

Abstract
West Nile virus (WNV) has had a major public health impact since its emergence in the Western Hemisphere; in 2012, nearly 3000 cases of WN neuroinvasive disease were identified in the United States. The underlying mechanisms of WN neurologic disease can only be studied to a limited extent in patients, but can be investigated in much greater detail in animal models. In this paper, we describe how we and others have employed a variety of electrophysiological and neurological techniques to study experimental WNV infections in hamsters and mice. The methods have included electrophysiological motor unit number estimation; optogenetic photoactivation of the spinal cord and electromyography; plethysmography; measurement of heart rate variability as an indication of autonomic nervous system dysfunction; and an assessment of spatial memory loss using the Morris water maze. These techniques provide a more refined assessment of disease manifestations in rodents than traditional measurements of weight loss and mortality, and should make it possible to identify targets for therapeutic intervention and to directly assess the effects of novel treatments.
Introduction
Some signs and symptoms in human subjects that may be tentatively associated with neurological involvement or that are clearly associated with West Nile neurological disease (WNND) can also be observed in mice or hamsters (Table 1), the two rodent species suitable for WNV investigations. These rodent models have been valuable for understanding the mechanisms of neurological signs and symptoms in human subjects and how they might be managed or treated.
Table 1.
Comparison of West Nile virus human neurological signs and symptomsa to those seen in mice and hamstersb (Adapted from (Morrey et al., 2004b)).
Not all subjects show all symptoms or signs.
Alignment with human symptoms were subjective and may not correlate exactly.
ND-Not determined
Most human WNV cases are subclinical, or develop a short-term febrile illness, which is referred to as WN fever (Bode et al., 2006; Hayes et al., 2005; Sejvar, 2007). Fever is often recognized to occur during viremia, but fever is also associated with generalized inflammation of the meninges. Interestingly, WNV-infected hamsters monitored continuously with radiotelemetry do not have a fever during the viremic phase, but can have a temperature spike at days 5-6 when viral induced meningitis is observed (Siddharthan et al., 2009; Wang et al., 2013a) (Table 1). These data suggest that WN fever in some cases might reflect neurological involvement, and not just the viremic phase. Having an animal model for WNV fever might provide an opportunity to investigate the cause of WNV-induced fever and the neurological implications in human subjects.
A small subset of WNV patients develops more serious neurologic deficits (Table 1). Patients can present with meningitis symptoms, which include neck stiffness and light sensitivity (Bouffard et al., 2004; Omalu et al., 2003; Sampson et al., 2000; Sejvar et al., 2003a; Steele et al., 2000; Weiss et al., 2001). Inflammation of the meninges can be observed in the rodent models (Ben-Nathan et al., 1995; Camenga et al., 1974; Hunsperger and Roehrig, 2006), which suggests that they also get disease signs of meningitis, but efforts to observe these signs have not been undertaken, except for perhaps the detection of fever associated with CNS infection as described above (Wang et al., 2013a).
Encephalitis as an infection of the brain is a more serious development of WNND (Table 1). WNV-infected neuronal cells have been observed postmortem in the brainstem, which contains many vital nerve connections for motor and sensory systems from the main part of the brain to the rest of the body, and in the cerebellum involved in motor control in addition some cognitive functions. In rodent models, tissues collected at the time of death do not typically contain abundant WNV-infected cells due to prior clearance by the immune system, so it is not possible to understand viral tropism and pathogenesis without sampling tissues throughout the course of disease development (Siddharthan et al., 2009; Tesh et al., 2005). Herein lies the value of rodent models in that they have been used in temporal studies to determine that the virus can infect many areas of the brain and spinal cord and subsequently affect neurological functions.
Some WNV patients complain of confusion or altered mental status (Carson et al., 2006) (Table 1). In a retrospective study with 54 persons about a year and a half after acute illness, the study cohorts scored below the 15 percentile on some cognitive tests as compared to normative controls. (Sejvar et al., 2008). Further human studies should be done to confirm these results, but rodent models could also help to identify neurological mechanisms of cognitive deficits. The greatest density of lesions in WNV-infected hamsters is observed in the area of the prefrontal cortex (PFC) (Siddharthan et al., 2009), which plays a critical role in cognition and executive functions in humans and rodents. Extensive studies in the rat model have revealed that sub-regions of the PFC control distinct components of cognitive executive function (Chudasama and Robbins, 2006; Dalley et al., 2004). Additional WNV-induced lesions are also observed in the limbic system particularly with the hippocampus (Hunsperger and Roehrig, 2006; Siddharthan et al., 2009) and thalamus (Ali et al., 2005; Davis et al., 2006). Lesions in these anatomical regions might affect cognitive function via disturbance of connections between the PFC and the limbic system. Behavioral assays in rodents coupled with virological and histological assays could elucidate the effect that WNV might have on cognitive and executive functions.
Some WNV patients describe symptoms that may reflect a loss of proprioception (Moon et al., 2005) (Table 1), which is a declining sense of the relative position of neighboring parts of the body. The cerebellum is involved in coordinating this communication to motor functions. Rodent models could possibly be useful for these investigations inasmuch as WNV can infect the cerebellum in rodents.
Some disease signs and symptoms of WNV encephalomyelitis are consistent with dysfunction of the autonomic nervous system, i.e., respiratory, cardiac, renal and gastrointestinal functions (Table 1). The most widely recognized WNV-induced disease sign controlled by autonomic function is respiratory distress (Betensley et al., 2004; Sejvar et al., 2005), which can result in respiratory failure with a poor prognosis (Sejvar et al., 2006). WNV-induced respiratory distress mechanisms have been extensively studied in rodents and are discussed below.
Cases of cardiac or renal involvement, although much less frequent, have been reported (Table 1). A case study of myocarditis has been reported in a patient having a confirmed case of WNV (Omalu et al., 2003). The patient developed cardiac arrhythmias and global myocardial dysfunction. Cardiac complications including arrhythmia are also described in a report of hospitalized patients with WNV disease (Bode et al., 2006). There are many reports of WNV-induced cardiac involvement in other mammalian (Lichtensteiger et al., 2003) and avian species (Gibbs et al., 2005). Electrocardiograms obtained from radiotelemetry in WNV-infected hamsters revealed some cardiac disturbances, but the implications on WNND have yet to be determined (Wang et al., 2011).
In regards to renal function, 22% of patients with WNV-induced paralysis developed bladder dysfunction (Saad et al., 2005). A case study report claimed to be the first report of urological sequelae in a patient with WNV; the patient also had respiratory distress requiring intubation (Shpall et al., 2003). Other more subtle autonomic-like dysfunctions may also occur in WNV neurological disease. For example, adrenal insufficiency, as detected by a corticotropin test, was identified in 70% patients with severe WNV disease (Abroug et al., 2006).
A central question is if autonomic dysfunction is due to direct damage of motor functions or to damage of neurons generally regulating sympathetic or parasympathetic functions. Rodent studies using heart rate variability as an indicator of autonomic function, electromyography of the intestine and diaphragm, nerve conduction velocity, electrocardiography, plethysmography, and immunofluorescence assays indicated that WNV does cause some autonomic dysfunction, but many of these dysfunctions are caused by direct damage to motor functions (Morrey et al., 2012; Wang et al., 2011, 2013a; Wang et al., 2013b).
Parkinsonism has been observed in 69% of WNV patients in one study (Sejvar et al., 2003a) (Table 1). Parkinson’s disease is a neurodegenerative disease caused by death of dopaminergic neurons in the substantia nigra. Two WNV patients have been described by neuroimaging procedures with heavy involvement of the substantia nigra, which correlated with Parkinsonism features of the patient (Bosanko et al., 2003). It is not known if rodents have Parkinsonism, but hamsters infected with WNV manifest front limb tremors (Morrey et al., 2004b). No studies have been done to correlate these tremors with histopathogensis of infection of the substantia nigra. The other alternative mechanism of tremors that could be addressed with rodent models is hyper-excitability of neurons or synapses. In the mouse model of amyotrophic lateral sclerosis, limb tremors were associated with an elevation of excitatory synapses (Sunico et al., 2011). Consistent with this finding is that the EMG amplitudes of motor neurons in the lumbosacral spinal cord stimulated by optogenetic photoactivation of some mice infected with WNV are much greater than the amplitudes of sham-infected mice (Wang et al., 2013b). Further rodent studies could be done to correlate tremors with hyper-excitability of motor neurons using this approach and to investigate the mechanism.
When performed, electrophysiological studies have been useful in identifying some motor deficits and rarely occurring seizures (Bagic et al., 2007). For example, electrophysiological assays of one study (Li et al., 2003) revealed severe denervation in a paralyzed patient, which was substantiated with abnormal MRI in the anterior lumbar spinal cord. However, clinical exams and electrophysiological tests, although necessary, have been inadequate to fully investigate physiological mechanisms of WNND, because the electrophysiological deficits could not be correlated with histopathological conditions as can be done in rodent models.
MRI have been useful in identifying spinal cord and cauda equine abnormalities that reflect the motor deficits of acute flaccid paralysis or extreme weakness (Leyssen et al., 2003; Petropoulou et al., 2005). As mentioned above, MRI has revealed heavy involvement of the substantia nigra in a WNV patient with Parkinsonism features (Bosanko et al., 2003). Other than these examples, MRI findings in patients with WNND are generally nonspecific (Petropoulou et al., 2005).
Fortunately, physiological and electrophysiological approaches in rodent models have been valuable for investigating mechanisms of motor function deficits of the spinal cord, neuro-respiratory deficits of the spinal cord and brainstem, autonomic dysfunction, and memory deficits. These experimental approaches will be reviewed, along with how these approaches have been used to evaluate therapeutic interventions.
Rodent WNV infections
The general features of WNV infection of rodents are thought to be similar to human infection. Peripheral injection of WNV in mice and probably hamsters results in accumulation of WNV-infected cells in the lymph nodes and spleens, which facilitates extra-neurologic replication, viremia, and exposure of all vascular tissues to the virus. Langerhans cells are likely vehicles for rapidly transporting the virus from the skin to these lymphatic tissues (Byrne et al., 2001; Diamond et al., 2003a; Johnston et al., 2000). The development of IgM or neutralizing antibodies beginning at days 3-5 for both rodents (Diamond et al., 2003a; Diamond et al., 2003b; Hunsperger and Roehrig, 2006; Morrey et al., 2007) and human subjects (2002; Busch et al., 2008) eventually removes the virus from the serum and from extra-neurological tissues, except for low-level persistent virus in kidneys of hamsters (Tesh et al., 2005; Tonry et al., 2005). There are conflicting reports as to whether there is persistent shedding of WNV RNA in the urine of persons (Gibney et al., 2011; Murray et al., 2010). In the hamster model, infectious viral titers decline to the limits of detection in the cerebrospinal fluid (CSF) by days 6 due to the appearance of WNV-specific neutralizing antibodies titers in the CSF (Morrey et al., 2004b; Morrey et al., 2007). Viral antigens are detected in mice and hamsters in the cerebral cortex, hippocampus, brainstem, and spinal cord (Hunsperger and Roehrig, 2006; Xiao et al., 2001), and histopathological lesions can be identified in coronal sections throughout the whole brain and spinal cord (Siddharthan et al., 2009).
The mechanisms of entry of the virus are uncertain, but according to rodent studies could involve hematogenous spread of infected cells across the blood brain barrier (BBB) (Hunsperger and Roehrig, 2009), permeabilization of the BBB (Wang et al., 2004), trans-cellular movement of virus from the luminal to apical sides of endothelial cells (Verma et al., 2009; Xu et al., 2012), trafficking of WNV-associated leukocytes across endothelial cells (Dai et al., 2008), and retrograde axonal infection (Hunsperger and Roehrig, 2006; Samuel et al., 2007).
The time in which the virus infects the human CNS with respect to the initial exposure to the virus is not known, but viral proteins and RNA appear in rodent CNS structures within 2-4 days after viral exposure (Hunsperger and Roehrig, 2006). Appearance of infectious virus in the cerebrospinal fluid of hamsters is a marker for infection of the CNS and occurs at day 4 after viral challenge (Morrey et al., 2007). Overt signs of disease in hamsters such as front limb tremors, diarrhea, difficulty walking, and paralysis are observed at 7 to 12 days after subcutaneous viral challenge (Morrey et al., 2004b; Xiao et al., 2001). Two laboratory-acquired human WNV infections indicates that febrile illness occurs at 3-4 days after viral exposure (2002), but the time of onset of WNND in human subjects after viral exposure is uncertain, except for a patient that developed clinical encephalitis 13 days after receiving transfusions of blood components, one of which was retrospectively positive for WNV (Macedo de Oliveira et al., 2004).
One outcome that is markedly different between rodent and human WNV infections is the mortality rate. Mortality rate in rodents can vary depending on the strain of virus, but rates with the New York strain and the 2002 strain WN02 are typically 60-90% (Morrey et al., 2004a; Morrey et al., 2008c; Oliphant et al., 2005). In contrast, the human mortality rate is <1% (Petersen and Marfin, 2002). Even though mortality may be a good endpoint for evaluating therapeutic agents when administered before or slightly after viral exposure and before the virus has infected the CNS, mortality may not be a suitable endpoint when evaluating therapeutics that are anticipated to treat neuropathological conditions of WNND. In light of the fact that WNV-infected people often present to their physicians with signs and symptoms reflective of WNND, the neurological models of WNV discussed below should be valuable, or even essential, in developing effective treatments for WNV.
Motor neuron function
WNV can cause poliomyelitis-like illness or acute flaccid paralysis in WNV-infected persons, which is histologically confirmed in the grey matter of the anterior spinal cord and in the brainstem of postmortem tissues (Doron et al., 2003; Fratkin et al., 2004; Jeha et al., 2003; Sejvar et al., 2005; Sejvar et al., 2003b). Similar histopathology occurs in WNV-infected hamsters (Morrey et al., 2008b; Samuel et al., 2007; Siddharthan et al., 2009; Xiao et al., 2001) and mice (Hunsperger and Roehrig, 2006) where the ventral cord has lymphocytic infiltration, perivascular cuffing, and neurophagia. Similar signs are documented with nearly all flavivirus encephalitides, i.e., Japanese encephalitis virus (JEV) (Johnson, 1987), tick-borne encephalitis (TBE) virus (Gelpi et al., 2005), and the murine Modoc virus (Leyssen et al., 2003). Observing histopathological changes in the central nervous system (CNS), however, does not necessarily cause or indicate the types of neurological deficits. For example, the spinal cord functions are vast and diverse, where the cord acts as a conduit for descending motor functions, as a conduit for ascending sensory information, and as a center for coordinating sensory/motor reflexes. Essentially, it is a conduit between the brain and nearly all other body functions. Therefore, histopathological damage to the spinal cord by WNV could affect a wide range of neurological disease phenotypes. Since WNV clearly causes motor function deficits in human subjects, human clinical procedures employed for evaluating WNND and other motor diseases have been adapted for measurement of motor functions in rodents infected with WNV.
In neurodegenerative diseases such as poliomyelitis (Ohka and Nomoto, 2001) and amyotrophic lateral sclerosis (Rashidipour and Chan, 2008; Shefner et al., 2006), the loss of motor neurons can be clinically detected by using electrophysiological motor unit number estimation (MUNE) (Dantes and McComas, 1991), where a motor unit consists of a motor neuron and all its associated muscle fibers. Since a presumptive use of MUNE in the human WNV infection appears to be a possible marker for muscle weakness and clinical recovery (Cao et al., 2005), the MUNE procedure was adapted for use in hamsters (Siddharthan et al., 2009). To perform the MUNE procedure, the rostral sciatic nerve is stimulated with incremental increases of voltage. The resulting M-wave depolarization and polarization voltages are recorded at the plantar aspect of the hind limb. As the stimulus is increased, more motor units are recruited or activated. The increased activation of motor units is detected by incremental jumps in the amplitude of the M-wave. The more incremental jumps that are detected, the more motor units the animal possesses. Recordings are obtained from the minimally detected voltage to the supra-maximal voltage (compound muscle action potential, CMAP) where no more incremental jumps are detected. The MUNE is calculated as the average voltage of the increments divided into the CMAP (Shefner et al., 2006; Shefner et al., 2002).
The use of MUNE procedure successfully identifies slight motor function deficits where there is visually no overt paresis or paralysis, where there is paresis, or where the level of MUNE suppression is greatest with overt paralysis (Siddharthan et al., 2009) (Figure 1). In this figure one can see the uninfected hamster #617 has normal detectable M-waves with incremental jumps in the amplitude of the M-wave, whereas the WNV-infected #663 hamster does not show these features. In a study investigating the progression of WNV-induced MUNE suppression, MUNE is suppressed beginning at day 9 after subcutaneous WNV challenge, and continues beyond day 92 (Siddharthan et al., 2009). To our knowledge this is the first animal model of WNV long-term neurological sequelae. Additionally, these studies reveal that reduced staining of cholineacetyltransferase in the motor neuron cell bodies strongly correlates with MUNE suppression at day 10, whereas the total number of neurons does not correlate, which suggests that loss the of motor neuron functions contributes more to motor deficits than simply death of neurons at this point of disease progression.
Figure 1.
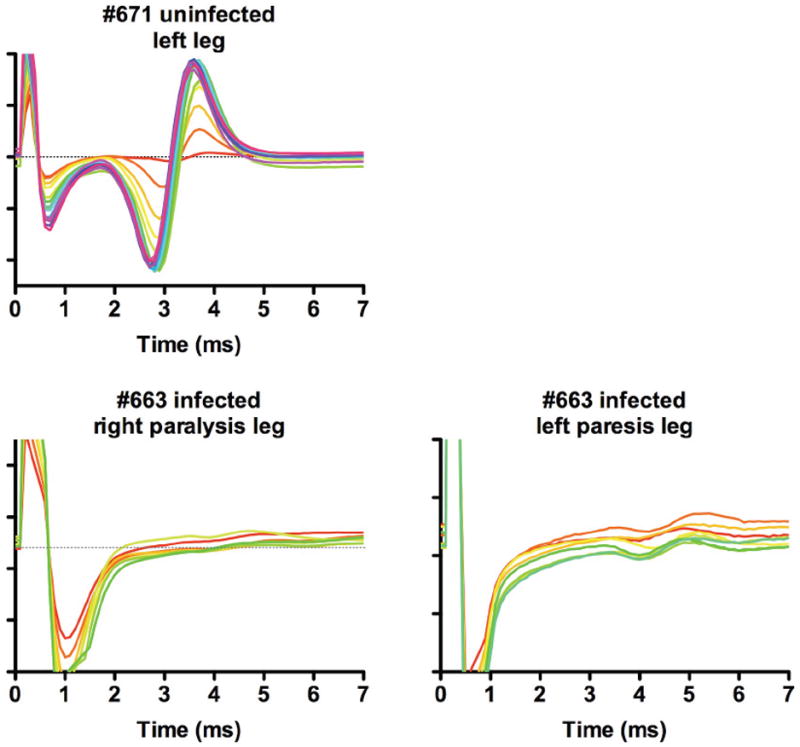
M-wave tracings from hind limbs of WNV-infected hamster with paresis or paralysis for calculation of MUNE (Siddharthan et al., 2009). Animal #671 was uninfected. Hamster (#663) was injected s.c. with WNV, which became paralyzed in the right limb and had paresis (limb weakness) on the left limb. MUNE in both limbs are graphically represented. The graphs are constructed such that the greater number of MUNE increments produces more of the color spectrum beginning with yellow, to orange, to red, and to blue.
To confirm that defective motor neurons, not axonal degeneration, are the likely cause of the MUNE suppression, nerve conduction velocity (NCV) is performed, which is a measurement of the velocity that action potentials travel through motor and sensory fibers. NCV is obtained in WNV-infected hamsters by measuring the time-delay between stimulation of the sciatic nerve to measurement of the EMG of the gastrocnemius muscle. The time-delay of demyelinated axons are slower than normal axons. An experiment with WNV-infected rodents demonstrated that axons or myelin sheaths are not degenerated, because the NCV is not slower in WNV-infected rodents (Wang et al., 2011). Therefore, therapeutic intervention should focus on treating motor neuron dysfunction and not demyelination.
The advantage of the MUNE procedure is that it successfully detects WNV-induced motor function deficits specifically in hamsters where other electrophysiological procedures, such as H-reflex (unpublished data), fail due to technical or biological limitations. The disadvantage of the MUNE procedure in WNV-infected hamsters is that it requires 1 to 2 hours to assay each hamster, and the detection of the incremental MUNE steps is subjective for each operator.
An optogenetics approach has also been employed to measure motor function deficits. Transgenic mice are used that express the light-gated ion channel, channelrhodopsin-2 (ChR2). Because the ChR2 gene is driven by the promoter of choline acetyltransferase (ChAT), the neurotransmitter for motor neurons, the ChR2 protein is expressed in the membranes of specifically spinal cord motor neurons (Wang et al., 2013b). When ChR2 is exposed to blue light, the ion channel opens for exchange of ions, which creates an action potential across the membrane. As with natural polarization signals, the action potential transfers through the axon to activate the motor plate of the respective muscle that the neuron innervates. For example, some motor neurons in the lumbosacral spinal cord innervate muscles served by the sciatic nerve. To establish the motor function deficit model, a cannula mount is surgically attached to the dorsal aspect of the spinal cord. To test the function of the motor neurons in this area, laser optical fibers are placed into the cannula, and pulses of blue laser light precisely activate motor neurons by opening the light-gated ChR2. When the lumbosacral-caudal equine of the cord is photoactivated in this way, electromyography (EMG) can be measured on the gastrocnemius or plantar aspect of the hind limbs to monitor the photoactivation of the motor neurons. From the data shown in Figure 2, the blue EMG signal is in exact registration with the optogenetic photoactivation in red (Wang et al., 2013b). The strength or amplitude of the EMG signal can be quantified with the root mean square (RMS) calculation, and will provide a suitable endpoint to measure therapeutic agents anticipated to treat motor function deficits caused by WNV.
Figure 2.
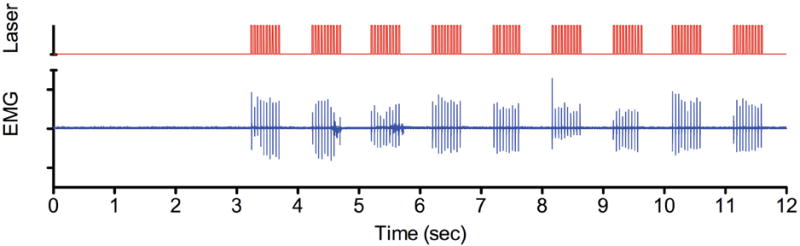
Optogenetic photoactivation of lumbosacral cord motor neurons. An uninfected ChAT-mhChR2-YFP transgenic mouse was implanted with a cannula mount. A fiber optic was inserted into the cannula mount and pulsed with blue-light to activate the neurons containing the ChR2. Red color indicates the photoactivation signal. Blue color indicates the EMG signal of the gastrocnemius muscle.
When optogenetic photoactivation is performed in transgenic mice infected intrathecally with WNV, the amplitudes of the EMGs are significantly suppressed compared to transgenic mice receiving sham infection (unpublished data). Although this optogenetics approach requires specialized laser and recording instrumentation committed to the ABSL-3 animal laboratory, the measurements are not subjective evaluations for individual operators as is the MUNE procedure. Moreover, the procedure requires 15 minutes for each animal as compared to MUNE that requires 1 to 2 hours per animal. As this procedure becomes refined to obtain longitudinal measurements, investigations on the mechanisms of pathogenesis and treatments for WNV-induced motor function deficits can be investigated. With this model in hand, one could draw on the extensive research and development of candidate drugs used to treat other motor deficit neurological diseases, such as for amyotrophic lateral sclerosis (ALS). For example, Table 2 lists some of the drugs that have been evaluated for ALS treatment (Morrison, 2002), and might in principle be evaluated for treatment of WNV-induced motor function deficits using the described optogenetic photoactivation model.
Table 2.
Treatments evaluated for amyotrophic lateral sclerosis (ALS) that might be suitable for treatment of motor function deficits of WNV. Reviewed in (Morrison, 2002).
| Antiglutamate agents | Calcium regulators |
|---|---|
| Riluzole | Verapamil |
| L-threonine | Nimodepine |
| Dextromethorphan | |
| Lamotrigine | Cholinergic system |
| Gabapentin | Pysostigimine |
| 3-4 diaminopyridine | |
| Neurotrophic agents | Tetrahydroaminoacridine |
| CNTF | |
| BDNF | Antioxidants |
| GDNF | Topiramate |
| IGF-1 | D-penicillamine |
| TRH | Selegiline |
| Xaliproden | N-acetyl cysteine |
| Vitamin E | |
| Immunomodulators | |
| Azathioprine | Mitochondrial function |
| Cyclophosphamide | Creatine |
| Cyclosporine |
Respiratory function
Respiratory distress is a serious outcome of WNND (Sejvar et al., 2005), which can result in respiratory failure with a poor prognosis (Sejvar et al., 2006). Hamster and mouse models have been used to validate that the respiratory distress is caused by neurological deficits (Morrey et al., 2012) and is the primary physiological mechanism of death for WNV and other viral encephalitides (Wang et al., 2013b). Respiratory deficits are measured in these rodent models by plethysmography (Morrey et al., 2012), oxygen saturation (SaO2) (Morrey et al., 2012), diaphragmatic electromyography (EMG) (Morrey et al., 2010), and optogenetic photoactivation of phrenic motor neurons in the cervical cord (Wang et al., 2013b). Respiratory deficits are further identified by challenging the infected animals with hypercapnia (7% CO2) (Wang et al., 2013b).
Representative tracings of whole body plethysmography are shown for mice (Figure 3). The principle is that as the rodent breathes in the sealed chamber, changes in voltage are recorded from pressure-sensitive transducers. Qualitatively, one can tell the difference in the tracings between sham-infected and WNV-infected mice, particularly if the animals are noticeably moribund (Figure 3). To quantitatively interpret the patterns, the shapes of the curves are mathematically described by 16 different algorithms with the apparatus used in a WNV study (minute volume, tidal volume, enhanced pause, end expiratory pause, end inspiratory pause, peak expiratory flow, peak inspiratory flow, frequency, inspiratory time, expiratory time, relaxation time, pause, time delay, specific airway resistance, specific airway conductance, mid-expiratory flow) (Morrey et al., 2012). Of the 8 parameters markedly affected by WNV infection, minute volume (MV) as a measure of lung capacity over time was the most unambiguous indicator of WNV-induced respiratory stress. The suppression of MV during development of neurological disease is also supported by reduced SaO2 as measured by pulse oximetry (Morrey et al., 2012); however, pulse oximetry is less accurate in mice and is not performed on hamsters due to the lack of sufficient tail for the application of a cuff.
Figure 3.
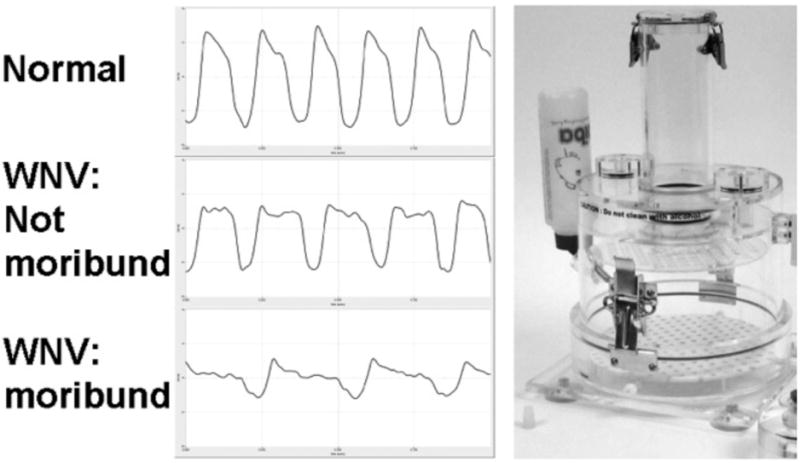
Representative minute volume (MV) plethysmography tracings in a normal C57BL/6 mouse, a not-moribund WNV-infected mouse, and a moribund WNV-infected mouse with lethargy. (right) Picture of plethysmography chamber.
The use of plethysmography facilitated the discovery that respiratory insufficiency is the likely physiological mechanism of death for a subset of arboviral encephalitides, including WNV (Wang et al., 2013b). Respiratory insufficiency is the only physiological readout that correlates strongly with WNV-induced mortality (Morrey et al., 2012) (Figure 4). No other disease parameters in WNV-infected rodents, i.e., cerebral edema, overt seizures, starvation or dehydration, cardiac abnormalities, paralysis, nose bleeding, front limb tremors, memory loss, or autonomic dysfunctions correlate with mortality (Morrey et al., 2004b; Morrey et al., 2008a; Morrey et al., 2008b; Siddharthan et al., 2009; Smeraski et al., 2011; Wang et al., 2011). Remarkably, respiratory insufficiency as measured by % normal MV caused by Japanese encephalitis virus (JEV), neuro-adapted Sindbis virus (NSV), North American tick-borne encephalitis Powassan virus (not shown) also correlates strongly with mortality (Wang et al., 2013b) (Figure 4). Of those animals that died, as marked by yellow circles in the figure, the MV was suppressed below 2 standard deviations of the values from sham-infected and normal mice. However, respiratory insufficiency is not associated with death of mice infected with Western equine encephalitis virus (WEEV), which suggests that respiratory insufficiency is the physiological mechanism of death for a subset of encephalitides, but not all. Since respiratory insufficiency is a good predictor as to which individuals may die, suppression of MV might be used as a trigger to employ therapies to prevent death, which otherwise might not be indicated.
Figure 4.
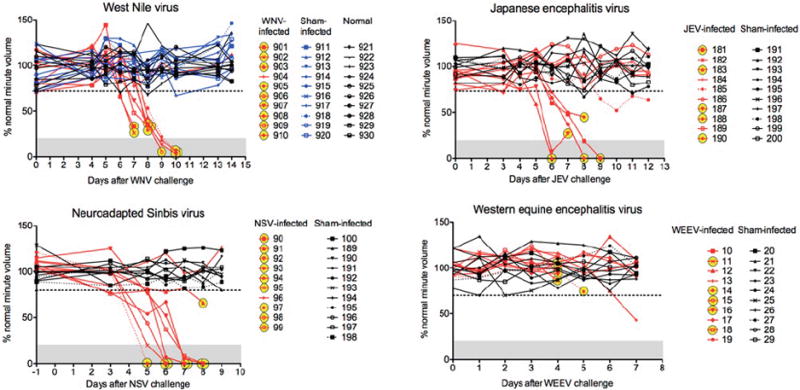
Correlation of mortality with suppressed minute volume (MV) in mice infected with WNV, Japanese encephalitis virus (JEV), neuroadapted Sindbis virus (NSV), and Western equine encephalitis virus (WEEV). To calculate the daily normal average MV for determining the percent normal value of each measurement, 10 normal mice for WNV, 10 sham-infected mice for JEV, 10 sham-infected mice for NSV, and 10 sham-infected mice for WEEV were used. The horizontal dotted lines were two standard deviations (2SD) from the mean of all percent normal values across all days. Yellow circles indicate those animals that died. (Adapted from Figure 1 in open-access publication (Wang et al., 2013b)).
EMG of the diaphragm has been valuable in establishing the neurological cause of respiratory insufficiency. Early in development of the procedure, electrodes attached to near the predicted motor plates of the diaphragm and exiting the dorsal skin of the animal allowed for measurements of EMG over time in alert, non-anesthetized hamsters (Morrey et al., 2010). This procedure is surgically involved, but has yielded data to indicate that WNV-infected mice develop diaphragmatic EMG suppression as compared to sham-infected animals.
The diaphragmatic EMG readout was further developed in anesthetized mice to eliminate variability of behavior in alert animals (Wang et al., 2013b). Bilateral vagotomy is performed on ventilated isoflorane-anesthetized mice to abolish mechanoreceptor feedback. In these mice, diaphragmatic EMG signals are not detected. The middle EMG tracing of each mouse in Figure 5 shows the absence of diaphragmatic EMG signals in vagotomized mice infected with WNV, WEEV, and sham. When the anesthetized mice are then exposed to hypercapnia at 7% CO2, the chemoreceptor cells in the medulla oblongata signal innervation of the diaphragm and are detected by EMG (bottom tracings, Figure 5). The WNV-infected mouse (#594) with confirmed respiratory insufficiency as detected by plethysmography does not show any EMG signal, as compared to sham- and WEEV-infected mice that had robust EMG signals of the diaphragm in response to hypercapnia. The loss of diaphragmatic EMGs for WNV is consistent with loss of plethysmography results (Figure 4). Essentially, anesthetized WNV-, POWV-, and NSV-infected animals, but not WEEV-infected animals, are not able to neurologically compensated for hypercapnia (Morrey et al., 2012).
Figure 5.
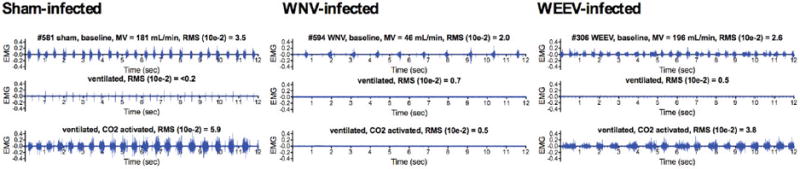
Effect of arboviral infections on diaphragmatic EMGs of mice challenged with hypercapnia. C57BL/6 mice were infected with sham, WNV, or WEEV. Plethysmography was performed daily to detect the virally infected mice having MV values below 2SD of the normal MV values. The diaphragmatic EMGs of these mice with respiratory insufficiencies were then measured in mice before (top EMG readings) and after intubation and vagotomy (middle EMG readings). The mice were challenged with 7% CO2 and the diaphragm EMGs were measured (bottom EMG readings). The animal numbers, MV values, and RMS values reflecting the amplitudes are all listed above the EMG readings. (Adapted from Figure 3 in open-access publication (Wang et al., 2013b))
Another respiratory neurological deficit in phrenic neurons is detected with the use of the same optogenetics transgenic mice expressing ChR2 in their spinal cords as employed to measure motor function deficits in Figure 2. Since the ChR2 is expressed from the choline acetyltransferase promoter, the function of phrenic neurons in the cervical cord controlling the innervation of the diaphragm can be monitored in infected mice (Wang et al., 2013b). When the cervical cord (C4-5) neurons are illuminated with fiber optics, EMG activation can be detected in the diaphragms of sham-infected mouse #170 (Figure 6). The amplitudes of the EMGs from transgenic mice infected subcutaneously with WNV (#157) are significantly suppressed compared to transgenic mice receiving sham, which correlates with a histological loss of the numbers of these neurons with orexin-1 receptor staining (Wang et al., 2013b). Therefore, development of plethysmography, diaphragmatic EMG, and optogenetic procedures reveals that WNV-infected mice die from respiratory insufficiency due to neurological deficits, and that therapeutic intervention strategies should target these deficits.
Figure 6.
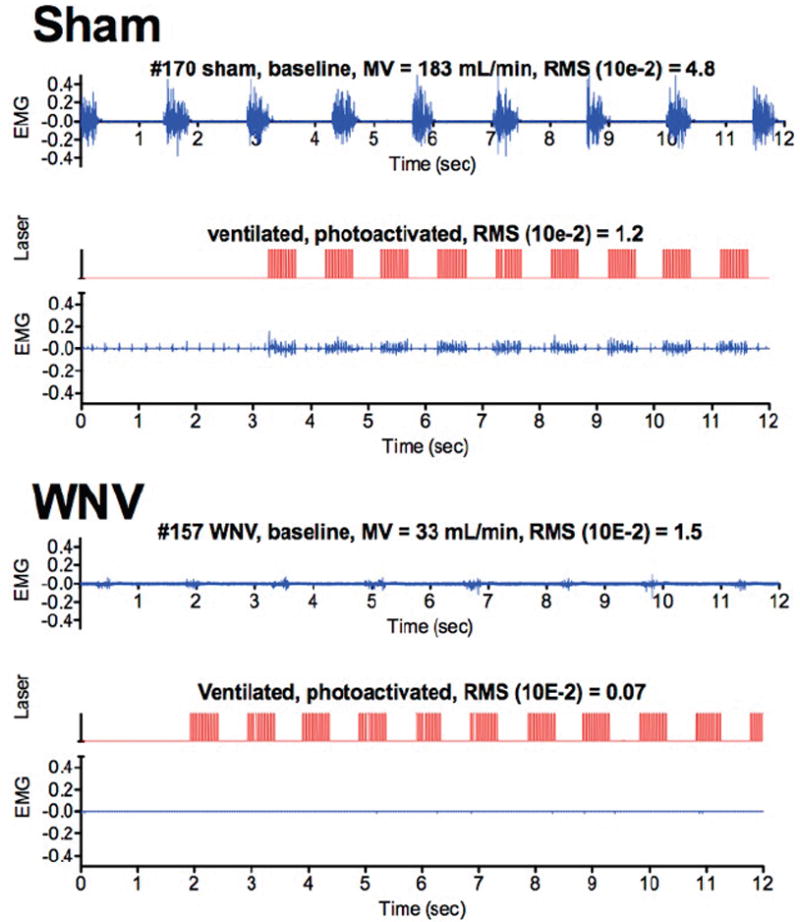
Effect of WNV infection on EMG of diaphragms of ChAT-mhChR2-YFP transgenic mice challenged with optogenetic photoactivation of C3-C4 cervical cord containing motor neurons innervating the diaphragm. Mice were infected with sham or WNV. Plethysmography was performed daily to detect the WNV-infected mice having MV values below 2SD of the normal MV values. The diaphragmatic EMGs of the mice with respiratory insufficiencies were then measured before (top EMG readings) and after intubation and vagotomy (not shown) to confirm the absence of EMG readings. An optical fiber was inserted between C3-C4 vertebrae (Morrey et al., 2008b). Diaphragmatic EMG was used to measure photoactivation of the phrenic neurons. The EMGs of the diaphragm (blue) were directly aligned with the photoactivation signals (red). (Adapted from Figure 1 in open-access publication (Wang et al., 2013b))
Autonomic function
Experimental procedures have been developed to monitor autonomic dysfunction in WNV-infected rodents (Wang et al., 2011), since human clinical studies and case reports have identified certain signs and symptoms that are reflective of autonomic dysfunctions. Heart rate variability (HRV) has been used as a well-accepted and widely-used indicator of autonomic function in human patients and in rodents (1996). Parasympathetic autonomic function affects heart rate by cardiopulmonary coupling, which is the neurological connectivity that causes the heart rate to increase during respiratory inspiration and causes it to decrease during expiration. This physiological event is referred to as respiratory sinus arrhythmia (Grossman and Taylor, 2007), and it results in more efficient cardiopulmonary function. Therefore, higher HRV is an indicator of healthy autonomic parasympathetic function. Conversely, reduced HRV is an indicator of unhealthy parasympathetic function.
The HRV is monitored in rodents infected with WNV using radiotelemetry chips (Wang et al., 2011). A midline dorsal incision is made along the spine, and a subcutaneous pocket is made to house the telemetric device. Two recording-leads subcutaneously tunneled toward the left and right clavicular regions are sutured to the pectoral muscles. Telemetry receivers on platforms under the cages are used to collect data for calculating the frequency and time domains, which are mathematical computations used to identify HRV. The frequency domain is analyzed with the power spectral densities of the heartbeats based on the fast Fourier transform. The time domains are based on the time of each beat between the ECG R peaks. From these data the mean R-R interval, and standard deviation of normal R-R intervals are calculated for each animal. During the development of WNND, the HRV is progressively reduced in hamsters, which suggests that WNV infection causes autonomic dysfunction in hamsters and possibly in infected people. It is currently not known at this time, however, the locations of neurological lesions that contribute to this autonomic dysfunction. Observations of similar radiotelemetry studies indicate that mice do not develop reduced HRV despite the development of fatal WNND, and that autonomic dysfunction is not the physiological cause of death (Wang et al., 2013b), whereas as discussed previously, respiratory insufficiency from lesions directly affecting respiratory function is likely the physiological cause of death. These data suggest that drugs affecting the autonomic nervous system are not likely to affect mortality, but might be beneficial for some limited autonomic dysfunctions.
Memory
Published clinical records and surveys indicate that some WNV-infected patients complain of memory problems (Carson et al., 2006; Cook et al., 2010; Gottfried et al., 2005). Rodent models with Theiler’s murine encephalomyelitis virus (Buenz et al., 2006) and Borna disease virus (Rubin et al., 1998) develop spatial memory loss, which is associated with infection in the hippocampus. To experimentally evaluate spatial memory in WNND, infected hamsters are evaluated in a Morris water maze (MWM) test. Motor function tests are first used to identify surviving animals that have normal motor functions before entering them into the MWM test, so as to not confound the memory results with their inability to swim normally (Smeraski et al., 2011). The MWM test consists of a circular water basin filled with cloudy water placed under a video surveillance camera. Swimming animals are trained to remember the position of a submersible platform on which they can anticipate resting. Fifty-six percent of infected hamsters spend more time in the quadrant of the submersible platform than the other three quadrants, as compared to 92% of hamsters treated with a WNV-specific antibody (hE16) to prevent infection (Smeraski et al., 2011), which substantiates the notion that WNV-infected persons can have memory deficits, and that these deficits can be investigated with the use of rodent models which may provide opportunities for therapeutic intervention.
Utility
Due to the specialization of the procedures described in this review, and that neurophysiological procedures are typically not found in ABSL-3 virology laboratories, the utility of these procedures are limited by most investigators. Nevertheless, new avenues of discovery in basic neurovirology, preclinical therapeutic development, and clinical applications for viral encephalitis are likely available to those willing to make the financial and personnel investments in these neurological approaches.
Plethysmography is very useful in detecting acute arbovirus-induced respiratory failure in rodents, which is likely the physiological mechanism of death. Commercially available instrumentation for rodents facilitates operation after sufficient training by the supplier. Other benefits of whole body plethysmography are the use of non-sedated mice and time of the procedure that takes <2 minutes per mouse. If multiple chambers are available, multiple mice can be measured simultaneously.
The utility for basic neurovirology is that plethysmography has been (Morrey et al., 2012; Wang et al., 2013b), and should be useful in identifying the neuro-anatomical location of lesions responsible for respiratory failure, and the physiological, molecular, and cellular mechanisms of death. In preclinical development, this basic knowledge of pathogenesis should provide targets for therapeutic intervention. It is also conceivable that early respiratory monitoring of viral infected patients might alert the physician to the onset of more serious complications of encephalitis. The disadvantage of plethysmography is that WNND is usually not fatal in human patients, so other assays are necessary to measure more common neurological deficits.
Since poliomyelitis-like disease and motor function deficits are well documented in some arbovirus-infected patients, tools to neurologically monitor motor function deficits in rodent models is important, if not necessary, to discover the physiological mechanisms of this deficit. Tools such as EMG and optogenetic photoactivation will be important to pre-clinically evaluate candidate therapeutics (Table 2). Since mortality is not a surrogate readout to monitor limb motor deficits (Morrey et al., 2010; Morrey et al., 2008b; Siddharthan et al., 2009), these neurological tools are probably essential for pre-clinical development of therapeutics. Such studies will also solidify the value of current clinical tests of motor function. Optogenetic photoactivation of motor neurons in the spinal cord is our favored experimental assay by us for measuring motor deficits responsible for limb weakness, paresis, or paralysis. The procedure essentially has two components: optogenetic stimulation and EMG readout. The main advantage of the optogenetics approach is the accuracy, exquisite sensitivity, and quantitative measurements of subclinical limb weakness to overt paralysis. EMGs are relatively straightforward to perform. The disadvantages are that the procedure requires transgenic mice expressing channelrhodopsin in motor neurons, surgical expertise, specialized training in optogenetics, and assembly of specialized instruments. The alternative for measuring motor deficits is motor unit number estimation (MUNE), which is multiple EMG measurements of limb muscle at sequentially different levels of voltage stimulation of the nerves innervating the muscle, but it is difficult to perform, subjective, employs custom-assembled instrumentation and software, and is best performed only in hamsters as opposed to mice.
Surgically implanted radiotelemetry chips have proven to be useful to experimentally monitor autonomic function by HRV, ECG cardiac function, temperature, and activity levels. They might be useful for measuring loss of circadian rhythm, but further studies are necessary to confirm loss of circadian rhythm. Chips designed to measure blood pressure, however, involve difficult surgical procedures that limit their utility. These basic physiological studies may help to investigate autonomic dysfunctions in patients and may serve to better clinically manage the disease using currently available clinical tests.
The electrophysiological and physiological approaches described herein have been valuable, and should be valuable in the future, to help elucidate physiological mechanisms of WNND and to identify clinically relevant endpoints for evaluating therapies. The authors are currently evaluating the efficacy of a neurotropic factor on motor deficits, and are planning the evaluation of antagonists to receptors of a respiratory regulatory protein using these procedures. Ultimately, the advancements described in this review should help with the development of future treatments and management of WNND and other arboviral encephalitides.
Highlights.
Neurological procedures can be used to investigate West Nile virus disease in rodent models.
Respiratory deficits are identified by plethysmography and EMG of the diaphragm.
Motor deficits are detected by motor unit number estimation and EMG of muscle.
Deficits of motor neurons are identified by optogenetic photoactivation.
Morris water maze testing reveals spatial memory deficits in hamsters surviving WNV infection.
Acknowledgments
The work was supported by Rocky Mountain Regional Centers of Excellence, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH) [U54 AI-065357 to J.D.M.], Virology Branch, NIAID, NIH, [HHSN272201000039I to J.D.M.], and Utah Agriculture Research Station [UTA00424 to J.D.M.].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Heart rate variability. Standards of measurement, physiological interpretation, and clinical use. Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Eur Heart J. 1996;17:354–381. [PubMed] [Google Scholar]
- Laboratory-acquired West Nile virus infections--United States, 2002. MMWR Morb Mortal Wkly Rep. 2002;51:1133–1135. [PubMed] [Google Scholar]
- Abroug F, Ouanes-Besbes L, Ouanes I, Nciri N, Dachraoui F, Najjar F. Adrenal insufficiency in severe West Nile Virus infection. Intensive Care Med. 2006;32:1636–1639. doi: 10.1007/s00134-006-0298-z. [DOI] [PubMed] [Google Scholar]
- Agamanolis DP, Leslie MJ, Caveny EA, Guarner J, Shieh WJ, Zaki SR. Neuropathological findings in West Nile virus encephalitis: a case report. Ann Neurol. 2003;54:547–551. doi: 10.1002/ana.10731. [DOI] [PubMed] [Google Scholar]
- Ali M, Safriel Y, Sohi J, Llave A, Weathers S. West Nile virus infection: MR imaging findings in the nervous system. AJNR Am J Neuroradiol. 2005;26:289–297. [PMC free article] [PubMed] [Google Scholar]
- Asnis DS, Conetta R, Teixeira AA, Waldman G, Sampson BA. The West Nile Virus outbreak of 1999 in New York: the Flushing Hospital experience. Clin Infect Dis. 2000;30:413–418. doi: 10.1086/313737. [DOI] [PubMed] [Google Scholar]
- Bagic A, Boudreau EA, Greenfield J, Sato S. Electro-clinical evolution of refractory non-convulsive status epilepticus caused by West Nile virus encephalitis. Epileptic Disord. 2007;9:98–103. doi: 10.1684/epd.2007.0056. [DOI] [PubMed] [Google Scholar]
- Ben-Nathan D, Huitinga I, Lustig S, van Rooijen N, Kobiler D. West Nile virus neuroinvasion and encephalitis induced by macrophage depletion in mice. Arch Virol. 1996;141:459–469. doi: 10.1007/BF01718310. [DOI] [PubMed] [Google Scholar]
- Ben-Nathan D, Maestroni GJM, Lustig S, Conti A. Protective effects of melatonin in mice infected with encephalitis viruses. Archives of Virology. 1995;140:223–230. doi: 10.1007/BF01309858. [DOI] [PubMed] [Google Scholar]
- Betensley AD, Jaffery SH, Collins H, Sripathi N, Alabi F. Bilateral diaphragmatic paralysis and related respiratory complications in a patient with West Nile virus infection. Thorax. 2004;59:268–269. doi: 10.1136/thorax.2003.009092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode AV, Sejvar JJ, Pape WJ, Campbell GL, Marfin AA. West Nile virus disease: a descriptive study of 228 patients hospitalized in a 4-county region of Colorado in 2003. Clin Infect Dis. 2006;42:1234–1240. doi: 10.1086/503038. [DOI] [PubMed] [Google Scholar]
- Bosanko CM, Gilroy J, Wang AM, Sanders W, Dulai M, Wilson J, Blum K. West nile virus encephalitis involving the substantia nigra: neuroimaging and pathologic findings with literature review. Arch Neurol. 2003;60:1448–1452. doi: 10.1001/archneur.60.10.1448. [DOI] [PubMed] [Google Scholar]
- Bouffard JP, Riudavets MA, Holman R, Rushing EJ. Neuropathology of the brain and spinal cord in human West Nile virus infection. Clin Neuropathol. 2004;23:59–61. [PubMed] [Google Scholar]
- Buenz EJ, Rodriguez M, Howe CL. Disrupted spatial memory is a consequence of picornavirus infection. Neurobiol Dis. 2006;24:266–273. doi: 10.1016/j.nbd.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Busch MP, Kleinman SH, Tobler LH, Kamel HT, Norris PJ, Walsh I, Matud JL, Prince HE, Lanciotti RS, Wright DJ, Linnen JM, Caglioti S. Virus and antibody dynamics in acute west nile virus infection. J Infect Dis. 2008;198:984–993. doi: 10.1086/591467. [DOI] [PubMed] [Google Scholar]
- Byrne SN, Halliday GM, Johnston LJ, King NJ. Interleukin-1beta but not tumor necrosis factor is involved in West Nile virus-induced Langerhans cell migration from the skin in C57BL/6 mice. J Invest Dermatol. 2001;117:702–709. doi: 10.1046/j.0022-202x.2001.01454.x. [DOI] [PubMed] [Google Scholar]
- Camenga DL, Nathanson N, Cole GA. Cyclophosphamide-potentiated West Nile viral encephalitis: relative influence of cellular and humoral factors. J Infect Dis. 1974;130:634–641. doi: 10.1093/infdis/130.6.634. [DOI] [PubMed] [Google Scholar]
- Cao NJ, Ranganathan C, Kupsky WJ, Li J. Recovery and prognosticators of paralysis in West Nile virus infection. J Neurol Sci. 2005;236:73–80. doi: 10.1016/j.jns.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Carson PJ, Konewko P, Wold KS, Mariani P, Goli S, Bergloff P, Crosby RD. Long-term clinical and neuropsychological outcomes of West Nile virus infection. Clin Infect Dis. 2006;43:723–730. doi: 10.1086/506939. [DOI] [PubMed] [Google Scholar]
- Chapa JB, Ahn JT, DiGiovanni LM, Ismail MA. West Nile virus encephalitis during pregnancy. Obstet Gynecol. 2003;102:229–231. doi: 10.1016/s0029-7844(03)00614-8. [DOI] [PubMed] [Google Scholar]
- Chudasama Y, Robbins TW. Functions of frontostriatal systems in cognition: comparative neuropsychopharmacological studies in rats, monkeys and humans. Biol Psychol. 2006;73:19–38. doi: 10.1016/j.biopsycho.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Cook RL, Xu X, Yablonsky EJ, Sakata N, Tripp JH, Hess R, Piazza P, Rinaldo CR. Demographic and clinical factors associated with persistent symptoms after West Nile virus infection. Am J Trop Med Hyg. 2010;83:1133–1136. doi: 10.4269/ajtmh.2010.09-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Wang P, Bai F, Town T, Fikrig E. Icam-1 participates in the entry of west nile virus into the central nervous system. J Virol. 2008;82:4164–4168. doi: 10.1128/JVI.02621-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalley JW, Cardinal RN, Robbins TW. Prefrontal executive and cognitive functions in rodents: neural and neurochemical substrates. Neuroscience and biobehavioral reviews. 2004;28:771–784. doi: 10.1016/j.neubiorev.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Dantes M, McComas A. The extent and time course of motoneuron involvement in amyotrophic lateral sclerosis. Muscle Nerve. 1991;14:416–421. doi: 10.1002/mus.880140506. [DOI] [PubMed] [Google Scholar]
- Davis LE, DeBiasi R, Goade DE, Haaland KY, Harrington JA, Harnar JB, Pergam SA, King MK, DeMasters BK, Tyler KL. West Nile virus neuroinvasive disease. Ann Neurol. 2006;60:286–300. doi: 10.1002/ana.20959. [DOI] [PubMed] [Google Scholar]
- Diamond MS, Shrestha B, Marri A, Mahan D, Engle M. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J Virol. 2003a;77:2578–2586. doi: 10.1128/JVI.77.4.2578-2586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Sitati EM, Friend LD, Higgs S, Shrestha B, Engle M. A critical role for induced IgM in the protection against West Nile virus infection. J Exp Med. 2003b;198:1853–1862. doi: 10.1084/jem.20031223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doron SI, Dashe JF, Adelman LS, Brown WF, Werner BG, Hadley S. Histopathologically proven poliomyelitis with quadriplegia and loss of brainstem function due to West Nile virus infection. Clin Infect Dis. 2003;37:e74–77. doi: 10.1086/377177. [DOI] [PubMed] [Google Scholar]
- Fratkin JD, Leis AA, Stokic DS, Slavinski SA, Geiss RW. Spinal cord neuropathology in human West Nile virus infection. Arch Pathol Lab Med. 2004;128:533–537. doi: 10.5858/2004-128-533-SCNIHW. [DOI] [PubMed] [Google Scholar]
- Gelpi E, Preusser M, Garzuly F, Holzmann H, Heinz FX, Budka H. Visualization of Central European tick-borne encephalitis infection in fatal human cases. J Neuropathol Exp Neurol. 2005;64:506–512. doi: 10.1093/jnen/64.6.506. [DOI] [PubMed] [Google Scholar]
- Gibbs SE, Ellis AE, Mead DG, Allison AB, Moulton JK, Howerth EW, Stallknecht DE. West Nile virus detection in the organs of naturally infected blue jays (Cyanocitta cristata) J Wildl Dis. 2005;41:354–362. doi: 10.7589/0090-3558-41.2.354. [DOI] [PubMed] [Google Scholar]
- Gibney KB, Lanciotti RS, Sejvar JJ, Nugent CT, Linnen JM, Delorey MJ, Lehman JA, Boswell EN, Staples JE, Fischer M. West nile virus RNA not detected in urine of 40 people tested 6 years after acute West Nile virus disease. The Journal of infectious diseases. 2011;203:344–347. doi: 10.1093/infdis/jiq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottfried K, Quinn R, Jones T. Clinical description and follow-up investigation of human West Nile virus cases. South Med J. 2005;98:603–606. doi: 10.1097/01.SMJ.0000155633.43244.AC. [DOI] [PubMed] [Google Scholar]
- Grossman P, Taylor EW. Toward understanding respiratory sinus arrhythmia: relations to cardiac vagal tone, evolution and biobehavioral functions. Biological psychology. 2007;74:263–285. doi: 10.1016/j.biopsycho.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Hayes EB, Sejvar JJ, Zaki SR, Lanciotti RS, Bode AV, Campbell GL. Virology, pathology, and clinical manifestations of West Nile virus disease. Emerg Infect Dis. 2005;11:1174–1179. doi: 10.3201/eid1108.050289b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsperger EA, Roehrig JT. Temporal analyses of the neuropathogenesis of a West Nile virus infection in mice. Journal of Neurovirology. 2006;12:129–139. doi: 10.1080/13550280600758341. [DOI] [PubMed] [Google Scholar]
- Hunsperger EA, Roehrig JT. Nocodazole delays viral entry into the brain following footpad inoculation with West Nile virus in mice. Journal of Neurovirology. 2009;15:211–218. doi: 10.1080/13550280902913255. [DOI] [PubMed] [Google Scholar]
- Jeha LE, Sila CA, Lederman RJ, Prayson RA, Isada CM, Gordon SM. West Nile virus infection: a new acute paralytic illness. Neurology. 2003;61:55–59. doi: 10.1212/01.wnl.0000073617.08185.0a. [DOI] [PubMed] [Google Scholar]
- Johnson RT. The pathogenesis of acute viral encephalitis and postinfectious encephalomyelitis. The Journal of infectious diseases. 1987;155:359–364. doi: 10.1093/infdis/155.3.359. [DOI] [PubMed] [Google Scholar]
- Johnston LJ, Halliday GM, King NJ. Langerhans cells migrate to local lymph nodes following cutaneous infection with an arbovirus. J Invest Dermatol. 2000;114:560–568. doi: 10.1046/j.1523-1747.2000.00904.x. [DOI] [PubMed] [Google Scholar]
- Khairallah M, Ben Yahia S, Ladjimi A, Zeghidi H, Ben Romdhane F, Besbes L, Zaouali S, Messaoud R. Chorioretinal involvement in patients with West Nile virus infection. Ophthalmology. 2004;111:2065–2070. doi: 10.1016/j.ophtha.2004.03.032. [DOI] [PubMed] [Google Scholar]
- Leis AA, Stokic DS. Neuromuscular Manifestations of Human West Nile Virus Infection. Curr Treat Options Neurol. 2005;7:15–22. doi: 10.1007/s11940-005-0002-6. [DOI] [PubMed] [Google Scholar]
- Leyssen P, Croes R, Rau P, Heiland S, Verbeken E, Sciot R, Paeshuyse J, Charlier N, De Clercq E, Meyding-Lamade U, Neyts J. Acute encephalitis, a poliomyelitis-like syndrome and neurological sequelae in a hamster model for flavivirus infections. Brain Pathol. 2003;13:279–290. doi: 10.1111/j.1750-3639.2003.tb00028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Loeb JA, Shy ME, Shah AK, Tselis AC, Kupski WJ, Lewis RA. Asymmetric flaccid paralysis: a neuromuscular presentation of West Nile virus infection. Ann Neurol. 2003;53:703–710. doi: 10.1002/ana.10575. [DOI] [PubMed] [Google Scholar]
- Lichtensteiger CA, Heinz-Taheny K, Osborne TS, Novak RJ, Lewis BA, Firth ML. West Nile virus encephalitis and myocarditis in wolf and dog. Emerg Infect Dis. 2003;9:1303–1306. doi: 10.3201/eid0910.020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey NP, Staples JE, Lehman JA, Fischer M. Surveillance for human West Nile virus disease - United States, 1999-2008. MMWR Surveill Summ. 2010;59:1–17. [PubMed] [Google Scholar]
- Macedo de Oliveira A, Beecham BD, Montgomery SP, Lanciotti RS, Linnen JM, Giachetti C, Pietrelli LA, Stramer SL, Safranek TJ. West Nile virus blood transfusion-related infection despite nucleic acid testing. Transfusion. 2004;44:1695–1699. doi: 10.1111/j.0041-1132.2004.04130.x. [DOI] [PubMed] [Google Scholar]
- Mazurek JM, Winpisinger K, Mattson BJ, Duffy R, Moolenaar RL. The epidemiology and early clinical features of West Nile virus infection. Am J Emerg Med. 2005;23:536–543. doi: 10.1016/j.ajem.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Moon TD, Nadimpalli A, Martin EB, Ortiz MA, Van Dyke RB. Balance and gait abnormalities of a child with West Nile virus infection. Pediatr Infect Dis J. 2005;24:568–570. doi: 10.1097/01.inf.0000164704.05076.1c. [DOI] [PubMed] [Google Scholar]
- Morrey JD, Day CW, Julander JG, Blatt LM, Smee DF, Sidwell RW. Effect of interferon-alpha and interferon-inducers on West Nile virus in mouse and hamster animal models. Antivir Chem Chemother. 2004a;15:101–109. doi: 10.1177/095632020401500202. [DOI] [PubMed] [Google Scholar]
- Morrey JD, Day CW, Julander JG, Olsen AL, Sidwell RW, Cheney CD, Blatt LM. Modeling hamsters for evaluating West Nile virus therapies. Antiviral Res. 2004b;63:41–50. doi: 10.1016/j.antiviral.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Morrey JD, Olsen AL, Siddharthan V, Motter NE, Wang H, Taro BS, Chen D, Ruffner D, Hall JO. Increased blood-brain barrier permeability is not a primary determinant for lethality of West Nile virus infection in rodents. J Gen Virol. 2008a;89:467–473. doi: 10.1099/vir.0.83345-0. [DOI] [PubMed] [Google Scholar]
- Morrey JD, Siddharthan V, Olsen AL, Wang H, Julander JG, Hall JO, Li H, Nordstrom JL, Koenig S, Johnson S, Diamond MS. Defining limits of treatment with humanized neutralizing monoclonal antibody for West Nile virus neurological infection in a hamster model. Antimicrob Agents Chemother. 2007;51:2396–2402. doi: 10.1128/AAC.00147-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrey JD, Siddharthan V, Wang H, Hall JO. Respiratory insufficiency correlated strongly with mortality of rodents infected with West Nile virus. PLoS ONE. 2012;7:e38672. doi: 10.1371/journal.pone.0038672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrey JD, Siddharthan V, Wang H, Hall JO, Motter NE, Skinner RD, Skirpstunas RT. Neurological suppression of diaphragm electromyographs in hamsters infected with West Nile virus. J Neurovirol. 2010;16:3198–3129. doi: 10.3109/13550284.2010.501847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrey JD, Siddharthan V, Wang H, Hall JO, Skirpstunas RT, Olsen AL, Nordstrom JL, Koenig S, Johnson S, Diamond MS. West Nile virus-induced acute flaccid paralysis is prevented by monoclonal antibody treatment when administered after infection of spinal cord neurons. J Neurovirol. 2008b;14:152–163. doi: 10.1080/13550280801958930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrey JD, Taro BS, Siddharthan V, Wang H, Smee DF, Christensen AJ, Furuta Y. Efficacy of orally administered T-705 pyrazine analog on lethal West Nile virus infection in rodents. Antiviral Res. 2008c;80:377–379. doi: 10.1016/j.antiviral.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison KE. Therapies in amyotrophic lateral sclerosis-beyond riluzole. Curr Opin Pharmacol. 2002;2:302–309. doi: 10.1016/s1471-4892(02)00169-8. [DOI] [PubMed] [Google Scholar]
- Murray K, Walker C, Herrington E, Lewis JA, McCormick J, Beasley DW, Tesh RB, Fisher-Hoch S. Persistent infection with West Nile virus years after initial infection. The Journal of infectious diseases. 2010;201:2–4. doi: 10.1086/648731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohka S, Nomoto A. Recent insights into poliovirus pathogenesis. Trends in microbiology. 2001;9:501–506. doi: 10.1016/s0966-842x(01)02200-4. [DOI] [PubMed] [Google Scholar]
- Oliphant T, Engle M, Nybakken GE, Doane C, Johnson S, Huang L, Gorlatov S, Mehlhop E, Marri A, Chung KM, Ebel GD, Kramer LD, Fremont DH, Diamond MS. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat Med. 2005;11:522–530. doi: 10.1038/nm1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omalu BI, Shakir AA, Wang G, Lipkin WI, Wiley CA. Fatal fulminant panmeningo-polioencephalitis due to West Nile virus. Brain Pathol. 2003;13:465–472. doi: 10.1111/j.1750-3639.2003.tb00477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen LR, Marfin AA. West Nile virus: a primer for the clinician. Ann Intern Med. 2002;137:173–179. doi: 10.7326/0003-4819-137-3-200208060-00009. [DOI] [PubMed] [Google Scholar]
- Petropoulou KA, Gordon SM, Prayson RA, Ruggierri PM. West Nile virus meningoencephalitis: MR imaging findings. AJNR American journal of neuroradiology. 2005;26:1986–1995. [PMC free article] [PubMed] [Google Scholar]
- Rashidipour O, Chan KM. Motor unit number estimation in neuromuscular disease. The Canadian journal of neurological sciences. 2008;35:153–159. doi: 10.1017/s0317167100008568. [DOI] [PubMed] [Google Scholar]
- Rubin SA, Yednock TA, Carbone KM. In vivo treatment with anti-alpha4 integrin suppresses clinical and pathological evidence of Borna disease virus infection. J Neuroimmunol. 1998;84:158–163. doi: 10.1016/s0165-5728(97)00249-x. [DOI] [PubMed] [Google Scholar]
- Saad M, Youssef S, Kirschke D, Shubair M, Haddadin D, Myers J, Moorman J. Acute flaccid paralysis: the spectrum of a newly recognized complication of West Nile virus infection. J Infect. 2005;51:120–127. doi: 10.1016/j.jinf.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Sampson BA, Ambrosi C, Charlot A, Reiber K, Veress JF, Armbrustmacher V. The pathology of human West Nile Virus infection. Hum Pathol. 2000;31:527–531. doi: 10.1053/hp.2000.8047. [DOI] [PubMed] [Google Scholar]
- Samuel MA, Wang H, Siddharthan V, Morrey JD, Diamond MS. Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc Natl Acad Sci U S A. 2007;104:17140–17145. doi: 10.1073/pnas.0705837104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sejvar JJ. The long-term outcomes of human West Nile virus infection. Clin Infect Dis. 2007;44:1617–1624. doi: 10.1086/518281. [DOI] [PubMed] [Google Scholar]
- Sejvar JJ, Bode AV, Marfin AA, Campbell GL, Ewing D, Mazowiecki M, Pavot PV, Schmitt J, Pape J, Biggerstaff BJ, Petersen LR. West Nile virus-associated flaccid paralysis. Emerg Infect Dis. 2005;11:1021–1027. doi: 10.3201/eid1107.040991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sejvar JJ, Bode AV, Marfin AA, Campbell GL, Pape J, Biggerstaff BJ, Petersen LR. West Nile Virus-associated flaccid paralysis outcome. Emerg Infect Dis. 2006;12:514–516. doi: 10.3201/eid1203.050643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sejvar JJ, Curns AT, Welburg L, Jones JF, Lundgren LM, Capuron L, Pape J, Reeves WC, Campbel GL. Neurocognitive and functional outcomes in persons recovering from West Nile virus illness. J Neuropsychol. 2008;2:477–499. doi: 10.1348/174866407x218312. [DOI] [PubMed] [Google Scholar]
- Sejvar JJ, Haddad MB, Tierney BC, Campbell GL, Marfin AA, Van Gerpen JA, Fleischauer A, Leis AA, Stokic DS, Petersen LR. Neurologic manifestations and outcome of West Nile virus infection. JAMA : the journal of the American Medical Association. 2003a;290:511–515. doi: 10.1001/jama.290.4.511. [DOI] [PubMed] [Google Scholar]
- Sejvar JJ, Leis AA, Stokic DS, Van Gerpen JA, Marfin AA, Webb R, Haddad MB, Tierney BC, Slavinski SA, Polk JL, Dostrow V, Winkelmann M, Petersen LR. Acute flaccid paralysis and West Nile virus infection. Emerg Infect Dis. 2003b;9:788–793. doi: 10.3201/eid0907.030129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shefner JM, Cudkowicz M, Brown RH., Jr Motor unit number estimation predicts disease onset and survival in a transgenic mouse model of amyotrophic lateral sclerosis. Muscle Nerve. 2006;34:603–607. doi: 10.1002/mus.20628. [DOI] [PubMed] [Google Scholar]
- Shefner JM, Cudkowicz ME, Brown RH., Jr Comparison of incremental with multipoint MUNE methods in transgenic ALS mice. Muscle Nerve. 2002;25:39–42. doi: 10.1002/mus.10000. [DOI] [PubMed] [Google Scholar]
- Shpall AI, Varpetian A, Ginsberg DA. Urinary retention in a patient with West Nile virus. Urology. 2003;61:1259. doi: 10.1016/s0090-4295(03)00119-5. [DOI] [PubMed] [Google Scholar]
- Siddharthan V, Wang H, Motter NE, Hall JO, Skinner RD, Skirpstunas RT, Morrey JD. Persistent West Nile virus associated with a neurological sequela in hamsters identified by motor unit number estimation. J Virol. 2009;83:4251–4261. doi: 10.1128/JVI.00017-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeraski CA, Siddharthan V, Morrey JD. Treatment of spatial memory impairment in hamsters infected with West Nile virus using a humanized monoclonal antibody MGAWN1. Antiviral Research. 2011;91:43–49. doi: 10.1016/j.antiviral.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele KE, Linn MJ, Schoepp RJ, Komar N, Geisbert TW, Manduca RM, Calle PP, Raphael BL, Clippinger TL, Larsen T, Smith J, Lanciotti RS, Panella NA, McNamara TS. Pathology of fatal West Nile virus infections in native and exotic birds during the 1999 outbreak in New York City, New York. Vet Pathol. 2000;37:208–224. doi: 10.1354/vp.37-3-208. [DOI] [PubMed] [Google Scholar]
- Sunico CR, Dominguez G, Garcia-Verdugo JM, Osta R, Montero F, Moreno-Lopez B. Reduction in the motoneuron inhibitory/excitatory synaptic ratio in an early-symptomatic mouse model of amyotrophic lateral sclerosis. Brain Pathol. 2011;21:1–15. doi: 10.1111/j.1750-3639.2010.00417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesh RB, Siirin M, Guzman H, Travassos da Rosa AP, Wu X, Duan T, Lei H, Nunes MR, Xiao SY. Persistent West Nile virus infection in the golden hamster: studies on its mechanism and possible implications for other flavivirus infections. J Infect Dis. 2005;192:287–295. doi: 10.1086/431153. [DOI] [PubMed] [Google Scholar]
- Tonry JH, Xiao SY, Siirin M, Chen H, da Rosa AP, Tesh RB. Persistent shedding of West Nile virus in urine of experimentally infected hamsters. Am J Trop Med Hyg. 2005;72:320–324. [PubMed] [Google Scholar]
- Verma S, Lo Y, Chapagain M, Lum S, Kumar M, Gurjav U, Luo H, Nakatsuka A, Nerurkar VR. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: Transmigration across the in vitro blood-brain barrier. Virology. 2009;385:425–433. doi: 10.1016/j.virol.2008.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Siddharthan V, Hall JO, Morrey JD. Autonomic nervous dysfunction in hamsters infected with West Nile virus. PLoS ONE. 2011;6:e19575. doi: 10.1371/journal.pone.0019575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Siddharthan V, Hall JO, Morrey JD. Autonomic deficit not cause of death in West Nile virus neurological disease. Clinical Autonomic Research. 2013a doi: 10.1007/s10286-013-0213-y. Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Siddharthan V, Kesler KK, Hall JO, Motter NE, Julander JG, Morrey JD. Fatal Neurological Respiratory Insufficiency Common Among Viral Encephalitides. The Journal of Infectious Diseases. 2013b doi: 10.1093/infdis/jit186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- Weiss D, Carr D, Kellachan J, Tan C, Phillips M, Bresnitz E, Layton M. Clinical findings of West Nile virus infection in hospitalized patients, New York and New Jersey, 2000. Emerg Infect Dis. 2001;7:654–658. doi: 10.3201/eid0704.010409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao SY, Guzman H, Zhang H, Travassos da Rosa AP, Tesh RB. West Nile virus infection in the golden hamster (Mesocricetus auratus): a model for West Nile encephalitis. Emerg Infect Dis. 2001;7:714–721. doi: 10.3201/eid0704.010420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Waeckerlin R, Urbanowski MD, van Marle G, Hobman TC. West Nile virus infection causes endocytosis of a specific subset of tight junction membrane proteins. PLoS ONE. 2012;7:e37886. doi: 10.1371/journal.pone.0037886. [DOI] [PMC free article] [PubMed] [Google Scholar]