

Abstract
Mannose-6-phosphate (M6P)-terminated oligosaccharides are important signals for M6P-receptor-mediated targeting of newly synthesized hydrolases from Golgi to lysosomes, but the precise structural requirement for the M6P ligand–receptor recognition has not been fully understood due to the difficulties in obtaining homogeneous M6P-containing glycoproteins. We describe here a chemoenzymatic synthesis of homogeneous phosphoglycoproteins carrying natural M6P-containing N-glycans. The method includes the chemical synthesis of glycan oxazolines with varied number and location of the M6P moieties and their transfer to the GlcNAc-protein by an endoglycosynthase to provide homogeneous M6P-containing glycoproteins. Simultaneous attachment of two M6P-oligosaccahrides to a cyclic polypeptide was also accomplished to yield bivalent M6P-glycopeptides. Surface plasmon resonance binding studies reveal that a single M6P moiety located at the low α-1,3-branch of the oligomannose context is sufficient for a high-affinity binding to receptor CI-MPR, while the presence of a M6P moiety at the α-1,6-branch is dispensable. In addition, a binding study with the bivalent cyclic and linear polypeptides reveals that a close proximity of two M6P-oligosaccharide ligands is critical to achieve high affinity for the CI-MPR receptor. Taken together, the present study indicates that the location and valency of the M6P moieties and the right oligosaccharide context are all critical for high-affinity binding with the major M6P receptor. The chemoenzymatic method described here provides a new avenue for glycosylation remodeling of recombinant enzymes to enhance the uptake and delivery of enzymes to lysosomes in enzyme replacement therapy for the treatment of lysosomal storage diseases.
Graphical Abstract
INTRODUCTION
The cation-independent and cation-dependent mannose 6-phosphorate (M6P) receptors, namely, CI-MPR and CD-MPR, are two P-type lectins essential for transporting newly synthesized hydrolases from the Golgi apparatus to lysosomes through their interactions with specific M6P-containing high-mannose oligosaccharide signals attached to the hydrolases.1,2 The larger M6P receptor, CI-MPR, also present on the surface of most mammalian cells, modulates the level of extracellular hydrolases secreted and the insulin-like growth factor II via receptor-mediated endocytosis. Defects in this receptor-mediated targeting processes would result in deficiency of lysosomal enzymes critical for the degradation of substrates stored in lysosomes, causing lysosomal storage diseases.3,4 So far, more than 50 different human lysosomal storage diseases, including Gaucher, Fabry, and Pompe diseases, have been identified, which affect 1 in 7000 newborns.4 Enzyme replacement therapy (ERT), via infusion of exogenous recombinant lysosomal enzymes, is currently the only available clinical method for the treatment of these inherited lysosomal storage disorders.5,6 Targeting those enzymes through the CI-MPR-mediated endocytosis and delivery to lysosomes represents a major strategy to enhance cellular uptake and overall efficiency in the ERT-based treatment of lysosomal storage diseases.1,2,5 For this purpose, several approaches to tag the enzymes with the M6P-oligosaccharide signals have been attempted, including chemical conjugation of synthetic or natural M6P-containing oligosaccharides and enzymatic phosphorylation to introduce M6P moieties.7–14 For example, conjugation of synthetic M6P oligosaccharides to the recombinant human acid α-glucosidase (rhGAA) through selectively oxidized sialic acid or galactose residues in the N-glycans of the enzyme led to M6P-modified rhGAA that showed an enhanced uptake and demonstrated up to 5-fold greater potency than the unmodified rhGAA in a Pompe disease mouse model.9–11 Despite these promising studies, chemical conjugation suffers from heterogeneity in linkage sites, potential instability of the conjugates, and introduction of unnatural linkers that may be immunogenic in humans. Moreover, as to the molecular recognition between CI-MPR receptor and M6P-glycoproteins, it is still not fully understood how the location of the M6P moiety in the oligosaccharide, the valency of the M6P ligands, and/or the oligosaccharide context affect the binding and affinity for the M6P receptor CI-MPR. A detailed structure–activity relationship study has been hampered by the difficulties in obtaining structurally well defined phosphorylated oligosaccharides and homogeneous M6P-containing glycoproteins relevant to the natural M6P ligands.12,14–17 As an effort to address the problem, we have launched a project aiming to synthesize homogeneous phosphoglycopeptides and phosphoglycoproteins carrying natural M6P-containing glycans and to use them as probes to decipher the ligand structural requirements for M6P receptor recognition. For this purpose, we sought to explore the chemoenzymatic method that we and other groups have previously developed, which takes advantage of the transglycosylation activity of a class of endo-β-N-acetylglucosaminidases (ENGases) and related glycosynthase mutants to ligate a preassembled oligosaccharide (in the form of oligosaccharide oxazoline) to a GlcNAc-peptide or -protein to form a homogeneous glycopeptide or glycoprotein.18–34 This synthetic strategy has been applied for synthesizing complex glycopeptides and for site-specific glycosylation remodeling of glycoproteins including therapeutic antibodies.20,25,26,35–41 However, it remains to be tested whether ENGases are efficient to recognize M6P-containing oligosaccharide oxazoline as donor substrate to synthesize M6P-containing glycopeptides or glycoproteins.42,43 We describe in this paper the chemical synthesis of an array of M6P-containing high-mannose-type N-glycans with varied number and location of the M6P moieties, the enzymatic transfer of the phosphoglycans, and the surface plasmon resonance (SPR) analysis of the binding of the synthetic M6P-containing glycoproteins and bivalent M6P-glycopeptides to the major M6P receptor, CI-MPR. We found that the Endo-A glycosynthase mutant, EndoA-N171A, instead of the wild-type enzyme, could efficiently transfer the synthetic M6P-glycan oxazolines to form glycoproteins carrying large natural M6P-containing N-glycans. Our SPR binding analysis reveals an unexpected selectivity of the binding of CI-MPR to the M6P-glycan isomers, indicating that the location of the M6P moiety in the oligosaccharide context is critical for CI-MPR recognition. The binding analysis with the bivalent glycopeptides shows a remarkable multivalent effect on the affinity of the ligands for the M6P receptor.
RESULTS AND DISCUSSION
Design
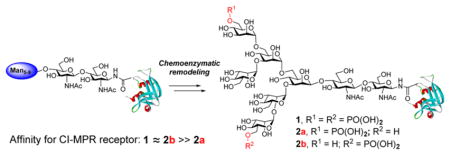
The major objective of this study is to elucidate the detailed structural requirement for the recognition of M6P-contatining glycoprotein ligands by the M6P receptor through chemoenzymatic synthesis and binding studies. The long-term goal is to develop a method that could be applied for glycosylation remodeling of recombinant enzymes, which usually contain few or no M6P terminal sugars, to specific M6P-tagged enzymes for an enhanced ERT for the treatment of lysosomal storage diseases. The overall design is depicted in Figure 1. Selected M6P-oligosaccharide oxazolines corresponding to natural M6P N-glycans with varied number and location of the phosphate groups are chemically synthesized and evaluated as substrates for enzymatic transglycosylation. A recombinant glycoprotein (e.g., a lysosomal enzyme) is deglycosylated by an ENGase. Then the respective M6P-glycan is transferred to the GlcNAc-protein acceptor by an appropriate ENGase or a glycosynthase mutant to yield a homogeneous M6P-containing glycoprotein. Binding studies of these synthetic M6P-containing glycoprotein ligands with the M6P receptor offer an opportunity to probe the detailed structural requirement of the site-specific M6P attachments for M6P receptor recognition.
Figure 1.

Chemoenzymatic glycosylation remodeling for synthesizing mannose-6-phosphorylated glycoproteins.
Chemical Synthesis of the Man6GlcNAc Glycan Oxazoline Carrying Two Terminal Mannose-6-phosphate (M6P) Moieties and Its Glycan Oxazoline Derivative
Our glycosylation remodeling strategy for the construction of homogeneous phosphorylated glycoproteins first requires the synthesis of the M6P-glycan oxazolines as potential substrates for enzymatic transglycosylation. Chemical synthesis of phosphate-containing N-glycans poses a new level of complexity in comparison with unmodified N-glycans. Recently, Chen and co-workers have reported an efficient synthesis of several M6P-containing and GlcNAc-M6P-containing oligosaccharides, employing a late-stage phosphorylation strategy to avoid the difficulties of protecting group manipulations in the presence of the labile benzyl-protected phosphodiesters.12,15 Thus, we decided to use a similar synthetic strategy to introduce the phosphate group at a late stage. The synthesis commenced with the glycosylation between the glycosyl donor (1)12 and the disaccharide acceptor (2),37 under the promotion of N-iodosuccinimide (NIS)/TfOH to provide the tetrasaccharide (3), in which the C6-OH group of the terminal mannose moiety was selectively protected with a TIPS group (Scheme 1). Regioselective benzylidene ring opening was achieved by treatment of 3 with Et3SiH/PhBCl2 to provide tetrasaccharide derivative (4) with a free OH at the C6 of the core β-mannose moiety. Then a trimannose moiety was installed at the C6-OH via glycosylation of 4 with the trimannosyl donor (5)12 under the promotion of NIS and AgOTf to give the heptasaccharide derivative (6). The α-mannoside linkage for the newly generated glycosidic bonds was determined by the relatively large coupling constants (over 170 Hz) between the C-1 and H-1 of all the α-mannosides [the coupling constants (1JCH) for the five α-mannosides appeared at 173.9, 172.2, 171.8, 171.7, and 171.6 Hz, respectively]. Conversion of the 2-azido group into the 2-acetamido group was achieved by treatment of 6 with AcSH to afford the key intermediate (7), where the two potential phosphorylation sites at the C6 of the outer mannose moieties were differentially protected with an acetyl and a TIPS group, respectively, to allow selective phosphorylation. To introduce two phosphate groups, the acetyl and TIPS groups were removed by treatment with MeONa in MeOH and TBAF, respectively, to give the diol (8), which was phosphorylated with dibenzyl N, N-diisopropylphosphoramidite, followed by oxidation with mCPBA to give the bis-phosphorylated derivative (9) in 87% yield in two steps. Global deprotection of the Bn groups via catalytic hydrogenolysis with Pd–C in MeOH and THF, followed by Pd(OH)2-C in MeOH and H2O,12 gave the free oligosaccharide (10) in quantitative yield. Finally, oxazoline ring formation was achieved in a single step by treatment of 10 with an excess amount of 2-chloro-1,3-dimethylimidazolinium chloride (DMC)22,44 in water in the presence of TEA to afford the glycan oxazoline (11) in 90% yield after gel filtration purification (Scheme 1). It was found that the one-step conversion of the phosphorylated glycan into the corresponding glycan oxazoline worked equally efficiently as for neutral N-glycans. The identity of the product was confirmed by MS and NMR (1H, 13C, and 31P) analysis.
Scheme 1. Synthesis of the Bis-M6P Heptasaccharide Oxazoline (11)a.

aReagents and conditions: (a) NIS, TfOH, MS4A, Et2O, −40 °C to rt, 76%. (b) Et3SiH, PhBCl2, 4 Å molecular sieves (MS4A), CH2Cl2, −78 °C, 85%. (c) 5, NIS, AgOTf, MS4A, 0 °C to rt, 97%. (d) AcSH, pyridine, CHCl3, rt, 86%. (e) NaOMe, MeOH, THF, rt. (f) TBAF, THF, rt, 75% in two steps. (g) (BnO)2PiPr2, tetrazole, MS4A, CH2Cl2 then mCPBA, 87%. (h) Pd–C, H2, THF, MeOH then Pd(OH)2–C, H2, MeOH, H2O, quant. (i) DMC, Et3N, H2O, rt, 90%.
To synthesize the two monophosphorylated glycan isomers (15a and 15b), selective deprotection of the C-6 hydroxyl groups on the outer mannose moiety on either arm was first performed, followed by site-specific phosphorylation (Scheme 2). Thus, treatment of 7 with a catalytic amount of MeONa in MeOH and THF resulted in selective removal of the acetyl group on the outer mannose located at the α-1,6-branch of the core. Then the phosphate group was introduced at the C-6 free OH through treatment of 12 with dibenzyl N, N-diisopropylphosphoramide followed by mCPBA to give the fully protected derivative (13). The TIPS group in 13 was removed with TBAF, followed by final global deprotection of the Bn groups via hydrogenolysis to provide the free glycan (14), which was converted into the glycan oxazoline (15a) by reaction with DMC in a single step. On the other hand, the installation of a phosphate at the C6 position in the outer mannose moiety at the α-1,3-branch was achieved by selective deprotection of the TIPS group on that position, followed by site-specific phosphorylation and phosphite oxidation to give 17. Hydrogenolysis of 17 with Pd–C in THF and MeOH and then with Pd(OH)2-C in MeOH and H2O gave the free monophosphorylated glycan (18). Interestingly, the O-acetyl group was simultaneously removed during the hydrogenolysis when Pd(OH)2–C was used as the catalyst, probably due to the basic conditions of Pd(OH)2. The result was confirmed by MS and NMR analysis of the product. Finally, treatment of the reducing oligosaccharide (18) with DMC/TEA in water gave the isomeric monophosphorylated glycan oxazoline (15b) (Scheme 2).
Scheme 2. Synthesis of the Monophosphorylated Heptasaccharide Oxazolines (15a and 15b)a.

aReagents and conditions: (a) NaOMe, MeOH, THF, rt, 86%. (b) (BnO)2PNiPr2, tetrazole, MS4A, CH2Cl2 then mCPBA, 79%. (c) TBAF, THF, rt, 55%. (d) Pd–C, H2, THF, MeOH, rt, 96%. (e) DMC, Et3N, H2O, rt, 88%. (f) TBAF, THF, rt, 80%. (g) (BnO)2PNiPr2, tetrazole, MS4A, CH2Cl2 then mCPBA, 75%. (h) Pd–C, H2, THF, MeOH, rt, then Pd(OH)2–C, H2, MeOH, H2O, 98%. (i) DMC, Et3N, H2O, rt, 98%.
For the synthesis of the bis-phosphorylated Man3GlcNAc core, the disaccharide (19)18,37 was glycosylated with the glycosyl donor (20)12 under the catalysis of TMSOTf to give the tetrasaccharide (21). The O-acetyl group at the C-2 of the glycosyl donor ensures the formation of the α-glycosidic linkage for the newly formed glycosidic bonds via neighboring group participation. Then the O-acetyl groups were changed to the permanent O-Bn protection groups to give 22, which was subsequently converted to the 2-acetamide derivative (23) by reduction of the azide group with AcSH in pyridine. Selective deprotection of the TIPS groups followed by phosphorylation/oxidation gave the fully protected, bis-phosphorylated derivative (25). Global deprotection of 25 by catalytic hydrogenolysis provided free tetrasaccharide 26, which was subjected to the single-step oxazoline formation reaction with DMC/TEA to afford the bis-phosphorylated Man3GlcNAc oxazoline (27) (Scheme 3).
Scheme 3. Synthesis of the Bis-M6P Tetrasaccharide Oxazoline (27)a.

aReagents and conditions: (a) TMSOTf, MS4A, CH2Cl2, −40 °C to rt, 60%. (b) NaOMe, MeOH, THF, rt. (c) NaH, BnBr, DMF, rt, 77% in two steps. (d) AcSH, pyridine, CHCl3, rt, 73%. (e) TBAF, THF, rt, 96%. (f) (BnO)2PNiPr2, tetrazole, MS4A, CH2Cl2 then mCPBA, 70%. (g) Pd–C, H2, THF, MeOH, rt, then Pd(OH)2–C, H2, MeOH, H2O, 97%. (h) DMC, Et3N, H2O, rt, 97%.
Synthesis of Phosphorylated Glycoproteins through Enzymatic Transglycosylation
We have previously shown that a glycosynthase mutant, EndoA-N171A, was able to transfer the sugar oxazoline of natural high-mannose-type N-glycans to a GlcNAc protein acceptor to form a homogeneous glycoprotein with the natural β-1,4-linkage for the newly generated glycosidic bond.20 The use of the glycosynthase mutant is important for the synthesis, as wild-type Endo-A leads to quick hydrolysis of high-mannose-type glycoprotein product. However, it remained to demonstrate whether synthetic phosphorylated high-mannose-type glycan oxazolines could serve as efficient substrates for the enzymatic transglycosylation. To test this point, we chose bovine ribonuclease B, a natural glycoprotein enzyme, as a model system for enzymatic glycosylation remodeling to produce M6P-containing glycosylated enzymes for enhanced M6P-receptor-mediated cellular uptake. Thus, the natural ribonuclease B (28), which contains heterogeneous nonphosphorylated Man5-Man9 glycan at the Asn-34 site, was deglycosylated by wild-type Endo-A to give the homogeneous GlcNAc-RNase (29).45 We found that all three phosphorylated heptasaccharide oxazolines (11, 15a, and 15b) containing one or two M6P moieties acted as good substrates of EndoA-N171A for transglycosylation. Thus, incubation of GlcNAc-RNase (29) and the bis-M6P-containing heptasaccharide oxazoline (11) (molar ratio of donor/acceptor, 6/1) with EndoA-N171A (0.1 μg/μL) in Tris buffer (100 mM, pH 7.1) at 30 °C for 2 h gave 72% of the transglycosylation product (30) (Scheme 4).
Scheme 4.

Enzymatic Glycosylation Remodeling To Synthesize Homogeneous M6P-Containing Glycoproteins
The newly formed glycoprotein was eluted earlier than the starting GlcNAc-RNase (29) under the reverse-phase HPLC (RP-HPLC) condition (see Experimental Section), which was readily purified by RP-HPLC. The identity of the glycoprotein product (30) was confirmed by ESI-MS (calculated, M = 15 222 Da; found, M = 15 224, deconvolution data). Similarly, the enzymatic reactions between GlcNAc-RNase (29) and monophosphorylated glycan oxazoline (15a or 15b) under the catalysis of EndoA-N171A were performed in a similar way as the synthesis of 30 to give the isomeric phosphorylated glycoproteins 31a and 31b, respectively. Again, the identity and purity of the glycoprotein products were confirmed by HPLC and ESI-MS analysis (see the Supporting Information). We found that the transformation of GlcNAc-RNase (29) to the phosphorylated glycoprotein products could be driven to 80–90% when additional sugar oxazoline was added to the reaction mixtures. The enzymatic reaction was readily performed on a milligram scale. Moreover, the excess phosphorylated glycan oxazoline substrate was recovered during RP-HPLC purification of the product with subsequent gel filtration in the form of free oligosaccharide, which was readily converted into the glycan oxazoline in a single step with DMC/Et3N, thus permitting the recycling of glycan oxazoline for transglycosylation. The transfer of the smaller bis-phosphorylated Man3GlcNAc oxazoline (27) to GlcNAc-RNase was carried out with wild-type Endo-A to give the corresponding phosphoglycoprotein (32) in 60% isolated yield. The phosphoglycoprotein (32) carrying the truncated phosphorylated Man3GlcNAc2 turned out to be a poor substrate for Endo-A hydrolysis, thus allowing for an accumulation of the product (32) (Scheme 4). Similar results with the truncated bis-phosphorylated Man3GlcNAc oxazoline (27) were observed and reported recently by Fairbanks and co-workers.43 In contrast, we found that wild-type Endo-A did not work on the larger phosphorylated glycan oxazolines (11, 15a, and 15b) for transglycosylation and led to very low yields of the transglycosylation products, mainly due to the rapid hydrolysis of the transglycosylation products by the wild-type enzyme.
Chemoenzymatic Synthesis of a Cyclic Polypeptide Carrying Two Phosphorylated N-Glycans
To demonstrate the feasibility of the chemoenzymatic method for introducing multiple phosphorylated glycans into polypeptides and also to probe the effects of the number and proximity of the M6P ligands on M6P receptor binding, we synthesized a cyclic polypeptide carrying two M6P-containing oligosaccharides. For this purpose, we chose the HIV-1 V1 V2 cyclic glycopeptide derived from HIV-1 strain ZM109 as a model, which we have previously identified as a neutralizing epitope of the broadly HIV-neutralizing antibody PG9.36 The cyclic peptide (33) containing two GlcNAc moieties at the N160 and N173 sites was synthesized by following the previously reported procedure.36 We first tested the enzymatic transfer of the bis-phosphorylated Man6GlcNAc glycan by EndoD-N322Q mutant, which was previously shown to be efficient to transfer of Man5GlcNAc glycan to GlcNAc-peptide substrate.36 Interestingly, the Endo-D glycosynthase mutant was unable to recognize the phosphorylated Man6GlcNAc oxazoline as a substrate for glycan transfer to the acceptor, probably due to the restricted substrate specificity of the Endo-D mutant. Then we examined EndoA-N171A for double transglycosylation and found that EndoA-N171A could efficiently achieve double glycosylation of the GlcNAc-peptide acceptor (33). When an excess amount of the phosphoglycan oxazoline (11) (5 mol equiv per GlcNAc acceptor) was used, a moderate yield of the cyclic glycopeptide 34 carrying two M6P-containing N-glycans was achieved (Scheme 5). In addition, the corresponding linear glycopeptide (35) was synthesized by reduction of the disulfide with DTT followed by alkylation of the free cysteine residues with iodoacetamide (Scheme 5). The corresponding linear glycopeptide was prepared for probing the cooperative effect of the two M6P-containing glycans on the affinity for M6P receptor. The cyclic and linear phosphoglycopeptides (34 and 35) were purified by HPLC, and their identity was confirmed by LC–MS analysis.
Scheme 5.

Synthesis of Double-Glycosylated M6P-Containing Glycopepides
Binding Studies with the CI-MPR
With the successful synthesis of the M6P-containing glycoproteins and glycopeptides, we performed a binding study with the cation-independent mannose phosphate receptor (CI-MPR) using SPR technology. The CI-MPR was immobilized on the chips and the different glycoforms were probed as analytes at 10 °C. As revealed by the binding experiments (Figure 2), the glycoprotein carrying a bis-M6P-Man6GlcNAc2 glycan (30) was efficiently recognized by the CI-MPR, with a deduced KD of 28.9 nM. Unexpectedly, the phosphorylated glycoprotein (31a) carrying a M6P at the α-1,6-branched arm of the glycan core did not show measurable binding affinity within the tested concentrations (15.6–1000 nM), suggesting that the affinity could be well above micromolar concentrations. In contrast, the other isomer (31b) carrying a M6P at the low α-1,3-branched arm of the core showed a high affinity for CI-MPR (KD = 61 nM), which was comparable to that of the glycoprotein (30) carrying two M6P moieties (at both the α-1,6 and α-1,3 arms). The difference in affinity of the two M6P isomers for the receptor was remarkable and unexpected. These results suggest that a single M6P moiety at the low α-1,3-branched arm of the core is sufficient to achieve the high affinity, and the M6P at the upper α-1,6-arm of the glycan core is dispensable for the binding to the receptor. On the other hand, the KD of the glycoprotein (32) carrying the unnatural bis-M6P Man3GlcNAc2 for the CI-MPR was 168 nM, which was about 5-fold weaker than that of the glycoprotein (30) carrying the larger natural M6P-containing N-glycan.
Figure 2.

SPR binding data of the CI-MPR with M6P-containing glycopeptides. Synthetic glycoproteins and glycopeptides analytes were flowed over an immobilized chip with 2-fold serial dilution of the highest concentration of 1 μM. The black line is the fitting curve. (a) Man6-2P-RNaseB, (b) Man6-1Pa-RNaseB, (c) Man6-2Pb-RNaseB, and (d) Man3-2P-RNaseB.
The binding of the cyclic glycopeptide carrying two M6P-phosphoglycans (34) revealed another important observation related to the multivalent effect (Figure 3). We found that the cyclic glycopeptide (35) showed very tight binding to CI-MPR (KD = 2.35 nM), the affinity of which was more than 10-fold higher than that of the best phosphoglycoprotein ligand (30) carrying one M6P-phosphoglycan. This result reveals a clear beneficial clustering effect in CI-MPR recognition. Interestingly, reducing the disulfide bond in the cyclic glycopeptide (34) to the corresponding linear glycopeptide (35) (KD = 46 nM) resulted in a significant (ca. 20-fold) decrease of the affinity for CI-MPR. It is known that receptor CI-MPR has three carbohydrate recognition sites, including two high-affinity sites located in domains 3 and 9 and a low-affinity site within domain 5.2,46,47 Our experimental data suggest that not only the number of the M6P moieties in the context but also the proximity of the M6P ligands is critical for achieving high-affinity binding to the receptor by simultaneous interactions at the (high-affinity) carbohydrate binding sites.
Figure 3.

SPR binding data of the CI-MPR with M6P-containing glycopeptides. The ZM109 sequence of HIV-1 V1 V2 loop of gp120 was used as a model peptide for installment of double glycan at N160 and N173 positions. Synthetic glycopeptides were flowed over an immobilized chip with 2-fold serial dilution of the highest concentration of 1 μM. The black line is the fitting curve. (a) Bis(Man6-2P)-cyclic glycopeptide (34) and (b) bis(Man6-2P)-linear glycopeptide (35).
CONCLUSION
An efficient chemoenzymatic method for the synthesis of homogeneous M6P-containing glycopeptides and M6P-glycoproteins is established. It is demonstrated that the endoglycosynthase mutant, EndoA-N171A, is capable of transferring preassembled M6P-containing high-mannose glycan oxazoline to a GlcNAc-protein to reconstitute homogeneous M6P-containing glycoproteins with all natural linkages, in contrast with typical chemical conjugations that often introduce unnatural linkages. We have also demonstrated that multiple M6P-containing glycans can be introduced in a polypeptide by simultaneous transglycosylations to obtain phosphorylated glycopeptides carrying two M6P-containing oligosaccharides. The binding studies reveal a previously undiscovered high-affinity M6P ligand, indicating that a single M6P moiety in the terminus of the α-1,3-branch of the oligomannose context is sufficient for a high-affinity binding (with a nanomolar affinity), while the presence of a M6P moiety at the α-1,6-branch of the core is dispensable. The successful synthesis of the M6P glycoform isomers and the bivalent M6P-glycopeptides, coupled with binding studies, made it possible to probe the detailed structural requirements of the glycoprotein ligands in M6P receptor recognition in terms of the precise location and the proximity of the M6P moieties in the context of the high-mannose N-glycans. The chemoenzymatic method described here provides an exciting new avenue for glycosylation remodeling of recombinant enzymes to enhance the cellular uptake and delivery of enzymes to lysosomes in ERT for the treatment of lysosomal storage diseases. Further studies along this line are in progress.
EXPERIMENTAL SECTION
Materials
The EndoA-N171A and EndoD-N322Q glycosynthase mutants were expressed following the previously reported procedures.20,48 Bovine pancreatic ribonuclease B was purchased from Sigma-Aldrich (St. Louis, MO). Recombinant IGF-II R (CI-MPR) was purchased from R&D Systems. TLC was performed using silica gel on aluminum plates (Sigma-Aldrich). Flash column chromatography was performed on silica gel 60 (230–400 mesh). SDS–PAGE was performed using 18% (w/v) gel. NMR spectra were recorded on a Bruker 500 MHz spectrometer. The chemical shifts were assigned in ppm. The ESI-MS spectra were measured on a Micromass ZQ-4000 single quadruple mass spectrometer and LQX linear ion trap mass spectrometer (Thermo Scientific). High-resolution (HR) mass spectra were collected with a JEOL AccuTOF-CS (ESI-TOF) equipped with an ESI source and a time-of-flight (TOF) detector. Analytical RP-HPLC was performed on a Waters 626 HPLC instrument with a C18 column (3.5 μm, 4.6 × 250 mm) at 40 °C. The column was eluted with a linear gradient of 24–33% aq MeCN containing 0.1% TFA for 30 min at the flow rate of 0.5 mL/min. Preparative HPLC was performed with a Waters 600 HPLC instrument and a Waters C18 column (5.0 μm, 10 × 250 mm). The column was eluted with a suitable gradient of MeCN–H2O containing 0.1% TFA at a flow rate of 4 mL/min.
Synthesis of Tetrasaccharide 3
A mixture of 1 (361.8 mg, 0.44 mmol), 2 (576.5 mg, 0.53 mmol), and 4 Å molecular sieves (MS4A) (1.20 g) in Et2O (12 mL) was stirred for 30 min and cooled at −40 °C. Then NIS (155.6 mg, 0.69 mmol) and TfOH (0.33 M in Et2O, 161 μL, 0.05 mmol) were added. The resulting mixture was stirred for 7 h under argon at −40 °C. After warming up to rt, the mixture was filtered through a Celite pad. The filtrate was diluted with EtOAc; washed with sat. aq NaHCO3, 5% aq Na2S2O3, and brine; dried over Na2SO4; and filtered. The filtrate was concentrated to dryness. The residue was subjected to flash chromatography on silica gel (hexane/EtOAc = 95/5 to 80/20) to give the tetrasaccharide (3) (621.6 mg, 76%). MALDI-TOF MS: calcd for C110H125N3O20SiNa [M + Na]+, 1858.85; found, 1858.85. 1H NMR (CDCl3, 500 MHz): δ 7.38–7.19 (m, 54H), 6.99 (t, 1H, J = 7.32 Hz), 5.44 (s, 1H), 5.34 (s, 1H), 5.22 (s, 1H), 5.04 (d, 1H, J = 10.4 Hz), 4.91 (d, 1H, J = 12.1 Hz), 4.90 (d, 1H, J = 10.7 Hz), 4.84 (d, 1H, J = 11.2 Hz), 4.79 (d, 1H, J = 11.7 Hz), 4.77 (d, 1H, J = 11.7 Hz), 4.67 (d, 1H, J = 11.7 Hz), 4.63–4.33 (m, 15H), 4.26 (m, 2H), 4.09 (1H, t, J = 9.6 Hz), 4.03–3.78 (m, 8H), 3.69–3.44 (m, 10H), 3.31 (t, 1H, J = 9.3 Hz), 3.20 (d, 1H, J = 9.3 Hz), 2.98 (dt, 1H, J = 9.3, 5.1 Hz), 1.07 (m, 3H), 1.03 (s, 18H). 13C NMR (CDCl3, 125 MHz): δ 139.12, 138.85, 138.46, 138.41, 138.38, 137.85, 137.71, 137.20, 136.82, 128.84, 128.54, 128.43, 128.37, 128.33, 128.25, 128.17, 128.13, 128.10, 128.06, 127.99, 127.92, 127.86, 127.74, 127.65, 127.61, 127.54, 127.47, 127.29, 127.23, 125.71, 101.32, 101.11, 100.37, 99.81, 97.52, 81.53, 79.65, 78.84, 78.52, 76.53, 75.16, 76.07, 75.00, 74.93, 74.71, 74.47, 74.28, 73.67, 73.43, 73.39, 73.00, 72.20, 72.05, 71.43, 70.78, 69.72, 68.38, 68.18, 67.05, 65.69, 62.29, 18.07, 18.03, 12.05.
Synthesis of Tetrasaccharide 4
A mixture of 3 (360.0 mg, 0.20 mmol) and MS4A (0.7 g) in CH2Cl2 (7 mL) was stirred for 30 min and cooled at −78 °C. Et3SiH (156 μL, 0.98 mmol) and PhBCl2 (127 μL, 0.98 mmol) were added. The resulting mixture was stirred for 2.5 h under argon at −78 °C. Et3N (273 μL, 1.96 mmol) was added and the resulting mixture was stirred for 15 min at −78 °C and then filtered through a Celite pad. The filtrate was diluted with CH2Cl2, washed with sat. aq NaHCO3 and brine, dried over Na2SO4, and filtered. The filtrate was concentrated to dryness. The residue was subjected to flash chromatography on silica gel (hexane/EtOAc = 97/3 to 80/20) to obtain alcohol 4 (305.0 mg, 85%). MALDI-TOF MS: calcd for C110H127N3O20SiNa [M + Na]+, 1860.86; found, 1860.87. 1H NMR (CDCl3, 500 MHz): δ 7.38–7.14 (m, 55H), 5.19 (s, 1H), 5.09 (s, 1H), 4.98 (d, 1H, J = 10.8 Hz), 4.92–4.89 (m, 3H), 4.80 (d, 1H, J = 11.5 Hz), 4.74 (1H, d, 11.5 Hz), 4.68–4.65 (m, 4H), 4.59–4.41 (m, 14H), 4.24 (d, 1H, J = 8.2 Hz), 4.17 (t, 1H, J = 9.6 Hz), 4.00 (s, 1H), 3.90–3.85 (m, 5 H), 3.75–3.69 (m, 3H), 3.64–3.42 (m, 9H), 3.26 (t, 1H, J = 10.0 Hz), 3.15 (m, 2H), 2.96 (m, 1H), 1.06 (m, 3H), 1.02 (s, 18H). 13C NMR (CDCl3, 125 MHz): δ 138.99, 138.80, 138.50, 138.40, 138.36, 138.08, 137.89, 137.71, 136.95, 128.47, 128.46, 128.42, 128.32, 128.25, 128.20, 128.01, 127.96, 127.93, 127.87, 127.84, 127.70, 127.64, 127.61, 127.46, 127.35, 127.22, 127.18, 101.31, 100.65, 100.26, 98.55, 81.46, 81.39, 79.46, 78.62, 75.53, 75.10, 74.97, 74.88, 74.73, 74.65, 74.50, 74.20, 73.77, 73.43, 73.36, 73.34, 72.88, 72.35, 72.10, 71.63, 70.69, 69.92, 65.80, 62.50, 61.84, 18.02, 17.97, 12.01.
Synthesis of Heptasaccharide 6
A mixture of 4 (514.9 mg, 0.28 mmol) and 5 (636.6 mg, 0.42 mmol) in Et2O (20 mL) containing MS4A (2.00 g) was stirred for 30 min and cooled at 0 °C. NIS (157.6 mg, 0.70 mmol) and AgOTf (14.4 mg, 0.06 mmol) were added. The resulting mixture was stirred for 3 h under argon at 0 °C to room temperature and then filtered through a Celite pad. The filtrate was diluted with EtOAc; washed with sat. aq NaHCO3, 5% aq Na2S2O3, and brine; dried over Na2SO4; and filtered. The filtrate was concentrated to dryness. The residue was subjected to flash chromatography on silica gel (hexane/EtOAc = 95/5 to 80/20) to obtain the heptasaccharide 6 (860.7 mg, 97%). MALDI-TOF: [M + Na]+ calcd for C193H214N3O36Si, M = 31 9946; found, 3199.44. 1H NMR (CDCl3, 500 MHz): δ 7.30–7.03 (m, 100H), 5.27 (s, 1H), 5.20 (s, 1H), 5.11 (s, 1H), 4.39–4.88 (m, 4H), 4.84 (d, 1H, J = 12.1 Hz), 4.83 (d, 1H, J = 11.2 Hz), 4.79 (d, 2H, 11.8 Hz), 4.74 (d, 1H, J = 12.1 Hz), 4.71 (s, 1H), 4.65 (d, 1H, J = 10.7 Hz), 4.58–4.55 (m, 4H), 4.52–4.32 (m, 31H), 4.25 (d, 1H, J = 11.8 Hz), 4.21–4.15 (m, 4H), 4.07–4.05 (m, 2H), 3.99 (t, 1H, J = 9.7 Hz), 3.92–3.85 (m, 11H), 3.81–3.78 (m, 4H), 3.71 (d, 2H, J = 15.6 Hz), 3.62–3.43 (m, 16H), 3.36 (d, 1H, J = 10.7 Hz), 3.18 (t, 1H, J = 9.0 Hz), 3.10 (m, 2H), 3.00 (d, 1H, J = 11.8 Hz), 1.95 (s, 3H), 1.20 (m, 3H), 0.99 (s, 27H). 13C NMR (CDCl3, 125 MHz): δ 170.90, 139.02, 138.88, 138.79, 138.74, 138.49, 138.39, 138.12, 137.87, 125.68, 128.54, 128.48, 128.36, 128.27, 128.18, 128.13, 128.06, 127.99, 127.88, 127.82, 127.69, 127.59, 127.46, 127.38, 127.33, 127.18, 127.09, 126.98, 126.84, 101.40, 100.80, 100.34, 100.11, 98.34, 98.17, 97.33, 81.50, 81.19, 80.32, 79.99, 79.85, 79.41, 79.12, 78.73, 78.33, 75.28, 75.12, 75.07, 75.00, 74.80, 74.75, 74.67, 74.56, 74.27, 74.14, 74.07, 73.75, 73.35, 73.21, 73.03, 72.96, 72.62, 72.41, 72.18, 72.04, 71.82, 71.60, 71.44, 71.01, 70.63, 70.24, 69.98, 69.85, 69.30, 68.13, 66.41, 65.51, 65.16, 63.42, 63.30, 62.28, 34.13, 32.77, 31.91, 31.42, 30.18, 30.02, 29.68, 29.43, 29.34, 29.23, 29.11, 27.56, 27.43, 27.32, 27.12, 24.90, 22.67, 20.82, 18.03, 17.97, 14.08, 11.99.
Synthesis of Heptasaccharide 7
A mixture of 6 (830.0 mg, 0.26 mmol), AcSH (4 mL), and pyridine (4 mL) in CHCl3 (4 mL) was stirred for 24 h at room temperature and then warmed at 60 °C and stirred for 24 h. The resulting mixture was concentrated and subjected to flash chromatography on silica gel (hexane/EtOAc = 90/10 to 50/50) to the acetamide (7) (719.6 mg, 86%). MALDI-TOF: [M + Na]+ calcd for C195H217NO37SiNa, 3215.48; found, 3215.48. 1H NMR (CDCl3, 500 MHz): δ 7.30–7.04 (m, 100H), 5.55 (d, 1H, J = 7.0 Hz), 5.22 (s, 1H), 5.20 (s, 1H), 5.12 (s, 1H), 5.00 (s, 1H), 4.99 (1H, s), 4.94 (d, 1H, J = 11.9 Hz), 4.89 (d, 2H, J = 10.7 Hz), 4.84–4.76 (m, 5H), 4.72 (s, 1H), 4.70 (d, 1H, J = 12.5 Hz), 4.65 (d, 1H, J = 10.8 Hz), 4.61 (d, 1H, 11.7 Hz), 4.53–4.30 (m, 26H), 4.24 (d, 1H, J = 11.6 Hz), 4.21–4.17 (m, 3H), 4.08–3.77 (m, 15H), 3.70–3.39 (m, 15H), 3.29 (1H, m), 3.20–3.15 (m, 2H), 1.94 (s, 3H), 1.42 (s, 3H), 1.22 (m, 3H), 0.99 (s, 27H). 13C NMR (CDCl3, 125 MHz): δ 170.90, 170.21, 139.18, 139.04, 138.79, 138.77, 138.68, 138.60, 138.53, 138.48, 138.3, 138.29, 138.12, 138.03, 137.93, 137.81, 128.55, 128.43, 128.29, 128.17, 128.09, 127.98, 127.85, 127.81, 127.70, 127.59, 127.45, 127.33, 127.30, 127.20, 127.08, 101.35, 100.66, 99.95, 99.18, 98.05, 96.73, 81.45, 80.24, 80.14, 80.01, 79.45, 79.20, 78.57, 78.21, 75.45, 75.10, 74.90, 74.72, 74.64, 74.35, 74.10, 73.74, 73.36, 73.28, 73.17, 73.10, 72.90, 72.65, 72.43, 72.15, 72.09, 71.81, 71.48, 71.10, 70.87, 70.03, 69.65, 69.27, 68.76, 66.58, 65.39, 63.37, 62.33, 56.32, 29.69, 23.11, 20.81, 18.02, 17.96, 11.99.
Synthesis of Diol 8
To a solution of 7 (111.7 mg, 0.035 mmol) in MeOH (3 mL) and THF (0.6 mL) was added NaOMe (25 wt % in MeOH, 8 μL, 0.035 mmol) at room temperature. The reaction mixture was stirred at room temperature for 7 h and then neutralized with Dowex and filtered. The filtrate was concentrated to dryness. To the solution of the residue in THF (3 mL) was added TBAF (1 M in THF, 175 μL, 0.18 mmol) at room temperature. The reaction mixture was stirred for 48 h at room temperature. The resulting mixture was diluted with EtOAc, washed with H2O and brine, dried over Na2SO4, and filtered. The filtrate was concentrated to dryness. The residue was subjected to flash chromatography on silica gel (hexane/EtOAc = 75/25 to 33/67) to obtain the diol 8 (77.7 mg, 75%). MALDI-TOF: [M + Na]+ calcd for C184H195NO36Na, 3017.34; found, 3017.33. 1H NMR (CDCl3, 500 MHz): δ 7.29–7.05 (m, 100H), 5.68 (d, 1H, J = 7.0 Hz), 5.24 (s, 1H), 5.16 (s, 1H), 5.05 (d, 1H, J = 7.7 Hz), 4.95–4.78 (m, 10H), 4.75 (d, 1H, J = 11.4 Hz), 4.69 (d, 1H, J = 11.4 Hz), 4.60–4.32 (m, 32H), 4.27 (d, 1H, J = 12.1 Hz), 4.13–4.09 (m, 2H), 4.01–3.52 (m, 36H), 3.43 (m, 2H), 3.20 (m, 2H), 1.45 (s, 3H). 13C NMR (CDCl3, 125 MHz): δ 170.34, 139.20, 138.79, 138.62, 138.58, 138.53, 138.48, 138.42, 138.38, 138.33, 138.30, 138.23, 138.16, 137.93, 137.87, 137.76, 128.56, 128.50, 128.40, 128.32, 128.26, 128.22, 128.09, 128.06, 128.03, 127.96, 127.91, 127.79, 127.75, 127.64, 127.59, 127.51, 127.43, 127.36, 127.30, 127.25, 127.20, 127.04, 101.20, 100.50, 100.05, 99.93, 99.09, 97.96, 96.80, 81.83, 80.02, 79.59, 79.24, 78.69, 78.32, 75.76, 75.47, 75.43, 75.21, 75.10, 75.07, 75.00, 74.92, 74.88, 74.80, 74.74, 74.71, 73.97, 73.39, 73.35, 73.16, 72.97, 72.71, 72.66, 72.59, 72.42, 72.39, 72.26, 72.22, 72.15, 72.07, 71.87, 71.73, 71.32, 70.97, 69.50, 69.30, 68.66, 66.74, 65.19, 62.19, 62.15, 56.75, 29.69, 23.12.
Synthesis of Bisphosphate 9
A mixture of diol 8 (74.0 mg, 0.024 mmol) and MS4A (0.2 g) in CH2Cl2 (2 mL) was stirred for 30 min. Tetrazole (3 wt % in MeCN, 440 μL, 0.14 mol) and (BnO)2PNiPr2 (42 μL, 0.12 mmol) were added. The resulting mixture was stirred for 2.5 h under argon at room temperature and then cooled at 0 °C for 10 min. mCPBA (77 wt %, 32.2 mg, 0.14 mmol) was added, and the reaction mixture was stirred at 0 °C for 1.5 h and then filtered through a Celite pad. The filtrate was diluted with CH2Cl2; washed with 5% aq Na2S2O3, sat. aq NaHCO3, and brine; dried over Na2SO4; and filtered. The filtrate was concentrated to dryness. The residue was subjected to flash chromatography on silica gel (hexane/EtOAc = 90/10 to 50/50) to obtain bisphosphate 9 (71.0 mg, 87%). MALDI-TOF: [M + Na]+ calcd for C212H221NO42P2Na, 3537.46; found, 3537.44. 1H NMR (CDCl3, 500 MHz): δ 7.33–7.02 (m, 120H), 5.71 (d, 1H, J = 7.2 Hz), 5.23 (s, 1H), 5.18 (s, 1H), 5.07 (s, 1H), 5.03–4.75 (m, 19H), 4.68 (d, 1H, J = 12.0 Hz), 4.60–4.33 (m, 30H), 4.29 (d, 1H, J = 12.1 Hz), 4.23 (d, 1H, J = 12.1 Hz), 4.16–3.45 (m, 40H), 3.35 (m, 1H), 3.28 (m, 1H), 3.15 (d, 1H, J = 12.1 Hz), 1.43 (3H, s). 13C NMR (CDCl3, 125 MHz): δ 170.28, 139.18, 138.78, 138.66, 138.53, 138.50, 138.37, 138.35, 138.26, 138.19, 138.16, 138.07, 138.01, 137.95, 137.86, 136.05, 135.99, 135.93, 135.89, 135.84, 128.62, 128.42, 128.40, 128.35, 128.31, 128.26, 128.15, 128.07, 128.00, 127.91, 127.88, 127.81, 127.77, 127.63, 127.58, 127.51, 127.44, 127.32, 127.23, 127.20, 127.14, 127.09, 127.03, 101.11, 100.52, 99.92, 99.26, 99.23, 98.24, 96.67, 81.70, 80.07, 80.00, 79.84, 79.46, 79.11, 78.77, 78.24, 75.44, 75.31, 75.20, 75.08, 74.90, 74.84, 74.72, 74.68, 74.66, 74.39, 73.91, 73.77, 73.62, 73.33, 73.25, 73.11, 72.70, 72.64, 72.50, 72.41, 72.08, 71.99, 71.80, 71.69, 71.48, 71.43, 71.10, 70.80, 69.44, 69.23, 69.19, 69.12, 69.08, 69.04, 66.45, 65.54, 56.05, 33.70, 31.91, 30.15, 29.69, 29.37, 26.69, 23.13, 22.69, 14.17, 14.10. 31P NMR (CDCl3, 162 MHz): δ −1.22, −1.40.
Synthesis of the Free Heptasaccharide Carrying Two Phosphates (10)
A mixture of 9 (68.0 mg, 0.019 mmol) and Pd–C (23 mg) in MeOH (1.5 mL) and THF (0.3 mL) was stirred under H2 atmosphere for 21 h. The reaction mixture was filtered through a Celite pad. The filtrate was concentrated to dryness. The mixture of the residue and Pd(OH)2–C in MeOH (1.5 mL) and H2O (1 mL) was stirred under H2 atmosphere for 21 h. The reaction mixture was filtered through a Celite pad. The filtrate was dissolved in H2O and lyophilized. The crude product was purified on a Sephadex LH-20 column by elution with H2O. Fractions containing the product were pooled and lyophilized to give the free heptasaccharide 10 (28.0 mg, quant.). HR-MS (ESI-TOF): [M – H]− calcd for C44H76NO42P, 1352.3323; found, 1352.3329. 1H NMR (D2O, 500 MHz): δ 5.24 (s, 1H), 5.09 (d, 1H, J = 2.1 Hz), 4.92 (d, 1H, J = 12.5 Hz), 4.88 (s, 1H), 4.76 (s, 1H), 4.72 (s, 1H), 4.65 (s, 1H), 4.55 (1H, d, J = 8.4 Hz), 4.10–3.47 (m, 38H), 3.41 (2H, d, J = 6.7 Hz), 3.38 (d, 1H, J = 6.7 Hz), 1.89 (s, 3H); 13C NMR (D2O, 125 MHz): δ 174.51, 102.61, 102.47, 101.04, 100.43, 100.27, 100.07, 95.58, 99.49, 95.26, 90.37, 81.59, 80.95, 80.90, 80.35, 79.90, 79.30, 78.86, 78.66, 74.71, 74.48, 73.54, 73.46, 72.36, 72.30, 72.21, 71.72, 71.66, 70.94, 70.88, 70.61, 70.51, 70.38, 70.29, 70.04, 69.68, 69.65, 67.18, 66.92, 66.71, 66.49, 66.37, 65.93, 65.79, 65.31, 64.97, 64.60, 62.64, 61.25, 60.33, 60.21, 53.95, 22.43, 22.10, 21.76. 31P NMR (D2O, 162 MHz): δ 0.73 (overlapped).
Synthesis of the Bis-M6P Heptasaccharide Oxazoline (11)
To a solution of free heptasaccharide 10 (28.0 mg, 0.021 mmol) in H2O (2.3 mL) were added Et3N (130 μL, 0.93 mmol) and 2-chloro-1,3-dimethylimidazolinium chloride (DMC) (52.4 mg, 0.31 mmol) at 0 °C. The reaction mixture was monitored by DIONEX HPAEC-PAD. Within 1 h, the HPAEC analysis indicated that the free oligosaccharide was converted into a new oligosaccharide that was eluted earlier than the reducing sugar under the HPAEC conditions. The product was purified by gel filtration on a Sephadex G-10 column that was eluted with 0.1% aq Et3N to afford compound 11 (27.8 mg, 90%) after lyophilization together with NaOH (2.6 mg). HR-MS (ESI-TOF): [M – H]− calcd for C44H75NO41P2, 1334.3217; found, 1334.3223. 1H NMR (D2O, 500 MHz): δ 6.10 (d, 1H, J = 7.1 Hz), 5.40 (s, 1H), 5.15 (s, 1H), 5,04 (1H, s), 4,95 (s, 1H), 4.92 (s, 1H), 4.75 (s, 1H), 4.38 (s, 1H), 4.21–3.64 (m, 40H), 3.42 (m, 1H), 2.07 (s, 3H). 13C NMR (D2O, 125 MHz): δ 170.18, 139.14, 139.06, 139.01, 138.80, 138.64, 138.56, 138.51, 138.49, 138.39, 138.30, 138.02, 137.97, 137.93, 137.82, 101.38, 100.81, 99.93, 99.20, 98.39, 98.26, 96.77, 81.46, 80.28, 80.04, 79.44, 78.54, 78.15, 75.48, 75.42, 75.38, 75.11, 75.03, 74.81. 74.74, 74.71, 74.67, 74.61, 74.56, 74.41, 74.21, 74.09, 73.73, 73.59, 73.27, 73.19, 73.05, 72.89, 72.62, 72.44, 72.19, 72.10, 72.07, 71.84, 71.73, 71.48, 71.26, 70.78, 69.65, 69.19, 68.82, 66.43, 65.30, 62.72, 62.31, 56.07, 29.69, 23.08, 18.03, 18.00, 17.97, 12.03, 12.00. 31P NMR (D2O, 162 MHz): δ 4.49 (overlapped).
Synthesis of Heptasaccharide 12
To a solution of 7 (320.0 mg, 0.10 mmol) in MeOH (5 mL) and THF (1 mL) was added NaOMe (25 wt % in MeOH, 23 μL, 0.10 mmol) at room temperature. The reaction mixture was stirred at room temperature for 7 h and then neutralized with Dowex and filtered. The filtrate was concentrated to dryness. The residue was subjected to flash chromatography on silica gel (hexane/EtOAc = 85/15 to 50/50) to obtain alcohol 12 (271.0 mg, 86%). MALDI-TOF: [M + Na]+ calcd for C193H215NO36SiNa, 3173.47; found, 3173.47. 1H NMR (CDCl3, 500 MHz): δ 7.31–7.05 (m, 100H), 5.67 (d, 1H, J = 7.1 Hz), 5.30 (s, 1H), 5.23 (s, 1H), 5.20 (s, 1H), 5.11 (s, 1H), 5.09–5.00 (m, 3H), 4.96–4.22 (m, 5H), 4.89 (d, 1H, J = 10.9 Hz), 4.86 (d, 1H, J = 10.5 Hz), 4.83–4.76 (4H, m), 4.73 (s, 1H), 4.70 (d, 1H, J = 11.9 Hz), 4.65 (d, 1H, J = 11.2 Hz), 4.59 (d, 1H, J = 11.9 Hz), 4.55–4.35 (m, 25H), 4.29 (d, 1H, J = 11.6 Hz), 4.24 (d, 1H, J = 11.6 Hz), 4.21–4.27 (m, 3H), 4.09–3.77 (m, 17H), 3.70–3.40 (m, 18H), 3.22–3.11 (m, 3H), 1.44 (3H, s), 1.21 (m, 3H), 1.00 (s, 18H). 13C NMR (CDCl3, 125 MHz): δ 170.24, 139.18, 138.82, 138.66, 138.59, 138.54, 138.45, 138.42, 138.39, 138.32, 138.30, 138.24, 138.18, 137.95, 137.89, 137.74, 128.60, 128.52, 128.44, 128.30, 128.29, 128.26, 128.10, 128.05, 127.96, 127.90, 127.78, 127.74, 127.67, 127.60, 127.52, 127.40, 127.38, 127.30, 127.26, 127.21, 127.05, 101.21, 100.53, 100.05, 99.98, 99.04, 97.98, 96.81, 81.82, 80.02, 79.60, 79.25, 78.70, 78.31, 75.77, 75.48, 75.43, 75.21, 75.10, 75.07, 75.00, 74.92, 74.88, 74.80, 74.74, 74.68, 73.97, 73.39, 73.34, 73.17, 72.97, 72.71, 72.66, 72.59, 72.41, 72.40, 72.28, 72.21, 72.14, 72.08, 71.88, 71.74, 71.35, 70.98, 69.45, 69.32, 68.67, 66.75, 65.20, 62.18, 62.15, 56.74, 29.70, 23.11, 18.01, 17.97, 11.99.
Synthesis of the Heptasaccharide Carrying a Monophosphate (13)
A mixture of 12 (153.0 mg, 0.05 mmol) and MS4A (0.35 g) in CH2Cl2 (3.5 mL) was stirred for 30 min. Tetrazole (3 wt % in MeCN, 505 μL, 0.17 mol) and (BnO)2PNiPr2 (49 μL, 0.15 mmol) were added. The resulting mixture was stirred for 2.5 h under argon at room temperature and then cooled at 0 °C for 10 min. mCPBA (38.1 mg, 0.17 mmol) was added and the reaction mixture was stirred at 0 °C for 1.5 h and then filtered through a Celite pad. The filtrate was diluted with CH2Cl2; washed with 5% aq Na2S2O3, sat. aq NaHCO3, and brine; dried over Na2SO4; and filtered. The filtrate was concentrated to dryness. The residue was subjected to flash chromatography on silica gel (hexane/EtOAc = 85/15 to 45/55) to obtain the monophosphate 13 (130.5 mg, 79%). MALDI-TOF: [M + Na]+ calcd for C207H228NO39PSiNa, 3433.53; found, 3433.52. 1H NMR (CDCl3, 500 MHz): δ 7.31–7.05 (m, 110H), 5.67 (d, 1H, J = 7.1 Hz), 5.23 (s, 1H), 5.20 (s, 1H), 5.11 (s, 1H), 5.05 (d, 1H, J = 7.6 Hz), 4.96 (d, 1H, J = 12.1 Hz), 4.93–4.84 (m, 5H), 4.82–4.76 (9H, m), 4.69 (d, 1H, J = 11.9 Hz), 4.65 (d, 1H, J = 11.1 Hz), 4.57–4.35 (m, 29H), 4.32 (d, 1H, J = 12.3 Hz), 4.28 (d, 1H, J = 11.8 Hz), 4.20 (t, 1H, J = 9.8 Hz), 4.09–3.77 (m, 18H), 3.74–3.49 (m, 16H), 3.45–3.40 (m, 2H), 3.30 (m, 1H), 3.18–3.11 (m, 2H), 1.43 (3H, s), 1.21 (m, 3H), 1.00 (s, 18H). 13C NMR (CDCl3, 125 MHz): δ 170.15, 139.05, 138.73, 138.55, 138.50, 138.45, 138.32, 138.30, 138.18, 138.12, 138.09, 138.01, 137.97, 137.80, 128.60, 128.42, 128.28, 128.10, 128.00, 127.85, 127.82, 127.75, 127.65, 127.52, 127.43, 127.30, 127.28, 127.22, 127.19, 127.05, 101.12, 100.45, 99.92, 99.26, 99.21, 98.11, 96.87, 96.78, 81.65, 80.13, 79.97, 79.45, 79.23, 78.80, 78.35, 75.89, 75.45, 75.32, 75.25, 75.01, 74.89, 74.70, 74.68, 74.61, 74.52, 74.27, 74.16, 73.90, 73.53, 73.33, 73.27, 73.14, 72.73, 72.68, 72.45, 72.15, 72.10, 72.05, 71.83, 71.77, 71.44, 71.13, 70.80, 70.10, 69.47, 69.26, 69.15, 66.65, 65.49, 63.34, 33.28, 29.79, 27.55, 24.73, 23.10, 20.84, 18.00, 17.97, 12.00. 31P NMR (CDCl3, 162 MHz): δ −1.23
Synthesis of the Free Heptasaccharide Carrying a Monophosphate (14)
To a solution of phosphate (114.0 mg, 0.033 mmol) in THF (2 mL) was added TBAF (1 M in THF, 334 μL, 0.33 mmol) at room temperature. The reaction mixture was stirred for 24 h at room temperature. The resulting mixture was diluted with EtOAc, washed with H2O and brine, dried over Na2SO4, and filtered. The filtrate was concentrated to dryness. The residue was subjected to flash chromatography on silica gel (hexane/EtOAc = 80/20 to 40/60) to obtain the de-TIPS product (59.7 mg, 55%). MALDI-TOF MS: [M + Na]+ calcd for C198H208NO39PNa, 3277.40; found, 3277.40. 1H NMR (CDCl3, 500 MHz): δ 7.31–7.05 (m, 110H), 5.69 (d, 1H, J = 7.1 Hz), 5.22 (s, 1H), 5.00 (d, 1H, J = 6.8 Hz), 4.96–4.92 (m, 4H), 4.87–4.74 (m, 6H), 4.69 (d, 1H, J = 12.1 Hz), 4.93–4.84 (m, 5H), 4.82–4.76 (9H, m), 4.69 (d, 1H, J = 11.8 Hz), 4.59–4.38 (m, 24H), 4.34 (d, 1H, J = 12.2 Hz), 4.28 (d, 1H, J = 11.8 Hz), 4.23 (d, 1H, J = 11.8 Hz), 4.18 (m, 1H), 4.09–3.96 (m, 4H), 3.94–3.65 (m, 17H), 3.61–3.44 (m, 13H), 3.31 (m, 1H), 3.20–3.17 (m, 2H), 2.26 (t, 1H, J = 6.7 Hz), 1.44 (3H, s). 13C NMR (CDCl3, 125 MHz): δ 170.18, 139.09, 138.80, 138.65, 138.50, 138.42, 138.35, 138.27, 138.20, 138.10, 138.09, 138.01, 137.96, 137.84, 128.64, 128.45, 128.27, 128.13, 128.10, 128.00, 127.89, 127.83, 127.77, 127.62, 127.51, 127.42, 127.30, 127.27, 127.20, 127.17, 127.04, 101.11, 100.56, 99.93, 99.25, 99.20, 98.10, 96.85, 96.80, 81.64, 80.10, 79.98, 79.43, 79.20, 78.77, 78.32, 75.91, 75.50, 75.30, 75.25, 75.02, 74.90, 74.72, 74.69, 74.62, 74.50, 74.27, 74.16, 73.91, 73.55, 73.32, 73.26, 73.12, 72.70, 72.63, 72.50, 72.13, 72.10, 72.01, 71.82, 71.70, 71.45, 71.11, 70.82, 70.03, 69.46, 69.27, 69.12, 66.67, 65.47, 63.40, 33.27, 29.80, 27.58, 24.73, 23.09, 20.84. 31P NMR (CDCl3, 162 MHz): δ −1.24
The de-TIPS product (56.2 mg, 0.017 mmol) was subjected to catalytic hydrogenation with Pd/C (20 mg) in MeOH (2.5 mL) and THF (0.5 mL) under a H2 atmosphere for 21 h. The reaction mixture was filtered through a Celite pad. The filtrate was concentrated to dryness. The residue was loaded on Sephadex LH-20 column and eluted with H2O. Fractions containing the product were pooled and lyophilized to give the free heptasaccharide with monophosphate 14 (20.7 mg, 96%). HR-MS (ESI-TOF): [M – H]− calcd for C44H75NO40P, 1272.3659; found, 1272.3674. 1H NMR (D2O, 500 MHz): δ 5.37 (s, 1H), 5.26 (s, 1H), 5.10 (d, 1H, J = 11.6 Hz), 5.07 (s, 1H), 4.92 (s, 1H), 4.88 (1H, s), 4.81 (s, 1H), 4.72 (1H, d, J = 8.0 Hz), 4.26 (m, 1H), 4.20–3.99 (m, 7H), 3.99–3.62 (m, 32H), 2.06 (s, 3H). 13C NMR (D2O, 125 MHz): δ 174.51, 102.48, 102.40, 101.02, 100.59, 100.52, 100.25, 100.03, 99.54, 95.44, 95.24, 90.38, 80.89, 80.44, 80.01, 78.56, 74.66, 74.52, 73.60, 73.47, 23.38, 7241, 72.36, 71.81 70.95, 70.62, 70.46, 70.27, 70.20, 70.07, 69.68, 69.15, 67.19, 66.90, 66.39, 65.74, 65.27, 64.30, 61.24, 61.15, 60.25, 53.93, 23.29, 22.10 (selected peaks). 31P NMR (D2O, 162 MHz): δ 0.79.
Synthesis of the Mono-M6P Heptasaccharide Oxazoline (15a)
To a solution of the free heptasaccharide 14 (7.4 mg, 5.8 μmol) in H2O (1 mL) were added Et3N (45 μL, 0.32 mmol) and 2-chloro-1,3-dimethylimidazolinium chloride (DMC) (17.5 mg, 0.11 mmol) at 0 °C. The reaction mixture was monitored by DIONEX HPAEC-PAD. Within 1 h, the HPAEC analysis indicated that the free oligosaccharide was converted into a new oligosaccharide that eluted earlier than the reducing sugar under the HPAEC conditions. The product was purified by gel filtration on a Sephadex G-10 column that was eluted with 0.1% aq Et3N to afford compound 15a (6.9 mg, 88%) after lyophilization together with NaOH (0.5 mg). HR-MS (ESI-TOF): [M – H]− calcd for C44H74NO38P, 1254.3554; found, 1254.3537. 1H NMR (D2O, 500 MHz): δ 6.10 (d, 1H, J = 7.2 Hz), 5.35 (s, 1H), 5.15 (s, 1H), 5.05 (s, 1H), 4,93 (s, 1H), 4.91 (s, 1H), 4.73 (s, 1H), 4.37 (1H, s), 4.21–3.67 (m, 39H), 3.58 (m, 1H), 3.43 (m, 1H), 2.08 (s, 3H). 13C NMR (D2O, 125 MHz): δ 168.73, 102.69, 102.42, 101.41, 100.86, 100.17, 99.90, 99.50, 80.26, 79.02, 78.80, 78.57, 78.04, 74.46, 73.46, 72.87, 71.15, 71.05, 70.72, 70.54, 70.34, 70.26, 70.16, 70.03, 69.63, 69.44,67.14, 66.96, 66.71, 66.26, 66.09, 65.94, 65.49, 65.25, 63.26, 61.81, 61.17, 13.12 (selected peaks). 31P NMR (D2O, 162 MHz): δ 4.51.
De-O-silylation of 7
To a solution of 7 (372.4 mg, 0.12 mmol) in THF (5 mL) was added TBAF (1 M in THF, 720 μL, 0.72 mmol). The reaction mixture was stirred for 48 h at room temperature and diluted with EtOAc, washed with H2O and brine, dried over Na2SO4, and filtered. The filtrate was concentrated to dryness. The residue was subjected to flash chromatography on silica gel (hexane/EtOAc = 85/15 to 40/60) to obtain alcohol 16 (264.0 mg, 75%). MALDI-TOF: [M + Na]+ calcd for C186H197NO37Na, 3059.35; found, 3059.35. 1H NMR (CDCl3, 500 MHz): δ 7.30–7.04 (m, 100H), 5.56 (d, 1H, J = 6.9 Hz), 5.53 (s, 1H), 5.23 (s, 1H), 5.15 (s, 1H), 5.12 (s, 1H), 5.01 (s, 1H), 5.00 (d, 1H, J = 12.1 Hz), 4.96 (1H, s), 4.93 (d, 1H, J = 11.9 Hz), 4.89 (d, 1H, J = 10.7 Hz), 4.87 (s, 1H), 4.85 (d, 1H, J = 11.9 Hz), 4.84 (d, 1H, J = 11.9 Hz), 4.81–4.77 (m, 3H), 4.75 (d, 1H, J = 12.1 Hz), 4.70 (d, 1H, J = 12.1 Hz), 4.60 (d, 1H, J = 11.9 Hz), 4.57–4.30 (m, 30H), 4.24 (d, 1H, J = 12.2 Hz), 4.17 (br s, 1H), 4.09–4.02 (m, 2H), 3.99 (d, 1H, J = 8.9 Hz), 3.95–3.82 (m, 13H), 3.77–3.65 (m, 7H), 3.61–3.44 (m, 13H), 3.31 (m, 1H), 3.24 (d, 1H, J = 11.9 Hz), 3.19 (t, 1H, J = 7.9 Hz), 2.37 (br s, 1H), 1.94 (s, 3H), 1.44 (3H, s). 13C NMR (CDCl3, 125 MHz): δ 170.90, 170.23, 139.20, 138.76, 138.65, 138.61, 138.53, 138.47, 138.34, 138.16, 138.13, 137.92, 137.80, 128.54, 128.46, 128.39, 128.29, 128.21, 128.17, 128.09, 127.97, 127.85, 127.82, 127.75, 127.63, 127.59, 127.43, 127.39, 127.35, 127.30, 127.14, 127.03, 101.22, 100.63, 100.04, 99.91, 99.19, 98.07, 96.76, 81.79, 80.00, 79.60, 79.48, 79.21, 78.58, 78.20, 76.14, 75.77, 75.57, 75.43, 75.19, 75.07, 75.00, 74.81, 74.70, 74.65, 74.39, 74.10, 73.72, 73.34, 73.10, 72.95, 72.70, 72.64, 72.58, 72.40, 72.20, 72.08, 71.83, 71.72, 71.43, 71.12, 70.83, 70.04, 69.46, 69.24, 68.73, 67.96, 67.42, 66.48, 65.48, 63.35, 62.12, 56.31, 33.25, 29.67, 25.59, 23.13, 20.81.
Synthesis of the Monophosphate 17
A mixture of alcohol 16 (234.0 mg, 0.08 mmol) and MS4A (0.5 g) in CH2Cl2 (5 mL) was stirred for 30 min. Then tetrazole (3 wt % in MeCN, 800 μL, 0.27 mol) and (BnO)2PNiPr2 (78 μL, 0.23 mmol) were added. The resulting mixture was stirred for 2 h under argon at room temperature and then cooled at 0 °C for 10 min. mCPBA (77 wt %, 60.4 mg, 0.27 mmol) was added, and the reaction mixture was stirred at 0 °C for 1 h and then filtered through a Celite pad. The filtrate was diluted with CH2Cl2; washed with 5% aq Na2S2O3, sat. aq NaHCO3, and brine; dried over Na2SO4; and filtered. The filtrate was concentrated to dryness. The residue was subjected to flash chromatography on silica gel (hexane/EtOAc = 80/20 to 55/45) to obtain the monophosphate 17 (183.7 mg, 72%). MALDI-TOF MS: [M + Na]+ calcd for C200H210NO40PNa, 3319.41; found, 3319.41. 1H NMR (CDCl3, 500 MHz): δ 7.30–7.04 (m, 110H), 5.57 (d, 1H, J = 9.3 Hz), 5.23 (s, 1H), 5.18 (s, 1H), 5.06 (1H, s), 5.02 (1H, s), 5.01–4.96 (m, 3H), 4.94–4.75 (m, 10H), 4.67 (d, 1H, J = 11.8 Hz), 4.60 (d, 1H, J = 11.8 Hz), 4.57–4.31 (m, 29H), 4.24 (d, 1H, J = 11.8 Hz), 4.20–3.82 (m, 23H), 3.78–3.44 (m, 18H), 3.35 (m, 1H), 3.27 (t, 1H, J = 8.1 Hz), 3.23 (d, 1H, J = 11.2 Hz), 1.93 (s, 3H), 1.42 (s, 3H). 13C NMR (CDCl3, 125 MHz): δ 170.89, 170.16, 139.17, 138.79, 138.68, 138.51, 138.45, 138.37, 138.27, 138.20, 138.12, 138.06, 138.00, 137.96, 137.85, 128.61, 128.41, 128.29, 128.16, 128.09, 128.02, 127.88, 127.82, 127.77, 127.63, 127.52, 127.45, 127.33, 127.29, 127.20, 127.16, 127.08, 101.11, 100.54, 99.92, 99.27, 99.22, 98.08, 96.87, 96.76, 81.68, 80.11, 80.01, 79.47, 79.20, 78.76, 78.24, 75.92, 75.45, 75.32, 75.20, 75.08, 74.88, 74.73, 74.70, 74.65, 74.51, 74.29, 74.13, 73.92, 73.61, 73.33, 73.25, 73.12, 72.71, 72.66, 72.49, 72.15, 72.09, 72.00, 71.79, 71.68, 71.44, 71.10, 70.83, 70.04, 69.44, 69.26, 69.05, 66.66, 65.46, 63.37, 33.26, 29.70, 27.59, 24.74, 23.09, 20.82. 31P NMR (CDCl3, 162 MHz): δ −1.32.
Synthesis of the Free Heptasaccharide with Monophosphate (18)
Compound 17 (160.0 mg, 0.049 mmol) was dissolved in MeOH (3 mL) and THF (0.5 mL) containing Pd–C (80 mg), and the mixture was stirred under a H2 atmosphere for 48 h. The reaction mixture was filtered through a Celite pad. The filtrate was concentrated to dryness. The residue was dissolved in MeOH (3 mL) and H2O (0.5 mL) containing Pd(OH)2–C (80 mg) and the mixture was stirred under a H2 atmosphere for 48 h. The reaction mixture was filtered through a Celite pad. The filtrate was dissolved in H2O and lyophilized. The residue was subject to gel filtration on a Sephadex LH-20 column which was eluted with H2O. Fractions containing the product were pooled and lyophilized to give the free heptasaccharide with monophosphate 18 (62.3 mg, quant.). HR-MS (ESI-TOF): [M – H]− calcd for C44H75NO40P, 1272.3659; found, 1272.3666. 1H NMR (D2O, 500 MHz): δ 5.41 (s, 1H), 5.26 (s, 1H), 5.11 (d, 1H, J = 12.6 Hz), 5.03 (s, 1H), 4.92(s, 1H), 4.88 (1H, s), 4.81 (s, 1H), 4.70 (1H, d, J = 8.0 Hz), 4.25 (m, 1H), 4.20–4.16 (m, 2H), 4.11–3.65 (m, 37H), 2.05 (s, 3H). 13C NMR (D2O, 125 MHz): δ 174.84, 102.64, 102.47, 101.02, 100.38, 100.15, 100.02, 99.50, 95.25, 90.43, 80.78, 80.18, 79.28, 78.64, 74.46, 73.47, 72.88, 72.36, 71.02 70.73, 70.54, 70.40, 70.31, 70.10, 69.65, 69.15, 67.18, 66.90, 66.76, 66.32, 65.93, 65.41 64.77, 64.66, 61.12., 60.19, 53.94, 22.40, 22.07 (selected peaks). 31P NMR (D2O, 162 MHz): δ 0.87.
Synthesis of the Mono-M6P Heptasaccharide Oxazoline (15b)
To a solution of the free heptasaccharide 18 (30.0 mg, 0.024 mmol) in H2O (2 mL) were added Et3N (148 μL, 1.06 mmol) and 2-chloro-1,3-dimethylimidazolinium chloride (DMC) (59 mg, 0.35 mmol) at 0 °C. The reaction mixture was monitored by DIONEX HPAEC-PAD. After 1.5 h, the HPAEC analysis indicated that the free oligosaccharide was converted into a new oligosaccharide that eluted earlier than the reducing sugar under the HPAEC conditions. The product was purified by gel filtration on a Sephadex G-10 column that was eluted with 0.1% aq Et3N to afford compound 15b (30.8 mg, 98%) after lyophilization together with NaOH (1.9 mg). ESI-MS: [M + H]+ calcd for C44H75NO38P, 1256.37; found, 1278.15 [M + Na]+. 1H NMR (D2O, 500 MHz): δ 6.10 (d, 1H, J = 7.2 Hz), 5.43 (s, 1H), 5.17 (s, 1H), 5.04 (1H, s), 4.94 (s, 2H), 4.81 (s, 1H), 4.39 (s, 1H), 4.21–3.67 (m, 40H), 3.43 (m, 1H), 2.08 (s, 3H). 13C NMR (D2O, 125 MHz): δ 168.73, 102.69, 102.42, 101.41, 100.86, 100.17, 99.90, 99.50, 80.26, 79.02, 78.80, 78.57, 78.04, 74.46, 73.46, 72.87, 71.15, 71.05, 70.72, 70.54, 70.34, 70.26, 70.16, 70.03, 69.63, 69.44, 67.14, 66.96, 66.71, 66.26, 66.09, 65.94, 65.49, 65.25, 63.26, 61.81, 61.17, 13.12 (selected peaks). 31P NMR (D2O, 162 MHz): δ 4.21.
Synthesis of Tetrasaccharide 21
A mixture of disaccharide 19 (375.5 mg, 0.46 mmol), imidate 20 (1291.3 mg, 1.84 mmol), and MS4A (1.8 g) in CH2Cl2 (18 mL) was stirred for 30 min and cooled at −40 °C. TMSOTf (0.45 M in CH2Cl2, 408 μL, 0.18, mmol) was added. The resulting mixture was stirred for 3 h under argon at −40 °C and then filtered through a Celite pad. The filtrate was diluted with CH2Cl2, washed with sat. aq NaHCO3 and brine, dried over Na2SO4, and filtered. The filtrate was concentrated to dryness. The residue was subjected to flash chromatography on silica gel (hexane/EtOAc = 100/0 to 90/10) to give the tetrasaccharide (21) (489.6 mg, 60%). MALDI-TOF: [M + Na]+ calcd for C109H139N3O220Si2Na, 1920.93; found, 1920.92. 1H NMR (CDCl3, 500 MHz): δ 7.34–7.11 (m, 45H), 5.41 (s, 1H), 5.33 (s, 1H), 5.07 (s, 1H), 4.99 (d, 1H, J = 11.8 Hz), 4.89 (d, 1H, J = 11.8 Hz), 4.87 (d, 1H, J = 11.8 Hz), 4.86 (d, 1H, J = 11.8 Hz), 4.83 (d, 1H, J = 11.8 Hz), 4.72–4.57 (m, 10H), 4.47 (d, 1H, J = 11.3 Hz), 4.45 (d, 1H, J = 11.8 Hz). 4.43 (d, 1H, J = 11.8 Hz), 4.39 (d, 1H, J = 11.8 Hz), 4.27 (d, 1H, J = 11.3 Hz), 4.25 (d, 1H, J = 8.3) Hz, 4.02–3.91 (m, 5H), 3.85 (dd, 1H, J = 9.9, 3.2 Hz), 3.84–3.56 (m, 11H), 3.50–3.45 (m, 3H), 3.34 (t, 1H, J = 9.0 Hz), 3.31 (m, 1H), 3.20 (m, 1H), 2.04 (s, 3H), 1.99 (s, 3H), 1.08–1.03 (m, 42H). 13C NMR (CDCl3, 125 MHz): δ 170.10, 170.08, 138.97, 138.87, 138.59, 138.55, 138.04, 137.91, 137.85, 137.78, 136.92, 128.52, 128.38, 128.35, 128.30, 128.21, 128.14, 128.04, 128.01, 127.94, 127.86, 127.75, 127.66, 127.62, 127.46, 127.41, 127.30, 127.26, 127.23, 100.91, 100.55, 99.19, 97.74, 80.68, 80.50, 78.20, 77.87, 77.84, 75.08, 75.02, 74.87, 74.78, 74.65, 74.42, 74.15, 73.72, 73.60, 73.53, 72.92, 71.79, 71.40, 70.79, 68.80, 68.52, 66.27, 65.80, 62.46, 62.22, 29.68, 20.88, 20.84, 18.03, 18.00, 17.97, 17.92, 12.04, 12.02.
Synthesis of Tetrasaccharide 22
To a solution of 21 (489.6 mg, 0.26 mmol) in MeOH (7.5 mL) and THF (2.5 mL) was added NaOMe (25 wt % in MeOH, 59 μL, 0.26 mmol) at room temperature. The reaction mixture was stirred at room temperature for 22 h and then neutralized with AcOH (15 μL, 0.26 mmol). The resulting mixture was concentrated to dryness in vacuo. The residue was dissolved in DMF (10 mL), and NaH was added at room temperature. The reaction mixture was stirred at room temperature for 30 min and then BnBr (123 μL, 1.03 mmol) was added. The mixture was stirred at room temperature for 2 h, diluted with EtOAc/hexane (1/1), washed with H2O and brine, dried over Na2SO4, and filtered. The filtrate was concentrated to dryness. The residue was subjected to flash chromatography on silica gel (hexane/EtOAc = 99/1 to 85/15) to obtain benzyl ether 22 (394.0 mg, 77%). MALDI-TOF MS: [M + Na]+ calcd for C119H147N3O20Si2Na, 2017.00; found, 2017.01. 1H NMR (CDCl3, 500 MHz): δ 7.35–7.11 (m, 55H), 5.17 (s, 1H), 4.94 (d, 1H, J = 12.2 Hz), 4.91 (d, 1H, J = 11.8 Hz), 4.89 (d, 1H, J = 11.8 Hz), 4.88 (s, 1H), 4.87 (d, 1H, J = 11.8 Hz), 4.82 (d, 1H, J = 11.8 Hz), 4.76 (d, 1H, J = 11.8 Hz), 4.71 (d, 1H, J = 11.8 Hz), 4.65–4.39 (m, 15H), 4.32 (d, 1H, J = 11.8 Hz), 4.23 (d, 1H, J = 7.9 Hz), 4.03–3.78 (m, 10H), 3.73–3.65 (m, 6H), 3.61–3.59 (m, 2H), 3.46–3.42 (m, 3H), 3.31–3.27 (m, 2H), 3.09 (m, 1H), 1.26 (s, 3H), 1.07–1.00 (m, 42H). 13C NMR (CDCl3, 125 MHz): δ 139.04, 138.92, 138.90, 138.79, 138.66, 138.55, 138.50, 138.16, 137.82, 136.87, 100.96, 100.4, 99.73, 98.26, 80.82, 80.22, 79.96, 79.62, 79.07, 75.90, 75.36, 75.1, 74.96, 74.87, 74.82, 74.79, 74.47, 74.40, 74.21, 73.90, 73.49, 73.45, 72.38, 72.28, 72.23, 71.38, 70.81, 68.60, 65.98, 65.93, 63.18, 62.68, 29.69, 18.06, 18.05, 18.00, 17.96, 12.01, 11.98.
Synthesis of Tetrasaccharide 23
A mixture of 22 (370.0 mg, 0.19 mmol), AcSH (2 mL), and pyridine (2 mL) in CHCl3 (2 mL) was stirred for 48 h at room temperature. The resulting mixture was concentrated and subjected to flash chromatography on silica gel (hexane/EtOAc = 95/5 to 75/25) to obtain acetamide 23 (271.3 mg, 73%). MALDI-TOF MS: [M + Na]+ calcd for C121H151NO21Si2Na, 2033.02; found, 2033.02; 1H NMR (CDCl3, 500 MHz): δ 7.34–7.12 (m, 55H), 5.49 (d, 1H, J = 8.1 Hz), 5.19 (s, 1H), 4.90 (d, 1H, J = 6.2 Hz), 4.89 (d, 1H, J = 11.8 Hz), 4.86 (d, 1H, J = 11.8 Hz), 4.86 (d, 1H, J = 11.8 Hz), 4.85 (d, 1H, 10.7 Hz), 4.75 (s, 1H), 4.74 (d, 1H, J = 11.8 HZ), 4.74 (d, 1H, J = 11.8 Hz), 4.62–4.39 (m, 16H), 4.33 (d, 1H, J = 11.8 Hz), 4.04 (t, 1H, J = 9.6 Hz), 4.00 (m, 2H), 3.91–3.66 (m, 17H), 3.51–3.47 (m, 2H), 3.35 (d, 1H, J = 10.5 Hz), 3.21 (m, 1H), 1.45 (s, 3H), 1.08–1.00 (m, 42H). 13C NMR (CDCl3, 125 MHz): δ 169.90, 139.21, 139.02, 138.87, 138.81, 138.64, 138.49, 138.44, 138.09, 138.04, 137.88, 128.37, 128.27, 128.19, 128.04, 127.82, 127.79, 127.73, 127.69, 127.62, 127.59, 127.50, 127.45, 127.39, 127.25, 127.21, 127.09, 126.73, 100.84, 99.86, 99.27, 98.08, 80.30, 80.00, 79.92, 79.13, 76.56, 75.90, 75.50, 75.32, 75.29, 75.20, 74.77, 74.68, 74.63, 74.57, 74.28, 73.41, 73.23, 73.06, 72.39, 72.29, 71.71, 70.59, 69.50, 66.18, 63.36, 62.45, 54.04, 29.69, 23.09, 18.25, 18.06, 18.04, 18.00, 17.95, 12.22, 11.99.
Synthesis of Tetrasaccharide 24
To a solution of acetamide 23 (247.8 mg, 0.12 mmol) in THF (5 mL) was added TBAF (1 M in THF, 985 μL, 0.99 mmol). The reaction mixture was stirred for 48 h at room temperature and diluted with EtOAc, washed with H2O and brine, dried over Na2SO4, and filtered. The filtrate was concentrated to dryness. The residue was subjected to flash chromatography on silica gel (hexane/EtOAc = 65/35 to 0/100) to obtain diol 24 (199.8 mg, 96%). MALDI-TOF: [M + Na]+ calcd for C103H111NO21Na, 1720.75; found, 1720.75. 1H NMR (CDCl3, 500 MHz): δ 7.35–7.10 (m, 55H), 5.24 (d, 1H, J = 7.52 Hz), 5.18 (s, 1H), 4.98 (s, 1H), 4.92–4.84 (m, 6H), 4.72 (m, 1H), 4.64–4.42 (m, 16H), 4.37 (t, 1H, J = 11.3 Hz), 4.11–4.05 (m, 2H), 3.95–3.88 (m, 3H), 3.84–3.81 (m, 2H), 3.76–3.54 (m, 15H), 3.45 (m, 1H), 3.29 (m, 1H), 1.51 (s, 3H). 13C NMR (CDCl3, 125 MHz): δ 170.31, 139.15, 138.67, 138.54, 138.46, 138.31, 138.14, 137.96, 137.76, 128.49, 128.47, 128.33, 128.29, 128.24, 128.14, 128.07, 128.03, 127.78, 127.59, 127.55, 127.53, 127.42, 127.38, 127.31, 126.76, 100.97, 100.27, 99.14, 98.04, 80.30, 79.94, 79.88, 78.68, 77.36, 75.71, 75.48, 75.34, 75.25, 75.18, 75.07, 74.99, 74.90, 74.81, 74.49, 73.71, 73.56, 73.11, 72.75, 72.72, 72.43, 72.37, 71.80, 70.76, 69.04, 66.75, 62.41, 61.93, 55.45, 29.68, 23.21.
Synthesis of Tetrasaccharide 25
A mixture of diol 24 (176.4 mg, 0.10 mmol) and MS4A (0.4 g) in CH2Cl2 (4 mL) was stirred for 30 min. Tetrazole (3 wt % in MeCN, 1.85 mL, 0.62 mmol) and (BnO)2PNiPr2 (175 μL, 0.52 mmol) were added. The resulting mixture was stirred for 2.5 h under argon at room temperature and then cooled at 0 °C for 10 min. mCPBA (77 wt %, 139.7 mg, 0.62 mmol) was added and the reaction mixture was stirred at 0 °C for 1 h and then filtered through a Celite pad. The filtrate was diluted with CH2Cl2; washed with 5% aq Na2S2O3, sat. aq NaHCO3, and brine; dried over Na2SO4; and filtered. The filtrate was concentrated to dryness. The residue was subjected to flash chromatography on silica gel (hexane/EtOAc = 70/30 to 40/60) to obtain bisphosphate 25 (144.4 mg, 63%). MALDI-TOF: [M + Na]+ calcd for C131H137NO27P2Na, 2240.88; found, 2240.87. 1H NMR (CDCl3, 500 MHz): δ 7.36–7.05 (m, 75H), 5.19 (d, 1H, J = 7.8 Hz), 5.11 (s, 1H), 5.10 (d, 1H, J = 11.8 Hz), 5.07–4.80 (m, 15H), 4.65 (d, 1H, J = 11.8 Hz), 4.63 (s, 1H), 4.62 (d, 1H, J = 11.8 Hz), 4.57–4.41 (m, 13H), 4.37 (d, 1H, J = 11.8 Hz), 4.34 (d, 1H, J = 11.8 Hz), 4.28 (d, 1H, J = 11.8 Hz), 4.26 (m, 1H), 4.17 (m, 1H), 4.05 (d, 2H, J = 7.0 Hz), 4.01–3.97 (m, 3H), 3.91–3.58 (m, 12H), 3.47 (m, 1H), 3.45 (d, 1H, J = 10.4 Hz), 3.32 (m, 1H), 1.49 (s, 3H). 13C NMR (CDCl3, 125 MHz): δ 170.13, 139.21, 138.60, 138.44, 138.40, 138.36, 138.31, 138.23, 138.10, 138.03, 137.94, 135.83, 128.68, 128.48, 128.45, 128.38, 128.31, 128.20, 128.05, 128.01, 127.92, 127.87, 127.82, 127.79, 127.74, 127.72, 127.67, 127.62, 127.56, 127.50, 127.42, 127.36, 127.30, 126.52, 100.19, 100.06, 99.19, 98.02, 80.62, 80.19, 79.87, 78.69, 77.89, 76.26, 75.63, 75.49, 75.17, 74.92, 74.81, 74.71, 74.61, 74.33, 74.05, 73.88, 73.37, 72.57, 72.30, 71.91, 71.80, 71.76, 70.84, 70.75, 69.30, 69.26, 69.16, 69.11, 69.05, 68.96, 68.92, 67.29, 67.25, 66.95, 66.37, 66.17, 56.41, 29.68, 23.27. 31P NMR (CDCl3, 162 MHz): δ −0.97, −1.28.
Synthesis of the Free Tetrasaccharide with Bisphosphate (26)
A mixture of 25 (114.0 mg, 0.051) and Pd–C (23 mg) in MeOH (3 mL) and THF (0.5 mL) was stirred under H2 atmosphere for 24 h. The reaction mixture was filtered through a Celite pad. The filtrate was concentrated to dryness. A mixture of the residue and Pd(OH)2–C in MeOH (3 mL) and H2O (1 mL) was stirred under H2 atmosphere for 21 h. The reaction mixture was filtered through a Celite pad. The filtrate was dissolved in H2O and lyophilized. The crude product was purified on a Sephadex LH-20 column by elution with H2O. Fractions containing the product were pooled and lyophilized to give the free heptasaccharide with bisphosphate 26 (43.3 mg, 97%). HR-MS (ESITOF): [M – H]− calcd for C26H46NO27P2, 866.1738; found, 866.1725. 1H NMR (D2O, 500 MHz): δ 5.18 (d, 1H, J = 3.2 Hz), 5.08 (s, 1H), 4.88 (s, 1H), 4.76 (s, 1H), 4.68 (m, 1H), 4.20 (s, 1H), 4.20–4.02 (m, 4H), 3.95–3.82 (m, 6H), 3.78–3.67 (m, 8H), 3.64–3.58 (m, 3H), 3.53–3.50 (m, 2H), 2.02 (s, 3H). 13C NMR (D2O, 125 MHz): δ 174.74, 102.64, 100.37, 99.94, 90.62, 80.62, 80.23, 74.68, 74.17, 72.44, 72.22, 71.68, 70.33, 70.11, 69.21, 66.60, 66.37, 66.03, 64.80, 64.39, 62.64, 60.27, 53.76, 22.42, 22.09 (selected peaks). 31P NMR (D2O, 162 MHz): δ 0.71 (overlapped).
Synthesis of the Bis-M6P Tetrasaccharide Oxazoline (27)
To a solution of the free tetrasaccharide (26) (20.0 mg, 0.023 mmol) in H2O (2 mL) were added Et3N (145 μL, 1.04 mmol) and 2-chloro-1,3-dimethylimidazolinium chloride (DMC) (58.5 mg, 0.35 mmol) at 0 °C. The reaction mixture was monitored by DIONEX HPAEC-PAD. Within 1 h, the HPAEC analysis indicated that the free oligosaccharide was converted into a new oligosaccharide that eluted earlier than the reducing sugar under the HPAEC conditions. The product was purified by gel filtration on a Sephadex G-10 column that was eluted with 0.1% aq Et3N to afford compound 27 (21.7 mg, 97%) after lyophilization together with NaOH (2.8 mg). HR-MS (ESI-TOF): [M – H]− calcd for C26H45NO26P2, 848.1632; found, 848.1664. 1H NMR (D2O, 500 MHz): δ 6.10 (d, 1H, J = 7.0 Hz), 5.10 (s, 1H), 4.95 (s, 1H), 4.90 (s, 1H), 4.79 (m, 1H), 4.39 (s, 1H), 4.21 (1H, s), 4.11–4.05 (m, 3H), 4.01–3.65 (m, 18H), 3.58 (m, 1H), 3.43 (1H, m), 2.08 (s, 3H). 13C NMR (D2O, 125 MHz): δ 161.68, 102.81, 100.47, 100.07, 90.62, 80.81, 80.46, 80.13, 74.17, 72.89, 72.25, 70.16 69.15, 66.17, 65.91, 63.24, 62.89, 60.27, 53.80, 22.10 (selected peaks). 31P NMR (D2O, 162 MHz): δ 4.00 (overlapped signals).
Synthesis of Bis-phosphoglycoprotein 30
A solution of oxazoline 11 (3.1 mg, 2.2 μmol) and GlcNAc-RNase (29) (5.0 mg, 0.36 μmol) in Tris buffer (100 mM, pH 7.1, 200 μL) was incubated with the Endo-A mutant (N171A) (20 μg) at 30 °C. The reaction was monitored by reverse-phase HPLC. After 2 h, the product was purified by HPLC to afford glycoprotein 30 (3.56 mg, 65%). Analytical RP-HPLC: tR = 26.4 min (gradient, 5–50% aq MeCN containing 0.1% TFA for 30 min; flow rate, 0.5 mL/min). ESI-MS: calcd, M = 15 222 Da; found (m/z), 1692.35 [M + 9H]9+, 1523.10 [M + 10H]10+, 1384.64 [M+ 11H]11+, 1269.65 [M+ 12H]12+. Deconvolution of the ESI-MS: M = 15 221.0 Da.
Synthesis of Phosphoglycoprotein 31a
A solution of oxazoline 15a (2.9 mg, 2.2 μmol) and GlcNAc-RNaseB (5.0 mg, 0.36 μmol) in Tris buffer (100 mM, pH 7.1, 200 μL) was incubated with the Endo-A mutant (N171A) (20 μg) at 30 °C. The reaction was monitored by reverse-phase HPLC. After 2 h, the product was purified by HPLC to afford glycoprotein 31a (3.82 mg, 70%). Analytical RP-HPLC: tR = 26.5 min (gradient, 5–50% aq MeCN containing 0.1% TFA for 30 min; flow rate, 0.5 mL/min); ESI-MS: calcd, M = 15 142 Da; found (m/z), 1514.86 [M + 10H]10+, 1377.21 [M + 11H]11+, 1262.47 [M + 12H]12+, 1165.46 [M + 13H]13+. Deconvolution of the ESI-MS: M = 15 139.0 Da.
Synthesis of Phosphoglycoprotein 31b
A solution of oxazoline 15b (2.9 mg, 2.2 μmol) and GlcNAc-RNaseB (5.0 mg, 0.36 μmol) in Tris buffer (100 mM, pH 7.1, 200 μL) was incubated with the Endo-A mutant (N171A) (20 μg) at 30 °C. The reaction was monitored by reverse-phase HPLC. After 2 h, the product was purified by HPLC to afford glycoprotein 31b (3.93 mg, 72%). Analytical RP-HPLC: tR = 26.5 min (gradient, 5–50% aq MeCN containing 0.1% TFA for 30 min; flow rate, 0.5 mL/min). ESI-MS: calcd, M = 15 142 Da; found (m/z), 1515.18 [M + 10H]10+, 1377.45 [M + 11H]11+, 1262.75 [M + 12H]12+, 1165.61 [M + 13H]13+. Deconvolution of the ESI-MS: M = 15 141.5 Da.
Synthesis of Phosphoglycoprotein 32
A solution of oxazoline 27 (2.1 mg, 2.2 μmol) and GlcNAc-RNase B (5.0 mg, 0.36 μmol) in Tris buffer (100 mM, pH 7.1, 200 μL) was incubated with Endo-A (20 μg) at 30 °C. After 2 h, another portion of oxazoline 27 (2.1 mg, 2.2 μmol) was added to reaction mixture. The reaction was monitored by reverse-phase HPLC. After 2 h, the product was purified by HPLC to afford glycoprotein 32 (3.2 mg, 60%). Analytical RP-HPLC: tR = 26.6 min (gradient, 5–50% aq MeCN containing 0.1% TFA for 30 min; flow rate, 0.5 mL/min). ESI-MS: calcd, M = 14 736 Da; found (m/z), 1638.38 [M + 9H]9+, 1474.66 [M + 10H]10+, 1340.43 [M + 11H]11+, 1229.16 [M + 12H]12+. Deconvolution of the ESI-MS: M = 14 736.6 Da.
Synthesis of Cyclic Phosphoglycopeptide 34
A solution of oxazoline 11 (4.59 mg, 3.2 μmol) and (GlcNAc)2-ZM109 (1.0 mg, 0.32 μmol) in Tris buffer (100 mM, pH 7.1, 170 μL) was incubated with the Endo-A mutant (N171A) (6.8 μg) at 30 °C. The reaction was monitored by reverse-phase HPLC. After 2 h, the product was purified by HPLC to afford glycopeptide 34 (0.90 mg, 48%). The glycopeptide was quantified with a standard solution of the starting material by measuring the UV absorbance at 280 nm. Analytical RP-HPLC: tR = 19.4 min (gradient, 5–50% aq MeCN containing 0.1% TFA for 30 min; flow rate, 0.5 mL/min). ESI-MS: calcd, M = 5832 Da; found (m/z), 1945.30 [M + 3H]3+, 1459.19 [M + 4H]4+, 1167.38 [M + 5H]5+. Deconvolution of the ESI-MS: M = 5832.5 Da.
Synthesis of Linear Glycopeptide 35
A solution of cyclic glycopeptide 34 (500 μg, 85 nmol) was dissolved in H2O (857 μL), followed by addition of DTT (100 mM, 212 μL) to reduce the disulfide bond. After 1 h, 1-iodopropan-2-one (250 mM, 26.8 μL) was added and the mixture was incubated for 1 h. The product was purified by HPLC to afford the acyclic phosphoglycopeptide 35 (268 μg, 53%). The acyclic glycopeptide was quantified with a standard solution of starting material by measuring the UV absorbance at 280 nm. Analytical RP-HPLC: tR = 20.8 min (gradient, 5–50% aq MeCN containing 0.1% TFA for 30 min; flow rate, 0.5 mL/min). ESI-MS: calcd, M = 5948 Da; found (m/z), 1487.99 [M + 4H]4+, 1190.55 [M + 5H]5+. Deconvolution of the ESI-MS: M = 5948.2 Da.
Surface Plasmon Resonance (SPR) Measurements
SPR measurements were performed on a Biacore T100 instrument (GE Healthcare). Recombinant human IGF-II R (CI-MPR) was purchased from R&D Systems. Approximately 9000 resonance units (RU) of CI-MPR was immobilized on a CM5 sensor chip in a sodium acetate buffer (25 μg/mL, pH 4.0) at 10 °C, using the amine coupling kit provided by the manufacturer. Mannose 6-phosphate containing glycoproteins or glycopeptides were determined at 10 °C under a flow rate of 10 μL/min. HBS-P+ buffer (10 mM HEPES, 150 mM NaCl, 0.05% surfactant P20, pH 7.4) was used as sample buffer and running buffer. Association was measured for 3 min and dissociation for 10 min at same flow rate (10 μL/min). The surface regeneration was performed by 0.5% SDS at a flow rate of 10 μL/min for 60 s. Synthetic glycoproteins and glycopeptide analytes flowed over an immobilized chip with 2-fold serial dilution of the highest concentration of 1 μM. Kinetic analyses were performed by global fitting of the binding data to a 1:1 Langmuir binding model using BIAcore T100 evaluation software, and the equilibrium constant KD was derived.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (NIH grant R01 GM080374 to L.X.W.). T.Y. was partially supported by a research fellowship from Daiichi Sankyo Co., Ltd., Tokyo, Japan.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b05762.
ESI-MS and HPLC profiles of glycoproteins 30, 31a,b, 32, 34, and 35 and 1H and 13C NMR spectra of 10, 11, 14, 15a,b, 18, and 26 (PDF)
NOTE ADDED AFTER ASAP PUBLICATION
This paper was published ASAP on August 16, 2016. The TOC, Figure 1, Schemes 1–5, and the Supporting Information files have been updated to show corrected structures of the sugar residues. The revised version was posted on August 30, 2016.
References
- 1.Ghosh P, Dahms NM, Kornfeld S. Nat Rev Mol Cell Biol. 2003;4:202. doi: 10.1038/nrm1050. [DOI] [PubMed] [Google Scholar]
- 2.Dahms NM, Olson LJ, Kim JJ. Glycobiology. 2008;18:664. doi: 10.1093/glycob/cwn061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coutinho MF, Prata MJ, Alves S. Mol Genet Metab. 2012;105:542. doi: 10.1016/j.ymgme.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 4.Klein AD, Futerman AH. Pediatr Endocrinol Rev. 2013;11(Suppl 1):59. [PubMed] [Google Scholar]
- 5.Lachmann RH. Curr Opin Pediatr. 2011;23:588. doi: 10.1097/MOP.0b013e32834c20d9. [DOI] [PubMed] [Google Scholar]
- 6.Sly WS. Mol Med. 2004;101:100. [PubMed] [Google Scholar]
- 7.Zhu Y, Li X, Kyazike J, Zhou Q, Thurberg BL, Raben N, Mattaliano RJ, Cheng SH. J Biol Chem. 2004;279:50336. doi: 10.1074/jbc.M409676200. [DOI] [PubMed] [Google Scholar]
- 8.Zhu Y, Li X, McVie-Wylie A, Jiang C, Thurberg BL, Raben N, Mattaliano RJ, Cheng SH. Biochem J. 2005;389:619. doi: 10.1042/BJ20050364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu Y, Jiang JL, Gumlaw NK, Zhang J, Bercury SD, Ziegler RJ, Lee K, Kudo M, Canfield WM, Edmunds T, Jiang C, Mattaliano RJ, Cheng SH. Mol Ther. 2009;17:954. doi: 10.1038/mt.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou Q, Stefano JE, Harrahy J, Finn P, Avila L, Kyazike J, Wei R, Van Patten SM, Gotschall R, Zheng X, Zhu Y, Edmunds T, Pan CQ. Bioconjugate Chem. 2011;22:741. doi: 10.1021/bc1005416. [DOI] [PubMed] [Google Scholar]
- 11.Zhou Q, Avila LZ, Konowicz PA, Harrahy J, Finn P, Kim J, Reardon MR, Kyazike J, Brunyak E, Zheng X, Patten SM, Miller RJ, Pan CQ. Bioconjugate Chem. 2013;24:2025. doi: 10.1021/bc400365a. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y, Chan YM, Wu J, Chen C, Benesi A, Hu J, Wang Y, Chen G. ChemBioChem. 2011;12:685. doi: 10.1002/cbic.201000785. [DOI] [PubMed] [Google Scholar]
- 13.McVie-Wylie AJ, Lee KL, Qiu H, Jin X, Do H, Gotschall R, Thurberg BL, Rogers C, Raben N, O’Callaghan M, Canfield W, Andrews L, McPherson JM, Mattaliano RJ. Mol Genet Metab. 2008;94:448. doi: 10.1016/j.ymgme.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song X, Lasanajak Y, Olson LJ, Boonen M, Dahms NM, Kornfeld S, Cummings RD, Smith DF. J Biol Chem. 2009;284:35201. doi: 10.1074/jbc.M109.056119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y, Chen G. J Org Chem. 2011;76:8682. doi: 10.1021/jo2010999. [DOI] [PubMed] [Google Scholar]
- 16.Franzyk H, Christensen MK, Jorgensen RM, Meldal M, Cordes H, Mouritsen S, Bock K. Bioorg Med Chem. 1997;5:21. doi: 10.1016/s0968-0896(96)00194-0. [DOI] [PubMed] [Google Scholar]
- 17.Berkowitz DB, Maiti G, Charette BD, Dreis CD, MacDonald RG. Org Lett. 2004;6:4921. doi: 10.1021/ol0479444. [DOI] [PubMed] [Google Scholar]
- 18.Li B, Zeng Y, Hauser S, Song H, Wang LX. J Am Chem Soc. 2005;127:9692. doi: 10.1021/ja051715a. [DOI] [PubMed] [Google Scholar]
- 19.Wang LX. Carbohydr Res. 2008;343:1509. doi: 10.1016/j.carres.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang W, Li C, Li B, Umekawa M, Yamamoto K, Zhang X, Wang LX. J Am Chem Soc. 2009;131:2214. doi: 10.1021/ja8074677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang LX, Huang W. Curr Opin Chem Biol. 2009;13:592. doi: 10.1016/j.cbpa.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang W, Yang Q, Umekawa M, Yamamoto K, Wang LX. ChemBioChem. 2010;11:1350. doi: 10.1002/cbic.201000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang W, Zhang X, Ju T, Cummings RD, Wang LX. Org Biomol Chem. 2010;8:5224. doi: 10.1039/c0ob00341g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Umekawa M, Li C, Higashiyama T, Huang W, Ashida H, Yamamoto K, Wang LX. J Biol Chem. 2010;285:511. doi: 10.1074/jbc.M109.059832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang W, Li J, Wang LX. ChemBioChem. 2011;12:932. doi: 10.1002/cbic.201000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giddens JP, Lomino JV, Amin MN, Wang LX. J Biol Chem. 2016;291:9356. doi: 10.1074/jbc.M116.721597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang LX, Amin MN. Chem Biol. 2014;21:51. doi: 10.1016/j.chembiol.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rising TW, Claridge TD, Davies N, Gamblin DP, Moir JW, Fairbanks AJ. Carbohydr Res. 2006;341:1574. doi: 10.1016/j.carres.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 29.Rising TW, Claridge TD, Moir JW, Fairbanks AJ. ChemBioChem. 2006;7:1177. doi: 10.1002/cbic.200600183. [DOI] [PubMed] [Google Scholar]
- 30.Heidecke CD, Ling Z, Bruce NC, Moir JW, Parsons TB, Fairbanks AJ. ChemBioChem. 2008;9:2045. doi: 10.1002/cbic.200800214. [DOI] [PubMed] [Google Scholar]
- 31.Rising TW, Heidecke CD, Moir JW, Ling Z, Fairbanks AJ. Chem - Eur J. 2008;14:6444. doi: 10.1002/chem.200800365. [DOI] [PubMed] [Google Scholar]
- 32.Parsons TB, Moir JW, Fairbanks AJ. Org Biomol Chem. 2009;7:3128. [Google Scholar]
- 33.Fairbanks AJ. Pure Appl Chem. 2013;85:1847. [Google Scholar]
- 34.Tomabechi Y, Squire MA, Fairbanks AJ. Org Biomol Chem. 2014;12:942. doi: 10.1039/c3ob42104j. [DOI] [PubMed] [Google Scholar]
- 35.Li H, Li B, Song H, Breydo L, Baskakov IV, Wang LX. J Org Chem. 2005;70:9990. doi: 10.1021/jo051729z. [DOI] [PubMed] [Google Scholar]
- 36.Amin MN, McLellan JS, Huang W, Orwenyo J, Burton DR, Koff WC, Kwong PD, Wang LX. Nat Chem Biol. 2013;9:521. doi: 10.1038/nchembio.1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ochiai H, Huang W, Wang LX. J Am Chem Soc. 2008;130:13790. doi: 10.1021/ja805044x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amin MN, Huang W, Mizanur RM, Wang LX. J Am Chem Soc. 2011;133:14404. doi: 10.1021/ja204831z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei Y, Li C, Huang W, Li B, Strome S, Wang LX. Biochemistry. 2008;47:10294. doi: 10.1021/bi800874y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zou G, Ochiai H, Huang W, Yang Q, Li C, Wang LX. J Am Chem Soc. 2011;133:18975. doi: 10.1021/ja208390n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang W, Giddens J, Fan SQ, Toonstra C, Wang LX. J Am Chem Soc. 2012;134:12308. doi: 10.1021/ja3051266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.During the present work, Fairbanks and co-workers reported the first example of using an ENGase-catalyzed transglycosylation for the synthesis of a phosphorylated glycoprotein carrying an unnatural M6P-Man3GlcNAc2 oligosaccharide (see ref 43). No receptor binding data was reported.
- 43.Priyanka P, Parsons TB, Miller A, Platt FM, Fairbanks AJ. Angew Chem, Int Ed. 2016;55:5058. doi: 10.1002/anie.201600817. [DOI] [PubMed] [Google Scholar]
- 44.Noguchi M, Tanaka T, Gyakushi H, Kobayashi A, Shoda SI. J Org Chem. 2009;74:2210. doi: 10.1021/jo8024708. [DOI] [PubMed] [Google Scholar]
- 45.Li B, Song H, Hauser S, Wang LX. Org Lett. 2006;8:3081. doi: 10.1021/ol061056m. [DOI] [PubMed] [Google Scholar]
- 46.Hancock MK, Yammani RD, Dahms NM. J Biol Chem. 2002;277:47205. doi: 10.1074/jbc.M208534200. [DOI] [PubMed] [Google Scholar]
- 47.Bohnsack RN, Song X, Olson LJ, Kudo M, Gotschall RR, Canfield WM, Cummings RD, Smith DF, Dahms NM. J Biol Chem. 2009;284:35215. doi: 10.1074/jbc.M109.056184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fan SQ, Huang W, Wang LX. J Biol Chem. 2012;287:11272. doi: 10.1074/jbc.M112.340497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.