

Abstract
The human microbiome encodes vast numbers of uncharacterized enzymes, limiting our functional understanding of this community and its effects on host health and disease. By incorporating information about enzymatic chemistry into quantitative metagenomics, we determined the abundance and distribution of individual members of the glycyl radical enzyme superfamily among the microbiomes of healthy humans. We identified many uncharacterized family members, including a universally distributed enzyme that enables commensal gut microbes and human pathogens to dehydrate trans-4-hydroxy-L-proline, the product of the most abundant human posttranslational modification. This ‘chemically-guided functional profiling’ workflow can therefore use ecological context to facilitate the discovery of enzymes in microbial communities.
Communities of microorganisms (microbiomes) occupy nearly every environment on Earth, and these complex assemblages carry out metabolic processes that affect surrounding habitats and organisms (1). For example, the human gut microbiome metabolizes non-digestible dietary components, produces essential vitamins and nutrients, and synthesizes metabolites that are linked to human disease (2, 3). Despite their importance, we have extremely limited knowledge of the specific biochemical reactions performed by microbiomes and the precise mechanisms by which this chemistry shapes microbial ecosystems (4).
This deficit stems from our incomplete understanding of the microbial enzymes that catalyze these chemical transformations. Collectively, the genomes of the organisms that comprise microbiomes (metagenomes) encode vast numbers of enzymes, most of which are uncharacterized. This issue complicates efforts to predict the metabolic activities present within these communities (functional profiling). For instance, 78–86% of genes in Human Microbiome Project (HMP) metagenomes cannot be assigned a metabolic function and ~50% cannot be given any annotation (4, 5). Moreover, genes that can be annotated are typically mapped to large enzyme superfamilies without considering that a single superfamily can catalyze many different chemical reactions and that as many as 80% of enzymes within a superfamily can be uncharacterized or misannotated (6, 7). Thus, functional profiling strategies that can accurately identify enzymes in microbiomes are needed, including both characterized enzymes and enzymes of unknown function that play important but unrecognized roles in these habitats.
The significance of this problem can be appreciated by considering the difficulties associated with studying the activities and roles of glycyl radical enzymes (GREs) in the human gut microbiome. GREs use protein-based radicals to accomplish challenging chemical transformations (Fig. 1A) (8), with the key glycine-centered radical installed posttranslationally by a radical S-adenosylmethionine enzyme (9). These enzymes participate in evolutionarily ancient, anaerobic primary metabolism, including carbohydrate utilization (pyruvate formate-lyase and related α-ketolyases, PFL) (Fig. 1B) and deoxyribonucleotide synthesis (class III ribonucleotide reductase) (8, 10). Previous metagenomic and metaproteomic studies have indicated that the glycyl radical enzymes (GREs) are one of the most abundant protein superfamilies in the human gut microbiome (11–13). Furthermore, activities of characterized GREs from gut microbes are strongly linked to human health. Production of trimethylamine (TMA) from choline (choline trimethylamine-lyase, CutC) (14) is associated with heart (15) and liver diseases (16). Decarboxylation of p-hydroxyphenylacetate gives p-cresol (p-hydroxyphenylacetate decarboxylase, HPAD) (17), which interferes with human drug metabolism and is elevated in children with autism (18, 19). Despite these intriguing connections to human biology, little is known about the abundance and distribution of different types of GREs in human microbiomes. Efforts to accurately identify these enzymes in microbiomes, including attempts to detect CutC in stool metagenomes (20), have been complicated by the high amino acid sequence similarities of GREs and the many superfamily members with unknown functions.
Figure 1. An overview of the glycyl radical enzyme (GRE) superfamily.
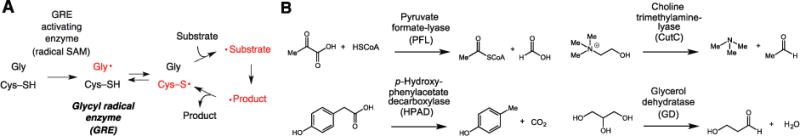
(A) Shared mechanistic features of GREs. SAM, S-adenosylmethionine. (B) Chemical reactions catalyzed by selected characterized GREs.
Here we show that integrating information about enzymatic chemistry into quantitative metagenomics can improve our ability to detect both known and uncharacterized members of enzyme superfamilies in microbiomes. Using a workflow that combines protein sequence similarity network (SSN) analysis with quantitative metagenomics, we first determined the abundance and distribution of individual members of the GRE superfamily in healthy human microbiomes. We identified and quantified biochemically characterized GREs as well as uncharacterized family members that are locally abundant, widespread, or unique in given body sites, prioritizing them for further study based on their ecological context. Employing this strategy, we discovered that the most abundant uncharacterized GRE in HMP stool metagenomes is a trans-4-hydroxy-L-proline dehydratase. This previously unknown enzyme is found in all subjects and thus likely plays a prominent role in the human gut microbiome.
Chemically-guided functional profiling incorporates an understanding of enzymatic activity into quantitative metagenomics
Our approach, which we call ‘chemically-guided functional profiling’ (Fig. 2), begins by identifying an enzyme superfamily of interest, comparing the amino acid sequences of all family members to one another, and then visualizing the resulting pairwise relationships as an SSN (21). Guided by an understanding of how amino acid residues of characterized family members contribute to their activities, mechanisms, and structures, we can construct an SSN that clusters together sequences of enzymes that likely share the same biochemical function. Importantly, this analysis can differentiate family members with distinct activities, regardless of whether or not an enzyme’s function is known.
Figure 2.
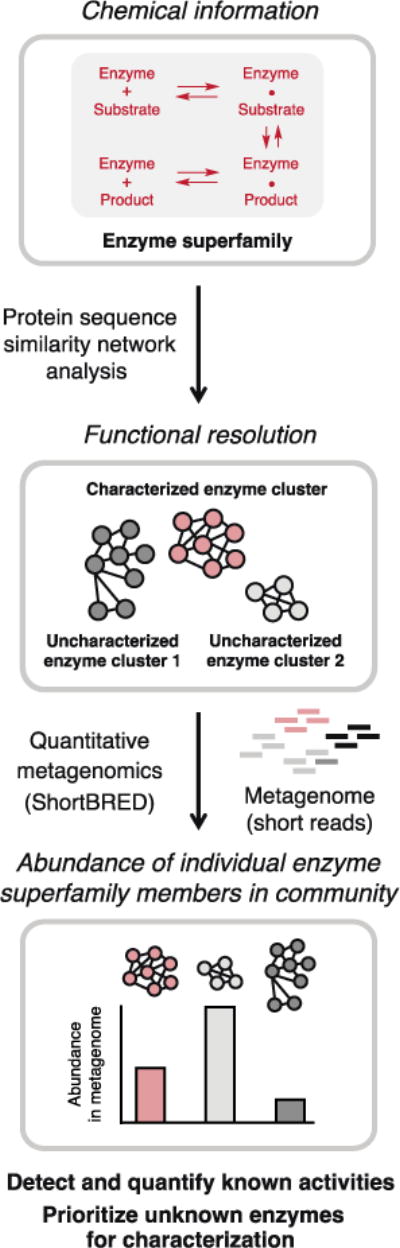
Chemically-guided functional profiling incorporates chemical information into metagenomic analyses to reveal the abundance and distribution of individual members of enzyme superfamilies in microbial communities.
The SSN is then used to interpret data generated by the quantitative metagenomic analysis tool ShortBRED (Short, Better Representative Extract Dataset) (22). Given the amino acid sequences of the enzyme superfamily as input, ShortBRED identifies sequence markers unique to similar family members and quantifies their relative abundance in raw metagenomic sequencing data with high specificity. Mapping the sequence markers and abundance data produced by ShortBRED back to the clusters of enzymes in the SSN then reveals the abundance of individual superfamily members in a microbial community, including enzymes of both known and unknown function. While SSN analysis has been used to study uncharacterized enzymes found in microbial genome sequencing projects (21, 23, 24), these efforts have not examined the presence of these enzymes in communities. Likewise, although sequences from assembled metagenomes have been incorporated into SSNs to expand the diversity of an enzyme superfamily (25), to our knowledge these networks have not been applied to large scale, quantitative metagenomic analyses.
Construction of an SSN for the GRE superfamily
We envisioned using this workflow to assess the distribution of the GRE superfamily across healthy human microbiomes. To begin our analysis, we used the web-based Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST) to build an SSN using 6,343 sequences from InterPro family IPR004184, which includes enzymes that have the so-called “PFL domain” and encompasses all functionally characterized GREs except for the phylogenetically distinct ribonucleotide reductases (26). Our initial network was constructed such that connected sequences shared an alignment score of at least 10−300. We iteratively refined this network by adding different percent identity filters, removing edges that did not meet each threshold to generate multiple SSNs (Fig. S1 and S2). By searching sequence databases and the literature, we then mapped biochemically characterized GREs onto each of these networks, using characteristic conserved active site amino acids known to be involved in substrate binding and catalysis to confirm our assignments (Fig. 3A). For instance, we annotated sequences that likely encode PFLs by looking for the catalytically essential active site Cys-Cys motif that is found in all GREs with this activity (>20 different proteins) (27).
Figure 3. Construction of a sequence similarity network (SSN) for the GRE superfamily.
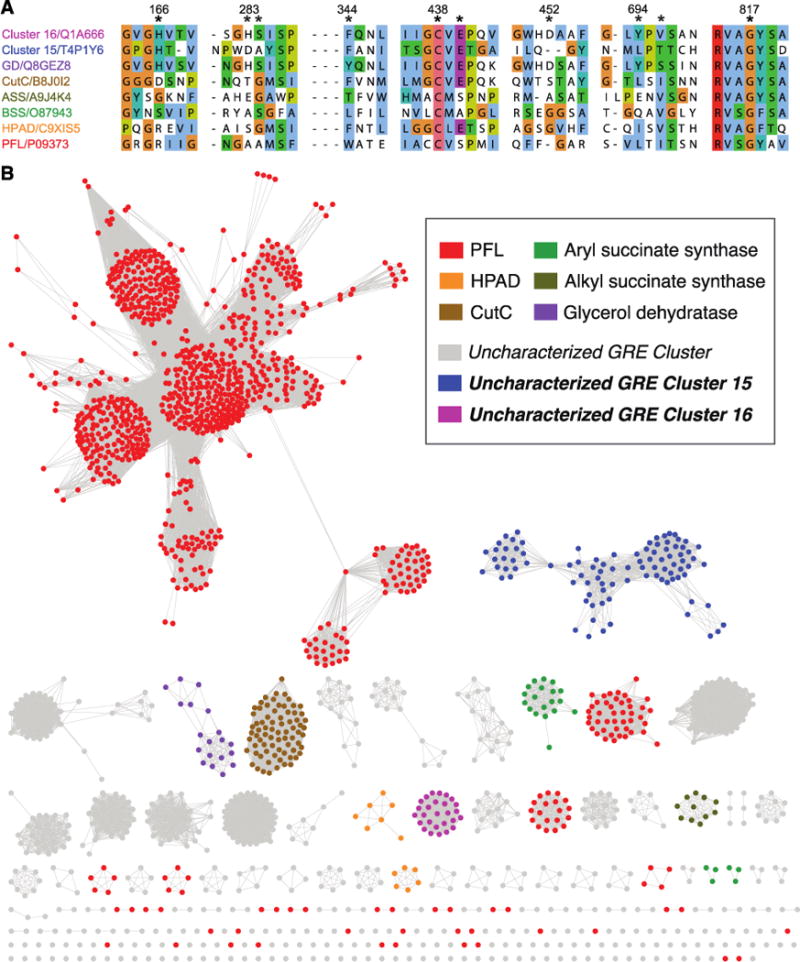
(A) Multiple sequence alignment of selected GREs. The regions shown contain residues that occupy the active sites of structurally characterized GREs and homology models of uncharacterized GREs. The residues at the positions marked with asterisks are conserved in different characterized GREs and are known to play roles in substrate binding or catalysis, making them useful for both identifying known GREs and revealing uncharacterized GREs with potentially distinct activities. Numbering corresponds to PD from Roseburia inulinivorans (uncharacterized GRE Cluster 16); accession numbers are from UniProt. (B) An SSN of the GRE superfamily (InterPro version 53.0; IPR004184, PFL domain) was constructed with an initial score of 10−300. The edge score was then refined such that nodes are connected by an edge if the pairwise sequence identity is ≥62% ID. Each of the 1843 nodes within the resulting SSN contains sequences with >95% amino acid identity.
Ultimately, we chose a minimum edge threshold for the SSN (62% ID) that separates GREs with biochemically verified activities into different clusters (Fig. 3B). Notably, this edge threshold also differentiates uncharacterized GREs that may possess disparate biochemical activities based on differences in predicted active site residues and genomic contexts. For example, at lower edge thresholds (e.g. 55% ID), glycerol dehydratase (GD) from Clostridium butyricum clusters with two uncharacterized GREs that are predicted to share only a subset of active site residues with GD (Fig. S3). While GD is encoded next to a 1,3-propanediol dehydrogenase (28), these other GREs are co-localized with additional genes predicted to encode microcompartment proteins, an aldehyde dehydrogenase, and a phosphate propanoyltransferase (Fig. S3). These distinct genomic contexts suggest that the activities of the uncharacterized GREs may differ from that of GD. At a minimum edge threshold of 62% ID, our SSN resolves these three enzymes into distinct clusters. Though we cannot know for certain that all of the GRE clusters in our SSN are isofunctional, the separation of these highly similar GREs indicates a strong likelihood that each cluster contains enzymes with the same biochemical activity. The presence of uncharacterized GREs reflects a larger trend within the SSN: 195 of the 241 clusters in the final SSN have no assignable biochemical function, suggesting that this enzyme superfamily contains substantial unexplored diversity.
Integrating the SSN with ShortBRED reveals the distribution and abundance of GREs in human microbiomes
With an SSN in hand, we used ShortBRED to profile the abundance of the entire GRE superfamily in 378 high-quality, first-visit metagenomes from healthy participants sequenced during the Human Microbiome Project (HMP) (5), focusing on six body sites: stool (reflective of the lower gastrointestinal tract), buccal mucosa (oral), supragingival plaque (oral), tongue dorsum (oral), anterior nares (skin), and posterior fornix (vaginal). These body sites range from aerobic (skin and vaginal) to microaerobic (oral) to anaerobic (gut) environments. ShortBRED-Identify first found unique protein sequence markers for highly similar GREs (85% amino acid identity) (Table S2). ShortBRED-Quantify then measured the abundance of each marker in the unassembled metagenomic reads. By tabulating the sequence markers belonging to each cluster of sequences in our SSN, we determined the abundance of each group of GREs within each metagenome. Finally, we normalized these abundance values using previously calculated average microbial genome sizes for each metagenomic sample (29).
This chemically-guided functional profiling workflow revealed the abundance and distribution of individual GRE clusters in microbiomes from healthy human subjects (Fig. 4A and Tables S3–S5). We detected sequences belonging to 75 of the 241 GRE clusters from our SSN, implying that the human host supports a wide range of GRE-mediated chemistry. We found GREs in all oral and stool metagenomes and a subset of samples from the other body sites. PFL is the most abundant family member in all GRE-containing samples, consistent with its role in anaerobic glucose metabolism (Fig. 4B). The presence of PFL in many facultative anaerobes and the existence of mechanisms for repairing oxygen-damaged PFL may explain its occurrence in both anaerobic and aerobic environments (30, 31). We observed a unique set of GREs in stool samples compared to the other body sites and identified significantly more GREs per microbial genome in this body site [p < 10−58, Kruskal-Wallis (KW); all p < 10−8, Dunn’s multiple comparisons test (DMC)]. Additionally, a larger number of distinct GRE clusters were located in the gut (75 vs. 5 – 15 for other body sites) (Fig. S4), indicating that this environment harbors a wider range of anaerobic metabolic processes.
Figure 4. Chemically-guided functional profiling of glycyl radical enzymes (GREs) in the human microbiome.
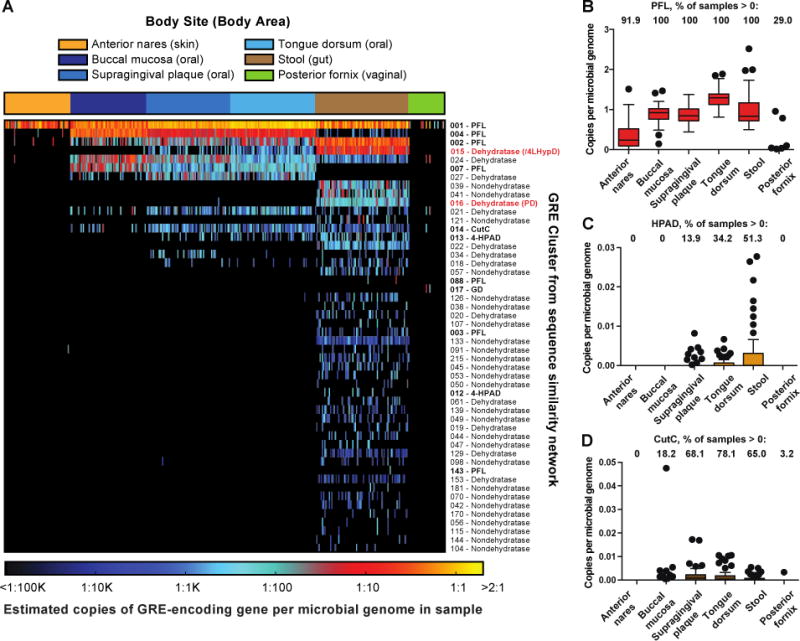
(A) Heatmap showing the abundance and distribution of the 50 most abundant GRE clusters in 378 Human Microbiome Project metagenomes from six body sites as quantified using ShortBRED. Biochemically characterized GRE clusters are shown in bold type, and GRE clusters characterized in this study are shown in red. Boxplots showing per-site abundance of (B) PFL, (C) HPAD, and (D) CutC across six body sites.
These results provide new insights about the ecological contexts of biochemically characterized GREs, including HPAD and CutC. While HPAD is found almost exclusively in stool samples (Fig. 4C), CutC is present with similar frequency in stool, supragingival plaque, buccal mucosa, and tongue dorsum samples (Fig. 4D and Table S5). Identifying this disease-linked enzyme in the oral microbiome is intriguing as periodontal disease and invasion of the GI tract by oral bacteria are associated with heart and liver diseases (32, 33). This finding, which could not have been predicted by the distribution of CutC in sequenced genomes (20), implies that the oral microbiome may be a reservoir for TMA-producing bacteria. Unlike PFL, HPAD and CutC are detected in only a subset of stool metagenomes, which is consistent with the observed variability in the levels of downstream metabolites p-cresol sulfate and trimethylamine-N-oxide in humans (14, 17) and could potentially contribute to interindividual differences in drug metabolism and disease susceptibility.
We also obtained information about the abundance of uncharacterized GREs in human microbiomes, and our data suggest that many unappreciated GRE-mediated activities exist in the human gut. GREs of unknown function represent nine out of the ten most abundant GRE clusters in stool metagenomes and, excluding PFL, outnumber characterized family members 63-fold. Interestingly, the ninth and tenth most abundant unknown GRE clusters were widely distributed in stool metagenomes (>50% of samples) but are both represented by a single sequence in the SSN. This observation serves as a reminder that proteins poorly represented in sequence databases may be widespread in biological habitats. This analysis also helped us to prioritize specific GREs for further study. We focused on the two most broadly distributed and abundant uncharacterized GREs in the human gut microbiome: Cluster 16, which is found in 96% of stool samples, is the third-most abundant GRE in stool metagenomes, and is enriched in this habitat relative to other body locations (p < 10−72, KW; all p < 10−15, DMC; Fig. 5A and Table S5); and Cluster 15, which is present in every stool sample, is the second-most abundant GRE in stool metagenomes, and is also enriched in the gut (p < 10−60, KW; all p < 10−11, DMC; Fig. 6A and Table S5).
Figure 5. Identification and characterization of propanediol dehydratase reveals amino acids involved in dehydration.
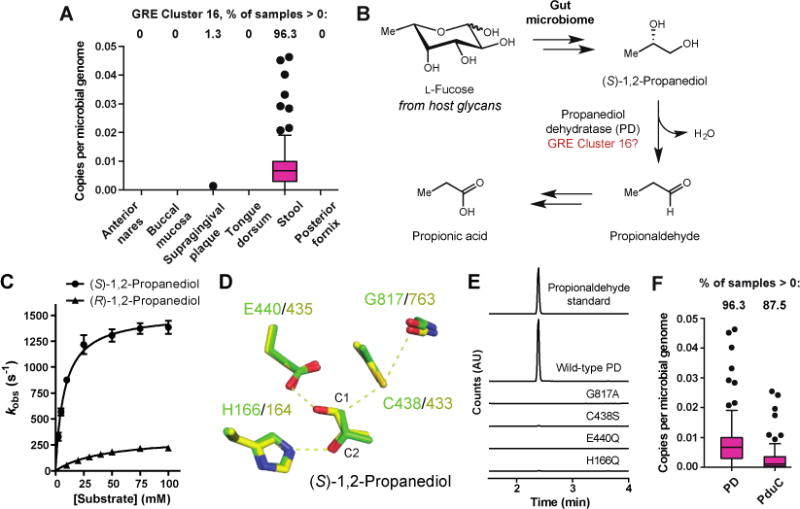
(A) Per-site abundance of GRE Cluster 16 across six body sites. (B) Hypothesized role of GRE Cluster 16 in L-fucose metabolism. (C) Kinetic analysis of PD. Error bars represent the mean ± standard deviation (SD) of three replicates. (D) Comparison of PD homology model (green) with GD crystal structure (yellow) identifies a characteristic set of active site residues required for dehydration. (E) Gas chromatography-mass spectroscopy (GC-MS) analysis of assays with wild-type PD or PD active site mutants and (S)-1,2-propanediol (time = 20 min). (F) Abundance of PD and B12-dependent propanediol dehydratase (PduC) in HMP stool metagenomes.
Figure 6. An abundant, uncharacterized GRE in the human gut is a trans-4-hydroxy-L-proline dehydratase (t4LHypD).
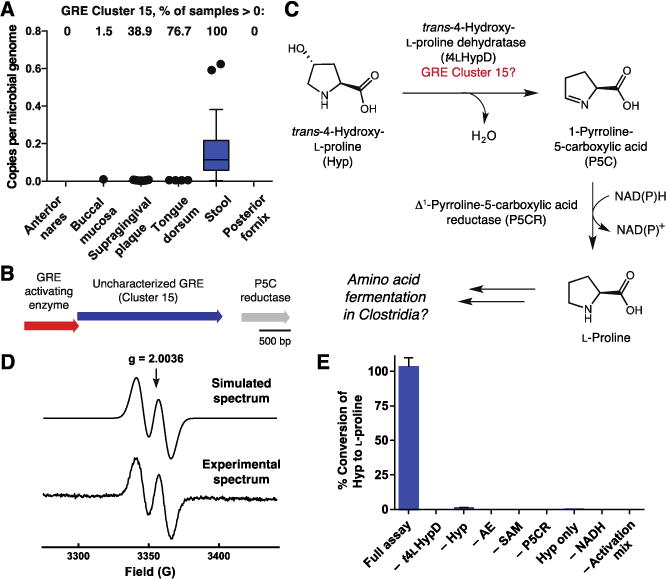
(A) Per-site abundance of GRE Cluster 15 across six body sites. (B) Conserved genomic context of GRE Cluster 15 in Clostridiales. (C) Hypothesized pathway for anaerobic Hyp metabolism involving uncharacterized GRE Cluster 15. (D) EPR spectrum of the glycine-centered radical of activated t4LHypD. An average of 0.51 ± 0.01 (mean ± SD) glycyl radical per t4LHypD monomer was observed with hyperfine coupling A = 1.44 mT. (E) LC-MS/MS detection of L-proline produced in vitro from Hyp by t4LHypD and P5C reductase (time = 1 h). Error bars represent the mean ± SD of three replicates. AE, t4LHypD activating enzyme.
The high abundance and wide distribution of these two GREs in metagenomes suggested they might play prominent functional roles in the healthy human gut. To investigate whether these genes were expressed in gut microbiomes, we applied our chemically-guided functional profiling workflow to analyze paired stool metagenomes and stool metatranscriptomes from eight healthy human subjects (34). Clusters 15 and 16 were present and transcribed in all samples (Fig. S5), indicating that these GREs are likely produced and active in the human gut. Collectively, these observations imply that these two enzymes perform core functions within the healthy human gut and are distinctive of this habitat. We therefore set out to characterize the biochemical functions of these GREs.
Characterization of Cluster 16 reveals a ubiquitous dehydratase motif within the GRE superfamily
We readily connected Cluster 16 to anaerobic L-fucose utilization, a microbial metabolic activity that plays an important role in maintaining gut microbial-host symbiosis. Human gut bacteria consume L-fucose derived from host glycans, producing beneficial short chain fatty acids like propionate as end products (35, 36). A key transformation required for bacteria to convert L-fucose to propionate is the dehydration of (S)-1,2-propanediol to propionaldehyde by B12-dependent propanediol dehydratase (37). The L-fucose metabolizing human gut bacterium Roseburia inulinivorans lacks this enzyme and instead encodes a member of GRE Cluster 16. This GRE was hypothesized to be a B12-independent propanediol dehydratase (PD) based on its co-localization with other fucose utilization genes in the R. inulinivorans genome and upregulation during growth on L-fucose (Fig. 5B) (38). However, when we began our study, the role of this GRE had not been biochemically validated.
We verified this proposal by characterizing R. inulinivorans PD and its activating enzyme (PD-AE) in vitro (Fig. S6). Electron paramagnetic resonance (EPR) spectroscopy showed that PD-AE could generate a glycine-centered radical on PD (Fig. S7). Gas chromatography-mass spectrometry (GC-MS) assays confirmed that activated PD converted (S)-1,2-propanediol to propionaldehyde (Fig. S8). Kinetic analyses showed a 26-fold difference in specificity for (S)- vs. (R)-1,2-propanediol (kcat = 1500 ± 100 s−1, Km = 8 ± 1 mM, kcat/Km = 1.9 ± 0.2 × 105 M−1 s−1 vs. kcat = 330 ± 40 s−1, Km = 44 ± 4 mM, kcat/Km = 7.5 ± 0.8 × 103 M−1 s−1), a stereochemical preference in accordance with PD’s proposed role in L-fucose metabolism (Fig. 5C). These findings agree qualitatively with a recently reported study of R. inulinivorans PD (39).
Identifying active site residues from PD that facilitate dehydration helped us to predict functions of additional uncharacterized GREs. We constructed a homology model of PD, docked both (S)- and (R)-1,2-propanediol into its active site, and compared these models to a crystal structure of the related GRE GD (Fig. 5D, Fig. S9) (40). Key active site amino acids from PD that are conserved in GD include: G817 and C438, the sites of the radical intermediates thought to initiate the reaction via hydrogen atom abstraction from C1 of the substrate; E440, a general base that may deprotonate the C1-hydroxyl group; and H166, which for GD is predicted computationally to protonate the departing C2-hydroxyl group (41). Our model and docking agree well with a recently reported crystal structure of PD bound to (S)-1,2-propanediol (root-mean-square deviation of 0.56 Å) (Fig. S9) (39). Site-directed mutagenesis experiments confirmed that these four residues are critical for activity (Fig. 5E). We therefore reason that this combination of amino acids, which is not found in GREs that perform other transformations, constitutes a ‘dehydratase motif’ that is predictive of enzyme function (Fig. S10). We uncovered this motif in 100 out of 195 uncharacterized clusters in the GRE SSN, indicating that dehydration is likely a widespread activity in this enzyme family (Fig. S11).
The discovery that PD was present at high abundance in 96% of the HMP stool metagenomes led us to investigate whether this enzyme or its B12-dependent counterpart propanediol dehydratase (PduC) was more abundant in the human gut microbiome. PduC was discovered in the 1960s, and certain gut pathogens, including Salmonella spp., use this enzyme to catabolize 1,2-propanediol to propionate (37, 38). Though these two enzymes catalyze the same dehydration reaction, they differ in their sensitivity to oxygen, making it unclear whether one type of enzyme would predominate in the largely anaerobic environment of the healthy human gut. We used ShortBRED to determine the abundance of PduC in the 80 HMP stool metagenomes analyzed above. Although we find that both dehydratases are widely distributed in human gut microbiomes (PD and PduC are present in 96% and 87% of stool samples, respectively), PD is significantly more abundant than PduC (p < 10−4, Mann-Whitney U test) (Fig. 5F). Furthermore, by examining the abundance of PD and PduC within each gut metagenome, we established that the median ratio of PD to PduC across all subjects was 5.2 to 1 (Fig. S12). This observation suggests that PD may make a greater contribution to propionate production from L-fucose in the healthy human gut. However, the presence of both enzymes indicates that this gut microbial metabolic process may also proceed under conditions of increased oxygen, such as during inflammation (42). Overall, this analysis demonstrates how chemically-guided functional profiling can provide insights into the ecology of enzymes that are well-characterized biochemically.
A prominent gut microbial GRE dehydrates trans-4-hydroxy-L-proline
Our analysis of dehydratases in the SSN revealed the characteristic dehydratase motif in sequences from Cluster 15, the most abundant uncharacterized GRE in the human gut (Fig. S11). However, inspection of multiple sequence alignments and a homology model of this enzyme uncovered additional predicted active site residues that differ from GD and PD, suggesting it might dehydrate a different substrate (Fig. 3, Fig. S13). Using sequences from Cluster 15 as search queries, we located this GRE in >850 sequenced bacterial and archaeal genomes deposited in the NCBI genome database, including prominent gut and oral commensals (Parabacteroides spp. and Clostridiales) as well as human pathogens like Clostridium difficile (>97% of sequenced isolates, NCBI database) (Fig. S14).
The genomic context of this putative dehydratase sheds light on its biochemical function. In the genomes of Clostridiales, the gene encoding this GRE is often clustered with genes encoding a GRE activating enzyme and a predicted Δ1-pyrroline-5-carboxylate (P5C) reductase (Fig. 6B). P5C reductase reduces P5C to L-proline as the final step in L-proline biosynthesis (43). Hypothesizing that these enzymes might participate in the same pathway, we considered the non-proteinogenic amino acid trans-4-hydroxy-L-proline (Hyp) as a potential substrate for the GRE (Fig. 6C). Dehydration of Hyp could generate P5C, which would be converted to L-proline by the P5C reductase. Many Clostridiales can use L-proline as an electron acceptor in amino acid fermentations (44). Interestingly, certain L-proline fermenting strains, including C. difficile, also use Hyp as an electron acceptor, but the enzymes that mediate this process have not been identified (45). Our proposed pathway would account for this metabolic activity and is consistent with the observation that expression of D-proline reductase, a key enzyme required for L-proline metabolism, is upregulated when C. difficile grows in the presence of Hyp (45).
In vitro characterization of the putative Hyp dehydratase (t4LHypD), its partner activating enzyme (t4LHypD-AE), and the co-localized P5C reductase from C. difficile 70-100-2010 confirmed this hypothesis (Fig. S15). We first used a spectrophotometric assay to verify that P5C reductase could interconvert P5C and L-proline (Fig. S16). EPR experiments then showed that t4LHypD-AE could install a glycine-centered radical on t4LHypD (51 ± 1% activation, Fig. 6D), establishing that these enzymes are an activating enzyme-GRE pair. Finally, incubation of activated t4LHypD, P5C reductase, NADH, and Hyp resulted in the full conversion of this amino acid to proline as detected by LC-MS/MS (Fig. 6E, Fig. S17). While each component of the full assay mixture was essential for production of proline, consumption of Hyp was still observed in assays lacking either P5C reductase or NADH (Fig. S17). This pattern of activity indicates that t4LHypD catalyzes the dehydration of Hyp to produce P5C and that this reaction does not require the presence of the downstream P5C reductase. t4LHypD displayed undetectable or greatly reduced activity toward other hydroxyproline stereoisomers based on the quantification of proline by LC-MS/MS in samples from end-point assays (Fig. S18). The kinetic parameters of t4LHypD further support the physiological relevance of this reaction (kcat = 45 ± 1 s−1, Km = 1.2 ± 0.1 mM, kcat/Km = 3.8 ± 0.3 × 104 M−1 s−1) (Fig. S19) (46). Likewise, experiments with sequenced Clostridiales isolates showed improved growth in Hyp-containing media and the accompanied consumption of Hyp only in strains encoding t4LHypD (Fig. S20).
Taken together, these experiments show that this abundant, universally distributed human gut microbial GRE is a Hyp dehydratase and define a pathway for anaerobic 4-hydroxyproline metabolism. The reaction performed by t4LHypD differs substantially from all other characterized hydroxyproline dehydratases, which accept 3-hydroxyproline. The hydroxyl group of 3-hydroxyproline is adjacent to the α-carbon of this amino acid, which has a relatively acidic proton (pKa ~ 29). In contrast, the hydroxyl substituent of 4-hydroxyproline cannot be readily eliminated using acid-base catalysis, as it is positioned between two carbon atoms that bear non-acidic protons (pKa ~ >40). The use of a radical enzyme provides an elegant solution to this chemical challenge.
The discovery of t4LHypD also reveals a previously unappreciated host-gut microbe metabolic interaction (Fig. 7). Many host and dietary proteins contain Hyp, including collagen, the most abundant host protein, and hydroxyproline-rich glycoproteins, the major proteinaceous component of higher plant and algal cell walls (47). In eukaryotes, Hyp is generated posttranslationally by prolyl 4-hydroxylases, members of the non-heme iron-dependent dioxygenase family (47). While C4-hydroxylation of L-proline is the most common posttranslational modification in the human proteome, it is rare in bacteria. Unlike the majority of posttranslational modifications, C4-hydroxylation of L-proline is considered to be irreversible by human metabolism. Instead, Hyp is oxidized to yield pyruvate and glyoxylate without forming L-proline (48). Remarkably, the actions of t4LHypD and P5C reductase allow bacteria to chemically ‘reverse’ proline hydroxylation. t4LHypD’s activity is also notable from an evolutionary perspective since Hyp formation requires molecular oxygen, a substrate that inactivates GREs and was not present during the evolution of ancestral GRE family members. t4LHypD therefore likely emerged after the oxygenation of Earth’s atmosphere in response to the evolution of this posttranslational modification in eukaryotic organisms.
Figure 7.
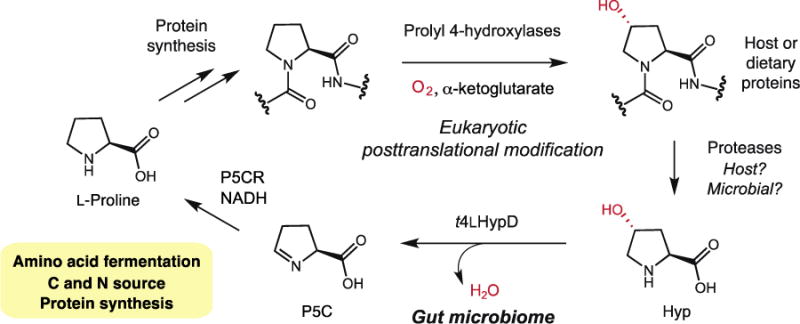
The intersection between gut microbial Hyp metabolism and host metabolism.
The universal distribution and high abundance of t4LHypD in stool metagenomes suggest that it plays a critical role in the healthy human gut. In addition to supporting microbial energy production, the conversion of Hyp to P5C and L-proline could supply the microbiome with sources of carbon and nitrogen. These products may also be further processed to provide amino acid building blocks for protein synthesis. Hyp metabolism might affect L-proline availability for the host, which is intriguing given this amino acid’s role in host cell stress responses and apoptosis (49). Gut microbes may liberate Hyp from collagen or collagen-derived peptides of host or dietary origin, affecting collagen homeostasis and Hyp availability (48). Finally, the distribution of t4LHypD in both gut commensals and human pathogens implies that Hyp utilization could contribute to colonization resistance or pathogenicity. Further experiments are needed to explore the many potential biological implications of this activity.
Conclusions
In summary, we have incorporated knowledge of enzymatic chemistry into quantitative metagenomics, designing and implementing a chemically-guided functional profiling strategy. Our analysis of the GRE superfamily in human microbiomes provided both new insights about GREs of known activity, including enzymes linked to human disease, and the ability to identify enzymes of unknown activity in these communities, revealing intriguing targets for further study. A combination of bioinformatic analyses and in vitro biochemical experiments proved critical for linking these highly abundant, uncharacterized sequences to corresponding microbial metabolic processes. In particular, the many questions raised by the activity and distribution of t4LHypD illustrate how enzyme discovery efforts can inspire hypothesis-driven microbiome research.
Chemically-guided functional profiling changes how we discover microbial enzymes by both facilitating their identification in complex multi’omics sequence datasets and prioritizing them for characterization based on their abundance, distribution, and expression in communities. The use of ecological context to guide characterization of unknown enzymes represents a striking departure from methods that have focused on targets present in sequenced organisms without considering their distributions in microbial habitats. This general strategy may be applied broadly to investigate the chemistry present in microbial communities. Our workflow can be used to profile metagenomes and metatrascriptomes obtained from any environment. Moreover, it can be readily extended to identify other types of enzymes, including the numerous enzyme superfamilies that have already been subjected to SSN analysis (26), provided that some superfamily members have been biochemically characterized. Further chemically-guided functional profiling could uncover novel metabolic interactions both within microbiomes and between microbes and hosts. For example, we are now poised to detect GREs present in patient populations, searching for known functions such as p-cresol and TMA production, as well as new metabolic activities that may influence disease progression. By expanding our knowledge of microbial enzymes and metabolism, this approach will advance progress toward a deeper mechanistic understanding of microbiomes.
Materials and Methods
Expanded materials and methods can be found in the Supplementary Materials.
Construction of GRE SSNs
SSNs were generated via the EFI-EST webtool (http://efi.igb.illinois.edu/efi-est/) (26) using IPR004184 (the pyruvate formate-lyase domain, version 53.0 of UniProt, accessed on October 9, 2015) as the input for option B with a minimum sequence length of 500 amino acids and no maximum length specified. Networks were subsequently generated with initial edge values of 10−50 or 10−300. The resulting representative node networks were visualized with Cytoscape 3.2 (50). Edge scores were further refined in Cytoscape, and additional details related to the process of refining the edge threshold can be found in the Supplementary Materials.
Quantification of enzyme abundances in metagenomes
ShortBRED was used to quantify the abundance of the GREs in metagenomes (22). All ShortBRED computations were performed on the Odyssey cluster supported by the Faculty of Arts and Science (FAS) Division of Science Research Computing Group at Harvard University. First, ShortBRED-Identify was used to find markers for all of the sequences from the GRE SSN. UniRef90 was used as the reference list (51), and the markers generated were specific to sequences in the SSN and were absent from UniRef90. ShortBRED-Identify was run with the default parameters, with the exception of the ‘–threads’ flag, which was increased to run effectively on the Odyssey cluster. With markers generated, ShortBRED-Quantify was then used to determine the abundance of the GREs in metagenomes generated as part of the HMP (5). We analyzed 378 high-quality, first-visit metagenomes from healthy human participants. The output from Short-BRED-Quantify was normalized to counts per microbial genome using previously computed average genome sizes for each sample (29). In addition to the HMP metagenomes, this analysis was repeated in the same manner with matched metagenomes and metatranscriptomes from eight individuals, except that the output was not normalized to counts per microbial genome (34). ShortBRED was also used to quantify the abundances of the B12-dependent diol dehydratases (IPR003206) in the HMP stool metagenomes in the same manner as the GREs, except that SSN analysis was not performed. This InterPro family contains the B12-dependent propanediol and glycerol dehydratases. Because both enzymes are known to dehydrate (S)-1,2-propanediol, we did not attempt to distinguish between them. Therefore our values represent upper limits for PduC abundance.
Code availability
The relevant scripts and instructions for performing ‘chemically-guided functional profiling’ with different SSNs or meta’omics datasets can be found online at http://scholar.harvard.edu/balskus/metagenomic-profiling.
Plasmid construction
The plasmids used in this study allowed for IPTG-inducible protein overexpression in Escherichia coli heterologous expression hosts. All plasmids were constructed using standard molecular biology techniques, including polymerase chain reaction, restriction enzyme digestion, ligation, Gibson Assembly, and site-directed mutagenesis. Primers were purchased from Integrated DNA Technologies and are listed in Table S1. All plasmid constructs were confirmed by DNA sequencing (Beckman Coulter Genomics). Genes encoding PD and PD-AE were amplified from R. inulinivorans DSM 16841 (DSMZ) and genes encoding t4LHypD, t4LHypD-AE, and P5C reductase were amplified from C. difficile 70-100-2010 (BEI Resources).
Protein overexpression and purification
All recombinant proteins used in this study were individually overexpressed in E. coli strains (BL21 (DE3) or BL21-CodonPlus(DE3)-RIL ΔproC::aac(3)IV), followed by purification by affinity chromatography for quantification of glycyl radical species by EPR, in vitro activity assays, and kinetics experiments. PD-AE was overexpressed in E. coli BL21 (DE3) co-transformed with pPH149 encoding E. coli IscSUA-HscBA-Fd genes (52). All purified proteins were rendered anoxic prior to assays by either sparging or through repeated vacuum-refill cycles with argon as the inert gas.
Glycyl radical generation and quantification by EPR spectroscopy
PD and t4LHypD were activated by their partner activating enzymes in the presence of S-adenosylmethionine and either 5-deazariboflavin or acriflavine, respectively. Glycyl radicals in activated samples were detected by EPR spectroscopy at 77 K and quantified using K2(SO3)2NO standards. Simulated spectra for glycyl radicals were obtained from experimental data using EasySpin (53), a MATLAB toolbox (MathWorks).
End-point enzymatic activity assays
PD and t4LHypD were first activated by their partner activating enzymes under the same conditions used for EPR studies. Activated GREs were incubated with their respective substrates under anaerobic conditions and at room temperature until quenching for product detection. Headspace GC-MS was used for the detection of propionaldehyde in PD activity assays. LC-MS/MS was used for the detection of proline in t4LHypD activity assays.
Coupled spectrophotometric assays for kinetics
The activity of PD was coupled to horse liver alcohol dehydrogenase (Sigma) and the activity of t4LHypD was coupled to P5C reductase for the reduction of respective products. Absorbance of NADH at 340 nm was recorded over time to calculate initial rates and kinetic parameters.
Growth experiments and metabolite analyses
Terrisporobacter glycolicus DSM 1288 (DSMZ), Clostridium sporogenes ATCC 15579 (ATCC), Clostridium difficile 70-100-2010 (BEI Resources), and Clostridium sticklandii DSM 519 (DSMZ) were grown at 37 °C under an atmosphere of 5% H2/95% N2. All media used for growth experiments in this study are modified from a previously reported phosphate and carbonate based medium with a minimal composition of amino acids (54). OD600 measurements of 5 mL cultures grown in Hungate tubes were taken until stationary phase. Hydroxyproline and proline content in spent media were quantified using LC-MS/MS.
Supplementary Material
| Overall statistics | Anterior nares | Buccal mucosa | Supragingival plaque | Tongue dorsum | Stool | Posterior fornix |
|---|---|---|---|---|---|---|
| Number of samples | 56 | 66 | 72 | 73 | 80 | 31 |
| Number of superclusters from SSN | 5 | 10 | 15 | 13 | 75 | 5 |
| Number of superclusters with median >0 (i.e., in >50% of samples) | 1 | 5 | 8 | 9 | 11 | 0 |
| Mean GRE copies/genome | 0.342 | 0.954 | 0.917 | 1.305 | 1.159 | 0.071 |
| Median GRE copies/genome | 0.239 | 0.971 | 0.893 | 1.322 | 1.094 | 0.000 |
| Std deviation GRE copies/genome | 0.317 | 0.236 | 0.205 | 0.210 | 0.357 | 0.250 |
| Statistics on characterized proteins | Anterior nares | Buccal mucosa | Supragingival plaque | Tongue dorsum | Stool | Posterior fornix |
|
| ||||||
| % of samples with PFL | 91.1 | 100.0 | 100.0 | 100.0 | 100.0 | 29.0 |
|
| ||||||
| Mean PFL copies/genome | 0.341 | 0.887 | 0.872 | 1.284 | 0.956 | 0.063 |
|
| ||||||
| Std deviation of PFL copies/genome | 0.318 | 0.220 | 0.203 | 0.209 | 0.362 | 0.218 |
|
| ||||||
| % of samples with dehydratases | 1.8 | 93.9 | 100.0 | 100.0 | 100.0 | 6.5 |
| Mean dehydratase copies/genome | 0.001 | 0.067 | 0.043 | 0.019 | 0.176 | 0.008 |
| Std deviation of dehydratases copies/genome | 0.005 | 0.089 | 0.021 | 0.022 | 0.120 | 0.033 |
|
| ||||||
| % of samples with t4LHypD | 0.0 | 1.5 | 38.9 | 76.7 | 100.0 | 0.0 |
|
| ||||||
| Mean t4LHypD copies/genome | 0.0000 | 0.0002 | 0.0010 | 0.0014 | 0.1429 | 0.0000 |
|
| ||||||
| Std deviation of t4LHypD copies/genome | 0.0000 | 0.0012 | 0.0018 | 0.0015 | 0.1165 | 0.0000 |
|
| ||||||
| % of samples with PD | 0.0 | 0.0 | 1.4 | 0.0 | 96.3 | 0.0 |
| Mean PD copies/genome | 0 | 0 | 1.91627E-05 | 0 | 0.0090 | 0 |
| Std deviation of PD copies/genome | 0 | 0 | 0.000162601 | 0 | 0.0097 | 0 |
|
| ||||||
| % of samples with CutC | 0.0 | 18.2 | 68.1 | 78.1 | 65.0 | 3.2 |
|
| ||||||
| Mean CutC copies/genome | 0.0000 | 0.0011 | 0.0017 | 0.0016 | 0.0007 | 0.0001 |
|
| ||||||
| Std deviation of CutC copies/genome | 0.0000 | 0.0059 | 0.0031 | 0.0026 | 0.0011 | 0.0006 |
|
| ||||||
| % of samples with HPAD | 0.0 | 0.0 | 13.9 | 34.2 | 51.3 | 0.0 |
| Mean HPAD copies/genome | 0.0000 | 0.0000 | 0.0004 | 0.0006 | 0.0028 | 0.0000 |
| Std deviation of HPAD copies/genome | 0.0000 | 0.0000 | 0.0013 | 0.0012 | 0.0056 | 0.0000 |
One sentence summary.
Integrating chemical knowledge and metagenomics reveals a gut microbial enzyme that processes a host-derived amino acid.
Acknowledgments
Financial support was provided by Harvard University, the Packard Fellowship for Science and Engineering (E.P.B.), the George W. Merck Fund (E.P.B.), the National Institutes of Health (U54DE023798) (C.H.), and the National Science Foundation (DBI-1053486 and EAGER 1453942) (C.H.). B.J.L. acknowledges support from the NSF GRFP (DGE1144152) and Y.W. acknowledges support from ASTAR Singapore. The computations in this paper were run on the Odyssey cluster supported by the FAS Division of Science, Research Computing Group at Harvard University. We thank J. Stubbe for helpful advice and feedback on this manuscript. We also thank the Klinman lab (UC Berkeley) for providing the plasmid pPH149, J. Nicoludis (Gaudet lab, Harvard University) for providing P1 phage lysate, J. Wang for assistance with GC-MS experiments, K. Chatman for assistance with LC-MS/MS experiments, and H. Nakamura for help with the synthesis of 5-deazariboflavin. A protocol that allows users to perform chemically-guided functional profiling with their own datasets is available online at http://scholar.harvard.edu/balskus/metagenomic-profiling.
Footnotes
References
- 1.Blaser MJ, Cardon ZG, Cho MK, Dangl JL, Donohue TJ, Green JL, Knight R, Maxon ME, Northen TR, Pollard KS, Brodie EL. Toward a predictive understanding of Earth’s microbiomes to address 21st century challenges. mBio. 2016;7:e00714–00716. doi: 10.1128/mBio.00714-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sekirov I, Russell SL, Antunes LCM, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90:859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- 3.Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host-gut microbiota metabolic interactions. Science. 2012;336:1262–1267. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- 4.Joice R, Yasuda K, Shafquat A, Morgan XC, Huttenhower C. Determining microbial products and identifying molecular targets in the human microbiome. Cell Metabolism. 2014;20:731–741. doi: 10.1016/j.cmet.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Franzosa EA, Hsu T, Sirota-Madi A, Shafquat A, Abu-Ali G, Morgan XC, Huttenhower C. Sequencing and beyond: integrating molecular ‘omics’ for microbial community profiling. Nat Rev Microbiol. 2015;13:360–372. doi: 10.1038/nrmicro3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schnoes AM, Brown SD, Dodevski I, Babbitt PC. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comp Biol. 2009;5:e1000605. doi: 10.1371/journal.pcbi.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selmer T, Pierik AJ, Heider J. New glycyl radical enzymes catalysing key metabolic steps in anaerobic bacteria. Biol Chem. 2005;386:981–988. doi: 10.1515/BC.2005.114. [DOI] [PubMed] [Google Scholar]
- 9.Wagner AF, Frey M, Neugebauer FA, Schäfer W, Knappe J. The free radical in pyruvate formate-lyase is located on glycine-734. Proc Natl Acad Sci USA. 1992;89:996–1000. doi: 10.1073/pnas.89.3.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reichard P. The evolution of ribonucleotide reduction. Trends Biochem Sci. 1997;22:81–85. doi: 10.1016/s0968-0004(97)01003-7. [DOI] [PubMed] [Google Scholar]
- 11.Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A, Takami H, Morita H, Sharma VK, Srivastava TP, Taylor TD, Noguchi H, Mori H, Ogura Y, Ehrlich DS, Itoh K, Takagi T, Sakaki Y, Hayashi T, Hattori M. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007;14:169–181. doi: 10.1093/dnares/dsm018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellrott K, Jaroszewski L, Li W, Wooley JC, Godzik A. Expansion of the protein repertoire in newly explored environments: human gut microbiome specific protein families. PLoS Comput Biol. 2010;6:e1000798. doi: 10.1371/journal.pcbi.1000798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kolmeder CA, de Been M, Nikkilä J, Ritamo I, Mättö J, Valmu L, Salojärvi J, Palva A, Salonen A, de Vos WM. Comparative metaproteomics and diversity analysis of human intestinal microbiota testifies for its temporal stability and expression of core functions. PLoS ONE. 2012;7:e29913. doi: 10.1371/journal.pone.0029913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Craciun S, Balskus EP. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci USA. 2012;109:21307–21312. doi: 10.1073/pnas.1215689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, DuGar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WHW, DiDonato JA, Lusis AJ, Hazen SL. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, Fearnside J, Tatoud R, Blanc V, Lindon JC, Mitchell SC, Holmes E, McCarthy MI, Scott J, Gauguier D, Nicholson JK. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci USA. 2006;103:12511–12516. doi: 10.1073/pnas.0601056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Selmer T, Andrei PI. p-Hydroxyphenylacetate decarboxylase from Clostridium difficile. A novel glycyl radical enzyme catalysing the formation of p-cresol. Eur J Biochem. 2001;268:1363–1372. doi: 10.1046/j.1432-1327.2001.02001.x. [DOI] [PubMed] [Google Scholar]
- 18.Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci USA. 2009;106:14728–14733. doi: 10.1073/pnas.0904489106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Persico AM, Napolioni V. Urinary p-cresol in autism spectrum disorder. Neurotoxicol Teratol. 2013;36:82–90. doi: 10.1016/j.ntt.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 20.Campo A Martínez-del, Bodea S, Hamer HA, Marks JA, Haiser HJ, Turnbaugh PJ, Balskus EP. Characterization and detection of a widely distributed gene cluster that predicts anaerobic choline utilization by human gut bacteria. mBio. 2015;6:e00042–00015. doi: 10.1128/mBio.00042-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Atkinson HJ, Morris JH, Ferrin TE, Babbitt PC. Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PLoS ONE. 2009;4:e4345. doi: 10.1371/journal.pone.0004345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaminski J, Gibson MK, Franzosa EA, Segata N, Dantas G, Huttenhower C. High-specificity targeted functional profiling in microbial communities with ShortBRED. PLoS Comput Biol. 2015;11:e1004557. doi: 10.1371/journal.pcbi.1004557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lohman JR, Ma M, Osipiuk J, Nocek B, Kim Y, Chang C, Cuff M, Mack J, Bigelow L, Li H, Endres M, Babnigg G, Joachimiak A, Phillips GN, Shen B. Structural and evolutionary relationships of “AT-less” type I polyketide synthase ketosynthases. Proc Natl Acad Sci USA. 2015;112:12693–12698. doi: 10.1073/pnas.1515460112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang H, Carter MS, Vetting MW, Al-Obaidi N, Patskovsky Y, Almo SC, Gerlt JA. A general strategy for the discovery of metabolic pathways: D-threitol, L-threitol, and erythritol utilization in Mycobacterium smegmatis. J Am Chem Soc. 2015;137:14570–14573. doi: 10.1021/jacs.5b08968. [DOI] [PubMed] [Google Scholar]
- 25.Brown SD, Babbitt PC. Inference of functional properties from large-scale analysis of enzyme superfamilies. J Biol Chem. 2012;287:35–42. doi: 10.1074/jbc.R111.283408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerlt JA, Bouvier JT, Davidson DB, Imker HJ, Sadkhin B, Slater DR, Whalen KL. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): a web tool for generating protein sequence similarity networks. BBA-Proteins Proteom. 2015;1854:1019–1037. doi: 10.1016/j.bbapap.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Becker A, Fritz-Wolf K, Kabsch W, Knappe J, Schultz S, Wagner AF Volker. Structure and mechanism of the glycyl radical enzyme pyruvate formate-lyase. Nat Struct Mol Biol. 1999;6:969–975. doi: 10.1038/13341. [DOI] [PubMed] [Google Scholar]
- 28.Raynaud C, Sarçabal P, Meynial-Salles I, Croux C, Soucaille P. Molecular characterization of the 1,3-propanediol (1,3-PD) operon of Clostridium butyricum. Proc Natl Acad Sci USA. 2003;100:5010–5015. doi: 10.1073/pnas.0734105100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nayfach S, Pollard KS. Average genome size estimation improves comparative metagenomics and sheds light on the functional ecology of the human microbiome. Genome Biol. 2015;16:1–18. doi: 10.1186/s13059-015-0611-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knappe J, Sawers G. A radical-chemical route to acetyl-CoA: the anaerobically induced pyruvate formate-lyase system of Escherichia coli. FEMS Microbiol Rev. 1990;6:383–398. doi: 10.1111/j.1574-6968.1990.tb04108.x. [DOI] [PubMed] [Google Scholar]
- 31.Wagner AFV, Schultz S, Bomke J, Pils T, Lehmann WD, Knappe J. YfiD of Escherichia coli and Y06I of bacteriophage T4 as autonomous glycyl radical cofactors reconstituting the catalytic center of oxygen-fragmented pyruvate formate-lyase. Biochem Biophys Res Comm. 2001;285:456–462. doi: 10.1006/bbrc.2001.5186. [DOI] [PubMed] [Google Scholar]
- 32.Mattila KJ, Nieminen MS, Valtonen VV, Rasi VP, Kesäniemi YA, Syrjälä SL, Jungell PS, Isoluoma M, Hietaniemi K, Jokinen MJ. Association between dental health and acute myocardial infarction. Brit Med J. 1989;298:779–781. doi: 10.1136/bmj.298.6676.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, Guo J, Le Chatelier E, Yao J, Wu L, Zhou J, Ni S, Liu L, Pons N, Batto JM, Kennedy SP, Leonard P, Yuan C, Ding W, Chen Y, Hu X, Zheng B, Qian G, Xu W, Ehrlich SD, Zheng S, Li L. Alterations of the human gut microbiome in liver cirrhosis. Nature. 2014;513:59–64. doi: 10.1038/nature13568. [DOI] [PubMed] [Google Scholar]
- 34.Franzosa EA, Morgan XC, Segata N, Waldron L, Reyes J, Earl AM, Giannoukos G, Boylan MR, Ciulla D, Gevers D, Izard J, Garrett WS, Chan AT, Huttenhower C. Relating the metatranscriptome and metagenome of the human gut. Proc Natl Acad Sci USA. 2014;111:E2329–E2338. doi: 10.1073/pnas.1319284111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr. 2002;22:283–307. doi: 10.1146/annurev.nutr.22.011602.092259. [DOI] [PubMed] [Google Scholar]
- 36.Reichardt N, Duncan SH, Young P, Belenguer A, McWilliam Leitch C, Scott KP, Flint HJ, Louis P. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J. 2014;8:1323–1335. doi: 10.1038/ismej.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daniel R, Bobik TA, Gottschalk G. Biochemistry of coenzyme B12-dependent glycerol and diol dehydratases and organization of the encoding genes. FEMS Microbiol Rev. 1998;22:553–566. doi: 10.1111/j.1574-6976.1998.tb00387.x. [DOI] [PubMed] [Google Scholar]
- 38.Scott KP, Martin JC, Campbell G, Mayer CD, Flint HJ. Whole-genome transcription profiling reveals genes up-regulated by growth on fucose in the human gut bacterium Roseburia inulinivorans. J Bacteriol. 2006;188:4340–4349. doi: 10.1128/JB.00137-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.LaMattina JW, Keul ND, Reitzer P, Kapoor S, Galzerani F, Koch DJ, Gouvea IE, Lanzilotta WN. 1,2-propanediol dehydration in Roseburia inulinivorans; structural basis for substrate and enantiomer selectivity. J Biol Chem. 2016;291:15515–15526. doi: 10.1074/jbc.M116.721142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Brien JR, Raynaud C, Croux C, Girbal L, Soucaille P, Lanzilotta WN. Insight into the mechanism of the B12-independent glycerol dehydratase from Clostridium butyricum: preliminary biochemical and structural characterization. Biochemistry. 2004;43:4635–4645. doi: 10.1021/bi035930k. [DOI] [PubMed] [Google Scholar]
- 41.Feliks M, Ullmann GM. Glycerol dehydratation by the B12-independent enzyme may not involve the migration of a hydroxyl group: a computational study. J Phys Chem B. 2012;116:7076–7087. doi: 10.1021/jp301165b. [DOI] [PubMed] [Google Scholar]
- 42.Campbell Eric L, Bruyninckx Walter J, Kelly Caleb J, Glover Louise E, McNamee Eóin N, Bowers Brittelle E, Bayless Amanda J, Scully M, Saeedi Bejan J, Golden-Mason L, Ehrentraut Stefan F, Curtis Valerie F, Burgess A, Garvey John F, Sorensen A, Nemenoff R, Jedlicka P, Taylor Cormac T, Kominsky Douglas J, Colgan Sean P. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity. 2014;40:66–77. doi: 10.1016/j.immuni.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deutch AH, Smith CJ, Rushlow KE, Krelschmer PJ. Escherichia coli Δ1-pyrroline-5-carboxylate reductase: gene sequence, protein overproduction and purification. Nucleic Acids Res. 1982;10:7701–7714. doi: 10.1093/nar/10.23.7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mead GC. The amino acid-fermenting Clostridia. J Gen Microbiol. 1971;67:47–56. doi: 10.1099/00221287-67-1-47. [DOI] [PubMed] [Google Scholar]
- 45.Jackson S, Calos M, Myers A, Self WT. Analysis of proline reduction in the nosocomial pathogen Clostridium difficile. J Bacteriol. 2006;188:8487–8495. doi: 10.1128/JB.01370-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bar-Even A, Noor E, Savir Y, Liebermeister W, Davidi D, Tawfik DS, Milo R. The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry. 2011;50:4402–4410. doi: 10.1021/bi2002289. [DOI] [PubMed] [Google Scholar]
- 47.Gorres KL, Raines RT. Prolyl 4-hydroxylase. Crit Rev Biochem Mol Biol. 2010;45:106–124. doi: 10.3109/10409231003627991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adams E, Frank L. Metabolism of proline and the hydroxyprolines. Annu Rev Biochem. 1980;49:1005–1061. doi: 10.1146/annurev.bi.49.070180.005041. [DOI] [PubMed] [Google Scholar]
- 49.Phang JM, Liu W, Zabirnyk O. Proline metabolism and microenvironmental stress. Annu Rev Nutr. 2010;30:441–463. doi: 10.1146/annurev.nutr.012809.104638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH. Uniprot Consortium, UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics. 2015;31:926–932. doi: 10.1093/bioinformatics/btu739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wecksler SR, Stoll S, Tran H, Magnusson OT, Wu SP, King D, Britt RD, Klinman JP. Pyrroloquinoline quinone biogenesis: demonstration that PqqE from Klebsiella pneumoniae is a radical S-adenosyl-L-methionine enzyme. Biochemistry. 2009;48:10151–10161. doi: 10.1021/bi900918b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stoll S, Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J Magn Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 54.Lovitt RW, Morris JG, Kell DB. The growth and nutrition of Clostridium sporogenes NCIB 8053 in defined media. J Appl Bacteriol. 1987;62:71–80. doi: 10.1111/j.1365-2672.1987.tb02382.x. [DOI] [PubMed] [Google Scholar]
- 55.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Mentjies P, Drummond A. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mitchell A, Chang HY, Daugherty L, Fraser M, Hunter S, Lopez R, McAnulla C, McMenamin C, Nuka G, Pesseat S, Sangrador-Vegas A, Scheremetjew M, Rato C, Yong SY, Bateman A, Punta M, Attwood TK, Sigrist CJA, Redaschi N, Rivoire C, Xenarios I, Kahn D, Guyot D, Bork P, Letunic I, Gough J, Oates M, Haft D, Huang H, Natale DA, Wu CH, Orengo C, Sillitoe I, Mi H, Thomas PD, Finn RD. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 2015;43:D213–D221. doi: 10.1093/nar/gku1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leppänen VM, Merckel MC, Ollis DL, Wong KK, Kozarich JW, Goldman A. Pyruvate formate lyase is structurally homologous to type I ribonucleotide reductase. Structure. 7:733–744. doi: 10.1016/S0969-2126(99)80098-7. [DOI] [PubMed] [Google Scholar]
- 60.Lehtiö L, Goldman A. The pyruvate formate lyase family: sequences, structures and activation. Protein Eng Des Sel. 2004;17:545–552. doi: 10.1093/protein/gzh059. [DOI] [PubMed] [Google Scholar]
- 61.Craciun S, Marks JA, Balskus EP. Characterization of choline trimethylamine-lyase expands the chemistry of glycyl radical enzymes. ACS Chem Biol. 2014;9:1408–1413. doi: 10.1021/cb500113p. [DOI] [PubMed] [Google Scholar]
- 62.Funk MA, Marsh ENG, Drennan CL. Substrate-bound structures of benzylsuccinate synthase reveal how toluene is activated in anaerobic hydrocarbon degradation. J Biol Chem. 2015;290:22398–22408. doi: 10.1074/jbc.M115.670737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Human Microbiome Project Consortium. A framework for human microbiome research. Nature. 2012;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bharadwaj VS, Dean AM, Maupin CM. Insights into the glycyl radical enzyme active site of benzylsuccinate synthase: a computational study. J Am Chem Soc. 2013;135:12279–12288. doi: 10.1021/ja404842r. [DOI] [PubMed] [Google Scholar]
- 65.Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modelling by MODELLER. Proteins. 1995;23:318–326. doi: 10.1002/prot.340230306. [DOI] [PubMed] [Google Scholar]
- 66.Alva V, Nam SZ, Söding J, Lupas AN. The MPI bioinformatics toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res. 2016;44:W410–W415. doi: 10.1093/nar/gkw348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lehtiö L, Grossmann JG, Kokona B, Fairman R, Goldman A. Crystal structure of a glycyl radical enzyme from Archaeoglobus fulgidus. J Mol Biol. 2006;357:221–235. doi: 10.1016/j.jmb.2005.12.049. [DOI] [PubMed] [Google Scholar]
- 68.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 69.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 70.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 71.Jacobson MP, Pincus DL, Rapp CS, Day TJF, Honig B, Shaw DE, Friesner RA. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 72.Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol. 2002;320:597–608. doi: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 73.Hildebrand A, Remmert A, Biegert A, Söding J. Fast and accurate automatic structure prediction with HHpred. Proteins. 2009;77:128–132. doi: 10.1002/prot.22499. [DOI] [PubMed] [Google Scholar]
- 74.Remmert M, Biegert A, Hauser A, Söding J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2012;9:173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 75.Kalnins G, Kuka J, Grinberga S, Makrecka-Kuka M, Liepinsh E, Dambrova M, Tars K. Structure and function of CutC choline lyase from human microbiota bacterium Klebsiella pneumoniae. J Biol Chem. 2015;290:21732–21740. doi: 10.1074/jbc.M115.670471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 77.Wei Y, Li B, Prakash D, Ferry JG, Elliott SJ, Stubbe J. A ferredoxin disulfide reductase delivers electrons to the Methanosarcina barkeri class III ribonucleotide reductase. Biochemistry. 2015;54:7019–7028. doi: 10.1021/acs.biochem.5b01092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci USA. 2003;100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Murib JH, Ritter DM. Decomposition of nitrosyl disulfonate ion. I. Products and mechanism of color fading in acid solution. J Am Chem Soc. 1952;74:3394–3398. [Google Scholar]
- 81.Carlson EE, Kiessling LL. Improved chemical syntheses of 1- and 5-deazariboflavin. J Org Chem. 2004;69:2614–2617. doi: 10.1021/jo049859f. [DOI] [PubMed] [Google Scholar]
- 82.Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. Highly selective hydrolytic kinetic resolution of terminal epoxides catalyzed by chiral (salen)coIII complexes. Practical synthesis of enantioenriched terminal epoxides and 1,2-diols. J Am Chem Soc. 2002;124:1307–1315. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 83.Nocek B, Chang C, Li H, Lezondra L, Holzle D, Collart F, Joachimiak A. Crystal structures of Δ1-pyrroline-5-carboxylate reductase from human pathogens Neisseria meningitidis and Streptococcus pyogenes. J Mol Biol. 2005;354:91–106. doi: 10.1016/j.jmb.2005.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kenklies J, Ziehn R, Fritsche K, Pich A, Andreesen JR. Proline biosynthesis from L -ornithine in Clostridium sticklandii: purification of Δ1-pyrroline-5-carboxylate reductase, and sequence and expression of the encoding gene. proC Microbiology. 1999;145:819–826. doi: 10.1099/13500872-145-4-819. [DOI] [PubMed] [Google Scholar]
- 85.Thiele B, Stein N, Oldiges M, Hofmann D. Direct analysis of underivatized amino acids in plant extracts by LC-MS-MS. Methods Mol Biol. 2012;828:317–328. doi: 10.1007/978-1-61779-445-2_25. [DOI] [PubMed] [Google Scholar]
- 86.Langrock T, García-Villar N, Hoffmann R. Analysis of hydroxyproline isomers and hydroxylysine by reversed-phase HPLC and mass spectrometry. J Chromatogr B. 2007;847:282–288. doi: 10.1016/j.jchromb.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 87.Toraya T. Cobalamin-dependent dehydratases and a deaminase: Radical catalysis and reactivating chaperones. Arch Biochem Biophys. 2014;544:40–57. doi: 10.1016/j.abb.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 88.Sandala GM, Smith DM, Radom L. Modeling the reactions catalyzed by coenzyme B12-dependent enzymes. Acc Chem Res. 2010;43:642–651. doi: 10.1021/ar900260c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.