

Abstract
Glycoengineered therapeutic antibodies and glycosite-specific antibody–drug conjugates (gsADCs) have generated great interest among researchers because of their therapeutic potential. Endoglycosidase-catalyzed in vitro glycoengineering technology is a powerful tool for IgG Fc (fragment cystallizable) N-glycosylation remodeling. In this protocol, native heterogeneously glycosylated IgG N-glycans are first deglycosylated with a wild-type endoglycosidase. Next, a homogeneous N-glycan substrate, presynthesized as described here, is attached to the remaining N-acetylglucosamine (GlcNAc) of IgG, using a mutant endoglycosidase (also called endoglycosynthase) that lacks hydrolytic activity but possesses transglycosylation activity for glycoengineering. Compared with in vivo glycoengineering technologies and the glycosyltransferase-enabled in vitro engineering method, the current approach is robust and features quantitative yield, homogeneous glycoforms of produced antibodies and ADCs, compatibility with diverse natural and non-natural glycan structures, convenient exploitation of native IgG as the starting material, and a well-defined conjugation site for antibody modifications. Potential applications of this method cover a broad scope of antibody-related research, including the development of novel glycoengineered therapeutic antibodies with enhanced efficacy, site-specific antibody–drug conjugation, and site-specific modification of antibodies for fluorescent labeling, PEGylation, protein cross-linking, immunoliposome formation, and so on, without loss of antigen-binding affinity. It takes 5–8 d to prepare the natural or modified N-glycan substrates, 3–4 d to engineer the IgG N-glycosylation, and 2–5 d to synthesize the small-molecule toxins and prepare the gsADCs.
INTRODUCTION
The interaction between the Fc domain of IgG antibodies and Fc receptors has an important role in antibody-dependent cell-mediated cytotoxicity (ADCC)1–3, complement-dependent cytotoxicity (CDC)4, anti-inflammatory activity5–7, pharmacokinetic half-life8,9, and immunogenicity10. These interactions are regulated by N-glycosylation of the IgG Fc domains11–14. Various glycoengineering technologies have emerged, aiming at enhancing the efficacy of therapeutic antibodies through N-glycosylation10. In addition to the approaches to in vivo N-glycoengineering through glycosyltransferase knockout/knock-in expression systems2,15,16 and in vitro treatment of antibodies with glycosyltransferases17,18, endoglycosidase-catalyzed N-glycosylation remodeling has become a powerful tool for the synthesis of homogeneous N-glycoproteins19–27. This chemoenzymatic method exploits the hydrolytic activity of wild-type (WT) endoglycosidases to cleave heterogeneous N-glycans and leave the innermost GlcNAc motifs on the glycoproteins. Next, endoglycosidase mutants28, also called glycosynthases—which lack hydrolytic activity but possess trans-glycosylation activity through the use of a homogeneous synthetic N-glycan oxazoline as the activated substrate—are used to assemble this intact N-glycan substrate on the GlcNAc-bearing glycoprotein generated by the aforementioned deglycosylation by the WT enzyme to accomplish the glycoengineering. Endo-β-N-acetylglucosaminidase from Streptococcus pyogenes (Endo-S), as one of these endoglycosidases, specifically recognizes the IgG Fc domain and has been used for efficient glyco-remodeling of therapeutic antibodies21–26. The glycan substrate specificity of Endo-S covers a wide range of natural and modified N-glycan structures. These unique features of Endo-S make it a perfect enzyme for in vitro IgG glycoengineering that is specific for the Fc domain and compatible with a diverse range of glycan substrates22. In addition, another endo-β-N-acetylglucosaminidase from S. pyogenes serotype M49, Endo-S2, and mutants thereof exhibit even more relaxed substrate specificity and are capable of acting on high-mannose, hybrid, and complex-type Fc N-glycans25,29.
Recently, we reported a one-pot chemoenzymatic method using azido-tagged N-glycopeptide precursors, and successive ‘click’ conjugation with alkyne-tagged small molecules for the synthesis of gsADCs26. The gsADC concept was originally developed using glycosyltransferase to introduce azido-tagged monosaccharides on external residues of IgG N-glycans, followed by small-molecule drug conjugation30–32. However, the limited substrate specificity of glycosyltransferases33 hampered their application in forming diverse structures for pharmaceutical screening. Besides its wide range of substrate structures, glyco-remodeling by Endo-S results in high yields, is cost-efficient for the semisynthesis of glycan substrates, generates homogeneous glycan–drug structures, and can be used to modify various IgGs. In addition, this glycosite-specific conjugation approach provides a new strategy for IgG labeling and modification with less influence on antigen-binding affinity as compared with random conjugation on lysine residues.
Comparison with other methods: advantages, limitations, and adaptations
This Endo-S-catalyzed in vitro glycoengineering approach could be directly used to modify thousands of commercial antibodies from different species (e.g., human, mouse, rabbit, goat) with various natural and non-natural N-glycans. In vitro glycoengineering circumvents the limitations of the in vivo glycoengineering technology, which requires expression optimization for each IgG and particular N-glycan in an engineered expression system by knockout/knock-in of key glycosyltransferases2,10,34. Other in vitro glycoengineering technologies using glycosyltransferases have limited substrate specificity, as the transferases recognize monosaccharide moieties on the sugar structure within a limited tolerance30–32. By contrast, Endo-S has a broad substrate scope and allows for a wider diversity of modifications on the nonreducing end. Moreover, it remains very challenging to prepare homogeneous glycoforms using in vivo technologies or the glycosyltransferase-catalyzed in vitro technology, because natural IgGs may carry highly heterogeneous N-glycans and some of them cannot be used as a substrate for certain glycosyltransferases. The current method removes all external N-glycans and remodels these glycans with well-defined synthetic substrates.
The ability to engineer antibody N-glycans in a site-specific manner enables specific functionalization on the Fc glycosite Asn297, which is highly conserved in various mammalian IgGs, and allows chemical modifications of this particular site. Compared with other site-specific conjugation strategies such as ThioMab technology35, unnatural amino acid embedding36, and the glycosyltransferase approach30,31, this method describes a simple procedure with lower technical requirements. Some key materials used in this method are already commercially available, including the endoglycosidases (Endo-M (endo-β-N-acetylglucosaminidase from Mucor hiemalis) and Endo-S), α-L-fucosidase, egg-yolk sialylglycopeptide (SGP), toxins, and linkers. More importantly, this method works efficiently in almost quantitative yields based on liquid chromatography–mass spectrometry (LC–MS) analysis and provides highly homogeneous end products.
Low efficiency in IgG defucosylation, a critical step in enhancing the ADCC of therapeutic antibodies, was a major limitation of the original version of this method, as it required a lengthy procedure (3 weeks). Recently, two better fucosidases were reported with markedly enhanced activity, allowing the digestion time to be shortened to 1 d23,25. We followed the literature25 and updated our current protocol to incorporate one of these. As compared with that for monosugar substrates in the glycosyltransferase approaches, the higher cost of total synthesis of complex N-glycan substrates in this method was another concern for its practical application in industry. To address this issue, we had previously optimized the extraction procedure for precursor SGP from egg yolk to the gram scale37; this optimized procedure can be used for semisynthesis of the substrates and to develop scaled-up procedures. Another challenge was that multiple purification steps are required for the antibody after deglycosylation, transglycosylation, and conjugation. We have applied the Endo-S enzymes to a magnetic nanoparticle solid support in order to simplify the procedure (data not shown).
Since the original method was published, we and other scientists have made substantial improvements, including the development of one-pot chemoenzymatic procedures26, use of new endoglycosidases25 and improved fucosidases23,25, identification of various N-glycan substrates23,26, the introduction of different types of modification chemistry24,37,38, and an optimized procedure for N-glycan extraction37, to broaden the adaptation of this method. We report here the updated protocol describing all required steps of this approach.
Applications
This protocol can be used for the synthesis of homogeneous glycoengineered therapeutic antibodies and gsADCs. Glycoengineered IgGs with optimized glycoforms attracted great interest from researchers and exhibited enhanced ADCC and/or CDC efficacy, especially for cancer treatment1,4,10. ADCs exploit the high binding affinity of the antibody to a particular antigen to deliver the conjugated toxin to cancer cells. As compared with the random conjugation on Lys or Cys amino acids of traditional ADCs, gsADCs have obvious advantages, such as convenient quality control, more consistent efficacy, and simplified pharmacokinetic detection.
This protocol is also useful for site-specific labeling or modification of IgG antibodies. Antibody labeling is a basic technology used in antigen-binding affinity assays, immunofluorescence microscopy, western blotting, FACS analysis, ELISA, and so on. However, labeling antibodies at random sites may also modify the antigen-binding fragment (Fab) and alter the antigen-binding efficacy of antibodies. The current method allows for specific labeling on the Fc fragment and therefore minimizes its influence on the Fab domains. Other site-specific modifications of antibodies, such as PEGylation, protein cross-linking, and immunoliposome formation, could also be achieved using this protocol.
Overview of the procedures
The main procedures of Endo-S-catalyzed synthesis of glycoengineered IgG and gsADCs are summarized in Figure 1. The procedures consist of the following key steps:
Preparation of deglycosylated IgG (2) bearing disaccharide Fucα1,6GlcNAc (2a) or monosaccharide GlcNAc (2b) from commercial therapeutic IgG (1). This is done by removal of the heterogeneous N-glycans from the specific Fc glycosite Asn297 of the native IgGs using Endo-S or Endo-S treatment combined with further defucosylation by the α-l-fucosidase C (AlfC) from Lactobacillus casei.
Semisynthesis of homogeneous N-glycopeptides (4a–f) as the N-glycan substrate precursors from an egg-yolk SGP (3).
One-pot chemoenzymatic synthesis of glycoengineered IgGs (5a–e) by transglycosylating the synthetic N-glycopeptide substrates (4a–f) onto Fucα1,6GlcNAc-IgG (2a) or GlcNAc-IgG (2b), catalyzed by a mutation (D233Q) of the Endo-S enzyme.
Site-specific conjugation of dibenzoazacyclooctyne (DBCO)-tagged small molecules (6a–d) to the azido-functionalized N-glycans of IgG (5a) via a copper-free ‘click’ reaction, yielding the gsADCs (7a–f).
Figure 1.
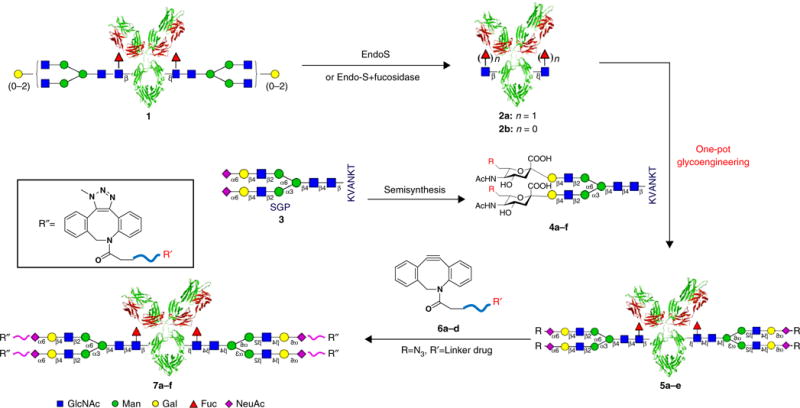
General scheme of chemoenzymatic synthesis of glycoengineered IgGs and gsADCs. EndoS, endo-β-N-acetylglucosaminidase from Streptococcus pyogenes; Fuc, fucose; Gal, galactose; GlcNAc, N-acetylglucosamine; KVANKT, Lys-Val-Ala-Asn-Lys-Thr; Man, mannose; NeuAc, N-acetylneuraminic acid.
The flowchart in Figure 2 provides an overview of the procedures. A simplified procedure for SGP extraction from egg yolks is provided in Box 1. Using SGP as the precursor, semisynthesis of non-natural SGP analogs carrying azido or alkyne functional groups is shown in Steps 1–17. N-glycan oxazolines are activated substrates of endoglycosynthases (endoglycosidase mutants) used for chemoenzymatic transglycosylation. From SGP or its analogs, the oxazoline substrates can be prepared via a one-pot reaction combined with N-glycan removal from the glycopeptides with Endo-M from Mucor hiemalis39 and successive oxazoline formation in aqueous solution40 (Steps 18–45). The in situ N-glycan oxazolines derived from SGP or its analogs can also be used directly in one-pot chemoenzymatic glycoengineering in the presence of preprepared GlcNAc-bearing IgG (2a or 2b) and Endo-S D233Q (Steps 74–85). The same chemoenzymatic transglycosylation reactions using purified N-glycan oxazolines are shown in Steps 63–73. Preparation of GlcNAc-IgG from deglycosylation of native IgG by WT Endo-S and α-L-fucosidase AlfC is described in Steps 46–62. The cloning, expression, and purification of the enzymes Endo-M, α-l-fucosidase AlfC, Endo-S WT, and its mutant D233Q are performed as described in the literature25,26,28 with modification as described in the Supplementary Results. Enzyme activity is critical to the deglycosylation and transglycosylation steps. We therefore describe the activity assays and unit definition for these enzymes in Box 2. DBCO-linked toxins (see Fig. 3 and Box 3 for their synthesis) are attached to the azido-tagged N-glycans of glycoengineered IgGs, via copper-free ‘click’ reaction, to yield the gsADCs (Steps 86–95). Through the combination of the glycosite-specific strategy and the random Lys conjugation, dual-payload ADC is achieved as described in Steps 96–108.
Figure 2.
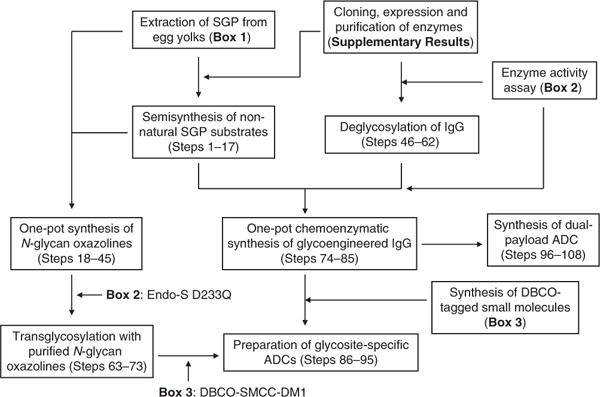
Flowchart of the preparation procedure for glycoengineered IgG and gsADCs.
Box 1. Extraction and purification of SGP ● TIMING 4 d.
SGP as a homogeneous N-glycan precursor is extracted from chicken egg yolk by successive procedures of defatting with ether, extraction with aqueous acetone, SPE with active carbon, and isolation with G-25 gel-filtration chromatography.
Take 200 eggs (purchased from local market). Open each shell and carefully separate the egg yolk from the egg-white liquid.
Put all the egg yolks in a 5-liter beaker. Add 1 liter of water and stir the mixture at RT for 30 min.
Lyophilize the mixture until dryness to obtain ~1.5 kg of egg-yolk powder. This step takes ~1 d.
Weigh 1.2 kg of egg-yolk powder from step 3 in a 10-liter beaker. Add 4 liters of tert-butyl methyl ether to the powder and stir at RT for 1 h. Filter the solid through a Buchner funnel and discard the liquid. Mix the filter cake with an additional 4 liters of tert-butyl methyl ether and repeat the stirring and filtration. Repeat this with 1 liter of tert-butyl methyl ether three times. This step takes ~2–3 h.
Stir the filter cake from step 4 with 4 liters of 70% acetone solution in a 10-liter beaker at RT for 30 min, filter the solid, and discard the liquid. Mix the filter cake with an additional 4 liters of 70% acetone solution and repeat the stirring and filtration. This step takes ~2 h.
-
Stir the filter cake from step 5 with 4 liters of 40% acetone solution in a 10-liter beaker at RT for 1 h. Filter the solid and collect the filtrate. Mix the filter cake with an additional 4 liters of 40% acetone solution and repeat the stirring and filtration. Concentrate the combined filtrate to 1 liter using a Rotavapor. This step takes ~3–5 h.
▲ CRITICAL STEP Set the water bath temperature of the Rotavapor below 35 °C (also for step 9). High temperatures, especially in combination with a low pH, may cause partial removal of the terminal sialic acid from SGP.
-
Prepare a column (10 × 10 cm, see EQUIPMENT section) filled with a mixture of 100 g of active carbon and 100 g of celite, prewashed with 1 liter of water. Flow the concentrated solution from step 6 through the column three times. This step takes ~1–2 h.
▲ CRITICAL STEP Avoid using a lathy column. This will cause high pressure and takes a long time to wash and elute.
Wash the column successively with 2 liters of pure water, 2 liters of 5% acetonitrile, and 2 liters of 10% acetonitrile. Elute the column two times with 2 liters of 25% acetonitrile. This step takes ~2–4 h.
Concentrate the combined elution solution from step 8 to a 500-ml volume using a Rotavapor. Lyophilize the residue to obtain 1.5 g of the crude SGP as a pale-yellow powder. This step takes ~1 d.
Dissolve the crude SGP from step 9 in 5 ml of water and centrifuge the solution at 3,200g at 4 °C for 30 min.
-
Take the supernatant from step 10 and load it onto a Sephadex G-25 column (2.6 × 80 cm). Elute the column with water. Collect the fractions containing SGP and lyophilize the combined fractions to obtain 0.5 g of SGP (purity >95%). This step takes ~1 d.
▲ CRITICAL STEP The crude SGP contains ~15% of a partial desialyl form with only one sialic acid on the nonreducing end of the SGP molecule. Gel-filtration with a G-25 column eluted with water or a 0.1 M acetic acid aqueous solution can separate the partial desialyl form from the pure SGP. However, if the crude SGP sample is overloaded or is in a large loading volume, the produced SGP may contain 5–15% of the mono desialyl form. In that case, repeated G-25 purification might be needed to obtain high-quality SGP.
? TROUBLESHOOTING
-
Monitor all the above processes with analytic HPLC. Also detect separation of SGP and its partial desialyl form with monosialic acid with HPAEC-PAD.
■ PAUSE POINT SGP can be stored at −20 °C for 6 months or at −80 °C for 12 months.
Box 2. Enzyme activity assay and unit definition ● TIMING 4–16 h.
The hydrolytic activities of the enzymes Endo-M, AlfC, and Endo-S against their corresponding substrates SGP, Fucα1,6GlcNAc-IgG, and native IgG are tested for unit definition. The transglycosylation activity of glycosynthase Endo-S D233Q is determined with GlcNAc-bearing IgG and an N-glycan oxazoline substrate (23).
The glycosidase activity assay and unit definition of Endo-M ● TIMING 4 h
-
Weigh 10 mg of SGP into a 1.5-ml centrifuge tube. Add 80 μl of 50 mM, pH 6.5 PB.
▲ CRITICAL STEP The SGP can be acidic after purification and may decrease the pH when mixed with PB. It is important to adjust the final pH of the solution to ~6.5 using 0.1 N NaOH.
Add 3.3 μl of 6.0 mg/ml Endo-M (freshly prepared; see Supplementary Methods).
Add additional 50 mM, pH 6.5 PB until the final volume of the assay reaction reaches 100 μl (final concentrations: SGP = 100 mg/ml, Endo-M 0.2 = mg/ml).
Incubate at 30 °C.
Take 2-μl samples from the reaction every 30 min and analyze the rate of SGP hydrolysis with analytic HPLC at each time point.
Take the final sample for HPLC detection after 4 h of incubation; this is when the SGP should be completely hydrolyzed.
-
Define that 1 unit of Endo-M hydrolyzes 10 mg of SGP in 100 μl of 50 mM, pH 6.5 PB within 4 h at 30 °C.
▲ CRITICAL STEP On the basis of this unit definition, the enzyme activity of fresh Endo-M should be no less than 50 units/mg. If this assay shows a lower enzyme activity, higher enzyme concentrations should be used in SGP hydrolysis.
The IgG defucosylation activity assay and unit definition of a-L-fucosidase AlfC ● TIMING 16 h
Prepare a solution of 1 mg of rituximab-GlcNAc(Fuc) in 50 μl of 50 mM, pH 7.5 Tris buffer in a 1.5-ml centrifuge tube.
Add 50 μg of AlfC (freshly prepared; see Supplementary Methods) to the tube (final concentrations: rituximab-GlcNAc(Fuc) = 20 mg/ml; AlfC = 1 mg/ml).
Incubate at 37 °C.
Take 2-μl aliquots from the reaction every 2 h and analyze the hydrolytic rate of rituximab-GlcNAc(Fuc) by LC–MS at each time point.
Take the final sample after 16 h of incubation; this is when the rituximab-GlcNAc(Fuc) should be completely defucosylated.
-
Define that 1 unit of AlfC hydrolyzes 1 mg of rituximab-GlcNAc(Fuc) in 50 μl of 50 mM, pH 7.5 Tris buffer within 16 h at 37 °C.
▲ CRITICAL STEP On the basis of this unit definition, enzyme activity of fresh AlfC should be no less than 20 unit/mg. If this assay shows a lower enzyme activity, higher enzyme concentrations should be used for IgG defucosylation.
The glycosidase activity assay and unit definition of wild-type Endo-S ● TIMING 1 h
Prepare a solution of 1-mg commercial Herceptin in 100 μl of 50 mM, pH 7.5 PB in a 1.5-ml centrifuge tube.
Add 5 μg of Endo-S (freshly prepared: see Supplementary Methods) to the tube (final concentrations: Herceptin = 10 mg/ml; Endo-S 0.05 = mg/ml).
Incubate at 37 °C.
Take 2-μl aliquots from the reaction after 5 min and analyze the hydrolytic rate of Herceptin with SDS-PAGE and LC–MS.
Herceptin deglycosylation should be complete within 5 min.
-
Define that 1 unit of WT Endo-S hydrolyzes 1 mg of Herceptin in 100 μl of 50 mM, pH 7.5 PB within 5 min at 37 °C.
▲ CRITICAL STEP On the basis of this unit definition, enzyme activity of fresh WT Endo-S should be no less than 200 unit/mg. If this assay shows a lower enzyme activity, higher enzyme concentrations should be used for IgG deglycosylation.
The transglycosylation activity assay and unit definition of Endo-S D233Q ● TIMING 2 h
-
1
Prepare a solution of 200 μg of Herceptin-GlcNAc(Fuc) in 36 μl of 50 mM, pH 7.4 Tris buffer in a 200-μl centrifuge tube.
-
2
Add 2 μl of 20 mM sialyl complex-type (SCT) N-glycan oxazoline (23, freshly prepared from PROCEDURE Step 44; final concentration = 1 mM).
-
3
Adjust the pH to 7.4 with 0.1 N HCl.
▲ CRITICAL STEP The glycan oxazoline is labile in an acidic environment, so it must be stabilized with ~6% NaOH after purification. When adding the oxazoline to the antibody solution, the pH increases, and adjustment with acid is required. Meanwhile, it is also important to avoid overadjustment, as the oxazoline is not stable at low pH. A tip for the pH adjustment in this step is to add a tiny amount (e.g., 1 μl) of HCl multiple times until the target pH value is reached.
-
4
Add 2 μl of 2 mg/ml Endo-S D233Q (freshly prepared: see Supplementary Methods) to the tube (final concentration = 0.1 mg/ml).
-
5
Incubate at 30 °C.
-
6
Take 2-μl aliquots from the reaction every 30 min and analyze the transglycosylation rate of Herceptin-GlcNAc(Fuc) with SDS-PAGE and LC–MS at each time point.
-
7
Take the final sample for analysis after 1 h of incubation; this is when the Herceptin should be completely transglycosylated.
-
8
Define that 1 unit of Endo-S D233Q transfers the SCT glycan (in a 1 mM concentration) onto 200 μg of Herceptin-GlcNAc(Fuc) in 40 μl of 50 mM, pH 7.4 Tris buffer at 30 °C with a 100% yield in 1 h.
▲ CRITICAL STEP On the basis of this unit definition, transglycosylation activity of fresh Endo-S D233Q should be no less than 250 unit/mg. If this assay shows a lower enzyme activity, higher enzyme concentrations should be used for IgG transglycosylation.
Figure 3.
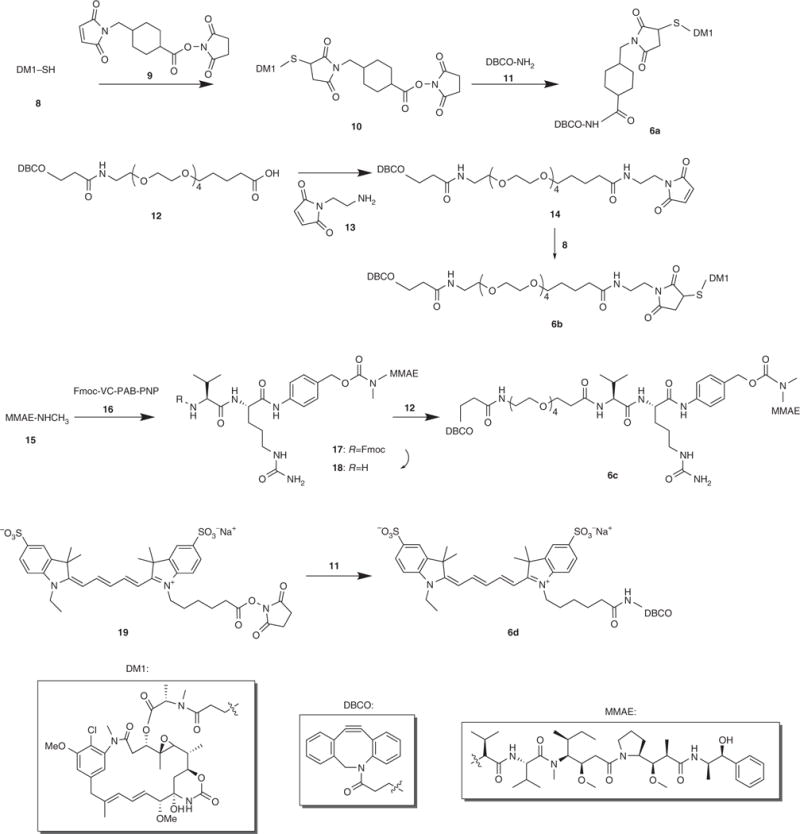
Synthesis of DBCO-tagged small molecules (Box 3).
Box 3. Synthesis of DBCO-tagged small molecules ● TIMING 1–4 d.
Synthesis of DM1-SMCC (10) ● TIMING 4 h
Weigh commercial DM1 (also called DM1-SH in Fig. 3) (8, 30.0 mg, 0.04 mmol, 1.0 equiv.) and N-succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) (9, 15 mg, 0.045 mmol, 1.1 equiv.) into a 25-ml round-bottom flask, dissolve them in a mixture of 10 ml of acetonitrile and 5 ml of 50 mM, pH 7.5 PB (vol/vol = 2:1), and purge with Ar.
Stir at RT, and monitor the reaction by taking a 1 μl aliquot of the reaction every 30 min for HPLC detection. After 2 h, the complete conversion of 8 should be observed.
Purify the crude product with preparative HPLC. Combine the fractions containing the target product.
-
Lyophilize the combined fractions to obtain compound 10 as a white powder (40.7 mg, yield = 95%).
■ PAUSE POINT Compound 10 can be stored at −20 °C for 3 months or at −80 °C for 6 months.
Synthesis of DBCO-SMCC-DM1 (6a) ● TIMING 4 h
Weigh DM1-SMCC (10, 5.4 mg, 5.0 μmol, 1.0 equiv.) in a 25-ml round-bottom flask. Add DBCO-NH2 (11, 1.5 mg, 5.5 μmol, 1.1 equiv.), Et3N (0.8 μl, 5.5 μmol, 1.1 equiv.), and 5 ml of acetonitrile.
Stir at RT, and monitor the reaction by taking a 1-μl aliquot of the reaction every 30 min for HPLC detection. After 2 h, the complete conversion of 10 should be observed.
Purify the crude product with preparative HPLC. Combine the fractions containing the target product.
-
Lyophilize the combined fractions to obtain compound 6a as a white powder (5.8 mg, yield = 94%).
■ PAUSE POINT Compound 6a can be stored at −20 °C for 6 months or at −80 °C for 12 months.
Synthesis of DBCO-PEG4-Maleimide (14) ● TIMING 2 d
Weigh commercial DBCO-PEG4-COOH (12, 5.8 mg, 10 μmol, 1.0 equiv.) in a 25-ml round-bottom flask. Add 2 ml of DMF.
Add N-(2-aminoethyl)maleimide (13, 7.2 mg, 30 μmol, 3.0 equiv.).
Add dicyclohexylcarbodiimide (DCC, 10.3 mg, 50 μmol, 5.0 equiv.) and pyridine (24 μl, 30.0 equiv.).
Stir the mixture at RT for 40 h, and purify the product with preparative HPLC.
-
Lyophilize the product fractions to yield compound 14 as a sticky oil (4 mg, yield = 57%).
■ PAUSE POINT Compound 14 can be stored at −20 °C for 6 months or at −80 °C for 12 months.
Synthesis of DBCO-PEG4-DM1 (6b) ● TIMING 4 h
Dissolve compound 14 (4.0 mg, 5.7 μmol, 1.0 equiv.) from step 5 of the previous section of this box in 5 ml of acetonitrile and 2.5 ml of 50 mM, pH 7.5 PB (vol/vol = 2:1).
Add DM1-SH (8, 4.6 mg, 6.27 μmol).
Purge the solution with argon, and stir it at RT for 2 h.
Purify the crude product with preparative HPLC. Combine the product fractions.
-
Lyophilize the combined fractions to obtain compound 6b as a white powder (5.3 mg, yield = 65%).
■ PAUSE POINT Compound 6b can be stored at −20 °C for 6 months or at −80 °C for 12 months.
Synthesis of Fmoc-VC-PAB-MMAE (17) ● TIMING 2 d
Weigh Fmoc-VC-PAB-PNP (40.0 mg, 52.2 μmol, 1.0 equiv.) and MMAE (56.0 mg, 78.0 μmol, 1.5 equiv.) in a 25-ml round-bottom flask. Add 8.0 ml of DMF.
Add hydroxybenzotriazole (HOBt, 1.6 mg, 10.4 μmol, 0.2 equiv.) and 120.0 μl of pyridine.
Stir at RT for 40 h, and purify the product with preparative HPLC.
-
Lyophilize the product fractions to obtain compound 17 as a sticky oil (58 mg, yield =3%).
■ PAUSE POINT Compound 17 can be stored at −20 °C for 6 months or at −80 °C for 12 months.
Synthesis of NH2-VC-PAB-MMAE (18) ● TIMING 2 h
Dissolve compound 17 (58 mg) from step 4 of the previous section of this box in 16 ml of acetonitrile. Add 4 ml of piperidine.
Stir at RT, and monitor the reaction by taking a 1-μl aliquot of the reaction every 30 min for HPLC detection. After 0.5 h, the complete conversion of 17 should be observed.
Remove the solvent and dissolve the crude product in 10 ml of 50% acetonitrile.
-
Purify the crude product with preparative HPLC and lyophilize the product fractions to obtain compound 18 as a white powder (46.6 mg, 96.2%).
■ PAUSE POINT Compound 18 can be stored at −20 °C for 6 months or at −80 °C for 12 months.
Synthesis of DBCO-PEG4-VC-PAB-MMAE (6c) ● TIMING 2 d
Weigh compound 12 (5.8 mg, 10 μmol, 1.0 equiv.) in a 25-ml round-bottom flask.
Add compound 18 (33.7 mg, 30 μmol, 3.0 equiv.) from step 4 of the previous section of this box.
Add 2 equiv. of DCC and 0.2 equiv. of HOBt.
Stir at RT for 40 h. Purify the crude product with preparative HPLC.
-
Lyophilize the product fractions to obtain compound 6c a white powder (13.5 mg, 80%).
■ PAUSE POINT Compound 6c can be stored at −20 °C for 6 months or at −80 °C for 12 months.
Synthesis of DBCO-Cy5 (6d) ● TIMING 6 h
Weigh 1 mg of Cy5-NHS (19, 1.3 μmol) and dissolve it in 100 μl of ddH2O and 100 μl of acetonitrile in a 1.5-ml centrifuge tube (tube 1).
Weigh 2 mg of DBCO-NH2 (11) and dissolve it in 200 μl of DMSO in another 1.5-ml centrifuge tube (tube 2).
Add 40 μl of the above solution (containing compound 11 (0.4 mg, 1.42 μmol, 1.1 equiv.)) from tube 2 to tube 1. Add 0.5 μl of Et3N (2.7 equiv.) to tube 1.
Stir at RT for 4 h. Purify the product by preparative HPLC.
-
Lyophilize the product fractions to obtain compound 6d as a dark blue powder (1.1 mg, 92%).
▲ CRITICAL STEP The compounds 19 and 6d are light-sensitive. Cover the reaction and purified product with aluminum foil.
■ PAUSE POINT Compound 6d can be stored at −20 °C for 6 months or at −80 °C for 12 months.
Experimental design
Before using this protocol for IgG glycoengineering, the availability of required materials and instruments must be confirmed. Follow Boxes 1–3 to prepare the key items for SGP synthesis, to test the activity of the required enzymes, and to prepare the small-molecule toxins for gsADCs. Analytical and preparative HPLC instruments are required for preparation of N-glycan substrates and toxins. LC–MS in a high mass range (2,000–5,000 Da) with deconvolution software for MS protein detection is very important for monitoring IgG glycoengineering and gsADC synthesis.
In critical steps, efficient monitoring and characterization of the reactions and products is critical to good results. The synthesis of SGP analogs and DBCO-linked small molecules is carried out and measured by analytic HPLC at time intervals (see Supplementary Results, Products 4a–f), and their purification is subject to preparative HPLC. N-glycan oxazoline formation in the presence of 2-chloro-1,3-dimethylimidazolinium chloride (DMC) is monitored with high-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD) and 1H NMR analysis (see Supplementary Results, products 22–26). This is to ensure a high conversion rate, which is particularly important for the sequential steps of chemoenzymatic transglycosylation using these in situ oxazolines via the one-pot strategy. SDS-PAGE and LC–MS are major tools for detection of IgG glycoengineering and gsADC conjugation (see Supplementary Results, products 5a–e and 7a–f). The band shift of IgG heavy chains after deglycosylation/glyco-remodeling on SDS-PAGE can roughly determine the efficiency of the enzymatic reaction. Moreover, LC–MS detection provides precise data on the intact IgG that can be used for monitoring the glycoengineering process (see examples in the Anticipated Results section).
When following this protocol, we suggest starting with a test scale of 100–200 μg of IgG, especially for precious antibody samples. The enzymatic transglycosylation (Steps 63–85) is the limiting step in the PROCEDURE, as the reaction yield is greatly dependent on the quality of N-glycan oxazolines, the enzyme activity, and the buffer pH and concentration. Optimization of these steps might be required to attain good results (see the Troubleshooting section for details) before scaling up to the milligram or higher scale.
MATERIALS
REAGENTS
Eggs (local market)
Tert-butyl methyl ether (Sinopharm Chemical Reagent, cat. no. 80144960) ! CAUTION It is toxic and flammable; avoid direct contact with skin and eyes. Wear gloves and eye protection and handle the compound in a fume hood.
Acetone (Sinopharm Chemical Reagent, cat. no. 10000418) ! CAUTION It is toxic and flammable; avoid direct contact with skin and eyes. Wear gloves and eye protection and handle the compound in a fume hood.
Active carbon (Sinopharm Chemical Reagent, cat. no. 10006619) ! CAUTION It is a dust powder. Wear a mask to avoid inhalation.
Celite (Sinopharm Chemical Reagent, cat. no. 20020662)
Sephadex G-25 (Sigma-Aldrich, cat. no. G2580)
HPLC-grade trifluoroacetic acid (TFA; J&K Chemical, cat. no. 134753) ! CAUTION It is highly toxic, volatile, and corrosive. Wear protective gloves, goggle, mask, and a lab coat.
HCl (hydrochloric acid; Sinopharm Chemical Reagent, cat. no. 10011018) ! CAUTION It is highly toxic, volatile, and corrosive. Wear protective gloves, goggle, mask, and a lab coat.
NaOH (sodium hydroxide; Sinopharm Chemical Reagent, cat. no. 10019718) ! CAUTION It is toxic and corrosive; avoid direct contact with skin and eyes. Wear gloves and eye protection and handle the compound in a fume hood.
2-Chloro-1,3-dimethylimidazolinium chloride (DMC; Sigma-Aldrich, cat. no. 529249)
β-1,4-Galactosidase (New England BioLabs, cat. no. P0730L)
Neuraminidase (Sigma-Aldrich, cat. no. N8271)
β-N-acetylglucosaminidase (New England BioLabs, cat. no. P0732L)
Sodium periodate (NaIO4; Sinopharm Chemical Reagent, cat. no. 80117316) ! CAUTION It is toxic and harmful to skin and eyes. Wear protective gloves, goggle, and a lab coat.
Glycerol (Sinopharm Chemical Reagent, cat. no. 10010618)
ESI-L low-concentration tuning mix (Agilent Technologies, part no. G1969-85000)
Acetic acid (Sinopharm Chemical Reagent, cat. no. 10000218) ! CAUTION It is toxic and corrosive. The irritant gas is harmful to the nose and eyes.
3-Azido-1-propanamine (J&K Chemical, cat. no. A2738)
Propargylamine (J&K Chemical, cat. no. 307008)
HPLC-grade acetonitrile (J&K Chemical, cat. no. 925301) ! CAUTION It is toxic, flammable, and corrosive; avoid direct contact with skin and eyes. Wear gloves and eye protection and handle the compound in a fume hood.
Herceptin (Trastuzumab) (Roche)
Rituximab (Rituxan; Roche)
DM1 (also called DM1-SH; N2′-deacetyl-N2′-(3-mercapto-1-oxopropyl)-maytansine; Levena Biopharma, cat. no. LN-T-4582) ! CAUTION It is highly toxic. Wear protective gloves, goggle, mask, and a lab coat.
MMAE (monomethyl auristain E; also called MMAE-NHCH3; Levena Biopharma, cat. no. LN-T-1458) ! CAUTION It is highly toxic. Wear protective gloves, goggle, mask, and a lab coat.
Fmoc-Val-Cit-para-aminobenzoic acid-para-nitrophenyl (Fmoc-Val-Cit-PAB-PNP; Levena Biopharma, cat. no. LN-L-3307)
Dibenzoazacyclooctyne (DBCO)-amine (Sigma-Aldrich, cat. no. 761540)
DBCO-PEG4-acid (Sigma-Aldrich, cat. no. 759902)
Sulfo-cyanine5 N-hydroxysuccinimidyl ester (Little-PA Sciences, cat. no. A04002)
Porous graphite carbon (PGC) cartridges (Hypercarb; Thermo Fisher Scientific, cat. no. 60106-303)
Isopropyl β-d-1 thiogalactopyranoside (Sangon Biotech, cat. no. A100487)
Ampicillin, sodium salt (Sangon Biotech, cat. no. A100339)
Kanamycin sulfate (Sangon Biotech, cat. no. A600286)
Imidazole (Sangon Biotech, cat. no. A500529)
Tris (hydroxymethyl) aminomethane (Sangon Biotech, cat. no. A100826)
Tween 20 (Sangon Biotech, cat. no. A100777)
N-Succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (Bide Pharmatech, cat. no. BD151232)
2-Hydroxyisoindoline-1,3-dione (Bide Pharmatech, cat. no. BD144580)
Glycine (Sinopharm Chemical Reagent, cat. no. 62011516)
Sodium cyanoborohydride (Sinopharm Chemical Reagent, cat. no. XW258956072)
Ethylenediamine (Sinopharm Chemical Reagent, cat. no. 10009518) ! CAUTION It is toxic, flammable, and corrosive; avoid direct contact with skin and eyes. Wear gloves and eye protection and handle the compound in a fume hood.
Maleic anhydride (Sinopharm Chemical Reagent, cat. no. 10013116)
DMSO (Sinopharm Chemical Reagent, cat. no. 30072418)
DMF (N,N-dimethylformamide; Sinopharm Chemical Reagent, cat. no. 81007718)
DCM (dichloromethane; Sinopharm Chemical Reagent, cat. no. 80047318) ! CAUTION It is toxic; avoid direct contact with skin and eyes. Wear gloves and eye protection and handle the compound in a fume hood.
Acetic anhydride (Sinopharm Chemical Reagent, cat. no. 10000318) ! CAUTION It is toxic; avoid direct contact with skin and eyes. Wear gloves and eye protection and handle the compound in a fume hood.
Sodium azide (Sinopharm Chemical Reagent, cat. no. 80115560) ! CAUTION It is highly toxic and explosive. Wear protective gloves, goggle, mask, and a lab coat.
Hydrazine hydrate (Sinopharm Chemical Reagent, cat. no. 80070418) ! CAUTION It is toxic, corrosive, and explosive. Wear protective gloves, goggle, mask, and a lab coat.
Dicyclohexylcarbodiimide (Sinopharm Chemical Reagent, cat. no. XW020080)
Azabenzene (Sinopharm Chemical Reagent, cat. no. 10018118)
1-Hydroxybenzotriazole (Sinopharm Chemical Reagent, cat. no. XW020054)
Piperidine (Sinopharm Chemical Reagent, cat. no. 80104216) ! CAUTION It is highly irritant to eyes and skin. Wear protective gloves, goggle, mask, and a lab coat.
EQUIPMENT
HPLC system (e.g., Beijing ChuangXinTongHeng LC3000 (analytical) instrument with an analytical C18 column (5 μm, 4.6 × 150 mm); Thermo Fisher Ultimate 3,000 (analytical) instrument with an analytical C18 column (5 μm, 4.6 × 150 mm); Beijing ChuangXinTongHeng LC3000 (preparative) instrument with a preparative C18 column (Waters, C18, optimum bed density (OBD), 5 μm, 19 × 250 mm))
Electron spray ionization mass spectrometer (ESI–MS) and liquid chromatography mass spectrometer (LC–MS) (liquid chromatography-time of flight (LC–TOF) instrument; Agilent, model no. 6230)
HPAEC-PAD system (Thermo Scientific, model no. ICS 5000+) with a PA100 anion exchange column (4 × 250 mm) and a guard column (4 × 50 mm)
NMR instrument (Varian-Mercury, model no. Plus 400 or 500)
Rotavapor (Scanvac; LaboGene, article no. 7.001.000.115)
Filter paper (Sinopharm Chemical Reagent, cat. no. 92410431S)
10-liter beaker (Sinopharm Chemical Reagent)
1-liter and 100-ml cylinders (Sinopharm Chemical Reagent)
1-liter, 500-ml, 250-ml, and 100-ml liquid storage bottles (Sinopharm Chemical Reagent)
50-ml and 15-ml Tubes (Jet Biofil, cat. nos. CFT011500, CFT011150)
10-ml, 5-ml, 2.0-ml, 1.5-ml, and 0.2-ml centrifuge tubes (Sanhe)
Buchner funnel (assorted sizes; Sinopharm Chemical Reagent)
Custom-ordered glass column (10 × 20 cm; Xiamei) for active carbon–celite mixture. Pack the mixed active carbon–celite (1:1) into the column until the column is half full (at the 10-cm mark).
Custom-ordered glass column (2.6 × 100 cm; Xiamei) for Sephadex G-25 beads. Swell the G-25 beads in water and pack the mixture into the column until the column is 80% full (at the 80-cm mark).
Fraction collector (Huxi Instruments, model no. BS-100A)
Centrifuge (Sorvall ST 8R; Thermo Fisher, cat. no. 75007214)
Amicon ultracentrifugal filter (30 kDa/10 kDa cutoff; Millipore, cat. no. UFC803024/801024)
Heat air gun (Atten Instrument, cat. no. AT-A822D)
REAGENT SETUP
Enzyme expression and purification
Endo-M, α-l-fucosidase AlfC, Endo-S WT, and its mutant D233Q are expressed in Escherichia coli and purified via a His-tag affinity column following the literature22,25,28, with modification as described in the Supplementary Methods. The enzymes can be stored in aliquots at −80 °C for up to 6 months.
Phosphate buffers
Na2HPO4 (A) and NaH2PO4 (B) were dissolved in pure water to obtain corresponding 0.2 M solutions (solution A and solution B), respectively. Mix these two solutions at specific vol/vol ratios to prepare phosphate buffers at different pH values: mix 67 ml of solution A and 33 ml of solution B to obtain a 0.2 M, pH 7.0 phosphate buffer; mix 86 ml of solution A and 14 ml of solution B to obtain a 0.2 M, pH 7.5 phosphate buffer; mix 94.7 ml of solution A and 5.3 ml of solution B to obtain a 0.2 M, pH 8.0 phosphate buffer; and mix 12.3 ml of solution A and 87.7 ml of solution B to obtain a 0.2 M, pH 6.0 phosphate buffer. Dilute these buffers with pure water to obtain diluted buffers in lower concentrations. These buffers can be stored at 4 °C for up to 2 months.
50 mM, pH 7.5 phosphate buffer containing 10% (vol/vol) DMSO
Dilute 100 ml of 0.2 M, pH 7.5 phosphate buffer with 260 ml of water and add 40 ml of DMSO. This buffer should be freshly prepared.
Tris buffers
Dissolve 121.14 g of Tris base in 900 ml of water, and adjust the pH to 8.0 with concentrated HCl. Add extra water until a total volume of 1 liter is reached, to make a 1.0 M, pH 8.0 Tris–HCl buffer stock. Dilute 50 ml of 1.0 M, pH 8.0 Tris–HCl with 900 ml of water and adjust the pH to 7.4 with concentrated HCl. Add extra water until a total volume of 1 liter is reached, to obtain a 50 mM, pH 7.4 Tris–HCl buffer. These Tris buffers can be stored at 4 °C for 2 months.
30 mM NaIO4 aqueous solution
Weigh 65 mg of sodium periodate and dissolve it in 10 ml of pure water to make a 30 mM NaIO4 aqueous solution. This solution should be freshly prepared and kept away from light.
5, 10, and 25% Acetonitrile
Mix 950 ml of water and 50 ml of acetonitrile to make 1 liter of 5% acetonitrile solution. Mix 900 ml of water and 100 ml of acetonitrile to make 1 liter of 10% acetonitrile solution. Mix 750 ml of water and 250 ml of acetonitrile to make 1 liter of 25% acetonitrile. These solutions should be freshly prepared.
5, 10, and 25% (vol/vol) Acetonitrile containing 1 mM NaOH
Dissolve 400 mg of sodium hydroxide in 10 ml of water to make a 1.0 M NaOH solution. This solution can be stored at 4 °C for 2 weeks. Mix 94.9 ml of water, 5 ml of acetonitrile, and 100 μl of 1.0 M NaOH to make a 5% acetonitrile solution containing 1 mM NaOH. Mix 89.9 ml of water, 10 ml of acetonitrile, and 100 μl of 1.0 M NaOH to make a 10% acetonitrile solution containing 1 mM NaOH. Mix 74.9 ml of water, 25 ml of acetonitrile, and 100 μl of 1.0 M NaOH to make a 25% acetonitrile solution containing 1 mM NaOH. These acetonitrile solutions should be freshly prepared.
Herceptin solution
Exchange the commercial Herceptin buffer with a 50 mM, pH 7.5 phosphate buffer and concentrate the sample with an Amicon ultracentrifugal filter (30 kDa cutoff). Prepare a stock Herceptin solution at a 38 mg/ml concentration in the 50 mM, pH 7.5 phosphate buffer. The sample can be stored at −80 °C for 12 months and at 4 °C for 1 month.
Rituximab solution
Exchange the commercial rituximab buffer with a 50 mM, pH 7.4 Tris buffer and concentrate the sample with an Amicon ultracentrifugal filter (30 kDa cutoff). Prepare a stock solution of rituximab at a 20 mg/ml concentration in the 50 mM, pH 7.5 Tris buffer. The solution can be stored at −80 °C for 12 months and at 4 °C for 1 month.
Glycine–HCl buffers
Dissolve 75.07 g of glycine in 900 ml of water and adjust the pH to 8.8 or 5.0 with concentrated HCl. Add extra water until the total volume reaches 1 liter to obtain 1.0 M, pH 8.8 or 5.0 glycine–HCl buffers. Mix 10 ml of 1.0 M, pH 5.0 glycine–HCl buffer with 85 ml of water and adjust the pH to 2.5 with concentrated HCl. Add extra water until the total volume reaches 100 ml to prepare a 100 mM, pH 2.5 glycine–HCl buffer. All these buffers can be stored at 4 °C for up to 1 month.
10% (vol/vol) Sulfuric acid in ethanol
Slowly add 50 ml of sulfuric acid to 450 ml of ethanol in an ice bath to obtain a 10% sulfuric acid solution in ethanol. The solution can be stored at room temperature (RT) ~25 °C for up to 6 months.
0.2% (vol/vol) Triethylamine (Et3N)
Add 4 ml of Et3N to 2 liters of water to prepare a 0.2% Et3N solution. This solution should be freshly prepared.
1% Glycerol
Add 100 μl of glycerol to 9.9 ml of water to prepare a 1% (vol/vol) glycerol solution. This solution can be stored at 4 °C for up to 1 month.
EQUIPMENT SETUP
HPLC
In our laboratory, analytical reverse-phase (RP)-HPLC is performed on a Beijing ChuangXinTongHeng LC3000 (analytic) instrument (HPLC-A) with a C18 column (5 μm, 4.6 × 150 mm) or a Thermo Fisher Ultimate 3000 instrument (HPLC-B) with a column (5 μm, 4.6 × 250 mm) at 40 °C. Preparative HPLC is performed on a Beijing ChuangXinTongHeng LC3000 (preparative) instrument (HPLC-C) with a preparative column (Waters, C18, OBD, 5 μm, 19 × 250 mm) at RT. The mobile phases consist of a water solution containing 0.1% TFA (solvent A) and an acetonitrile solution containing 0.1% TFA (solvent B). The elution conditions are as follows: method A—HPLC-A, a linear elution gradient of 98:2 to 10:90 (solvent A/B) over 30 min at a flow rate of 1 ml/min; method B—HPLC-A, a linear elution gradient of 98:2 to 89:11 (solvent A/B) in 10 min and then 89:11 to 10:90 (solvent A/B) over an additional 20 min at a flow rate of 1 ml/min; method C—HPLC-B, a linear elution gradient of 98:2 to 10:90 (solvent A/B) over 30 min at a flow rate of 1 ml/min; method D—HPLC-C, a suitable gradient of aqueous acetonitrile containing 0.1% TFA at a flow rate of 10 ml/min.
ESI–MS and LC–MS
In our lab, the ESI–MS spectra are measured on an Agilent 6230 LC–TOF MS spectrometer. Analyze the small molecules using a short guard column and elute with 70% methanol containing 0.1% formic acid. Record the mass spectra of small molecules in the mass range of 200–3,000 or 600–2,000 m/z under high-resolution MS mode (standard 3,200 m/z, 4 GHz). Key source parameters are as follows: drying nitrogen gas flow of 11 liters/min; nebulizer pressure of 40 psi; gas temperature of 350 °C; fragmenter voltage of 175 V; skimmer voltage of 65 V; and capillary voltage of 4,000 V. Measure the antibodies and ADCs with an Agilent C-18 column (3.5 μm, 50 × 2.1 mm) at 55 °C. Elute the column with an isocratic gradient of 5% acetonitrile (buffer B) and 95% water containing 0.1% formic acid (buffer A) for the first 10 min, a linear gradient of 5–95% acetonitrile for an additional 10 min, and an isocratic mobile phase of 95% acetonitrile for another 10 min at a flow rate of 0.2 ml/min.
Perform the mass spectra measurement of antibodies under the extended mass range mode (high 20,000 m/z, 1 GHz) and collect the data in the mass range of 800–5,000. Key source parameters are as follows: drying nitrogen gas flow of 11 liters/min; nebulizer pressure of 60 psi; gas temperature of 350 °C; fragmenter voltage of 400 V; skimmer voltage of 65 V; and capillary voltage of 5,000 V. Deconvolute the multiple charged peaks of the antibody using the Agilent MassHunter Bioconfirm software (deconvolution for protein, Agilent Technology) with a deconvolution range of 100–200 kDa; set the other parameters to default values for the protein deconvolution. Calibrate the TOF over the range of 0–5,000 m/z using Agilent ESI calibration mix solution before analysis. The peak of MS 922 is the internal standard for calibration.
HPAEC-PAD
In our lab, the HPAEC spectra are performed on an ICS 5000+ system (Thermo Scientific) with a PA100 anion exchange column (4 × 250 mm) and a guard column (4 × 50 mm) at 30 °C. The mobile phases consist of a water phase (solvent A), a 1.0 M sodium acetate aqueous solution (solvent B), and a 0.2 M sodium hydroxide aqueous solution (solvent C). Using this system (or one very similar), elute the column with an isocratic mobile phase of 50/0/50 (solvent A/B/C) for the first 2 min and then a linear gradient of 50/0/50 to 35/15/50 (solvent A/B/C) for an additional 20 min at a flow rate of 1 ml/min.
Sephadex G-25 gel-filtration
Swell 100 g of G-25 beads in 400 ml of water and pack the mixture into a custom-ordered glass column (2.6 × 100 cm, Xiamei) until the column is 80% full (at the 80-cm mark). The G-25 column is eluted with 3 column volumes of water, 0.2% triethylamine aqueous solution, or other suitable solvents by gravity. The fractions are collected by an automatic fraction collector instrument (BS-100A). For SGP and analog detection, we take 0.5-μl samples from each fraction tube and test them on a TLC plate stained with 10% sulfate acid in ethanol after drying with a heat gun. The fractions containing SGP product will develop black spots on the TLC plate after staining.
NMR
NMR spectra were measured on a Varian Mercury Plus 400 or 500 instrument.
PROCEDURE
Semisynthesis of aldehyde SGP (CHO-SGP, 20) ● TIMING 4 h
-
1|
Weigh 100 mg of SGP from Box 1 in a 15-ml tube and add 5 ml of 0.2 M pH 7.0 phosphate buffer. Stir within an ice bath for 15 min.
-
2|
Prepare an aqueous solution of 30 mM NaIO4.
-
3|
Add 5 ml of 30 mM NaIO4 solution (final concentration: 15 mM) to the SGP tube from Step 1. Incubate at 0 °C within an ice bath for 15 min. Monitor the reaction with HPLC (Supplementary Results).
▲ CRITICAL STEP Higher temperature and higher concentration of NaIO4 might cause further oxidation of other sugars.
-
4|
Immediately purify the reaction mixture by gel-filtration with a G-25 column (Equipment Setup). Elute the column with water and collect the fractions. Combine the fractions containing the target product.
▲ CRITICAL STEP If the reaction mixture is not immediately subjected to gel-filtration, 40 μl of 1% glycerol should be added to quench the excessive NaIO4. Otherwise, further oxidation side reactions will occur.
-
5|
Lyophilize the collected fractions to obtain a white powder (20; yield 94%; Fig. 4).
PAUSE POINT The CHO-SGP can be stored at −20 °C for 1 month or at −80 °C for 6 months.
Figure 4.
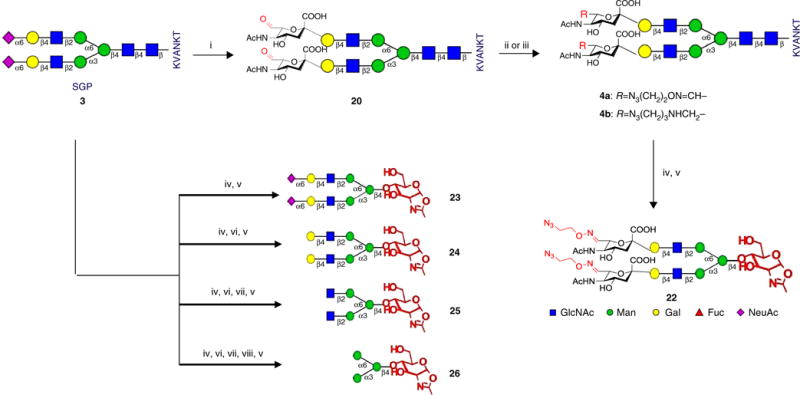
Semisynthesis of N-glycan oxazolines (Steps 1–44). (i) 15 mM NaIO4, PB, pH 7.0; (ii) N3(CH2)2ONH3+Cl− (21a), PB, pH 7.0; (iii) N3(CH2)3NH2, NaCNBH3, PB pH 6.0, MeOH; (iv) Endo-M, PB, pH 6.2–6.5; (v) DMC, Et3N; (vi) neuraminidase, PB, pH 5.0; (vii) β-1,4-galactosidase, PB, pH 6.5; (viii) β-N-acetylglucosaminidase, PB, pH 6.5.
Synthesis of azido-oxime-SGP (4a) ● TIMING 7 h
-
6|
Prepare the in situ CHO-SGP (20) following Steps 1–3. Add 40 μl of 1% glycerol to quench the remaining NaIO4.
-
7|
Place the 15-ml tube in an ice bath.
-
8|
Weigh 24.2 mg (174.5 μmol, 5 equiv.) of O-(2-azidoethyl)-hydroxylamine hydrochloride (21a; see Supplementary Methods for the synthetic procedure) and add it to the solution from Step 6.
-
9|
Stir within an ice bath for 4 h and monitor the reaction with HPLC (Supplementary Results).
-
10|
Immediately purify the reaction mixture by gel-filtration with a G-25 column (see Box 1, step 10 for column information). Elute the column with water.
-
11|
Lyophilize the collected fractions to obtain a white powder (4a; yield 92%).
■ PAUSE POINT The SGP analog 4a can be stored at −20 °C for 3 months or at −80 °C for 12 months.
Semisynthesis of azido-amine-SGP (4b) ● TIMING 5 h
-
12|
Weigh 10 mg (3.65 μmol) of CHO-SGP (purified; from Step 5) in a 2.0-ml tube, and add 0.5 ml of cold 50 mM, pH 6.0 phosphate buffer and 0.5 ml of cold methanol (4 °C).
-
13|
Add 3-azido-1-propanamine (10.8 μl, 109.5 μmol, 30 equiv.) and adjust the pH to 6.0 with acetic acid.
-
14|
Add NaCNBH3 (4.6 mg, 73 μmol, 20 equiv.).
▲ CRITICAL STEP It is important to weigh and add NaCNBH3 quickly, as it is labile in air.
-
15|
Stir within an ice bath for 3 h and monitor with HPLC (Supplementary Results).
-
16|
Purify the product with preparative HPLC and lyophilize the collected fractions to obtain a white powder (yield = 80%).
■ PAUSE POINT The SGP analog 4b can be stored at −20 °C for 3 months or at −80 °C for 12 months.
Synthesis of azido-or alkyne-functionalized-SGP (4c–f) ● TIMING 4–8 h
-
17|
Use in situ or purified CHO-SGP (20) as the starting material for the synthesis of 4c–f (see structures in Fig. 5 and characterization data in the Supplementary Results) according to Steps 6–11 for oxime linkage and according to Steps 12–16 for amine linkage.
Figure 5.
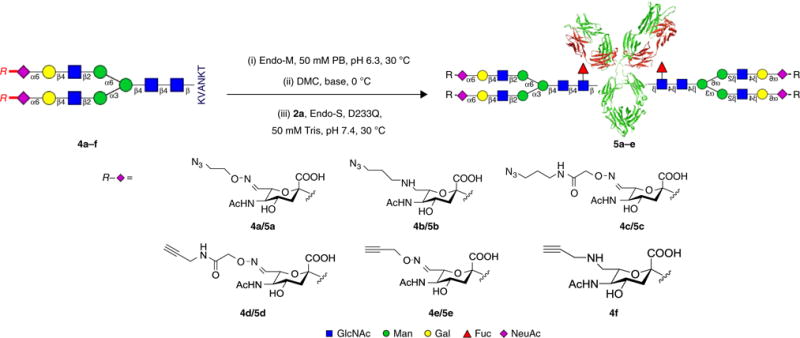
One-pot chemoenzymatic synthesis of glycoengineered Herceptin 5a–e with in situ oxazolines derived from SGP analogs 4a–e (Steps 74–85).
Semisynthesis of azido-oxime-Sia2Gal2GlcNAc2Man3GlcNAc oxazoline (22) as the endoglycosidase substrate ● TIMING 8 h
-
18|
Weigh 50 mg (17.2 μmol) of azido-oxime-SGP (4a) from Step 11 into a 1.5-ml centrifuge tube.
-
19|
Add 230 μl of 50 mM, pH 6.25 phosphate buffer. Adjust the pH to 6.25 with 0.1 N NaOH.
▲ CRITICAL STEP Compound 4a was purified from preparative HPLC with mobile phases containing 0.1% TFA; it is acidic because of remaining TFA after lyophilization. When dissolving it in a buffer, the pH of the mixture solution might be lower than the buffer pH and extra NaOH must be added to adjust the final pH to the expected value.
-
20|
Add 21 μl of 6 mg/ml Endo-M to the tube from Step 18 (final concentration of Endo-M: 0.5 mg/ml).
▲ CRITICAL STEP Determine the enzymatic activity of Endo-M before the reaction according to the Box 2 protocol. If the Endo-M activity is lower than desired, extend the reaction time or increase the enzyme concentration to complete the hydrolysis of SGP analog 4a.
-
21|
Incubate the mixture at 30 °C for 4 h. Monitor the reaction with HPLC until the hydrolysis of SGP analog 4a is complete (Supplementary Results).
-
22|
Place the solution into an ice bath.
-
23|
Add 108 μl (774 μmol, 45 equiv.) of Et3N to the residue from Step 20.
-
24|
Weigh 43.6 mg (258 μmol, 15 equiv.) of DMC and add it to the mixture.
▲ CRITICAL STEP The reaction must be performed under basic conditions. After adding all materials, confirm that the reaction pH is >11. If the pH is <11, the conversion rate might decrease and more Et3N will be required.
-
25|
Incubate the reaction mixture in an ice bath for 30 min. Monitor the reaction with 1H NMR.
▲ CRITICAL STEP The monitoring is important to ensure the high conversion rate of this reaction. Take 30-μl aliquots from the reaction mixture and lyophilize the sample to dryness. Then re-dissolve the residue in 500 μl of D2O for 1H NMR analysis. 1H NMR analysis of the oxazoline should indicate a particular proton signal of oxazoline-H1 at 6.0 p.p.m. as a doublet peak with a coupling constant of ~7.2 Hz (Supplementary Results).
? TROUBLESHOOTING
-
26|
Purify the mixture with gel-filtration through a G-25 column (2.6 × 80 cm) pre-equilibrated with 0.2% Et3N in ddH2O. Elute the column with 0.2% Et3N in ddH2O.
-
27|
Collect the fractions containing the products and add 2 mg of NaOH to the combined fractions.
! CAUTION The Et3N and NaOH are used to stabilize the oxazoline. Make sure that the gel-filtration continues to run at a pH value >10. If the pH is <10, oxazoline may partially decompose during processing.
-
28|
Lyophilize the fractions to obtain 35 mg of a white solid powder (containing 2 mg of NaOH, yield 91%).
? TROUBLESHOOTING
■ PAUSE POINT The oxazoline (22) can be stored at −20 °C for 2 months or it can be frozen at −80 °C for 6 months.
One-pot semisynthesis of N-glycan oxazolines (26–23) as endoglycosidase substrates: synthesis of Man3GlcNAc oxazoline (26) ● TIMING 4 d
-
29|
Dissolve SGP (20 mg, 7.0 μmol) in phosphate buffer (50 mM, pH 6.5, 80 μl) in a 1.5-ml tube. Adjust the pH to 6.5 with 0.1 N NaOH.
-
30|
Add 20 μl of 2.5 mg/ml Endo-M (final concentration 0.5 mg/ml) to the tube and incubate the mixture at 30 °C for 2–3 h. Monitor the reaction with HPLC until the SGP is completely hydrolyzed (Supplementary Results).
▲ CRITICAL STEP Determine the enzymatic activity of Endo-M before the reaction according to the Box 2 protocol. If the Endo-M activity is lower than desired, extend the reaction time or increase the enzyme concentration to complete SGP hydrolysis.
-
31|
Adjust the pH to 5.0 with 0.1 N HCl and add 2 μg of neuraminidase.
-
32|
Incubate at 37 °C for 12 h and monitor the reaction by HPAEC until the terminal sialic acid is completely hydrolyzed (Supplementary Results).
-
33|
Adjust the pH to 6.5 with 0.2 M Na2PO4.
-
34|
Add 8 U (unit definition from manufacturer) of β-1,4-galactosidase to hydrolyze the terminal galactose.
-
35|
Incubate at 37 °C for 12 h and monitor the reaction by HPAEC until the terminal galactose is completely hydrolyzed (Supplementary Results).
-
36|
Add 0.5 U (unit definition from manufacturer) of β-N-acetylglucosaminidase to hydrolyze the GlcNAc residues at the nonreducing end of the glycan.
-
37|
Incubate at 37 °C for 12 h and monitor the reaction by HPAEC until the terminal GlcNAc is completely hydrolyzed.
-
38|
Place the reaction mixture into an ice bath.
-
39|
Add Et3N (44 μl, 315 μmol, 45 equiv.) and DMC (17.7 mg, 105 μmol, 15 equiv.) to the tube.
▲ CRITICAL STEP The reaction must be performed under basic conditions. After adding all materials, check whether the reaction has a pH >11. If the pH is <11, the conversion rate might decrease and more Et3N will be required.
-
40|
Incubate in an ice bath for 30 min. Take a 1-μl aliquot for HPAEC analysis (Supplementary Results) and take a 5-μl aliquot for 1H NMR detection.
▲ CRITICAL STEP The monitoring is important to ensure a high conversion rate for this reaction. HPAEC could separate the free glycan from the oxazoline through chromatography. 1H NMR of the oxazoline should indicate a particular proton signal for oxazoline-H1at 6.0 p.p.m. as a doublet peak with a coupling constant of ~7.2 Hz. (Supplementary Results).
? TROUBLESHOOTING
-
41|
Purify the crude product by solid-phase extraction (SPE) with a PGC cartridge. Flow the sample through the PGC cartridge twice. Wash the cartridge successively with 1 ml of water, 1 ml of 5% MeCN containing 1 mM NaOH, and 1 ml of 10% MeCN containing 1 mM NaOH. Elute the cartridge with 1 ml of 25% MeCN containing 1 mM NaOH.
▲ CRITICAL STEP The NaOH is used to stabilize the oxazoline. Make sure that the SPE continues to run at a pH value >11. If the pH is <11, oxazoline might partially decompose during processing.
-
42|
Lyophilize the eluent to obtain a white powder (4 mg, 85%).
? TROUBLESHOOTING
■ PAUSE POINT The oxazoline (26) can be stored at −20 °C for 2 months or it can be frozen at −80 °C for 6 months.
Synthesis of GlcNAc2Man3GlcNAc oxazoline (25) ● TIMING 3 d
-
43|
Repeat Steps 29–35, and then perform the oxazoline formation reaction and purify the product by following Steps 38–42.
Synthesis of Gal2GlcNAc2Man3GlcNAc oxazoline (24) ● TIMING 2 d
-
44|
Repeat Steps 29–32, and then perform the oxazoline formation reaction and purify the product by following Steps 38–42.
Synthesis of Sia2Gal2GlcNAc2Man3GlcNAc oxazoline (23) ● TIMING 1 d
-
45|
Repeat Steps 29 and 30, and then perform the oxazoline formation reaction and purify the product according to Steps 38–42.
Preparation of Herceptin-Fucα1,6GlcNAc (2a) ● TIMING 4 h
-
46|
Prepare a solution of 38 mg of Herceptin in 1.0 ml of a 50 mM, pH 7.5 phosphate buffer (PB).
-
47|
Add 1 μl of 2 mg/ml WT Endo-S (final concentration: 2 μg/ml).
▲ CRITICAL STEP Determine the enzymatic activity of WT Endo-S before the reaction according to the Box 2 protocol. If the Endo-S activity is lower than desired, extend the reaction time or increase the enzyme concentration to complete deglycosylation.
-
48|
Incubate at 37 °C for 2 h. Monitor the reaction with LC–MS and SDS-PAGE until the deglycosylation is complete (Supplementary Results).
-
49|
To purify the deglycosylated Herceptin (2a) with an affinity column of protein A beads, first precondition the protein A column (3 ml) with 25 ml of glycine–HCl buffer (100 mM, pH 2.5) and pre-equilibrate with 50 ml of PB (50 mM, pH 8.0).
-
50|
Load the reaction mixture from Step 47 and flow it through the preconditioned protein A column three times.
-
51|
Wash the column with PB (50 mM, pH 8.0, 50 ml) containing 0.01% Tween 20.
-
52|
Wash the column with PB (50 mM, pH 6.0, 50 ml) containing 0.01% Tween 20.
-
53|
Wash the column with glycine–HCl (20 mM, pH 5.0, 30 ml) containing 0.01% Tween 20.
-
54|
Elute the column with glycine–HCl (100 mM, pH 2.5, 20 ml), followed by immediate neutralization to a pH value of ~7.5 with glycine–HCl (1 M, pH 8.8).
-
55|
Combine the IgG fractions and repurify them with the protein A affinity column, repeating Steps 49–54.
▲ CRITICAL STEP Complete removal of WT Endo-S is important. If the purified deglycosylated IgG contains WT Endo-S, the glycoengineered IgG product of the chemoenzymatic transglycosylation will be hydrolyzed by the remaining WT Endo-S in the next step.
? TROUBLESHOOTING
-
56|
Exchange the sample buffer with a 50 mM, pH 7.4 Tris–HCl buffer and concentrate the sample with an Amicon ultracentrifugal filter (30 kDa cutoff). Prepare the stock solution of 2a at a 20 mg/ml concentration in the 50 mM, pH 7.4 Tris–HCl buffer. Using deglycosylated IgG (2a) as the starting material, glycoengineered IgG (5a–e; Fig. 5), gsADC (7a–f; Fig. 1), and dpADC (28; Fig. 6) were prepared.
■ PAUSE POINT The 2a stock can be stored at 4 °C for 4 weeks or it can be frozen at −80 °C for 6 months.
Figure 6.
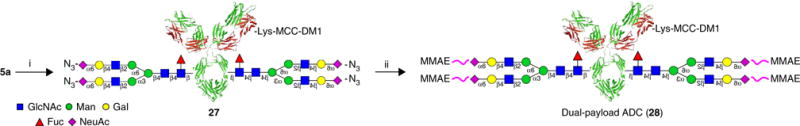
Synthesis of dual-payload ADC (28) (Steps 96–108). (i) DM1-SMCC 10, PB, pH 7.5; (ii) 6c, 50 mM PB, pH 7.5.
Preparation of rituximab-GlcNAc (2b) ● TIMING 1 d
-
57|
Prepare a solution of 20 mg of rituximab in 1.0 ml of a 50 mM, pH 7.5 Tris buffer.
-
58|
Add 1 μl of 2 mg/ml WT Endo-S (final concentration: 2 μg/ml).
-
59|
Add 50 μl of 20 mg α-L-fucosidase AlfC (final concentration: 1 mg/ml).
▲ CRITICAL STEP Determine the enzymatic activity of WT Endo-S and AlfC according to the Box 2 protocol before the reaction. If the enzyme activity is lower than desired, extend the reaction time or increase the enzyme concentration to complete deglycosylation.
-
60|
Incubate at 37 °C for 16 h and monitor the reaction with LC–MS until the defucosylation is complete (see Anticipated Results section; Fig. 7).
-
61|
Purify the IgG with a protein A affinity column according to Steps 49–54 (twice).
? TROUBLESHOOTING
-
62|
Combine the product fractions. Exchange the sample buffer with a 50 mM, pH 7.4 Tris–HCl buffer and concentrate the sample with an Amicon ultracentrifugal filter (30 kDa cutoff). Prepare the stock solution of 2b at a 20 mg/ml concentration in the 50 mM, pH 7.4 Tris–HCl buffer.
■ PAUSE POINT The 2b stock can be stored at 4 °C for 4 weeks or it can be frozen at −80 °C for 6 months.
Figure 7.
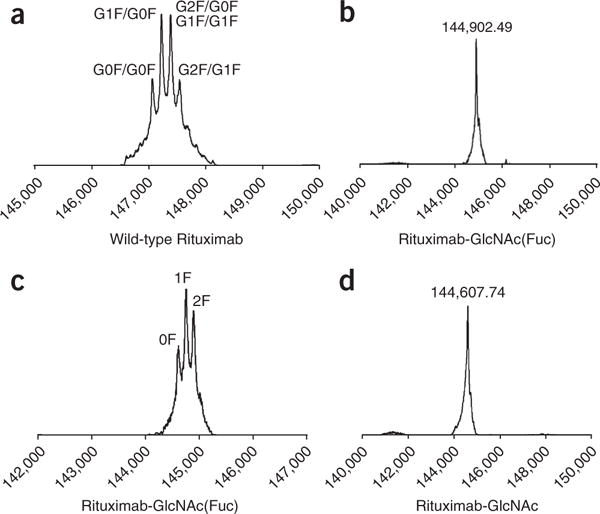
LC–MS profiles (deconvolution data) from IgG deglycosylation monitoring. (a) Native rituximab. (b) Rituximab after treatment with WT for 5 min. (c) Results after defucosylation of Fucα1,6GlcNAc-rituximab with AlfC for 4 h. the MS peak at 144,894.20 was designated as 2F (two fucoses); the MS peak at 144,749.13 was designated as 1F (one fucose); the MS peak at 144,602.67 was designated as 0F (no fucose). (d) Results after defucosylation of Fucα1,6GlcNAc-rituximab with AlfC for 16 h.
Chemoenzymatic synthesis of glycoengineered Herceptin 5a with purified azido-tagged N-glycan oxazoline (22) ● TIMING 5 h
-
63|
Weigh 23 mg of azido-oxime-SCT-oxazoline (22) (from Step 27, containing ~6% NaOH) in a 1.5-ml centrifuge tube and dissolve it with 200 μl of water to make a stock solution at a concentration of 50 mM.
-
64|
Add 500 μl of 50 mM, pH 7.4 Tris buffer to another 1.5-ml centrifuge tube.
-
65|
Add 20 μl of 50 mM 22 from the stock solution prepared in Step 63 to the tube from Step 64. Vortex for 15 s.
-
66|
Adjust the pH to 7.4 with 0.1 N HCl.
▲ CRITICAL STEP The glycan oxazoline is stabilized with ~6% NaOH after purification. When adding the oxazoline to the antibody solution, the pH increases and adjustment with acid is required. Meanwhile it is also important to avoid overadjustment, as the oxazoline is not stable at low pH. A tip for the pH adjustment in this step is to add a tiny amount (e.g., 1 μl) of HCl multiple times, until the target pH value is reached.
-
67|
Add 250 μl of 20 mg/ml Herceptin-GlcNAc(Fuc) (2a) (the stock solution from Step 56).
-
68|
Add 6.3 μl of 16 mg/ml Endo-S D233Q, and gently vortex for 5 s.
▲ CRITICAL STEP Determine the enzymatic activity of Endo-S D233Q before the reaction by following the Box 2 protocol. If the enzyme activity is lower than desired, extend the reaction time or increase the enzyme concentration to complete transglycosylation.
-
69|
Add additional 50 mM, pH 7.4 Tris–HCl buffer until the final volume is 1 ml (final concentrations: IgG: 5 mg/ml; oxazoline: 1mM; D233Q: 0.1 mg/ml).
-
70|
Incubate the mixture at 30 °C for 2 h and monitor the reaction with SDS-PAGE and LC–MS analysis (see Anticipated Results section; Fig. 8a,b).
? TROUBLESHOOTING
-
71|
Immediately purify the product by gel-filtration with a protein-A affinity column by following Steps 49–54.
▲ CRITICAL STEP It is better to purify the sample immediately after the reaction to avoid possible nonenzymatic reactions caused by long reaction times even under low temperature.
-
72|
Combine the product fractions. Exchange the sample buffer with a 50 mM, pH 7.4 Tris–HCl buffer and concentrate the sample with an Amicon ultracentrifugal filter (30 kDa cutoff). Prepare a stock solution of 5a at a 10 mg/ml concentration in the 50 mM, pH 7.4 Tris–HCl buffer.
■ PAUSE POINT The 5a stock can be stored at 4 °C for 4 weeks or it can be frozen at −80 °C for 6 months.
Figure 8.
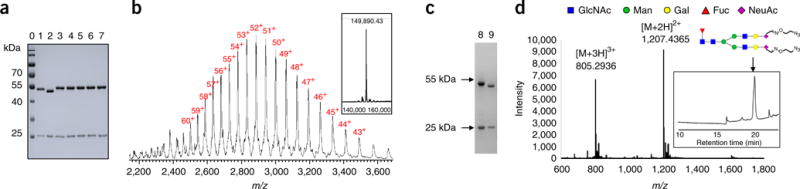
SDS-PAGE and LC–MS characterization of glycoengineered Herceptin (5a–e) bearing non-natural N-glycans26. (a) SDS-PAGE analysis of 5a–e. Lane 0: marker, lane 1: commercial Herceptin, lane 2: Herceptin-Fucα1,6GlcNAc (2a), lanes 3–7, 5a–e; (b) LC–MS profiles of 5a. The multiple charged m/z data are labeled with charge numbers. The deconvolution mass spectrum is shown in the embedded box. (c) SDS-PAGE analysis of PNGase-F digestion of 5a. Lane 8: 5a, Lane 9: 5a after PNGase-F digestion; (d) MS profile of the released non-natural glycan (shown in glycan symbol) from 5a by PNGase-F digestion. The ion flow chromatography of the PNGase-F digested sample is shown in the embedded box, and the peak for the glycan is marked with an arrow.
General procedure for chemoenzymatic synthesis of glyco-remodeled IgGs with purified N-glycan oxazoline (23–26) ● TIMING 5 h
-
73|
Prepare the stock solutions of the corresponding oxazolines (23–26; from Steps 29–45), IgG-GlcNAc, or IgG-Fucα1,6GlcNAc. Then, follow Steps 63–72 to prepare the glycoengineered IgGs (see Supplementary Results for more examples).
Preparation of 5a by one-pot procedure (Fig. 5) ● TIMING 7 h
-
74|
Weigh 10 mg of SGP non-natural derivative 4a in a 1.5-ml tube.
-
75|
Follow Steps 17–20 to hydrolyze the azido-glycan from 4a.
-
76|
Place the solution in an ice bath.
-
77|
Add 11 μl of 500 mg/ml NaOH aqueous solution, 8.8 mg of DMC, and water until the final volume is 100 μl.
▲ CRITICAL STEP For the one-pot chemoenzymatic procedure, we choose NaOH as the base to replace Et3N, because in the next step the enzymatic reaction with the Endo-S mutant is more compatible with neutralized sodium salts than triethyl ammonium salts. It is also important to control the pH (should be >11). If the pH is <11, oxazoline formation might be incomplete and it will require more NaOH and DMC to complete the oxazoline formation.
-
78|
Incubate in an ice bath for 30 min and monitor the reaction with 1H NMR analysis.
▲ CRITICAL STEP 1H NMR monitoring on oxazoline formation is important for the one-pot procedure. If incomplete oxazoline formation occurs in this step, enzymatic transglycosylation using this oxazoline in situ might be influenced.
? TROUBLESHOOTING
-
79|
Add 2.5 ml of Tris buffer (50 mM, pH 7.4) and adjust the pH to 7.4 with 0.1 N HCl.
▲ CRITICAL STEP The reaction solution contains NaOH, which may increase the pH of the Tris buffer. Therefore, extra HCl is required to control the pH value.
-
80|
Add 1.25 ml of 20 mg/ml Herceptin-Fucα1,6GlcNAc (2a).
-
81|
Add 50 μl of 10 mg/ml Endo-S D233Q and add extra Tris buffer (50 mM, pH 7.4) until the final volume is 5 ml (final concentrations: IgG: 5 mg/ml; glycan oxazoline: 1 mM; Endo-S D233Q: 0.1 mg/ml).
-
82|
Incubate the reaction mixture at 30 °C for 2 h. Monitor the reaction with SDS-PAGE and LC–MS.
? TROUBLESHOOTING
-
83|
Immediately purify the products by gel-filtration with a protein-A affinity column by following Steps 49–54.
-
84|
Repeat Step 72 to prepare the stock solution of 5a.
General procedure for one-pot chemoenzymatic synthesis of glycoengineered IgGs 5b–e ● TIMING 7 h
-
85|
Follow Steps 74–84 and replace the SGP analog 4a with 4b–e, respectively, to prepare the glycoengineered IgGs (see Supplementary Results for more examples).
Synthesis of gsADC 7a ● TIMING 1 d
-
86|
Add 1 ml of 10 mg/ml azido-oxime-Herceptin (5a) from Step 84 to a 15-ml centrifuge tube.
-
87|
Add 8 ml of 50 mM, pH 7.5 PB and 870 μl of DMSO. Gently vortex the tube for 15 s.
-
88|
Prepare a 10 mM solution of DBCO-SMCC-DM1 (6a; presynthesized according to Box 3) in DMSO by dissolving 5.8 mg of 6a in 470 μl of DMSO.
▲ CRITICAL STEP The stock solution of 6a can be stored at –20 °C for 6 months.
-
89|
Add 130 μl of 6a solution from Step 88. Gently vortex the tube for 15 s.
-
90|
Shake and incubate at 30 °C for 12 h, and then incubate at 4 °C for an additional 12 h.
-
91|
Monitor the reaction with SDS-PAGE and LC–MS until the conjugation is complete.
? TROUBLESHOOTING
-
92|
Purify the mixture by gel-filtration with a protein-A affinity column by following Steps 49–54.
-
93|
Combine the product fractions. Exchange the sample buffer with a 50 mM, pH 7.4 Tris–HCl buffer and concentrate the sample with an Amicon ultracentrifugal filter (30 kDa cutoff). Prepare a stock solution of 7a at a 1 mg/ml concentration in a 50 mM, pH 7.4 Tris–HCl buffer.
■ PAUSE POINT The 7a stock solution can be stored at 4 °C for 2 weeks or it can be frozen at −80 °C for 2 months.
-
94|
Lyophilize the 7a stock solution from Step 93 to obtain a white powder (yield >90%).
■ PAUSE POINT The lyophilized 7a powder can be stored at −80 °C for 6 months.
General procedure synthesis of gsADCs 7b–f ● TIMING 1 d
-
95|
Conjugate the azido-functionalized glycoengineered IgG 5a and b with DBCO-tagged molecules 6a–d separately, and follow Steps 86–94 to prepare the gsADCs 7b–f (see Supplementary Results and ref. 26 for more details).
Synthesis of dual-payload ADC 28 ● TIMING 1 d
-
96|
Add 0.5 ml of 10 mg/ml azido-Herceptin (5a) from Step 72 to a 15-ml centrifugal tube.
-
97|
Exchange the sample buffer with a 50 mM, pH 7.5 PB and concentrate the sample with an Amicon ultracentrifugal filter (30 kDa cutoff). Add extra 50 mM, pH 7.5 PB until the total volume is 4.5 ml.
▲ CRITICAL STEP The Tris buffer used in the 5a solution from Step 72 contains the amino group that is reactive with the active ester of 10. Exchange to a nonamine buffer is important for this conjugation.
-
98|
Add 0.5 ml of DMSO to the mixture and vortex gently.
-
99|
Add 53 μl of 10 mg/ml DM1-SMCC (10, 0.53 mg, 15.0 equiv.) in DMSO solution.
-
100|
Incubate at 37 °C for 1 h. Monitor the reaction by LC–MS to determine the drug–antibody ratio (DAR). Control the DAR value to keep it between 2.5 and 3.5.
-
101|
Immediately purify the mixture by gel-filtration with a protein-A affinity column according to Steps 49–54.
▲ CRITICAL STEP The purification procedure must be performed immediately once the DAR value is in the desired range. If you do not purify the ADC immediately, the conjugation reaction will keep running and the DAR may increase.
-
102|
Combine the product fractions and concentrate the sample by centrifugation at 3,200g at 4 °C for 30 min. Prepare a solution of 5 ml of ADC 27 at 1 mg/ml in a 50 mM, pH 7.5 PB containing 10% DMSO.
-
103|
Add 65 μl of 10 mM 6c in DMSO solution to the solution of 27 from Step 102.
-
104|
Incubate the mixture at 30 °C for 12 h.
-
105|
Monitor the reaction with SDS-PAGE and LC–MS.
? TROUBLESHOOTING
-
106|
Purify the mixture by gel-filtration with a protein-A affinity column by following Steps 49–54.
-
107|
Combine the product fractions. Exchange the sample buffer with a 50 mM, pH 7.4 Tris–HCl buffer and concentrate the sample with an Amicon ultracentrifugal filter (30 kDa cutoff). Prepare a stock solution of ADC 28 at a 1 mg/ml concentration in 50 mM, pH 7.4 Tris–HCl buffer.
■ PAUSE POINT The compound 28 stock solution can be stored at 4 °C for 2 weeks or it can be frozen at −80 °C for 2 months.
-
108|
Lyophilize the compound 28 stock solution from Step 107 to obtain a white powder (yield >90%). The lyophilized compound 28 powder can be stored at −80 °C for 6 months.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
TABLE 1.
|Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| Box 1, Step 11 | Low purity of SGP | (1) Unknown impurities without UV absorbance (2) Terminal sialic acid was partially removed |
(1) Purify again with preparative RP-HPLC (2) Control the pH (>7) and temperature (<35 °C) in all procedures. Redo gel-filtration |
| Box 1, Step 11 | Low yield of SGP | (1) Low extraction rate from egg-yolk powder (2) Low elution rate from SPE |
(1) In Box 1, step 6, extract with more 40% acetone and monitor with HPLC (2) Elute with more 25% MeCN |
| Step 25, 40 and 78 | Incomplete oxazoline formation | (1) Low pH (2) Loss of reactivity of DMC |
(1) Add more base (Et3N or NaOH) (2) Use fresh DMC |
| Steps 28 and 42 | Low purity of oxazolines | Contains impurities derived from DMC, GlcNAc-peptide, and salts | Perform PGC SPE first, and then G-25 gel-filtration. Repeat the procedure |
| Steps 28 and 42 | Decomposition of oxazoline | (1) Contains acidic impurities: DMC, Et3N·HCl (2) NaOH is not enough (3) Other acidic items are lyophilized in the lyophilizer |
(1) Purify the sample again (2) Add more NaOH for stabilization (3) Remove other acidic samples |
| Steps 55 and 61 | Endo-S remains | Endo-S binds IgG on the protein A resin | (1) Wash with pH-gradient buffers (2) Add 0.1–0.5% Tween 20 to the wash buffers |
| Steps 70 and 82 | Nonenzymatic reaction | Glycation of glycan oxazoline onto IgG amino acids | (1) Avoid high pH. Control the pH (<7.5) (2) Control the concentration of oxazolines (<2.5 mM) (3) Control the reaction time (<3h) |
| Steps 70 and 82 | Low yield of transglycosylation | (1) Low enzyme activity (2) Oxazolines have partially decomposed (3) Product hydrolysis by remaining WT Endo-S |
(1) Use fresh Endo-S D233Q (2) Control the quality of purified or in situ oxazolines (3) Purify the deglycosylated IgG again before transglycosylation |
| Steps 91 and 105 | Incomplete ‘click’ conjugation | Poor aqueous solubility of DBCO-tagged small molecules | Increase the concentration of DMSO, or add glycerol to improve the solubility |
● TIMING
Steps 1–17, semisynthesis of SGP analogs: 20–24 h
Steps 18–45, one-pot synthesis of N-glycan oxazolines: 11 d
Steps 46–62, preparation of deglycosylated IgG: 4 h + 1 d
Steps 63–73, preparation of glycoengineered IgG using purified oxazolines: 10 h
Steps 74–85, one-pot chemoenzymatic glycoengineering of IgG: 14 h
Steps 86–95, preparation of gsADCs: 2 d
Steps 96–108, preparation of dual-payload ADC: 1 d
Box 1, extraction of SGP from egg yolks: 4 d
Box 2, enzyme activity assays: 4–16 h
Box 3, synthesis of DBCO-tagged small molecules: 1–4 d
ANTICIPATED RESULTS
The procedures in this protocol describe the chemoenzymatic synthesis of glycoengineered IgGs and gsADCs, catalyzed by a glycosynthase Endo-S D233Q using purified or in situ N-glycan oxazolines as substrates. Non-natural N-glycans containing azido or alkyne function groups on terminal sialic acids can serve as the enzyme substrates for IgG glycoengineering. The glyco-remodeled IgGs bearing azido-tagged N-glycan enable site-specific conjugation via a copper-free ‘click’ reaction for preparation of gsADCs. The SGP extraction and semisynthesis of SGP analogs are described in Box 1 and Steps 1–16. One-pot synthesis and purification of N-glycan oxazolines are described in Steps 17–44. DBCO-tagged small molecules are synthesized as described in Box 3. The characterization data of these compounds are available in the Supplementary Results.
WT Endo-S can efficiently hydrolyze the N-glycans from the IgG Fc domain by cleaving the glycosidic bond of the GlcNAcβ1,4GlcNAc motif (Figs. 1 and 7). As monitored by both SDS-PAGE (Fig. 8a) and LC–MS (Fig. 7), native heterogeneous glycoforms of G0F, G1F, and G2F are completely removed after 5 min of treatment with Endo-S. SDS-PAGE shows an ~2-kDa lower band as compared with the native IgG heavy-chain band (Fig. 8a), and LC–MS showed a major MS of Fucα1,6GlcNAc-IgG (Fig. 7b). Further defucosylation with AlfC was monitored only by LC–MS (Figs. 3d and 7c) because the mass loss after defucosylation is too small to detect by SDS-PAGE. After 4 h of digestion with AlfC, Fucα1,6GlcNAc-IgG partially released the fucose moiety, as LC–MS (Fig. 7c) showed the MS data of intermediates containing two fucoses (2F, MS = 144,894.20), one fucose (1F, MS = 144,749.13), and no fucose (0F, MS = 144,602.67). The MS differences between 2F/1F/0F are ~145–147 Da, which is in close agreement with the calculated MS deduction of fucose (146 Da). Figure 7d shows complete defucosylation after 16 h, as only the 0F MS was detected.
Next, the Endo-S D233Q mutant—which lacks hydrolytic activity but maintains transglycosylation activity, using synthetic N-glycan oxazolines as donor substrates—was used to assemble the natural or non-natural N-glycan onto the GlcNAc-bearing IgG (2a or 2b) with purified N-glycan oxazolines or via the one-pot reaction with in situ oxazolines (Fig. 2). Fig. 8 shows the SDS-PAGE and LC–MS characterization of an example of glycoengineered Herceptin 5a. After chemoenzymatic transglycosylation, the heavy-chain bands for 5a–e shifted to an ~2-kDa-higher position as compared with that of deglycosylated Herceptin (2a), suggesting successful glycoengineering (Fig. 8a). LC–MS analysis indicates the deconvolution MS of 5a at 149,890.43, which is in good accordance with the mass increase of the attached azido-glycan. To ensure the correct linkage of the newly attached glycan, 5a was digested with PNGase-F, a commercial enzyme that cleaves the intact N-glycan at the innermost GlcNAc from the Asn297 residue of the glycosites (Supplementary Results). As shown in the SDS-PAGE gel of Figure 8c, the heavy-chain band of PNGase-F-treated 5a completely shifted to a lower position, suggesting the complete release of N-glycan without any nonenzymatic glycation being observed. Furthermore, LC–MS analysis of the resulting Herceptin-Asp297 (see literature data26) and azido-glycan (Fig. 8d) clearly verifies the enzymatic product that links the N-glycan to the GlcNAc motif of 2a.
The preparation of gsADCs (7a–f) was carried out via the copper-free ‘click’ reaction41,42 with azido-Herceptin (5a or 5b) and DBCO-tagged toxins (6a–d). The increased mass resulting from conjugation of gsADC is confirmed by SDS-PAGE (Fig. 9a) and LC–MS (Fig. 9b). To validate the site-specific conjugation, 7c was treated with PNGase-F for deglycosylation (Supplementary Results). Figure 9c shows the heavy-chain bands of 7c at a lower position. This band migrates to the same position as a deglycosylated commercial Herceptin, demonstrating the specific N-glycan conjugation of Herceptin. LC–MS analysis of the released glycan–drug complex (Fig. 9d) displays a series of charged m/z data in close agreement with the calculated molecular weight. These data solidly demonstrate that the glycosite-specific conjugation method can be successfully applied to ADC preparation.
Figure 9.
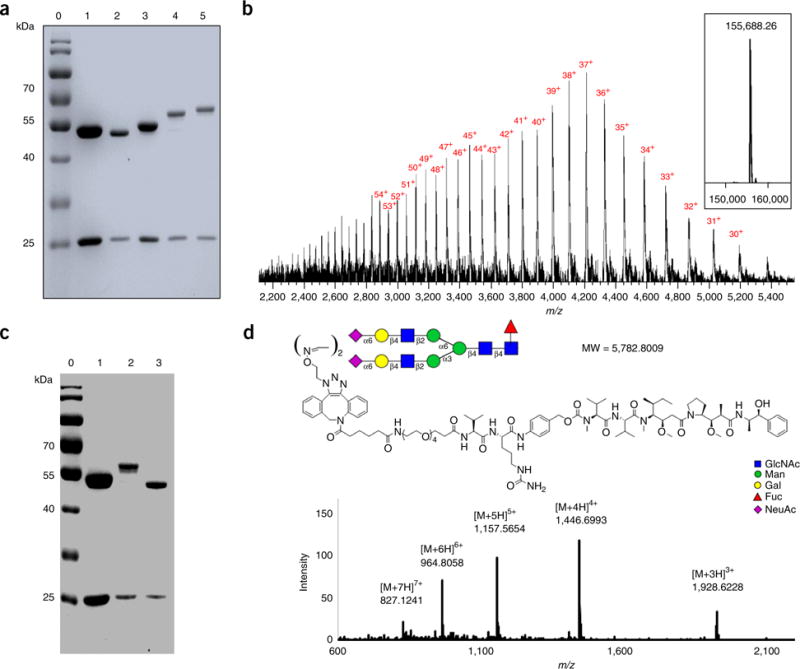
SDS-PAGE and LC–MS characterization of gsADC (7) (ref. 26). (a) SDS-PAGE analysis of 7b and 7c. Lane 0: marker, lane 1: commercial Herceptin, lane 2: Herceptin-Fucα1,6GlcNAc (2a), lane 3: glycoengineered Herceptin (5a), lanes 4 and 5: gsADC (7b and 7c). (b) LC–MS profiles for 7b. The multiple charged m/z data are labeled with charge numbers. The deconvolution mass spectrum is shown in the embedded box. (c) SDS-PAGE analysis of PNGase-F digestion of 7c. Lane 0: marker, lane 1: commercial Herceptin, lane 2: 7c, lane 3: 7c after PNGase-F digestion. (d) MS profile of the glycan–drug complex (shown with glycan symbol along with MMAE structure) released from 7c by PNGase-F digestion.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NNSFC, nos. 21372238 and 21572244 to W.H.), the SIMM Institute Fund (CASIMM0120153004 to W.H.), and the ‘Personalized Medicines: Molecular Signature-based Drug Discovery and Development’ Strategic Priority Research Program of the Chinese Academy of Sciences (grant no. XDA12020311 to W.H.).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS F.T. and W.H. designed the research and developed the methods; F.T. performed the experiments; F.T. and W.H. wrote the manuscript; and L.-X.W. developed the original IgG glycoengineering approach and revised the manuscript.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Shields RL, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. 2002;277:26733–26740. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 2.Yamane-Ohnuki N, et al. Establishment of FUT8 knockout Chinese hamster ovary cells: an ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol Bioeng. 2004;87:614–622. doi: 10.1002/bit.20151. [DOI] [PubMed] [Google Scholar]
- 3.Shinkawa T, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. 2003;278:3466–3473. doi: 10.1074/jbc.M210665200. [DOI] [PubMed] [Google Scholar]
- 4.Gramer MJ, et al. Modulation of antibody galactosylation through feeding of uridine, manganese chloride, and galactose. Biotechnol Bioeng. 2011;108:1591–1602. doi: 10.1002/bit.23075. [DOI] [PubMed] [Google Scholar]
- 5.Anthony RM, et al. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science. 2008;320:373–376. doi: 10.1126/science.1154315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anthony RM, Wermeling F, Karlsson MC, Ravetch JV. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc Natl Acad Sci USA. 2008;105:19571–19578. doi: 10.1073/pnas.0810163105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 8.Liu L. Antibody glycosylation and its impact on the pharmacokinetics and pharmacodynamics of monoclonal antibodies and Fc-fusion proteins. J Pharm Sci. 2015;104:1866–1884. doi: 10.1002/jps.24444. [DOI] [PubMed] [Google Scholar]
- 9.Goetze AM, et al. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology. 2011;21:949–959. doi: 10.1093/glycob/cwr027. [DOI] [PubMed] [Google Scholar]
- 10.Beck A, Reichert JM. Marketing approval of mogamulizumab: a triumph for glycoengineering. MAbs. 2012;4:419–425. doi: 10.4161/mabs.20996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sondermann P, Szymkowski DE. Harnessing Fc receptor biology in the design of therapeutic antibodies. Curr Opin Immunol. 2016;40:78–87. doi: 10.1016/j.coi.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 12.DiLillo DJ, Ravetch JV. Fc-Receptor interactions regulate both cytotoxic and immunomodulatory therapeutic antibody effector functions. Cancer Immunol Res. 2015;3:704–713. doi: 10.1158/2326-6066.CIR-15-0120. [DOI] [PubMed] [Google Scholar]
- 13.Brandsma AM, Jacobino SR, Meyer S, ten Broeke T, Leusen JH. Fc receptor inside-out signaling and possible impact on antibody therapy. Immunol Rev. 2015;268:74–87. doi: 10.1111/imr.12332. [DOI] [PubMed] [Google Scholar]
- 14.Ferrara C, et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc Natl Acad Sci USA. 2011;108:12669–12674. doi: 10.1073/pnas.1108455108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamane-Ohnuki N, Satoh M. Production of therapeutic antibodies with controlled fucosylation. MAbs. 2009;1:230–236. doi: 10.4161/mabs.1.3.8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Umana P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat Biotechnol. 1999;17:176–180. doi: 10.1038/6179. [DOI] [PubMed] [Google Scholar]
- 17.Warnock D, et al. In vitro galactosylation of human IgG at 1 kg scale using recombinant galactosyltransferase. Biotechnol Bioeng. 2005;92:831–842. doi: 10.1002/bit.20658. [DOI] [PubMed] [Google Scholar]
- 18.Raju TS, Briggs JB, Chamow SM, Winkler ME, Jones AJ. Glycoengineering of therapeutic glycoproteins: in vitro galactosylation and sialylation of glycoproteins with terminal N-acetylglucosamine and galactose residues. Biochemistry. 2001;40:8868–8876. doi: 10.1021/bi010475i. [DOI] [PubMed] [Google Scholar]
- 19.Li B, Song H, Hauser S, Wang LX. A highly efficient chemoenzymatic approach toward glycoprotein synthesis. Org Lett. 2006;8:3081–3084. doi: 10.1021/ol061056m. [DOI] [PubMed] [Google Scholar]
- 20.Huang W, et al. Glycosynthases enable a highly efficient chemoenzymatic synthesis of N-glycoproteins carrying intact natural N-glycans. J Am Chem Soc. 2009;131:2214–2223. doi: 10.1021/ja8074677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodfellow JJ, et al. An endoglycosidase with alternative glycan specificity allows broadened glycoprotein remodelling. J Am Chem Soc. 2012;134:8030–8033. doi: 10.1021/ja301334b. [DOI] [PubMed] [Google Scholar]
- 22.Huang W, Giddens J, Fan SQ, Toonstra C, Wang LX. Chemoenzymatic glycoengineering of intact IgG antibodies for gain of functions. J Am Chem Soc. 2012;134:12308–12318. doi: 10.1021/ja3051266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin CW, et al. A common glycan structure on immunoglobulin G for enhancement of effector functions. Proc Natl Acad Sci USA. 2015;112:10611–10616. doi: 10.1073/pnas.1513456112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parsons TB, et al. Optimal synthetic glycosylation of a therapeutic antibody. Angew Chem Int Ed Engl. 2016;55:2361–2367. doi: 10.1002/anie.201508723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li TZ, Tong X, Yang Q, Giddens JP, Wang LX. Glycosynthase mutants of endoglycosidase S2 show potent transglycosylation activity and remarkably relaxed substrate specificity for antibody glycosylation remodeling. J Biol Chem. 2016;291:16508–16518. doi: 10.1074/jbc.M116.738765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng T, et al. One-pot N-glycosylation remodeling of IgG with non-natural sialylglycopeptides enables glycosite-specific and dual-payload antibody-drug conjugates. Org Biomol Chem. 2016;14:9501–9518. doi: 10.1039/c6ob01751g. [DOI] [PubMed] [Google Scholar]
- 27.Priyanka P, Parsons TB, Miller A, Platt FM, Fairbanks AJ. Chemoenzymatic synthesis of a phosphorylated glycoprotein. Angew Chem Int Ed Engl. 2016;55:5058–5061. doi: 10.1002/anie.201600817. [DOI] [PubMed] [Google Scholar]
- 28.Umekawa M, et al. Mutants of Mucor hiemalis endo-beta-N-acetylglucosaminidase show enhanced transglycosylation and glycosynthase-like activities. J Biol Chem. 2008;283:4469–4479. doi: 10.1074/jbc.M707137200. [DOI] [PubMed] [Google Scholar]
- 29.Sjogren J, et al. EndoS and EndoS2 hydrolyze Fc-glycans on therapeutic antibodies with different glycoform selectivity and can be used for rapid quantification of high-mannose glycans. Glycobiology. 2015;25:1053–1063. doi: 10.1093/glycob/cwv047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Fang T, Boons GJ. Preparation of well-defined antibody-drug conjugates through glycan remodeling and strain-promoted azide-alkyne cycloadditions. Angew Chem Int Ed Engl. 2014;53:7179–7182. doi: 10.1002/anie.201402606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Q, et al. Site-specific antibody-drug conjugation through glycoengineering. Bioconjug Chem. 2014;25:510–520. doi: 10.1021/bc400505q. [DOI] [PubMed] [Google Scholar]
- 32.Zhu Z, et al. Site-specific antibody-drug conjugation through an engineered glycotransferase and a chemically reactive sugar. MAbs. 2014;6:1190–1200. doi: 10.4161/mabs.29889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tumbale P, Jamaluddin H, Thiyagarajan N, Acharya KR, Brew K. Screening a limited structure-based library identifies UDP-GalNAc-specific mutants of alpha-1,3-galactosyltransferase. Glycobiology. 2008;18:1036–1043. doi: 10.1093/glycob/cwn083. [DOI] [PubMed] [Google Scholar]
- 34.Li H, et al. Optimization of humanized IgGs in glycoengineered Pichia pastoris. Nat Biotechnol. 2006;24:210–215. doi: 10.1038/nbt1178. [DOI] [PubMed] [Google Scholar]
- 35.Junutula JR, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26:925–932. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 36.Zimmerman ES, et al. Production of site-specific antibody-drug conjugates using optimized non-natural amino acids in a cell-free expression system. Bioconjug Chem. 2014;25:351–361. doi: 10.1021/bc400490z. [DOI] [PubMed] [Google Scholar]
- 37.Sun B, et al. A simplified procedure for gram-scale production of sialylglycopeptide (SGP) from egg yolks and subsequent semi-synthesis of Man3GlcNAc oxazoline. Carbohydr Res. 2014;396:62–69. doi: 10.1016/j.carres.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 38.Maki Y, Okamoto R, Izumi M, Murase T, Kajihara Y. Semisynthesis of intact complex-type triantennary oligosaccharides from a biantennary oligosaccharide isolated from a natural source by selective chemical and enzymatic glycosylation. J Am Chem Soc. 2016;138:3461–3468. doi: 10.1021/jacs.5b13098. [DOI] [PubMed] [Google Scholar]
- 39.Yamamoto K, Kadowaki S, Fujisaki M, Kumagai H, Tochikura T. Novel specificities of Mucor hiemalis endo-beta-N-acetylglucosaminidase acting complex asparagine-linked oligosaccharides. Biosci Biotechnol Biochem. 1994;58:72–77. doi: 10.1271/bbb.58.72. [DOI] [PubMed] [Google Scholar]
- 40.Noguchi M, Tanaka T, Gyakushi H, Kobayashi A, Shoda S. Efficient synthesis of sugar oxazolines from unprotected N-acetyl-2-amino sugars by using chloroformamidinium reagent in water. J Org Chem. 2009;74:2210–2212. doi: 10.1021/jo8024708. [DOI] [PubMed] [Google Scholar]
- 41.Ning XH, Guo J, Wolfert MA, Boons GJ. Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast huisgen cycloadditions. Angew Chem Int Ed Engl. 2008;47:2253–2255. doi: 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baskin JM, et al. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci USA. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.