003; Wzorek et al., 2017), would not grossly impair protein folding. Whereas OmpLA consists solely of a 12-stranded β barrel (Snijder et al., 1999), EspP is a member of the autotransporter superfamily that contains a large N-terminal extracellular (“passenger”) domain in addition to a C-terminal 12-stranded β barrel domain (Dautin and Bernstein, 2007; Fan et al., 2016). Although the passenger domain is released by proteolytic cleavage following its translocation across the OM, the two domains are initially connected by a segment that passes through the β barrel pore (Barnard et al., 2007; Dautin et al., 2007; Barnard et al., 2012). Available evidence suggests that the α-helical linker is embedded inside a partially folded β barrel domain in the periplasm (Ieva et al., 2008). We found that lipid-facing arginine residues located near the middle of the β barrel strongly affected the biogenesis of both proteins. Even though the mutations did not affect the insertion-competence of OmpLA, they created a significant kinetic delay in the insertion of the protein into the OM that led to its eventual degradation by DegP. Similar mutations impaired the assembly of EspP, but did not affect the rapid targeting of the protein to the Bam complex. Although one class of mutants failed to integrate into the OM and were slowly degraded, their relatively prolonged interaction with the Bam complex was deleterious. Remarkably, a second class of mutants were integrated slowly into the OM but remained immune to degradation. These results suggest that the sequence of OMPs is constrained by the inability of DegP to eliminate at least some proteins that assemble slowly from forming stable interactions with the Bam complex.
RESULTS
DegP degrades OmpLA mutants that assemble slowly
Using an unbiased selection of proteins that are in the current knowledge base of bacterial OMP structures, we confirmed that ionizable residues are highly underrepresented on the lipid-facing surfaces of OMPs (Fig. S1). Although outward-facing ionizable residues are uncommon, previous studies have shown that they are often located near the extracellular surface (Slusky and Dunbrack, 2013) and at least in one case play an important role in protein assembly (Arunmanee et al., 2016). The observed charge asymmetry is consistent with evidence obtained in cell free assays that the placement of an ionizable residue near the middle of a lipid bilayer incurs a higher energetic cost (Hessa et al., 2007; Moon and Fleming, 2011).
We hypothesized that the introduction of ionizable residues onto the lipid-facing surface of OMPs might perturb their integration into the OM but would not grossly affect their final folded conformations. To test this idea, we initially introduced single arginine residues that are situated at different depths in the bilayer into the ~30 kD OmpLA protein. We chose to make arginine substitutions because the guanidinium functional group of this amino acid has an immeasurably high pKa and is therefore always charged (Harms et al., 2011). This strategy avoids any complications that might result from a non-uniform side chain charge state. We initially confirmed that the OmpLA mutants all fold into the native conformation in vitro and possess enzyme activity (Fig. S2). To examine protein biogenesis in vivo, MC4100 were transformed with a derivative of plasmid pTRC99a encoding either wild-type OmpLA or one of the arginine mutants under the control of a trc promoter and grown in minimal medium. Cells were subjected to pulse-chase labeling following the addition of IPTG, and immunoprecipitations were conducted using an anti-OmpLA antiserum.
Interestingly, we observed a highly position-dependent effect of the arginine mutations on the fate of OmpLA. Like wild-type OmpLA, five mutants that contain an arginine located near the periplasmic or extracellular side of the OM were completely stable for 30 min (Fig. 1A; Fig. 2A, gels 1–6). Because we expected that the integration of OmpLA into the OM would protect it from protease digestion, we investigated the localization of the protein by permeabilizing radiolabeled cells with lysozyme/EDTA and adding proteinase K (PK). We found that a fraction of both the wild-type and mutant proteins was resistant to protease treatment after pulse labeling and that almost all of the protein was resistant after a 1 min chase (Fig. S3). These results indicate that OmpLA is rapidly integrated into the OM and that the mutations do not measurably affect the kinetics of insertion. In contrast, several mutants that contain an arginine located closer to the middle of the bilayer (G212R, A210R, L208R) were slowly degraded (Fig. 1A; Fig 2A, gels 7–9; Fig. 2B). This observation is striking given that one of the mutations (G212R) is located on the same β strand immediately adjacent to another mutation (Y214R) that does not affect stability. The finding that alanine mutations at positions 212 or 208 or a serine mutation at position 210 did not destabilize the protein (Fig. S4) indicates that the presence of the lipid-exposed ionizable residue rather than the loss of the native residue led to degradation. Although the same degradation pattern was observed in an isogenic strain that lacks OmpT, the only known general protease in the E. coli OM (data not shown), all of the variants were stabilized in JMR352, a strain that lacks DegP (Fig. 3A, lanes 1–5; Fig. 3B). In light of previous work, these results suggest that lipid-facing ionizable residues located near the middle of the OmpLA β barrel create an energetic penalty for membrane integration and thereby lead to the retention and degradation of the protein in the periplasm.
Fig. 1.

Location of arginine mutations in OmpLA and EspP. A.–B. The crystal structures of OmpLA (PDB:1QD5; Snijder et al., 1999) and the EspP β barrel (PDB: 2QOM; Barnard et al., 2007) and the position of the arginine substitutions that were analyzed in this study are shown. Red: arginine substitutions that blocked insertion of the protein into the OM in wild-type cells; yellow: arginine substitutions that delayed membrane insertion; green: arginine substitutions that did not affect protein assembly.
Fig. 2.

Introduction of lipid-facing arginine residues near the middle of the OmpLA β barrel destabilizes the protein in wild-type E. coli. A. MC4100 transformed with pMDG2 (Ptrc-ompLA) or a pMDG2 derivative encoding the indicated OmpLA mutant were subjected to pulse-chase labeling after the addition of IPTG. Immunoprecipitations were then conducted using an anti-OmpLA antiserum. B. The percentage of the pulse-labeled protein that remained at each time point is shown.
Fig. 3.

OmpLA arginine variants are stable and gradually acquire PK resistance in the absence of DegP. JMR352 transformed with pMDG2 or a pMDG2 derivative encoding the indicated OmpLA mutant were subjected to pulse-chase labeling after the addition of IPTG. The OM was permeabilized and half of each sample was treated with PK. Immunoprecipitations were then conducted using an anti-OmpLA antiserum. The percentage of the protein that remained and that was resistant to PK digestion at each time point is shown. Because it was not possible to distinguish proOmpLA from mature OmpLA in some of the pulse-labeled samples, the protein observed in untreated cells after a 2 min chase was defined as 100%.
Consistent with the hypothesis that surface exposed arginine residues do not significantly impair protein folding, further analysis provided evidence that the OmpLA L208R, A210R and G212R mutants remain insertion competent for a prolonged period. To determine the location of the stabilized mutant proteins in JMR352, a portion of the cells that were radiolabeled in the experiment described above were permeabilized and treated with PK. Although only a small fraction of each variant was PK-resistant at early time points, the level of PK-resistant protein increased steadily (Fig. 3A, gels 2–4, lanes 6–10; Fig. 3C). By 20 min, the level of PK-resistance of the L208R and A210R mutants approached that of the wild-type protein. These results suggested that OmpLA variants that contain a lipid-facing arginine near the middle of the β barrel slowly insert into the OM in the absence of DegP. Consistent with this conclusion, the radiolabeled mutant proteins became sensitive to PK digestion when we permeabilized cells and solubilized membranes by adding the detergent n-dodecyl-D-maltoside (DDM) following a 20 min chase (Fig. 4A). Likewise, when we sonicated cells and isolated a membrane fraction, PK degraded the A210R and G212R mutants after the addition of DDM (Fig. 4B). Even in the presence of detergent, however, the protease removed only a ~1 kD fragment from wild-type OmpLA (as well as the L208R mutant that was associated with isolated membranes). The results suggest that while wild-type OmpLA inserts into the OM in a compact, protease-resistant conformation, the mutant proteins fold less stably. Indeed wild-type OmpLA was resistant to SDS denaturation and migrated rapidly on SDS-PAGE unless heated to >70° C, but the arginine variants unfolded at all temperatures (Fig. S5).
Fig. 4.

The PK resistance of OmpLA arginine mutants in a degP- strain is due to membrane integration. JMR352 transformed with pMDG2 or a pMDG2 derivative encoding the indicated OmpLA mutant were pulse labeled and subjected to a 20 min chase after the addition of IPTG. In (A) the OM was then permeabilized and equal portions of each sample were left untreated, treated with PK or treated with PK after the addition of DDM. In (B) cells were disrupted by sonication. After a portion of the total cell extract (T) was set aside, membranes (Memb) were separated from soluble proteins (Sol) by centrifugation. Equal portions of the membrane fractions were then left untreated, treated with PK or treated with PK after the addition of DDM. Immunoprecipitations were conducted using an anti-OmpLA antiserum.
We next wished to determine whether DegP could influence the fate of the OmpLA arginine mutants without degrading them. To this end we shifted MC4100 to 28° C, a temperature at which DegP functions primarily as a chaperone (Speiss et al., 1999), and permeabilized cells following pulse-chase labeling. Although the L208R mutant was still degraded by DegP at the lower temperature (data not shown), the A210R and G212R mutants were relatively stable (Fig. 6A). Neither mutant, however, exhibited the slow acquisition of PK resistance that was observed at 37° C. This finding cannot be explained by an inherent cold-sensitivity of membrane integration, because a slow increase in PK resistance was observed in JMR352 cells incubated at 28° C (Fig. 6B). Taken together, the results strongly suggest that DegP functions as a slow-acting “gatekeeper” that captures β barrel proteins that are not rapidly integrated into the OM, but does not degrade them immediately. Consistent with mathematical modeling (Costello et al., 2016), client proteins can presumably undergo multiple cycles of binding and release, but are eventually degraded through a stochastic process if they remain accessible to DegP for prolonged periods. Importantly, contrary to current models, DegP may not recognize substrates based on features that indicate insertion incompetence, but rather on their continued presence in the periplasm.
Fig. 6.

The introduction of a lipid-facing arginine residue near the extracellular or periplasmic side of the EspP β barrel does not significantly affect assembly. AD202 transformed with pRLS5 (Ptrc-espP) or a pRLS5 derivative encoding the indicated EspP mutant were subjected to pulse-chase labeling after the addition of IPTG. Immunoprecipitations were then conducted using an antiserum generated against an EspP C-terminal peptide. The percentage of the passenger domain that was released from the β domain by proteolytic cleavage at each time point is shown.
Integration-defective EspP mutants form a prolonged interaction with the Bam complex
To determine the generalizability of the results described above, we examined the effect of a similar set of lipid-facing arginine substitutions on the biogenesis of a second OM protein. We chose a member of the autotransporter superfamily produced by E. coli O157:H7 (EspP) for these experiments because its assembly in vivo has been extensively characterized. EspP is similar to OmpLA in that it contains a 12-stranded ~30 kD β barrel domain (Barnard et al., 2007), but it is more complex in that it also contains a large extracellular (“passenger”) domain connected to the β barrel domain by an α helical linker that traverses the β barrel pore. Available evidence suggests that the EspP β barrel domain begins to fold in the periplasm and incorporates the linker in a hairpin conformation (Ieva et al., 2008). Although the mechanism of passenger domain secretion is unknown, the initiation of translocation requires at least partial integration of the β barrel domain into the OM (Pavlova et al., 2013). Following completion of the passenger domain translocation reaction (which proceeds in a C- to N-terminal direction), the protein is cleaved into discrete passenger and β barrel domains in an intra-barrel cleavage reaction that requires the correct alignment of catalytic residues (Dautin et al., 2007; Ieva and Bernstein, 2009; Junker et al., 2009; Barnard et al., 2012). Like OmpLA, the free EspP β barrel domain folds into a stable structure that is highly resistant to thermal denaturation and that migrates rapidly on SDS-PAGE (Barnard et al., 2007).
We found that lipid exposed arginine residues exerted an effect on EspP assembly that, like the effect they exerted on OmpLA assembly, correlated with their depth in the lipid bilayer. To determine the effect of individual mutations, AD202 (MC4100 ompT::kan) were transformed with a plasmid encoding either wild-type EspP or an arginine variant under the control of the trc promoter. An ompT- strain was used to prevent degradation of the EspP passenger domain following its exposure on the cell surface. Cells were grown in minimal medium and subjected to pulse-chase radiolabeling following the addition of IPTG, and immunoprecipitations were conducted using an anti-peptide antiserum generated against the extreme C-terminus of EspP. The assembly of four mutants containing a lipid-facing arginine residue located close to the periplasmic or extracellular side of the β barrel domain (A1117R, Y1125R, W1149R and Y1175R; see Fig. 1B) was monitored by following the proteolytic maturation of the ~135 kD precursor form of the protein (proEspP) in which the two domains are covalently linked. As shown in Fig. 6, the mutations produced only a very subtle delay in the accumulation of the cleaved β domain. Because the mutations are located on the surface far from the intra-barrel cleavage site they presumably did not delay proteolytic maturation per se but rather an earlier stage of assembly.
Interestingly, while the presence of a lipid-exposed arginine residue located closer to the middle of the β barrel strongly impaired EspP assembly, we observed two distinct mutant phenotypes. We introduced single arginine substitutions on the same β strand as two mutations that did not significantly affect EspP biogenesis (A1117R and Y1125R; Fig. 1B). In addition to monitoring the effect of the mutations on the proteolytic maturation of proEspP, we assessed their effect on the initiation of passenger domain translocation (which results in the exposure of proEspP on the cell surface) by treating half of the cells harvested at each time point with PK. We found that the G1123R mutation dramatically slowed proteolytic maturation. Whereas 50% of the wild-type proEspP molecules were cleaved in <1 min and 90% were cleaved in 5 min, cleavage of the same fraction of the mutant protein required ~10 min and 30 min, respectively (Fig. 7A, gels 1–2; Fig. 7B). A long delay in the initiation of passenger domain translocation paralleled the delay in cleavage (Fig. 7C). The results imply that the mutation delays the initiation of passenger domain translocation and thereby indirectly affects later stages in the assembly pathway including proteolytic processing. Remarkably, none of the G1123R mutant protein was degraded despite its slow integration into the OM (Fig. 7D). Furthermore, the free β barrel domain showed the same resistance to SDS denaturation as its wild-type counterpart and therefore folded into a native state (Fig. 7E). In contrast, the L1121R mutation completely blocked exposure of the passenger domain and all subsequent assembly steps, and the presence of the I1119R mutation blocked the assembly of ~90% of the protein (Fig. 7A, gels 3–4; Fig. 7B). Unlike the G1123R mutant, however, these mutants were slowly degraded (Fig. 7D). Although the L1121R and I1119R mutants were stabilized by lowering the incubation temperature to 28° C or by producing them in a degP- strain, neither the suppression of DegP protease activity nor its complete elimination affected the efficiency of assembly (Figs. S6 and S7).
Fig. 7.

The introduction of a lipid facing arginine residue near the middle of the EspP β barrel delays or blocks membrane integration. A. AD202 transformed with pRLS5 (Ptrc-espP) or a pRLS5 derivative encoding the indicated EspP mutant were subjected to pulse-chase labeling after the addition of IPTG. Half of the cells were treated with PK, and immunoprecipitations were conducted using an antiserum generated against an EspP C-terminal peptide. The percentage of the passenger domain that was released from the β domain by proteolytic cleavage or surface exposed at each time point is plotted in (B) and (C). The percentage of the radiolabeled protein that remained at each time point is shown in (D). E. Cell membranes isolated from AD202 that produced wild-type EspP or the G1123R mutant were heated at the indicated temperature in SDS-PAGE sample buffer and the free β barrel domain was detected by Western blot.
The overall pattern of the results strongly suggests that lipid facing arginine residues impose a position-dependent energy cost on the integration of the EspP β barrel domain that parallels the cost imposed on the integration of OmpLA. Consistent with the idea that the mutations impact the energetics of insertion, combining two of the mutations described above that did not significantly affect EspP biogenesis (either Y1125R and W1149R or W1149R and Y1175R) produced an additive effect. Like the G1123R mutation, the double mutations slowed the initiation of passenger domain translocation considerably but otherwise did not affect protein biogenesis or sensitivity to degradation (Fig. S8). Furthermore, the finding that alanine substitutions at positions 1123, 1121 and 1119 did not affect EspP assembly (Fig. S9) confirms that our results are attributable to the presence of an unfavorably positioned ionizable residue rather than the loss of the native residue. Interestingly, while arginine mutations located near the middle of both the EspP β barrel domain and OmpLA strongly impaired assembly, the mutations exerted distinct effects on the two proteins. Unlike the OmpLA L208R, A210R and G212R mutants, the EspP G1123R mutant integrated slowly into the OM in degP+ cells in a native conformation and remained immune to degradation. In contrast, the L1121R and I1119R mutants, which contain an ionizable residue located progressively closer to the middle of the β barrel, never integrated into the OM (or integrated into the OM very inefficiently) even in the absence of DegP. Given that the fate of EspP is exquisitely sensitive to subtle changes in the position of the arginine residue, it is much more likely that the two mutations block insertion per se rather than induce gross misfolding.
To further investigate the properties of the EspP mutants, we next wished to determine if the mutations affect the targeting of the protein to the OM. To this end we exploited the observation that interactions between the EspP β barrel domain and cellular factors can be monitored by site-specific photocrosslinking (Ieva et al., 2011). AD202 transformed with a plasmid encoding wild-type EspP or an arginine variant and pDULE-Bpa were subjected to pulse-chase labeling, and the photoactivatable amino acid analog p-benzoylphenylalanine (Bpa) was incorporated at residue 1214 by amber suppression (Farrell et al., 2005). Half of the cells were UV irradiated, and half of the irradiated samples were treated with PK.
Consistent with previous results (Ieva et al., 2011), a ~160 kD polypeptide that contains proEspP covalently linked to BamD was immunoprecipitated with both anti-EspP and anti-BamD antisera from UV irradiated cells that produced the wild-type protein (Fig. 8A, top, lanes 1–4; Fig. S10). A similar crosslinking pattern was observed in cells that produced either the EspP G1123R or the EspP Y1125R/W1149 variant, but the crosslinking product persisted at later time points (Fig. 8A, bottom, lanes 1–4; Fig. S11). The detection of the crosslinking product even at the 0 min time point indicates that the mutant proteins were targeted to the Bam complex at essentially the same rate as wild-type EspP. The wild-type proEspP-BamD crosslinking product was completely sensitive to PK digestion because passenger domain translocation is normally initiated soon after the β barrel domain binds to the Bam complex (Fig. 8A, top, lanes 5–8) (Pavlova et al., 2013). In contrast, a significant fraction of the proEspP G1123R-BamD and proEspP Y1125R/W1149R-BamD crosslinking products was resistant to PK digestion at all time points (Fig. 8A, bottom, lanes 5–8; Fig. S11, lanes 5–8). These findings imply that the mutations do not affect the targeting of EspP to the Bam complex, but rather slow a subsequent step required for the full integration of the β barrel domain into the OM and the initiation of passenger domain translocation.
Fig. 8.

EspP mutants that contain a lipid facing arginine interact stably with the Bam complex. A and B. AD202 transformed with pDULE-Bpa and a derivative of pRI22 encoding wild-type EspP or EspP (G1123R), EspP (L1121R) or EspP (I1119R) with an amber mutation at position 1214 were subjected to pulse-chase labeling after the addition of IPTG. One aliquot of cells was UV-irradiated while an equal aliquot was left untreated. In part (A), PK was added to half of each sample. Immunoprecipitations were then conducted using the indicated antisera. The non-irradiated samples from part (A) are shown in Fig. S9.
Interestingly, the pro form of both EspP L1121R and EspP I1119R also formed persistent crosslinks with BamD (Fig. 8B). This observation implies that although the mutants are slowly degraded, they reach the Bam complex prior to proteolysis. It is unclear if the mutant proteins transiently dissociate from the Bam complex and then bind to DegP or if they are recognized by DegP while still engaged with the Bam complex. In any case, the finding that the G1123R and Y1125R/W1149R variants are immune to degradation strongly suggest that they form a unique protective interaction with the Bam complex or that they adopt a different conformation than the L1121R and I1119R mutants after they are targeted to the OM. Furthermore, given that the crosslinking of the EspP β barrel domain to Bam complex subunits is highly position-dependent (Ieva et al., 2011), the finding that residue 1214 of the L1121R and I1119R mutants was crosslinked specifically to BamD suggests that the mutations did not drastically alter the conformation of the protein.
Lipid-exposed arginine residues in EspP create dominant-negative phenotypes
Because the results of our crosslinking analysis strongly suggested that EspP mutants whose assembly is delayed or impaired form a relatively stable interaction with BamD, we hypothesized that the production of these mutants might impede the binding of other OMPs to the Bam complex and therefore be deleterious. To test this idea, we initially examined the growth of cells transformed with plasmids that encode the mutants under the control of the trc promoter on LB agar. Although production of the mutant proteins did not affect the growth of wild-type cells, we observed strong growth defects in DPR959 (Ricci et al., 2012), a strain that synthesizes a reduced amount of BamA. Unlike cells that contained a plasmid encoding wild-type EspP, cells that contained a plasmid encoding the G1123R, L1121R or I1119R mutant did not grow in the presence of 100 μM IPTG, and cells that contained a plasmid encoding the Y1125R/W1149R mutant exhibited only limited growth (Fig. 9A–B). Furthermore, the low level of expression that results from the leakiness of the trc promoter was sufficient to cause growth defects even in the absence of IPTG: cells that contained a plasmid encoding the G1123R mutant formed slightly mucoid colonies and cells that contained a plasmid encoding the L1121R or I1119R mutant formed small, abnormal colonies. To further explore the strongest phenotypes, we next introduced the L1121R or I1119R mutation into a plasmid that expresses only the EspP β barrel domain and a short fragment of the passenger domain (EspPΔ5) under the control of the tightly regulated rhaB2 promoter (Cardona and Valvano, 2005). Due to a mutation in the Shine-Dalgarno sequence, genes cloned into this plasmid are expressed at only a moderate level. Overnight cultures of AD202 harboring the plasmids were diluted and grown in LB after the addition of rhamnose. Although cells that expressed the mutant proteins initially grew as well as control cells, strong growth defects appeared after ~10 generations (Fig. 9C). Similar delayed growth defects have been observed following the depletion of BamA and BamD (Wu et al., 2005; Malinverni et al., 2006) and are presumably triggered when the concentration of OMPs falls below a critical level and the integrity of the OM is compromised. The finding that constitutive synthesis of the EspPΔ5 mutants in overnight cultures led to a dramatic reduction in the steady-state level of several endogenous OMPs confirmed that they inhibit the activity of the Bam complex (Fig. S12).
Fig. 9.

EspP mutants that contain a lipid facing arginine near the middle of the β barrel inhibit Bam complex function. A and B. DPR959 transformed with pRLS5 or a pRLS5 derivative encoding the indicated EspP mutant were incubated at 37° C on LB agar plates containing ampicillin (100 μg/ml) and either no IPTG or 100 μM IPTG. C. AD202 transformed with pSCrhaB2 (vector), pJH207 (PrhaB2-espPΔ5) or a pJH207 derivative encoding the indicated EspPΔ5 mutant were inoculated into LB containing trimethoprim (50 μg/ml) at OD550=0.0001. The growth of each culture at 37° C was monitored at OD550. D. AD202 transformed with pRLS5 and a derivative of pJH207 encoding wild-type EspPΔ5 or the indicated EspPΔ5 mutant were subject to pulse-chase labeling after the addition rhamnose and IPTG. Half of the cells were treated with PK, and immunoprecipitations were conducted using an antiserum generated against an EspP N-terminal peptide.
To obtain direct evidence that the L1121R and I1119R mutants hinder OM protein assembly, we examined the effect of expressing the mutant forms of EspPΔ5 on the biogenesis of full-length EspP. AD202 transformed with a plasmid encoding EspP under the control of the trc promoter and a plasmid encoding an EspPΔ5 derivative under the control of the rhaB2 promoter were subject to pulse-chase labeling 20 min after the addition of rhamnose. Half of the cells were treated with PK, and immunoprecipitations were conducted with an antiserum against an N-terminal EspP peptide that recognizes only the full-length protein. Consistent with our hypothesis, the EspP passenger domain was efficiently secreted and released from the β barrel domain in cells that produced wild-type EspPΔ5, but almost no exposure of the passenger domain or proteolytic maturation was observed in cells that produced one of the EspPΔ5 mutants (Fig. 9D). Presumably because the targeting of EspP to the OM was impaired, the protein was slowly degraded.
Taken together, the results of our phenotypic analysis corroborate the conclusion that the EspP G1123R, L1121R, I1119R and Y1125R/W1149R mutants form long-lived interactions with the Bam complex. The observation that the L1121R and I1119R mutants are especially deleterious implies that DegP degrades them too slowly to compensate for their inability to progress through the assembly pathway and integrate into the OM.
DISCUSSION
In this study we demonstrate that the sequence of bacterial OM proteins is constrained not only by their need to insert into the OM in a native state, but also by functional properties of the assembly and quality control machineries. We found that OmpLA mutants that contain specific lipid-facing arginine residues are slowly degraded by DegP. The mutations did not seem to strongly impair the folding of the protein because slow membrane integration was observed in a degP- strain. The membrane insertion was blocked, however, under conditions that favored the binding of client proteins to DegP over their degradation. Although we have not been able to determine if the OmpLA mutants interact with the Bam complex, these results suggest that DegP can recognize OMPs based simply on their persistence in the periplasmic space rather than on the detection of a terminally misfolded state and that OMPs are therefore under a strong selective pressure to integrate into the membrane within a specific time window. The results also suggest that DegP degrades substrates in a stochastic fashion that enables them to undergo repeated cycles of binding and release. This property of DegP presumably evolved to tolerate variation in the rate of OMP assembly and to avoid the premature degradation of proteins that are on pathway. Examination of a second protein, EspP, demonstrated how the Bam complex also constrains the sequence of OM proteins. Some lipid-facing arginine residues were highly deleterious because they delayed or blocked EspP assembly but did not affect targeting or the ability of the protein to form a stable interaction with the Bam complex until it was integrated into the OM or degraded by the slow acting quality control system. In essence, OMPs need to assemble on a time scale that enables them to bypass periplasmic quality control systems without significantly impeding the activity of the Bam complex.
Consistent with the results of in vitro studies (Hessa et al., 2007; Moon and Fleming, 2011), our results suggest that the presence of a lipid-facing ionizable residue imposes a position-dependent energy cost on the integration of a protein into a biological membrane. Single lipid-facing arginine residues introduced near the periplasmic or extracellular surface of either the OmpLA or EspP β barrel had no effect or a minimal effect on assembly whereas double mutations or single mutations located closer to the middle of the β barrel exerted a progressively greater effect. Although it is conceivable that the I1119R and L1121R mutations affect EspP folding, the observed correlation between the location of lipid-facing arginine residues and assembly defects strongly suggests that the mutations simply create an energy barrier that is too high to be overcome even when the protein is stabilized (and a factor that might compete with the Bam complex for binding is eliminated) in a degP- strain. It is unclear why the specific properties of OmpLA and EspP mutants that contain an arginine near the middle of the β barrel differed. Nevertheless, it is striking that the introduction of a single arginine residue onto a surface that has ~50 hydrophobic residues exerts such a strong effect on assembly. Consistent with biophysical studies (Surrey and Jahnig, 1995; Kleinschmidt and Tamm, 1996; Danoff and Fleming, 2017), the results raise the possibility that β barrels are not inserted into the OM in a single step as fully folded entities, but are rather inserted in a segmental or stepwise fashion that magnifies the effect of a single surface-exposed ionizable residue.
While lipid-exposed arginine residues located near the middle of the OmpLA or EspP β barrel create an especially strong assembly defect, there is presumably an evolutionary selection against many other residues that create similar but less dramatic kinetic effects. We have found that in addition to residues that impose an energetic barrier to membrane insertion, residues that perturb folding kinetics even slightly are disfavored. The residue that corresponds to G1066 in EspP, for example, is one of the most highly conserved residues in the autotransporter superfamily presumably because the introduction of even a subtle alanine mutation at this position delays the membrane integration of the β barrel by ~1 min by minimally altering its folding (Pavlova et al., 2013). Otherwise, the G1066A mutation exerts no discernable effect on EspP assembly.
Interestingly, our analysis of the EspP arginine mutants also revealed two distinct types of interactions between the Bam complex and unintegrated client proteins. In one type of interaction (illustrated by the I1119R and L1121R mutants) the client protein can form a prolonged interaction with the Bam complex, but remains susceptible to degradation by DegP. In the second type of interaction (illustrated by the G1123R mutant and double arginine mutants), the client protein also forms a prolonged interaction with the Bam complex but is completely protected from degradation and is slowly inserted into the OM in a native conformation. The stability of the protein indicates that it is resistant to degradation not only by DegP, but also by other proteases that have recently been shown to degrade assembly-defective mutants of another OMP (LptD) that have engaged the Bam complex (Soltes et al., 2017). Although it is unclear whether the two binding modes reflect differences in the conformation of the Bam complex or the substrate, it is tempting to speculate that they represent two different stages of the assembly process. At the first stage client proteins might form an initial interaction with the Bam complex but only become “committed” to insert into the OM at the second stage. At this second stage the Bam complex would catalyze the membrane insertion of client proteins, but energetic barriers created by specific mutations might slow the reaction.
Finally, although many proteins are subject to strong selective pressures to fold to a global free-energy minimum quickly (Levinthal, 1968; Anfinsen, 1973; Debes et al., 2013), the EspP G1123R and double arginine mutants are the first examples of variants that are disfavored because they directly inhibit an essential cellular process by reaching the native state slowly. As our results suggest, these mutants as well as the EspP I1119R and L1121R mutants should provide a useful tool for modulating the activity of the Bam complex and examining the consequences of acutely perturbing OMP assembly. The EspP I1119R and L1121R mutants may be especially useful because they do not integrate (or integrate poorly) into the OM and therefore can serve as dominant-negative inhibitors whose production blocks OMP assembly without rapidly affecting the composition of the OM. Furthermore, by strongly inhibiting the activity of the Bam complex within minutes, the expression of the arginine mutants overcomes a major drawback of BamA and BamD depletions. Effective depletion requires growing cultures for long periods of time, during which cells might undergo dramatic physiological changes that cloud the interpretation of experimental results.
EXPERIMENTAL PROCEDURES
Bacterial strains, growth conditions and antisera
The E. coli K-12 strains used in these experiments were MC4100 [araD139 Δ(argF-lac)169 λ-e14-flhD5301 Δ(fruK-yeiR)725(fruA25) relA1 rpsL150 rbsR22 Δ(fimB-fimE)632(::IS1) deoC1], AD202 (MC4100 ompT::kan) (Akiyama and Ito, 1990), JMR352 (MC4100 degP::Tn10) (Rizzitello et al., 2001), DPR959 (MC4100 arar/ bamA101) (Ricci et al., 2012) and HDB155 (JMR352 ompT::kan). Cultures were grown at 37° C in M9 medium containing 0.2% glycerol and all of the L-amino acids except methionine and cysteine (40 μg/ml) unless otherwise noted. Ampicillin (100 μg/ml), tetracycline (5 μg/ml) and trimethoprim (50 μg/ml) were added as needed. Rabbit polyclonal antisera generated against an EspP N-terminal peptide, EspP, BamA and BamD C-terminal peptides, an OmpC peptide, and Ffh have been described (Poritz et al., 1990; Szabady et al., 2005; Pavlova et al., 2013; Yap and Bernstein, 2013). A polyclonal antiserum against OmpA was obtained from P.C. Tai. A rabbit polyclonal antiserum was generated against the OmpLA extracellular loop peptide NH2-CQTSDLNKEAIASYDWAE-COOH.
Plasmid construction and site-directed mutagenesis
Plasmids pRLS5, which encodes full-length espP under the control of the trc promoter, pRI22, which encodes His-tagged espP under the control of the lac promoter, and pDULE-Bpa have been described (Farrell et al., 2005; Szabady et al., 2005; Ieva and Bernstein, 2009). To make plasmid pJH207, which encodes espPΔ5 under the control of the rhaB2 promoter, plasmid pJH110 (Pavlova et al., 2013) was digested with Nde I and Hind III and the ~1.0 kB fragment was cloned into the cognate sites of pSCrhaB2 (Cardona and Valvano, 2005). The E. coli pldA gene was amplified by PCR using the primers 5′-GAATTCATGCGGACTCTGCAGGGCTGG-3′ and 5′-AAGCTTGCATTCTTAAAACAAATCGTTTAGCATAACCCCCAC-3′ and genomic DNA from strain MG1655 and cloned into the pCR2.1-TOPO vector using the TOPO TA cloning kit (Invitrogen). The resulting plasmid was then digested with EcoR I and Hind III and the fragment containing the pldA gene was ligated into pTRC99a digested with the same enzymes to create pMDG2. Missense and amber mutations were introduced into espP by site-directed mutagenesis using the QuikChange II Site-Directed Mutagenesis Kit (Agilent) and mutations were introduced into pldA using a similar PCR-based method.
Pulse-chase labeling and photocrosslinking
Overnight cultures were washed and diluted into fresh M9 at OD550=0.02. When the cultures reached OD550=0.2, EspP or OmpLA synthesis was induced by the addition of 10 μM IPTG (for experiments in which cells contained pRLS5 or one of its derivatives) 100 μM IPTG (for experiments in which cells contained pMDG2 or one of its derivatives) or 200 μM IPTG (for experiments in which cells contained a pRl22 derivative). After 30 min pulse-chase labeling and photocrosslinking were performed essentially as described (Ieva and Bernstein, 2009). Radiolabeled cells were pipetted over ice and collected by centrifugation. In experiments on EspP, cells were resuspended in cold M9 salts, and half of each sample was treated with PK as described (Ieva and Bernstein, 2009). In experiments on OmpLA, cells were generally resuspended in spheroplast buffer (33 mM Tris pH 8.0, 40% sucrose) and permeabilized by incubating them at 0° C with 100 μg/ml lysozyme and 2 mM EDTA for 20 min. Half of each sample was then treated with PK. DDM (1.0%) was added prior to PK treatment where noted. Proteins in all samples were collected by TCA precipitation. Immunoprecipitations were performed as described (Ieva et al., 2008) and proteins were resolved by SDS-PAGE on 8–16% minigels (Life Technologies). Radioactive proteins were detected using a Fuji BAS-2500 phosphorimager. Percent surface exposure and percent passenger domain cleavage were calculated as described (Peterson et al., 2010). The percent protein remaining at N min was defined as [(proEspP + free β barrel domain)/(proEspP + free β barrel domain at 2 min)] x 100. The signal was normalized to take the difference in the number of radioactive amino acids in the two forms of the protein into account.
Dual expression of espPΔ5 and full-length espP
Overnight cultures of AD202 transformed with pRLS5 and a derivative of pJH207 were washed and diluted into fresh M9 at OD550=0.02. When the cultures reached OD550=0.2, EspPΔ5 synthesis was induced by the addition of 0.2% rhamnose. After 15 min, full-length EspP synthesis was then induced by the addition of 5 μM IPTG. After another 5 min, pulse-chase labeling and PK treatment were performed as described above.
Cell fractionation
Cultures were grown and radiolabeled as described above. At each time point 10 ml aliquots were pipetted over ice. Cells were then collected by centrifugation (3000 x g, 10 min, 4°C), resuspended in 1 mL TBS (25 mM Tris pH 7.4, 137 mM NaCl, 3 mM KCl) and disrupted using a Misonix 3000 sonicator (microtip, level 4). Unbroken cells were pelleted (3000 x g, 5 min, 4°C), and the supernatant was defined as the “total” cell extract. Subsequently 900 μl of each cell extract was centrifuged in a Beckman TLA100.2 rotor (100,000 x g, 30 min, 4°C). The supernatant was removed and defined as the “soluble” fraction. The pellet (“membrane” fraction) was then resuspended in 600 μl TBS. Equal portions of the membrane fraction were left untreated, treated with PK, or treated with DDM and PK as described above and then TCA precipitated.
Heat modifiability assays
Cultures (50 ml) were grown in M9 medium. When the cultures reached OD550=0.2 the synthesis of EspP or OmpLA was induced by the addition of IPTG. After 30 min cells were collected by centrifugation and resuspended in TBS at 10 OD units/ml. A membrane fraction was then isolated as described above. The membrane pellet was resuspended in TBS at 40 OD units/ml. Aliquots were then mixed with 2x Laemmli sample buffer and heated to a temperature between 25° C and 90° C for 15 min. Either the free EspP β barrel domain or full-length OmpLA was detected by Western blot.
Western blotting
Proteins were transferred to nitrocellulose and probed with an appropriate primary antibody. Antibody-antigen complexes were then detected using IRDye 680LT-labeled goat anti-rabbit IgG and an Odyssey near infrared imaging instrument (Licor).
Supplementary Material
Fig. 5.

OmpLA arginine mutants do not acquire PK resistance in wild-type E. coli at low temperature. MC4100 (A) or JMR352 (B) transformed with pMDG2 or a pMDG2 derivative encoding the indicated OmpLA mutant were shifted to 28° C and subjected to pulse-chase labeling after the addition of IPTG. The OM was permeabilized and half of each sample was treated with PK. Immunoprecipitations were then conducted using an anti-OmpLA antiserum.
Acknowledgments
We would like to thank Tom Silhavy for kindly providing E. coli strains. This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health Grants R01 GM079440 (to KGF) and T32 GM008403 (for AMP), and National Science Foundation Grant MCB1412108 (to KGF). AMP is a recipient of a National Science Foundation Graduate Research Fellowship (DGE 1232825).
Footnotes
AUTHOR CONTRIBUTIONS
AMP, KGF and HDB conceived and designed the study; JHP, AMP and HDB performed the experiments; all authors analyzed and interpreted the data; AMP, KGF and HDB wrote the manuscript.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
References
- Akiyama Y, Ito K. SecY protein, a membrane-embedded secretion factor of E. coli, is cleaved by the OmpT protease in vitro. Biochem Biophys Res Commun. 1990;167:711–715. doi: 10.1016/0006-291x(90)92083-c. [DOI] [PubMed] [Google Scholar]
- Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- Arunmanee W, Pathania M, Solovyova AS, Le Brun AP, Ridley H, Baslé A, van den Berg B, Lakey JH. Gram-negative trimeric porins have specific LPS binding sites that are essential for porin biogenesis. Proc Natl Acad Sci USA. 2016;113:E5034–5043. doi: 10.1073/pnas.1602382113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakelar J, Buchanan SK, Noinaj N. The structure of the β-barrel assembly machinery complex. Science. 2016;351:180–186. doi: 10.1126/science.aad3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard TJ, Dautin N, Lukacik P, Bernstein HD, Buchanan SK. Autotransporter structure reveals intra-barrel cleavage followed by conformational changes. Nat Struct Mol Biol. 2007;14:1214–1220. doi: 10.1038/nsmb1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard TJ, Gumbart J, Peterson JH, Noinaj N, Easley NC, Dautin N, et al. Molecular basis for the activation of a catalytic asparagine residue in a self-cleaving bacterial autotransporter. J Mol Biol. 2012;415:128–142. doi: 10.1016/j.jmb.2011.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell. 2010;40:238–252. doi: 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Cardano ST, Valvano MA. An expression vector containing a rhamnose-inducible promoter provides tightly regulated gene expression in Burkholderia cenocepacia. Plasmid. 2005;54:219–228. doi: 10.1016/j.plasmid.2005.03.004. [DOI] [PubMed] [Google Scholar]
- CastilloKeller M, Misra R. Protease-deficient DegP suppresses lethal effects of a mutant OmpC protein by its capture. J Bacteriol. 2003;185:148–154. doi: 10.1128/JB.185.1.148-154.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain AK, Bowie JU. Asymmetric amino acid compositions of transmembrane β-strands. Protein Sci. 2004;13:2270–2274. doi: 10.1110/ps.04777304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlson ES, Werner JN, Misra R. Differential effects of yfgL mutation on Escherichia coli outer membrane proteins and lipopolysaccharide. J Bacteriol. 2006;188:7188–7194. doi: 10.1128/JB.00571-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Henning U. A periplasmic protein (Skp) of Escherichia coli selectively binds a class of outer membrane proteins. Mol Microbiol. 1996;19:1287–1294. doi: 10.1111/j.1365-2958.1996.tb02473.x. [DOI] [PubMed] [Google Scholar]
- Costello SM, Plummer AM, Fleming PJ, Fleming KG. Dynamic periplasmic chaperone reservoir facilitates biogenesis of outer membrane proteins. Proc Natl Acad Sci USA. 2016;113:E4794–E4800. doi: 10.1073/pnas.1601002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danoff EJ, Fleming KG. Novel kinetic intermediates populated along the folding pathway of the transmembrane β-barrel OmpA. Biochemistry. 2017;56:47–60. doi: 10.1021/acs.biochem.6b00809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dautin N, Barnard TJ, Anderson DE, Bernstein HD. Cleavage of a bacterial autotransporter by an evolutionarily convergent autocatalytic mechanism. EMBO J. 2007;26:1942–1952. doi: 10.1038/sj.emboj.7601638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dautin N, Bernstein HD. Protein secretion in Gram-negative bacteria via the autotransporter pathway. Annu Rev Microbiol. 2007;61:89–112. doi: 10.1146/annurev.micro.61.080706.093233. [DOI] [PubMed] [Google Scholar]
- Debes C, Wang M, Caetano-Anolles G, Grater F. Evolutionary optimization of protein folding. PLoS Comput Biol. 2013;9:e1002861. doi: 10.1371/journal.pcbi.1002861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echenique-Rivera H, Brunelli B, Scarselli M, Taddei AR, Pizza M, Arico B, Serruto D. A naturally occurring single-residue mutation in the translocator domain of Neisseria meningitidis NhhA affects trimerization, surface localization, and adhesive capabilties. Infect Immun. 2011;79:4308–4321. doi: 10.1128/IAI.00198-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan E, Chauhan N, Udatha DB, Leo JC, Linke D. Type V secretion systems in bacteria. Microbiol Spectr. 2016;4 doi: 10.1128/microbiolspec.VMBF-0009-2015. [DOI] [PubMed] [Google Scholar]
- Farrell IS, Toroney R, Hazen JL, Mehl RA, Chin JW. Photo-cross-linking interacting proteins with a genetically encoded benzophenone. Nat Methods. 2005;2:377–384. doi: 10.1038/nmeth0505-377. [DOI] [PubMed] [Google Scholar]
- Fleming KG. A combined kinetic push and thermodynamic pull as driving forces for outer membrane protein sorting and folding in bacteria. Philos Trans R Soc B. 2015;370:20150026. doi: 10.1098/rstb.2015.0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X, Wang R, Ma J, Liu Y, Ezemaduka AN, Chen PR, Fu X, Chang Z. DegP primarily functions as a protease for the biogenesis of β-barrel outer membrane proteins in the Gram-negative bacterium Escherichia coli. FEBS J. 2014;281:1226–1240. doi: 10.1111/febs.12701. [DOI] [PubMed] [Google Scholar]
- Gessmann D, Chung YH, Danoff EJ, Plummer AM, Sandlin CW, Zaccai NR, Fleming KG. Outer membrane β-barrel protein folding is physically controlled by periplasmic lipid head groups and BamA. Proc Natl Acad Sci. 2014;111:5878–5883. doi: 10.1073/pnas.1322473111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Li H, Dong H, Zeng Y, Zhang Z, Paterson NG, et al. Structural basis of outer membrane protein insertion by the BAM complex. Nature. 2016;531:64–69. doi: 10.1038/nature17199. [DOI] [PubMed] [Google Scholar]
- Hagan CL, Kim S, Kahne D. Reconstitution of outer membrane protein assembly from purified components. Science. 2010;328:890–892. doi: 10.1126/science.1188919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Zheng J, Wang Y, Wang X, Liu Y, Sun C, et al. Structure of the BAM complex and its implications for biogenesis of outer-membrane proteins. Nat Struct Mol Biol. 2016;23:192–196. doi: 10.1038/nsmb.3181. [DOI] [PubMed] [Google Scholar]
- Harms MJ, Schlessman JL, Sue GR, García-Moreno B. Arginine residues at internal positions in a protein are always charged. Proc Natl Acad Sci. 2011;108:18954–18959. doi: 10.1073/pnas.1104808108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessa T, Meindl-Beinker NM, Bernsel A, Kim H, Sato Y, Lerch-Bader M, et al. Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature. 2007;450:1026–1030. doi: 10.1038/nature06387. [DOI] [PubMed] [Google Scholar]
- Ieva R, Bernstein HD. Interaction of an autotransporter passenger domain with BamA during its translocation across the bacterial outer membrane. Proc Natl Acad Sci USA. 2009;106:19120–19125. doi: 10.1073/pnas.0907912106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieva R, Skillman KM, Bernstein HD. Incorporation of a polypeptide segment into the β-domain pore during the assembly of a bacterial autotransporter. Mol Microbiol. 2008;67:188–201. doi: 10.1111/j.1365-2958.2007.06048.x. [DOI] [PubMed] [Google Scholar]
- Ieva R, Tian P, Peterson JH, Bernstein HD. Sequential and spatially restricted interactions of assembly factors with an autotransporter β domain. Proc Natl Acad Sci USA. 2011;108:E383–E391. doi: 10.1073/pnas.1103827108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackups R, Liang J. Interstrand pairing patterns in beta-barrel membrane proteins: the positive-outside rule, aromatic rescue, and strand registration prediction. J Mol Biol. 2005;354:979–993. doi: 10.1016/j.jmb.2005.09.094. [DOI] [PubMed] [Google Scholar]
- Jimenez-Morales D, Liang J. Pattern of amino acid substitutions in transmembrane domains of β-barrel membrane proteins for detecting remote homologs in bacteria and mitochondria. PLoS One. 2011;6:e26400. doi: 10.1371/journal.pone.0026400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker M, Bensingi RN, Clark PL. Vectorial transport and folding of an autotransporter virulence protein during outer membrane secretion. Mol Microbiol. 2009;71:1323–1332. doi: 10.1111/j.1365-2958.2009.06607.x. [DOI] [PubMed] [Google Scholar]
- Kim S, Malinverni JC, Sliz P, Silhavy TJ, Harrison SC, Kahne D. Structure and function of an essential component of the outer membrane protein assembly machine. Science. 2007;317:961–964. doi: 10.1126/science.1143993. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt JH, Tamm LK. Folding intermediates of a beta-barrel membrane protein. Kinetic evidence for a multi-step membrane insertion mechanism. Biochemistry. 1996;35:12993–13000. doi: 10.1021/bi961478b. [DOI] [PubMed] [Google Scholar]
- Krojer T, Sawa J, Schäfer E, Saibil HR, Ehrmann M, Clausen T. Structural basis for the regulated protease and chaperone function of DegP. Nature. 2008;453:885–890. doi: 10.1038/nature07004. [DOI] [PubMed] [Google Scholar]
- Levinthal C. Are there pathways for protein folding? J Chim Phys. 1968;65:44–45. [Google Scholar]
- Lipinska B, Fayet O, Baird L, Georgopoulos C. Identification, characterization, and mapping of the Escherichia coli htrA gene, whose product is essential for bacterial growth only at elevated temperatures. J Bacteriol. 1989;171:1574–1584. doi: 10.1128/jb.171.3.1574-1584.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu ZX, Zhao XS. Periplasmic quality control in biogenesis of outer membrane proteins. Biochem Soc Trans. 2015;43:133–138. doi: 10.1042/BST20140217. [DOI] [PubMed] [Google Scholar]
- Malinverni JC, Werner J, Kim S, Sklar JG, Kahne D, Misra R, Silhavy TJ. YfiO stabilizes the YaeT complex and is essential for outer membrane protein assembly in Escherichia coli. Mol Microbiol. 2006;61:151–164. doi: 10.1111/j.1365-2958.2006.05211.x. [DOI] [PubMed] [Google Scholar]
- Misra R, Peterson A, Ferenci T, Silhavy TJ. A genetic approach for analyzing the pathway of LamB assembly into the outer membrane of Escherichia coli. J Biol Chem. 1991;266:13592–13597. [PubMed] [Google Scholar]
- Moon CP, Fleming KG. Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers. Proc Natl Acad Sci USA. 2011;108:10174–10177. doi: 10.1073/pnas.1103979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita S, Masui C, Suzuki T, Dohmae N, Akiyama Y. Protease homolog BepA (YfgC) promotes assembly and degradation of β-barrel membrane proteins in Escherichia coli. Proc Natl Acad Sci USA. 2013;110:E3612–E3621. doi: 10.1073/pnas.1312012110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noinaj N, Kuszak AJ, Gumbart JC, Lukacik P, Chang H, Easley NC, Lithgow T, Buchanan SK. Structural insight into the biogenesis of β-barrel membrane proteins. Nature. 2013;501:385–390. doi: 10.1038/nature12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noinaj N, Kuszak AJ, Balusek C, Gumbart JC, Buchanan SK. Lateral opening and exit pore formation are required for BamA function. Structure. 2014;22:1055–1062. doi: 10.1016/j.str.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova O, Peterson JH, Ieva R, Bernstein HD. Mechanistic link between β barrel assenbly and the initiation of autotransporter secretion. Proc Natl Acad Sci USA. 2013;110:E938–E947. doi: 10.1073/pnas.1219076110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JH, Tian P, Ieva R, Dautin N, Bernstein HD. Secretion of a bacterial virulence factor is driven by the folding of a C-terminal segment. Proc Natl Acad Sci USA. 2010;107:17739–17744. doi: 10.1073/pnas.1009491107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer AM, Fleming KG. BamA alone accelerates outer membrane protein folding in vitro through a catalytic mechanism. Biochemistry. 2015;54:6009–6011. doi: 10.1021/acs.biochem.5b00950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poritz MA, Bernstein HD, Strub K, Zopf D, Wilhelm H, Walter P. An E. coli ribonucleoprotein containing 4.5S RNA resembles mammalian signal recognition particle. Science. 1990;250:1111–1117. doi: 10.1126/science.1701272. [DOI] [PubMed] [Google Scholar]
- Ricci DP, Hagan CL, Kahne D, Silhavy TJ. Activation of the Escherichia coli β-barrel assembly machine (Bam) is required for essential components to interact porperly with substrate. Proc Natl Acad Sci USA. 2012;109:3487–3491. doi: 10.1073/pnas.1201362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzitello AE, Harper JR, Silhavy TJ. Genetic evidence for parallel pathways of chaperone activity in the periplasm of Escherichia coli. J Bacteriol. 2001;183:6794–6800. doi: 10.1128/JB.183.23.6794-6800.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouviere PE, Gross CA. SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev. 1996;10:3170–3182. doi: 10.1101/gad.10.24.3170. [DOI] [PubMed] [Google Scholar]
- Sawa J, Heuck A, Ehrmann, Clausen T. Molecular transformers in the cell: lessons learned from the DegP protease-chaperone. Curr Opin Struct Biol. 2010;20:253–258. doi: 10.1016/j.sbi.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Sklar JG, Wu T, Gronenberg LS, Malinverni JC, Kahne D, Silhavy TJ. Lipoprotein SmpA is a component of the YaeT complex that assembles outer membrane proteins in Escherichia coli. Proc Natl Acad Sci USA. 2007;104:6400–6405. doi: 10.1073/pnas.0701579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slusky JSG, Dunbrack RL., Jr Charge asymmetry in the proteins of the outer membrane. Bioinformatics. 2013;29:2122–2128. doi: 10.1093/bioinformatics/btt355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder HJ, Ubarretxena-Belandia I, Blaauw M, Kalk KH, Verheij HM, Egmond MR, Dekker N, Dijkstra BW. Structural evidence for dimerization-regulated activation of an integral membrane phospholipase. Nature. 1999;401:717–721. doi: 10.1038/44890. [DOI] [PubMed] [Google Scholar]
- Soltes GR, Martin NR, Park E, Sutterlin HA, Silhavy TJ. Distincitve roles for periplasmic proteases in the maintenance of essential outer membrane protein assembly. J Bacteriol. 2017 doi: 10.1128/JB.00418-17. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiess C, Beil A, Ehrmann M. A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell. 1999;97:339–347. doi: 10.1016/s0092-8674(00)80743-6. [DOI] [PubMed] [Google Scholar]
- Strauch KL, Johnson K, Beckwith J. Characterization of degP, a gene required for proteolysis in the cell envelope and essential for growth of Escherichia coli at high temperature. J Bacteriol. 1989;171:2689–2696. doi: 10.1128/jb.171.5.2689-2696.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surrey T, Jähnig F. Kinetics of folding and membrane insertion of a beta-barrel membrane protein. J Biol Chem. 1995;270:28199–28203. doi: 10.1074/jbc.270.47.28199. [DOI] [PubMed] [Google Scholar]
- Szabady RL, Peterson JH, Skillman KM, Bernstein HD. An unusual signal peptide facilitates late steps in the biogenesis of a bacterial autotransporter. Proc Natl Acad Sci USA. 2005;102:221–226. doi: 10.1073/pnas.0406055102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmschneider MB, Sansom MS. Amino acid distributions in integral membrane protein structures. Biochim Biophys Acta. 2001;1512:1–14. doi: 10.1016/s0005-2736(01)00299-1. [DOI] [PubMed] [Google Scholar]
- Voulhoux R, Bos MP, Geurtsen J, Mols M, Tommassen J. Role of a highly conserved bacterial protein in outer membrane protein assembly. Science. 2003;299:262–266. doi: 10.1126/science.1078973. [DOI] [PubMed] [Google Scholar]
- Walton TA, Sandoval CM, Fowler CA, Pardi A, Sousa MC. The cavity-chaperone Skp protects its substrate from aggregation but allows independent folding of substrate domains. Proc Natl Acad Sci. 2009;106:1772–1777. doi: 10.1073/pnas.0809275106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb CT, Heinz E, Lithgow T. Evolution of the b-barrel assembly machinery. Trends Microbiol. 2012;20:612–620. doi: 10.1016/j.tim.2012.08.006. [DOI] [PubMed] [Google Scholar]
- Weski J, Ehrmann M. Genetics analysis of 15 protein folding factors and proteases of the Escherichia coli cell envelope. J Bacteriol. 2012;194:3225–3233. doi: 10.1128/JB.00221-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SH, von Heijne G. How translocons select transmembrane helices. Annu Rev Biophys. 2008;37:23–42. doi: 10.1146/annurev.biophys.37.032807.125904. [DOI] [PubMed] [Google Scholar]
- Wickner S, Maurizi MR, Gottesman S. Posttranslational quality control: folding, refolding, and degrading proteins. Science. 1999;286:1888–1893. doi: 10.1126/science.286.5446.1888. [DOI] [PubMed] [Google Scholar]
- Wimley W, White S. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, Kahne D. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell. 2005;121:235–245. doi: 10.1016/j.cell.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Wzorek JS, Lee J, Tomasek D, Hagan CL, Kahne DE. Membrane integration of an essential β-barrel protein prerequires burial of an extracellular loop. Proc Natl Acad Sci USA. 2017;114:2598–2603. doi: 10.1073/pnas.1616576114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap MN, Bernstein HD. Mutations in the Escherichia coli ribosomal protein L22 selectively suppress the expression of a secreted bacterial virulence factor. J Bacteriol. 2013;195:2991–2999. doi: 10.1128/JB.00211-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.