

Abstract
We analyzed 28 thymic epithelial tumors (TETs) using next-generation sequencing and identified a missense mutation (chromosome 7 c.74146970T>A) in GTF2I at high frequency in type A thymomas, a relatively indolent subtype. In a series of 274 TETs, we detected the GTF2I mutation in 82% of type A and 74% of type AB thymomas but rarely in the aggressive subtypes, where recurrent mutations of known cancer genes have been identified. Therefore, GTF2I mutation correlated with better survival. GTF2I β and δ isoforms were expressed in TETs, and both mutant isoforms were able to stimulate cell proliferation in vitro. Thymic carcinomas carried a higher number of mutations than thymomas (average of 43.5 and 18.4, respectively). Notably, we identified recurrent mutations of known cancer genes, including TP53, CYLD, CDKN2A, BAP1 and PBRM1, in thymic carcinomas. These findings will complement the diagnostic assessment of these tumors and also facilitate development of a molecular classification and assessment of prognosis and treatment strategies.
Tumors originating from thymic epithelial cells are the most common primary neoplasms of the mediastinum, but they are rare, with an incidence of only 0.32 per 100,000 people every year worldwide1. According to the 2004 WHO (World Health Organization) classification, TETs are subgrouped into thymic carcinomas and thymomas; the latter are further classified into types A, AB, B1, B2 and B3 according to their histological features2 (Supplementary Fig. 1). In several series, WHO classification is an independent prognostic factor3. Types A and AB thymomas have the best prognosis, with 10-year survival rates close to 100%, whereas thymic carcinomas are the most aggressive TETs, with a 50% 10-year survival rate4. Thymomas are frequently associated with paraneoplastic syndromes, especially myasthenia gravis, which is observed in 40% of patients5. Surgery is the mainstay of treatment, and prognosis is strongly determined by stage at diagnosis and the completeness of tumor resection2. Nonresectable and metastatic TETs are candidates for chemotherapy, which achieves tumor responses in 60–80% of patients4. However, chemotherapy is not curative in patients with metastatic disease, and the development of targeted therapies has been hampered by the insufficient characterization of the genetic abnormalities of TETs4.
We applied next-generation sequencing technologies to study the genetic aberrations of TETs (Supplementary Tables 1 and 2) and identified a highly recurrent mutation of GTF2I, which encodes TFII-I, in the more indolent thymomas. Conversely, mutations of well-characterized cancer genes were more common in the more aggressive histologies.
RESULTS
Copy number aberrations
Large copy number aberrations, affecting an entire chromosome arm (arm-level copy number aberrations), were present in more aggressive histotypes, in particular thymic carcinoma and types B3 and B2, but were uncommon in the more indolent types A and AB thymomas. We identified two major clusters of TETs according to their arm-level copy number aberrations, the first encompassing most of types A and AB tumors and the second including mainly the aggressive histotypes (Supplementary Fig. 2). Overall, the most frequent arm-level copy number losses involved chromosomes 6 (6p, 26%; 6q, 29%), 3p (22%) and 13q (18%). The most frequent arm-level copy number gains affected chromosomes 1q (55%), 7 (7p, 20%; 7q, 15%) and 20p (17%; Supplementary Figs. 3 and 4). Of particular interest were focal copy number aberrations that appeared to be within the significant peaks identified using GISTC, an analysis aimed at identifying significant regions of copy number aberrations possibly driving cancer growth (Supplementary Fig. 5 and Supplementary Table 3). These included focal amplification of the BCL2 locus, which correlated with increased expression of BCL2 transcripts according to RNA-seq data (Supplementary Fig. 6).
Somatic mutations in coding regions
Exome sequencing identified 722 somatic single nucleotide variations (SNVs) and 68 insertions/deletions (indels) in the coding regions of 28 TETs. We identified an average of 28 mutations per sample (range, 3–94; Supplementary Table 4). Thymic carcinomas had a significantly higher number of mutations than thymomas (Mann-Whitney U P = 0.001; Fig. 1a). Moreover, we observed mutations in several cancer genes in more than one case of thymic carcinoma, but they were usually single events in thymomas (Fig. 1b). We designed a custom 197–cancer gene panel to sequence 52 TETs (Supplementary Table 5), including 26 of the 28 tumors already characterized by whole exome sequencing. The two methods were highly correlated (χ2 P < 0.001; Supplementary Table 6).
Figure 1.
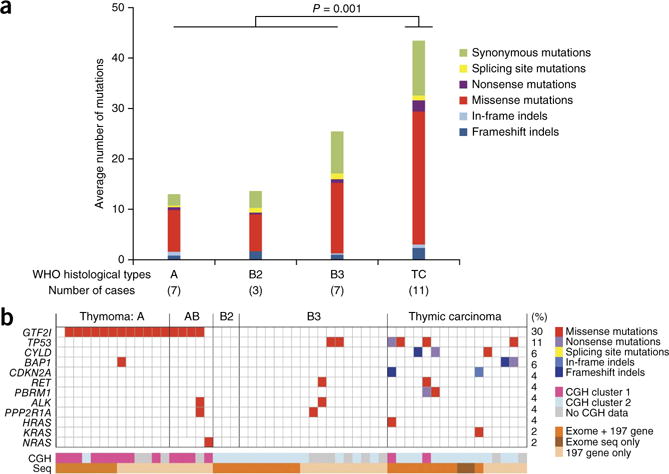
Overview of somatic mutations in TETs. (a) Average number of mutations (SNVs and indels) by WHO histotypes as detected by exome sequencing (n = 28). More mutations were observed in thymic carcinomas than in thymomas (Mann-Whitney U P = 0.001). Type B3 thymomas had more mutations than types A and B2, but this difference was not significant. (b) Mutations detected by exome sequencing or 197-gene assay (n = 54). At the bottom, the two tracks report the CGH clusters to which the cases belong and whether the samples have been sequenced by exome sequencing and/or 197-gene assay (Seq).
In thymomas, we observed frequent recurrent mutations in only one gene, GTF2I. This mutation was strikingly prevalent in the A and AB histotypes (Fig. 1b), and all the GTF2I-mutated cases presented the same single nucleotide change, T>A, at the same position on chromosome 7, 74146970 (Supplementary Fig. 7a). This mutation was not previously described as a polymorphism in the dbSNP137 and ESP5400 databases or as a somatic mutation in tumors (COSMIC database). The missense mutation of GTF2I leads to a leucine-to-histidine substitution and deleteriously alters the TFII-I protein structure and/or function according to SIFT and PolyPhen-2 predictions (Supplementary Table 4). The mutation affects the second conserved TFII-I repeat domain of the protein in proximity to its DNA binding site. Sanger sequencing confirmed the presence of mutations in tumors but not in normal DNA in all the mutated cases detected by exome sequencing (Supplementary Fig. 7b). The mutation was found in GTF2I but not its pseudogenes GTF2IP1 and LOC10093631 (Supplementary Note, Supplementary Tables 7–9 and Supplementary Figs. 8 and 9).
Prevalence of the GTF2I mutation in TETs
We assessed the frequency of the GTF2I mutation in a total of 274 TETs (270 tumors and 4 cell lines; Supplementary Tables 1 and 2). We evaluated tumors rich in cancer cells (>50%) for GTF2I mutation using Sanger sequencing (199 TETs). We further sequenced GTF2I mutations in 250 TET samples using a deep sequencing approach, which also included 78 tumors relatively rich in non-neoplastic thymocytes (cancer cells <50%). We sequenced a total of 172 cases using both Sanger and deep sequencing and found good concordance in mutation detection (Supplementary Note and Supplementary Table 2). We observed a GTF2I mutation (chromosome 7 c.74146970T>A) in 119 of the 270 TETs evaluated (43.4%), which was present most commonly in type A (82%) and AB (74%) thymomas (78% overall; Fig. 2a). The frequency of mutation progressively decreased in more aggressive histological types to only 8% in thymic carcinomas (3/36). We observed more mutations in the early stages (I–II, 57%) than in the advanced stages (III–IV) of disease (19%, χ2 P < 0.001). In a binomial logistic regression model that included WHO histotype, stage and completeness of resection, only histotype significantly predicted the presence of GTF2I mutation (P < 0.001; Supplementary Table 10).
Figure 2.

GTF2I mutation in TETs. (a) Frequency of GTF2I mutation by WHO histotype (combining the results of all analytic platforms). The number of patients sequenced is given in parenthesis next to each WHO histotype. (b) Kaplan-Meier survival curve demonstrating a more favorable outcome in GTF2I-mutated (MUT, blue line) than in wild-type (WT, red line) TETs (log-rank test P < 0.001; 83 and 121 evaluable patients, respectively).
Survival data were available for 204 patients (median follow-up of 39.4 months, 95% confidence interval (CI) 30.3–48.5 months). Patients with tumors bearing GTF2I mutations had a better prognosis than those bearing wild-type GTF2I (96% compared to 70% 10-year survival, respectively; log-rank P < 0.001; Fig. 2b), reflecting the higher mutation frequency in less aggressive tumors. Notably, all three of the patients with thymic carcinoma carrying GTF2I mutation were alive, with a median follow-up time of 27.6 months (95% CI 0–70 months) (Supplementary Fig. 10a). In patients with thymomas, there was a more favorable outcome in those with tumors carrying mutated GTF2I than in those carrying wild-type GTF2I (96% compared to 88% 10-year survival; log-rank P = 0.057; Supplementary Fig. 10b). Ki67 staining of histotypes rich in epithelial tumor cells (A, B3 and thymic carcinoma) demonstrated no substantial differences in proliferation between thymomas with or without GTF2I mutations in types A (2% compared to 3%, respectively) and B3 thymomas (13% compared to 11%, respectively). The three thymic carcinomas with GTF2I mutation had a lower proliferation rate than those with wild-type GTF2I (Ki67 staining, 5% compared to 14%; Supplementary Fig. 10c). Two of these thymic carcinomas were squamous cell and one was an undifferentiated carcinoma; all of them were positive for KIT immunohistochemistry. In models of multivariate survival analysis that included only two covariates, both GTF2I status and WHO classification were prognostic factors independent of disease stage. In contrast, GTF2I status and WHO classification were dependent on each other. Combining stage, WHO classification and GTF2I status in the same model, only stage was an independent prognostic factor (Supplementary Table 11). None of the four cell lines analyzed (Ty82, T1682, T1889 and IU-TAB1) had a GTF2I mutation (Supplementary Table 2).
Expression of the GTF2I mutation
RNA-seq analysis demonstrated expression of the GTF2I mutation in all thymomas evaluated (five type A and two type AB thymomas). Both the mutated and wild-type GTF2I alleles were expressed. The median number of reads covering the mutation locus was 1,114, and the mutated allele was present in an average of 47% (range, 44–49%) of the total GTF2I reads. Sanger sequencing of the cDNA confirmed the presence of the mutant GTF2I allele in the transcripts of all these samples. There are five known isoforms of GTF2I6 that differ by alternative splicing of exons 10 and 12. Using transcriptome sequencing, we found that exon 10 was expressed with very few reads, whereas exon 12 was expressed with approximately half of the reads relative to the neighboring exons in both the mutated and wild-type samples (Fig. 3a). Indeed, according to Cufflinks estimates, the expression of the β and δ isoforms was significantly higher than that of the other three isoforms (Kruskal-Wallis P < 0.0001, Dunn’s post hoc P < 0.0001 for both β and δ; Supplementary Fig. 11a). These results suggest that the β and δ isoforms are predominantly expressed in TETs. Using RT-PCR and specific primers designed for the β or δ isoform, we were able to confirm expression of the two isoforms and detected the T>A mutation in both of them. The observed T>A mutation resulted in p.Leu404His in the β isoform and p.Leu383His in the δ isoform of TFII-I.
Figure 3.
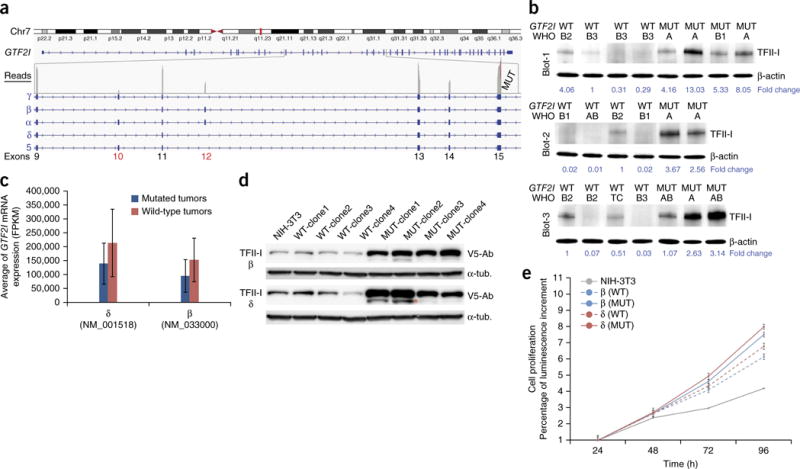
Expression of GTF2I isoforms and their role in cell proliferation. (a) There are five known isoforms of TFII-I (α, β, γ, δ and 5) that differ by alternative splicing of exons 10 and 12. In a case of type A thymoma with GTF2I mutation (MUT), the estimated gene expression status is depicted at the exon level. Exon 10 was silent, and exon 12 was expressed at about half the level of the other exons (gray bars in the read count). The absence of exon 10 limits the expression to the β and δ isoforms. Chr7, chromosome 7. (b) TFII-I expression was tested in 21 frozen tumors using antibodies to TFII-I by western blot analysis. The tumors were divided into three separate blots (Blot-1, Blot-2 and Blot-3). Every blot contains wild-type (WT) and mutant (MUT) cases in order to facilitate the comparison. TFII-I protein expression was higher in MUT than WT tumors. For each tumor, the histotype is indicated, and a loading control with β-actin is included. Normalized quantification of TFII-I protein expression is reported at the bottom of the blots. (c) The average of FPKM (fragments per kilobase of exon per million fragments mapped) values, an estimator of mRNA expression, was higher in wild-type (n = 7) than mutated (n = 5) tumors for both the β and δ GTF2I isoforms for tumors sequenced with HiSeq2000. (d) Using a lentiviral vector, WT and MUT β and δ isoforms were ectopically expressed in NIH-3T3 cells. Transfected β and δ isoforms of TFII-I were visualized using antibody to V5 (V5-Ab). The loading control was α-tubulin (α-tub.). In all clones tested, the expression of TFII-I–mutated isoforms was consistently higher than that of the wild-type isoforms. The extra lower molecular weight band indicated by the orange asterisk might be the result of TFII-I degradation during sample preparation. (e) NIH-3T3 cells carrying the mutated β and δ isoforms (solid lines) exhibit a higher proliferation rate than those carrying the wild-type isoforms (dashed lines). Experiments were repeated three times, and all error bars indicate the s.d.
We analyzed gene expression from RNA-seq data (Supplementary Table 12; unsupervised clustering of expression is shown in Supplementary Fig. 11b). Tumors with GTF2I mutations tended to cluster together in a group rich in types A and AB thymomas, similar to what we observed in the comparative genomic hybridization (CGH) results.
Functional characterization of the GTF2I mutation
The GTF2I mutation alters a residue within the amino acid sequence RILLAKE that may represent a noncanonical destruction box resembling the destruction box (RXXLXX[LIVM]) found in cyclins, PLK1 and Securin7. The RILLAKE>RILHAKE alteration may render TFII-I unrecognizable by the protein degradation machinery8. Notably, mutant tumors showed higher TFII-I expression than wild-type tumors at the protein level (Fig. 3b) but not the mRNA level (Fig. 3c). To understand the biological role of the GTF2I mutation and the elevated protein expression in mutant tumors, we created GTF2I cDNAs encoding the p.Leu404His and p.Leu383His β and δ isoforms by site-directed mutagenesis. We stably introduced pLenti6.3/V5-DEST expression vectors carrying the wild-type and mutated isoforms into NIH-3T3 cells (Fig. 3d). All β and δ isoforms accelerated cell proliferation in comparison to mock-transfected cells. Both the β and δ mutant isoforms increased cell proliferation more than their wild-type counterparts (Fig. 3e). In contrast, we observed no relevant differences in soft-agar colony formation between cells transfected with mutant or wild-type GTF2I (Supplementary Fig. 12). TFII-I can bind the FOS promoter and stimulate cell proliferation9. Indeed, tumors with GTF2I mutation tend to express more FOS mRNA than wild-type tumors (Supplementary Fig. 13a).
The mutant clones (both the β and δ isoforms) exhibited higher levels of TFII-I protein than wild-type clones (Fig. 3d). We inhibited protein synthesis using cycloheximide and observed a slightly slower degradation of mutant compared to wild-type TFII-I (Supplementary Fig. 13b). These results indicate that the p.Leu404His and p.Leu383His alterations may augment TFII-I expression post-transcriptionally, which may in turn accelerate cell proliferation by upregulation of cell cycle–control proteins10. Immunohistochemistry with a pan–TFII-I antibody showed higher expression of the protein in the nucleus of mutant cells. Type A thymomas without GTF2I mutation presented weak staining or were negative for TFII-I expression (Supplementary Fig. 13c).
Fusion genes in TETs
We investigated the presence of fusion genes using RNA-seq data and two independent algorithms: FusionMap11 and DeFuse12. We confirmed all the predicted fusions using RT-PCR followed by Sanger sequencing (Online Methods). We identified fusion transcripts in 7 of the 25 tumors evaluated, including the TY82 thymic carcinoma cell lines known to carry the BRD4-NUT fusion (Supplementary Fig. 14 and Supplementary Table 13). In these tumors, the number of fusion genes ranged from 1 to 16. There was an average of one fusion transcript in each case (range, 0–16). One B2 thymoma contained a remarkably high number of fusion transcripts (16 fusions) compared to the other samples. Although fusions involving GTF2I have been described recently in angiofibromas13, none of the fusion transcripts observed in TETs involved the GTF2I sequence.
DISCUSSION
We performed whole exome sequencing of TETs and identified a remarkably high frequency of GTF2I mutations. The frequency of this mutation was highest in types A and AB thymomas (78%), which are relatively indolent tumors, and gradually declined in the more aggressive histological types (8% in thymic carcinomas). The GTF2I mutation observed in TETs affected the same nucleotide on chromosome 7 c.74146970T>A in all cases, which causes a leucine-to-histidine substitution (p.Leu383His and p.Leu404His in the δ and β TFII-I isoforms, respectively). TFII-I is a multifunctional protein involved in the transcriptional regulation of several genes that control cell proliferation (c-FOS), cell cycle (cyclin D1) and developmental processes. It binds specifically to several DNA sequence elements and mediates growth factor signaling14. Although the deletion of a region on chromosome 7 that contains the GTF2I locus has been associated with Williams-Beuren syndrome15, very little is known about the role of TFII-I in human tumors. The size of Gtf2i heterozygous knockout mice is reduced because of slower growth, whereas Gtf2i-null mice show embryonic lethality15. Because TFII-I controls cell proliferation and the cell cycle in vitro, a role in cancer cell growth control has been hypothesized.
The structure of TFII-I encompasses six helix-loop-helix–like domains, each one containing a conserved I repeat, a nuclear localization domain, a DNA binding domain and an N terminus leucine zipper domain necessary for dimerization (Fig. 4a). The leucine-to-histidine alteration occurs in the conserved I repeat of the second TFII-I domain and substitutes a hydrophobic residue with a positively charged amino acid. We employed molecular modeling to simulate the structural alterations of TFII-I induced by the p.Leu404His alteration (Fig. 4b). Comparison to wild-type TFII-I indicated that the substitution of leucine to histidine changes neither the global nor the local conformation at the leucine site. We noticed that p.Leu404His forms a salt bridge with Glu400, which is absent in Leu404 (Fig. 4b). It is possible that the p.Leu404His alteration will disrupt the hydrophobic interaction of Leu404 with either a hydrophobic site in the interacting partner protein(s) or within TFII-I, which has not yet been crystallized. However, the three-dimensional (3D) structure of the fifth I repeat of mouse TFII-I has been solved by nuclear magnetic resonance spectroscopy16. More recently, the structure of the third human repeat of TFII-I has been deposited in 3D Macromolecular Structures (NCBI database) by the same group. The alignment of TFII-I repeat sequences suggests a similar structure between species. The altered amino acid is not conserved within the repeat sequences, confirming our hypothesis of a minor structural change in the domain structure. However, the high degree of conservation between species suggests an important role of this residue in protein function, which is in line with SIFT prediction. TFII-I contains three destruction boxes and is ubiquitinated and degraded in response to radiation-induced DNA damage8. Sequence analysis of the TFII-I mutant site shows that the mutation may alter a noncanonical destruction box of the RXXL motif. Therefore the leucine-to-histidine substitution may alter the recognition of this destruction box by the protein degradation machinery, leading to stabilization of the protein, as reflected by the increased TFII-I protein expression in GTF2I-mutant tumors and transfected cells. Further studies of other potential mechanisms of the mutation on TFII-I expression are warranted. The presence of exactly the same nucleotide substitution in GTF2I is a unique and newly discovered observation in TETs, which points to a critical role for GTF2I in TET biology. Although the relevance of the chromosome 7 c.74146970T>A mutation was shown by the accelerated proliferation of NIH-3T3 cells, the mutation had no prominent impact on NIH-3T3 cell transformation, indicating that mutant GTF2I may act as a cell growth–promoting rather than a strong transforming oncogene. Given that mutant GTF2I promotes NIH-3T3 cell proliferation (Fig. 3), the lower Ki67 index found in the three thymic carcinomas with GTF2I mutation may appear contradictory. However, the NIH-3T3 model has limitations because it is an immortalized fibroblast cell line, and the comparison between thymic carcinomas with mutant and wild-type GTF2I needs to take into account the fact that in these aggressive tumors, several additional genomic aberrations could accelerate cell proliferation more than GTF2I mutation. Moreover, the small sample size (three thymic carcinomas with GTF2I mutation) may not be representative of the true function of mutant GTF2I. The experiments in NIH-3T3 cells are useful to demonstrate that the GTF2I mutation is functional, but they do not elucidate its function in the context of thymic tumors that contain additional genomic mutations and a different epigenetic environment. Further studies are under way to verify the biological effect of mutant GTF2I during thymic tumor development.
Figure 4.

Structure of TFII-I protein and distributions of genomic alterations in TETs histotypes. (a) Schematic illustration of TFII-I domains, the I-repeat region and the position of the leucine-to-histidine change. The TFII-I repeats are as follows: six helix-loop-helix–like domains (purple boxes); the DNA binding domain basic region (BR, orange bar); the nuclear localization signal (NLS, green bar); the leucine zipper domain (LZ, gray bars) and the mutation locus (M, red arrow). (b) Model of TFII-I with p.Leu404His (blue) obtained after a 3-ns molecular dynamics simulation superimposed with wild-type TFII-I (tan). The residue Glu400 and the mutated residue p.Leu404His on TFII-I are highlighted. (c) Relative aberration frequencies according to WHO histotypes: GTF2I mutations (blue), arm-level copy number aberrations (orange) and number of mutations excluding GTF2I (purple).
The higher frequency of GTF2I mutation in types A and AB tumors suggests that the mutation could help identify a subset of TETs with favorable prognosis. Remarkably, only 2 patients (out of 83, 1 with type B3 and 1 with type B2) with GTF2I mutation died of tumor progression, in contrast to 26 of the 121 (21%) patients who had tumors harboring wild-type GTF2I; this difference in survival was statistically significant (P < 0.0001). Our finding is of particular interest, especially in view of the challenge represented by the histological classification of these rare tumors3,17. Our findings will need to be confirmed in an independent cohort of well-annotated TETs.
Thymic carcinomas are the most aggressive form of TETs2. Our data support the clinical dichotomy between thymomas and thymic carcinomas at the molecular level. Overall, the mutation frequency, copy number aberrations and expression profiles to some extent parallel the WHO classification. Thymic carcinomas are richer in mutations and copy number aberrations than thymomas; indeed, recurrent mutations of KIT have been reported in the literature in about 10% of thymic carcinomas18–20. In our series, the most common mutations of thymic carcinomas were observed in well-known cancer genes, i.e., TP53, CYLD and CDKN2A. CYLD is a tumor suppressor gene that is emerging as a frequently mutated gene in several types of cancer, and CYLD mutations have been detected in 19% of thymic carcinomas21–23.
Our data demonstrate an increasing number of mutations and copy number aberrations from type A to thymic carcinoma, with the exception of GTF2I mutations (Fig. 4c). Therefore, two models can be hypothesized. In the first model, GTF2I mutation is present exclusively in a distinct subgroup of TETs that have a favorable prognosis independent of their histological appearance. In the second model, GTF2I mutation is necessary for the founder tumor clone and is subsequently lost during the clonal evolution to more aggressive histotypes. GTF2I mutation might be necessary for the first steps of this multistep process and may become superfluous in the presence of more damaging mutations, such as mutations in TP53 or CYLD, and GTF2I-mutant clones might be outnumbered in more aggressive tumors during clonal evolution. The heterogeneity of TETs that is frequently described in histological sections derived from different regions of the same tumor suggests that heterogeneity within the tumor may be responsible for this clonal evolution.
In conclusion, our analysis provides a new perspective of the molecular aberrations that differentiate indolent thymomas from more aggressive thymomas and thymic carcinomas. The recurrent GTF2I mutations, which occur with a very high frequency in indolent tumors, represent a marker of favorable prognosis that may be useful in the classification of these rare tumors.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Samples
Tumor samples of 286 patients were collected from four different institutions: the National Cancer Institute (Bethesda, Maryland, USA), Pisa University Hospital (Pisa, Italy), Padua University Hospital (Padua, Italy) and Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Clinico Humanitas (Rozzano, Italy). Primary patient characteristics are summarized in Supplementary Table 2. All patients selected for this study were enrolled in protocols approved at the participating institutions, and written informed consent for genome profiling (including array comparative genomic hybridization and sequencing) was obtained from all study participants.
Nucleic acid extraction
Samples of thymic epithelial tumors were collected during surgical procedures or through an image-guided tumor biopsy. The collected specimens were immediately frozen in liquid nitrogen. Samples were embedded in optimal cutting temperature compound, and 8-μm slices were cut using a cryostat. A pathologist (J.R.-C.) evaluated the slices after hematoxylin and eosin staining in order to select regions rich with tumor cells for macrodissection. Samples were annotated with the pathologist’s estimation of tumor cellularity. DNA and RNA were extracted at the same time from the selected tumor portion using the All Prep RNA/DNA kit (Qiagen).
From formalin-fixed paraffin-embedded (FFPE) blocks, 4- and 10-μm slices were cut using a microtome. 4-μm slides were stained with hematoxylin and eosin, and a pathologist (G.F.) confirmed the tumor diagnosis and selected the tumor material. 10-μm slides were deparaffinized using Histochoice Clearing Agent (Sigma-Aldrich) and rehydrated through alcohol series. Thereafter, the stained and the 10-μm slides from the same block were matched, and the selected regions rich in tumor cells were macrodissected by scraping from the rehydrated slides. Samples were annotated with the pathologist’s estimation of tumor cellularity. DNA was extracted using the DNeasy Blood and Tissue kit (Qiagen) according to the vendor’s protocol but with an extended proteinase K digestion of at least 16 h at 70 °C.
Patient blood samples (5 ml) were collected in EDTA tubes and frozen at −80 °C. DNA was extracted from the whole blood using the QiAamp DNA Blood Maxi kit (Qiagen).
Array CGH
Tumors were chosen for array CGH (aCGH) depending on the availability of frozen material and on their tumor cell content; only samples rich in cancer cells (>80%) were selected. aCGH was performed in 65 cases; patient and sample characteristics are summarized in Supplementary Table 2. aCGH was performed as previously described24. The reference human genome was NCBI version 37.1. Data were analyzed using Nexus 7 (Biodiscovery Inc.) according to the following pipeline. A systematic correction was applied to the data in order to limit the wave-like artifacts due to genomic regions rich in GC nucleotides. The bias estimations were determined using a linear model that took into account the percentage of GC content and the length of the fragments. Bias estimations were then subtracted from the log2 ratio of the probes. Thereafter, probes were recentered through normalization to the median log2 ratio of the diploid regions that were determined sample by sample. Segmentation was performed using a rank segmentation algorithm according to the following settings: a significance threshold of 5.0 × 10−6, maximum contiguous probe spacing of 1,000 kb and a minimum of ten probes per segment. Sex chromosomes were removed from the analysis. The presence of copy number aberrations, which are candidate drivers of the tumor growth, was assessed using the GISTIC algorithm25. Regions with a Q bound lower than 0.25 and a G score higher than 1 were considered significant. GISTIC peaks related to germline copy number variations were filtered out (Supplementary Table 3). The Toronto database of genomic variants26 was adopted to define regions of germline copy number variations; if a GISTIC peak was fully mapped in one of these regions, the peak was removed from the list of significant results. Chromosome arm-level copy number aberrations were defined when more than 80% of a chromosome arm (p or q) was covered by copy number gains or losses. For example, chromosome 1q is 100,313,968 bp long; a single copy number gain of a portion of chromosome 1q longer than 80,251,174 bp (80% of 100,313,968) will be classified as an arm-level copy number gain of chromosome 1q. Also, an arm-level copy number gain of chromosome 1q will be described if the sum of the lengths of three regions of copy number gains mapped on 1q exceeds 80,251,174 bp. As previously described24, the 80% cutoff for the definition of chromosome arm-level copy number aberration was chosen based on the frequency distribution of the length of copy number aberrations. A hierarchical cluster of tumors was built using complete linkage of their chromosome arm-level copy number aberrations.
Transcriptome sequencing
Samples were selected for transcriptome sequencing if their RNA integrity number was >8, the hematoxylin and eosin staining demonstrated a proportion of cancer cells >80% and copy number aberrations larger than 5 Mb were detected using aCGH in order to safely exclude germline copy number variations that are usually (99%) shorter than 2.3 Mb27. Type A thymomas were exceptions because they typically do not present copy number aberrations. Type A thymomas were safely included after pathology review, as they always present a scant thymocyte component (non-neoplastic precursors of lymphocytes). Transcriptome sequencing was conducted at the National Cancer Institute sequencing facility according to the Illumina mRNA sample preparation kit. Briefly, using poly-T–bound magnetic beads, poly-A mRNAs were captured from total RNA. First- and second-strand cDNAs were serially synthetized. Overhanging fragment ends were repaired using T4 DNA and Klenow DNA polymerases, and adaptors were linked using T4 DNA Ligase. Ligation products were run on an agarose gel, and the 200-bp band was excised and used for DNA extraction. cDNA libraries were generated from the purified products and subsequently validated with the 2100 Agilent bioanalyzer (Agilent). According to the instructions, cDNA libraries were hybridized to a flow cell, amplified, linearized and denatured using Illumina Cluster Station in order to generate pair-end clusters ready for sequencing. The Genome Analyzer II or HiSeq2000 was used for sequencing (Supplementary Table 14). The read lengths and numbers of flow cells used for each tumor are reported in Supplementary Table 15.
Transcriptome sequencing data analysis workflow
Human genome hg19 was chosen as a reference, and only sequences mapped to human chromosomes 1–22, X, Y and M were retained. FASTQ files were obtained directly from the sequencing machine programmed to automatically trim the adaptors and barcode sequences. The reads were mapped to the reference genome using TopHat28. The quality of the RNA sequencing results was assessed using CollectRnaSeqMetrics available in Picard tools and with FASTQC software (Supplementary Table 14).
Estimation of gene expression
Gene expression was estimated from the mapped reads using the Cufflinks algorithm. Supplementary Table 12 reports the FPKM values of the transcripts annotated with the RefSeq database. The log10 transformation of the FPKM + 1 values was used to cluster thymic epithelial tumors. Samples were clustered using a hierarchical cluster and uncentered Pearson correlation. Two samples with high duplication rate and the cell lines were removed from the cluster analysis.
Prediction and validation of fusion transcripts
Candidate fusion transcripts were identified from the FASTQ files using two independent algorithms: FusionMap11 and DeFuse12. FusionMap with MONO version 2.10.8 was downloaded from the authors’ website. The following parameters were used according to previous reports11: MinimalFusionAlignmentLength = 25, FusionReportCutoff = 1 and NonCanonicalSpliceJunctionPenalty = 4. Moreover, at least 20 seed reads were required to support the predicted candidates.
DeFuse software was available online, and fusion candidates were identified and filtered as previously described12. The predicted fusion transcripts identified by both methods were evaluated using the BLAT tool from the UCSC website. Predicted fusions were excluded from the candidate list if one fusion arm had multiple possible alignments with an identity >95% or if they overlapped a region of human chained self-alignment29 or a region annotated with segmental duplications30, repeat maskers31, interrupted repeats31 and simple repeat32. The fusion transcripts included in this filtered list of candidates were validated using RT-PCR and Sanger sequencing. In brief, reverse transcription was performed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems). PCR primers were designed on opposite sides of the fusion junction and were tagged with M13 forward and reverse primer sequences. The reactions were carried out using Taq DNA polymerase (Invitrogen) and two negative controls: cDNA from the normal thymus of two unrelated subjects was included for each fusion candidate in order to exclude nonspecific amplification. Amplicons were run on an agar gel, and if the predicted size band was detected, PCR products underwent ExoSAP-IT (USB) purification and Sanger sequencing. Fusions were considered validated if the forward and reverse sequences were uniquely mapped to the predicted fusion transcript and if at least one of them spanned the junction sequence. The confirmed fusion transcripts were visualized using Circos-0.64 (Supplementary Table 12 and Supplementary Fig. 14).
Exome sequencing
Samples were included in the exome sequencing analysis if they fulfilled the following criteria: (i) tumor and normal DNA from the same patient were available; (ii) the tumor sample selected for DNA extraction presented at least 80% of tumor cells in hematoxylin and eosin–stained slides from the same specimen; and (iii) aCGH analysis revealed the presence of copy number aberrations larger than 5 Mb. Type A thymomas were included based on pathology review only, as they usually do not present copy number aberrations but are rich in epithelial cells. Exonic sequences were enriched using different capture-based platforms, as specified in Supplementary Table 15. Exome capture procedures were performed according to the respective vendors’ instructions. Exon-enriched libraries were subsequently paired-end sequenced using Illumina’s Genome Analyzer-II or HiSeq2000 as specified in Supplementary Table 15.
Exome sequencing data analysis workflow
Raw FASTQ sequence reads were first mapped to human genome 19 (USCS) using Novoalign, and then local realignment was performed around indels using the Genome Analysis Tool Kit (GATK). The duplicated reads were removed using the Picard tool, and base quality score recalibration was performed using GATK. Using VarScan2 (ref. 33), somatic mutations were identified by comparing the tumor and normal bam files of each patient. The detected somatic SNVs and indels were annotated using snpEff. Predicted SNVs and indels were further filtered and accepted if all of the following requirements were met: (i) there were at least four reads carrying the mutation in the tumor bam file; (ii) mutations present in more than 20% of the reads mapped to the mutation locus; (iii) there were at least eight reads covering the mutation locus in the normal bam file; and (iv) there were no more than 2% of reads carrying the mutation in the normal bam file. In order to further reduce the false positive calls, mutations identified as germline in at least one different patient blood control were filtered out. The somatic mutations of the coding regions identified using exome sequencing are reported in Supplementary Table 4. These mutations have been further annotated using Annovar34 with NCBI dbSNP build 137 data, the COSMIC database, SIFT35 and PolyPhen-2 (ref. 36) scores.
Confirmation and resequencing of the selected mutated genes
We validated the exome sequencing data using independently prepared libraries for high-depth sequencing on MiSeq sequencers (Illumina) using a custom panel of 197 genes. DNAs were extracted from samples with at least 80% cancer cells and fragmented by sonication. Then, indexed DNA libraries were prepared by three successive steps of end repair, A tailing and adaptor ligation to the DNA fragments. In subsequent PCR amplification steps, primers containing a flow cell attachment site (P5), sequencing primer sites for index read (Index SP) and application read 2 (Rd2 SP), unique 6-bp indices (Index) and a second flow cell attachment site (P7) were incorporated. The indexed libraries were then pooled in groups of up to 12, target enriched (Agilent) and sequenced. Sequence data were processed with our in-house variant calling pipeline, which includes BWA alignment37, GATK local realignment38, Strelka somatic variant calling39, SnpEff and SnpSift variant annotation40.
Sanger sequencing of the GTF2I locus
Sanger sequencing is able to detect mutations if they are present in a substantial percentage of cells (15–20%). This limits the possibility to detect GTF2I mutations if the gene and the pseudogene sequences were amplified at the same time with nonspecific primers because the mutation would be present in only 1:6 (17%) of the amplicons if heterozygous. In order to design specific primers for GTF2I, we took advantage of the nucleotide difference in exon 15 (C in the gene and T in the pseudogenes). A forward primer (Supplementary Table 16) was designed with its last 3′ base covering the C nucleotide that is specific for the GTF2I gene. The reverse primer anneals to both the GTF2I and pseudogene sequences. These primers were tagged with M13 forward and reverse sequences. DNA containing exclusively the GTF2I sequence or the pseudogene sequences was used for the optimization of the PCR conditions. The plasmids containing exclusively GTF2I exon 15 or the pseudogene sequences were generated during the TopoTA cloning experiments. With a melting temperature of 62.5 °C, only the GTF2I sequence was amplified. PCR was performed using Taq DNA Polymerase (Invitrogen) with 1.5 mM MgCl2 and 200 nM of forward and reverse primers according to the following amplification steps: step 1: 94 °C for 1 min; step 2: 94 °C for 30 s, 62.5 °C for 30 s, 72 °C for 45 s (35 times); step 3: 72 °C for 7 min. The amplicons were purified using ExoSap-IT (USB) and sequenced according to the Sanger method with M13 forward and M13 reverse primers. The chromosome 7 74146970 locus was inspected for mutations on both strands using Mac Vector. Using this optimization, we sequenced tumors with at least 50% cancer cells.
Sequencing of RNA from GTF2I and its pseudogenes
Primers were designed in order to selectively amplify the transcripts of GTF2I or its pseudogenes. Primers specific for GTF2I were located on its exon 10 and on the junction of exons 16 and 17 (Supplementary Table 16). Primers specific for the pseudogenes were templated on their exon 1 and on the exon 5 and 6 junction (reverse primers have the same sequence). All the primers were flanked with M13 primer sequences. Tumor RNA was converted into cDNA using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Pseudogenes and the GTF2I fragment were amplified by PCR using Taq DNA Polymerase (Invitrogen) according to the following program: step 1: 94 °C for 1 min; step 2: 94 °C for 30 s, 55 °C for 30 s, 72 °C for 45 s (35 times); step 3: 72 °C for 7 min. Amplicons of 503 bp (δ) and 566 bp (β) were verified with a run on a 1.2% agarose gel and then purified from unincorporated nucleotides and residual primer using ExoSAP-IT (USB). PCR products were sequenced using M13 primers and Sanger technology.
TopoTA cloning for detection of the T>A mutation in GTF2I and its pseudogenes
Primers able to simultaneously amplify the genomic DNA of GTF2I and its pseudogenes were designed (Supplementary Table 16). Forward and reverse primers were purchased from Integrated DNA Technologies in order to amplify a fragment of 218 bp containing C or T, a signature that distinguishes GTF2I from its pseudogenes, and the T>A mutation locus. PCR was performed using Taq DNA Polymerase (Invitrogen) according to the following amplification steps: step 1: 94 °C for 1 min; step 2: 94 °C for 30 s, 55 °C for 30 s, 72 °C for 45 s (35 times); step 3: 72 °C for 7 min. PCR amplicons were cloned into a pCR4-TOPO vector using the TopoTA Cloning kit for Sequencing (Invitrogen) according to the vendor’s instruction. Escherichia coli DH5α bacteria were transformed with the plasmid and plated on an LB-agar Petri dish with 100 μg ml−1 kanamycin selection and incubated overnight at 37 °C. Colonies selected for sequencing were resuspended in 5 ml of LB medium with 100 μg ml−1 kanamycin and grown overnight at 37 °C in a shaking incubator. DNA was extracted using the QIAprep Spin Miniprep kit (Qiagen) and plasmid sequenced using M13 forward and reverse primers.
GTF2I deep sequencing
Using PCR amplification followed by direct deep sequencing, the chromosome 7 c.74146970T>A mutation of GTF2I was sequenced in 250 samples (Supplementary Table 16). Forward and reverse primers were tailed with Illumina Adaptor tags for downstream next- generation sequencing using the BioMark HD System (Fluidigm) and Access Array IFC chips and kits (Fluidigm). Additionally, PCR products were indexed using an 8-mer oligo barcode. DNA was sequenced using 500-cycle MiSeq Reagent kits V2 (Illumina) and the MiSeq Benchtop Sequencer (Illumina).
GTF2I deep sequencing data analysis
In order to avoid potential alignment problems arising from the presence of the two pseudogenes homologous to GTF2I in the genome, we developed a new algorithm to avoid the alignment step altogether. We took advantage of the fact that the sequence of the primers used in the target selection and library preparation of the DirectSeq protocol are present at the 5′ end of each (valid) sequencer read. Only reads with a perfect match to the first 10 nt of any of the DirectSeq primers were retained for further analysis. Depending on the relative position (Supplementary Fig. 9 and Supplementary Tables 9 and 12) with respect to the recognized primer sequence, the nucleotide N1 discriminating between gene and pseudogene and the nucleotide N2 discriminating between variant and wild type were identified. Across the entire data set, the number of all possible combinations was counted for N1 and N2 for each primer. This count included nucleotide combinations not fitting the combinations CT (gene/WT), CA (gene/mutant), TT (pseudogene/WT) or TA (pseudogene/mutant) expected in the gene/pseudogene, with the WT/variant model as controls. These noncanonical combinations typically made up less than 1% of the reads associated with the primer, which is in line with the typical error rate of Illumina sequencers. Subsequently, we used this number as an estimate for the read error rate R as long as this estimate exceeded R > 0.5%, otherwise R = 0.5% was used as the noise estimate. A variant was called if the number of reads compatible with a mutated gene exceeded the number of reads expected as a result of read errors by at least 5 × R, i.e.,
P values for the association of the mutation status with WHO classification and stage were estimated using a χ2 test using a flat distribution as the null model.
Survival and statistical analyses
The Kaplan-Meier method was used to generate survival curves. Disease-related survival (DRS) was calculated from the date of the first histological diagnosis to the date of death due to tumor progression. DRS was chosen instead of the overall survival because, given the expected long survival, especially in the most indolent thymomas, and the advanced age of many patients, death was often not related to the tumor. Survival curves were compared by the log-rank test. A Cox proportional hazard model was initially built for the univariate analysis, which included WHO groupings (A, AB, B1 compared to B2, B3, thymic carcinoma), stage (I–II compared to III–IV), completeness of resection (R0 compared to R1–R2) and GTF2I mutation status. Subsequently, multivariate analysis was performed including prognostic factors found in the univariate analysis (P < 0.1). A binomial logistic regression model was built to correlate the presence of GTF2I mutation with WHO histotype, stage and completeness of resection. All tests were two tailed, considered significant if P < 0.05 and performed using SPSS version 20.
Structural model and molecular dynamics of TFII-I
A structural model of TFII-I was based on the solution structure of TFII-I (Brookhaven Protein Data Bank 2DN4). TFII-I was energy minimized using the consistent valence force field AMBER 10.0 simulation package41. The cutoff for nonbonded interaction energies was set to ∞ (no cutoff); other parameters were set to default. The dielectric constant was set at ε = 4 to account for the dielectric shielding found in proteins. The minimization was conducted in two steps: the first step used the steepest descent minimization for 5,000 cycles and then conjugate gradient minimization was used until the average gradient fell to <0.01 kcal M−1.
Using the energy-minimized structure of TFII-I as the initial model, 3-ns molecular dynamics (MD) simulations with a distant-dependent dielectric constant were conducted by using the SANDER module of the AMBER 10.0 simulation package41 with the PARM98 force-field parameter. MD simulations were performed using 0.001-ps time steps with temperature set at 300 °K. The SHAKE algorithm41 was used to keep all bonds involving hydrogen atoms rigid. Temperature and pressure coupling algorithms42 were used to maintain constant temperature and pressure. Electrostatic interactions were calculated with the Ewald particle mesh method43, and a dielectric constant at 1Rij and a nonbonded cutoff of 14 Å were used to the approximate electrostatic interactions and van der Waals interactions. Structural analyses were done using the SYBYL X.1 (Tripos International) molecular modeling program. The structural model figure was generated using UCSF Chimera, San Francisco.
Ectopic expression of the GTF2I mutation
pEBB plasmids containing GTF2I β (NM_033000.2) and δ (NM_001518.3) isoforms were purchased from Addgene. The GTF2I sequence in the plasmid was sequenced, and two synonymous mutations and one nonsynonymous mutation were identified in both isoforms. The nonsynonymous mutations were corrected using site-directed mutagenesis. Similarly, the chromosome 7 c.74146970T>A mutation was introduced in the plasmid using the QuikChange Site-Directed Mutagenesis kit (Agilent) according to the vendor’s protocol. The primers to introduce the mutation were designed using primer X (Supplementary Table 16). The β p.Leu404His and δ p.Leu383His mutated isoforms were generated in the pEBB plasmid. The mutated and wild-type GTF2I isoforms were first moved into a donor vector (pDONR221vector) through a recombinase reaction (Gateway BP Clonase II Enzyme mix, Invitrogen) and subsequently moved into a lentiviral vector (pLenti6.3/V5-DEST Gateway Vector kit, Invitrogen) using Gateway LR Clonase II Enzyme mix (Invitrogen) according to the vendor’s instructions. The pLenti6.3/V5-DEST plasmids with wild-type and mutated GTF2I isoforms were transfected into NIH-3T3 cells (purchased from ATCC and tested for mycoplasma contamination every 3 months) using Lipofectamine LTX (Invitrogen). NIH-3T3 cells were grown in DMEM (Gibco, Invitrogen) supplemented with 50 U ml−1 penicillin, 50 U ml−1 streptomycin (Invitrogen) and 10% heat-inactivated fetal bovine serum (Invitrogen) and grown in a 37 °C incubator with humidified 5% CO2 atmosphere. P-BABEpuro vector containing HRASV12H was purchased from Addgene and used as positive control for soft-agar assay44. Stable clones were selected using 8 μg ml−1 blasticidin or 1 μg ml−1 puromycin when appropriate (Gibco, Invitrogen). For each GTF2I isoform (both wild type and mutated), four independent stable pool transfectants were obtained. Stable ectopic TFII-I expression was confirmed by western blot analysis using anti-V5 (R960-25, Invitrogen, 1:5,000), and anti–α-tubulin (T6199, Sigma-Aldrich, 1:5,000) as a loading control.
Cell proliferation and soft agar assay
HRASV12H and mock-transfected NIH-3T3 cells were the positive44 and negative controls, respectively. 1,000 cells per well were plated in 96-well plates and tested for cell proliferation using a luminescent method (CellTiter-Glo Luminescent Cell Viability Assay, Promega) at 24, 48, 72 and 96 h. For wild-type and mutated GTF2I isoforms, four different stable pools were included in the experiments. Each experiment was replicated at least three times, and the average cell proliferation was calculated for both wild-type and mutated β and δ GTF2I isoforms. Soft agar assay was performed as previously described45 using 5,000 cells for each well of a six-well plate. Experiments were performed at least three times, and averages were calculated from the results of four distinct pools of each GTF2I variant. Expression of endogenous TFII-I was tested by western blot analysis using anti-TFII-I (4562, Cell Signaling, 1:1,000) in frozen primary thymic epithelial tumors for which GTF2I mutation status was available.
Cycloheximide assay
HeLa cells were purchased from ATCC and tested every 3 months for mycoplasma contamination. HeLa cells were transfected with pLenti6.3/V5-DEST containing β and δ isoforms with and without the mutation. Cells plated in six-well dishes were treated with cycloheximide at a 100 μg ml−1 concentration in order to inhibit protein synthesis. Cells were harvested at fixed time points, and protein was extracted and tested for the amount of TFII-I by western blot analysis. Anti-V5 was used to determine TFII-I concentration, with anti–α-tubulin used as a loading control.
FOS expression using RT-PCR
cDNA prepared from frozen tumors with or without GTF2I mutation was used to determine FOS expression using RT-PCR. TaqMan gene expression assay primers were purchased from Applied Biosystems. GAPDH was used as the endogenous control for mRNA expression. Real-time PCRs were operated on the ABI 7900HT fast real-time PCR system (Applied Biosystems). Fold changes in mRNA expression were calculated by the 2 − ΔCt method46.
Immunohistochemistry
Immunohistochemistry was performed as previously described47 using 4-μm slides cut from FFPE tumors. Anti-Ki67 (M7240, Dako, 1:50) and anti–TFII-I (clone 3E2, EMD Millipore, 1:100) were used. A pathologist (G.F.) evaluated the slides, taking into account the percentage of stained cells.
Supplementary Material
Acknowledgments
This work was supported by a US National Institutes of Health National Cancer Institute intramural program and the Georgetown University Lombardi Cancer Center. We thank A. Proietti for her help in reviewing pathology slides.
Footnotes
URLs. Human genome hg19, http://genome.ucsc.edu; Picard tools, http://picard.sourceforge.net; FASTQC software, http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc; Cufflinks algorithm, http://cufflinks.cbcb.umd.edu; RefSeq database, http://www.ncbi.nlm.nih.gov/refseq/; FusionMap with MONO version 2.10.8, http://www.omicsoft.com/fusionmap; DeFuse software, http://compbio.bccrc.ca/software/defuse; BLAT tool, http://genome.ucsc.edu/cgi-bin/hgBlat; Circos-0.64, http://circos.ca; Novoalign, http://www.novocraft.com/main/index.php; SnpEff, http://snpeff.sourceforge.net; COSMIC database, http://cancer.sanger.ac.uk/cancergenome/projects/cosmic; PrimerX, http://www.bioinformatics.org/primerx.
Accession codes. Array CGH and next-generation sequencing data have been deposited in GEO and are available with the accession codes GSE55852 and GSE57892, respectively.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
I.P., P.S.M., Y.W. and G.G. performed study design and writing. I.P., J.G., Y.J.Z., S.B. and S.D. performed data analysis. I.P., R.L.W., M.P., C.L. and K.J.K. performed genomic assays. I.-K.K., K.-S.P. and D.V. performed in vitro assays. M.L., G.F., P.A.Z., F.C., A.F., F.R., J.R.-C. and G.G. provided samples and collected clinical data. I.P., P.S.M., Y.W. and G.G. managed the project.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.de Jong WK, et al. Thymic epithelial tumours: a population-based study of the incidence, diagnostic procedures and therapy. Eur J Cancer. 2008;44:123–130. doi: 10.1016/j.ejca.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Travis WD, Brambilla E, Muller-Hermelink HK, Harris CC. Pathology and Genetics: Tumors of The Lung, Pleura, Thymus and Heart. IARC Press; Lyon, France: 2004. [Google Scholar]
- 3.Zucali PA, et al. Reproducibility of the WHO classification of thymomas: practical implications. Lung Cancer. 2013;79:236–241. doi: 10.1016/j.lungcan.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly RJ, Petrini I, Rajan A, Wang Y, Giaccone G. Thymic malignancies: from clinical management to targeted therapies. J Clin Oncol. 2011;29:4820–4827. doi: 10.1200/JCO.2011.36.0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okumura M, et al. Immunological function of thymoma and pathogenesis of paraneoplastic myasthenia gravis. Gen Thorac Cardiovasc Surg. 2008;56:143–150. doi: 10.1007/s11748-007-0185-8. [DOI] [PubMed] [Google Scholar]
- 6.Roy AL. Biochemistry and biology of the inducible multifunctional transcription factor TFII-I: 10 years later. Gene. 2012;492:32–41. doi: 10.1016/j.gene.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.King RW, Glotzer M, Kirschner MW. Mutagenic analysis of the destruction signal of mitotic cyclins and structural characterization of ubiquitinated intermediates. Mol Biol Cell. 1996;7:1343–1357. doi: 10.1091/mbc.7.9.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desgranges ZP, et al. Inhibition of TFII-I–dependent cell cycle regulation by p53. Mol Cell Biol. 2005;25:10940–10952. doi: 10.1128/MCB.25.24.10940-10952.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grueneberg DA, et al. A multifunctional DNA-binding protein that promotes the formation of serum response factor/homeodomain complexes: identity to TFII-I. Genes Dev. 1997;11:2482–2493. doi: 10.1101/gad.11.19.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashworth T, Roy AL. Phase specific functions of the transcription factor TFII-I during cell cycle. Cell Cycle. 2009;8:596–605. doi: 10.4161/cc.8.4.7728. [DOI] [PubMed] [Google Scholar]
- 11.Ge H, et al. FusionMap: detecting fusion genes from next-generation sequencing data at base-pair resolution. Bioinformatics. 2011;27:1922–1928. doi: 10.1093/bioinformatics/btr310. [DOI] [PubMed] [Google Scholar]
- 12.McPherson A, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol. 2011;7:e1001138. doi: 10.1371/journal.pcbi.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arbajian E, et al. A novel GTF2I/NCOA2 fusion gene emphasizes the role of NCOA2 in soft tissue angiofibroma development. Genes Chromosom Cancer. 2013;52:330–331. doi: 10.1002/gcc.22033. [DOI] [PubMed] [Google Scholar]
- 14.Roy AL. Biochemistry and biology of the inducible multifunctional transcription factor TFII-I. Gene. 2001;274:1–13. doi: 10.1016/s0378-1119(01)00625-4. [DOI] [PubMed] [Google Scholar]
- 15.Enkhmandakh B, et al. Essential functions of the Williams-Beuren syndrome–associated TFII-I genes in embryonic development. Proc Natl Acad Sci USA. 2009;106:181–186. doi: 10.1073/pnas.0811531106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doi-Katayama Y, et al. Solution structure of the general transcription factor 2I domain in mouse TFII-I protein. Protein Sci. 2007;16:1788–1792. doi: 10.1110/ps.072792007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verghese ET, et al. Interobserver variation in the classification of thymic tumours— a multicentre study using the WHO classification system. Histopathology. 2008;53:218–223. doi: 10.1111/j.1365-2559.2008.03088.x. [DOI] [PubMed] [Google Scholar]
- 18.Girard N, Mornex F, Van Houtte P, Cordier JF, van Schil P. Thymoma: a focus on current therapeutic management. J Thorac Oncol. 2009;4:119–126. doi: 10.1097/JTO.0b013e31818e105c. [DOI] [PubMed] [Google Scholar]
- 19.Petrini I, et al. Expression and mutational status of c-kit in thymic epithelial tumors. J Thorac Oncol. 2010;5:1447–1453. doi: 10.1097/JTO.0b013e3181e96e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ströbel P, et al. Thymic carcinoma with overexpression of mutated KIT and the response to imatinib. N Engl J Med. 2004;350:2625–2626. doi: 10.1056/NEJM200406173502523. [DOI] [PubMed] [Google Scholar]
- 21.Chapman MA, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ley TJ, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bignell GR, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petrini I, et al. Copy number aberrations of BCL2 and CDKN2A/B identified by array-CGH in thymic epithelial tumors. Cell Death Dis. 2012;3:e351. doi: 10.1038/cddis.2012.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beroukhim R, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci USA. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 27.Ostrovnaya I, Nanjangud G, Olshen AB. A classification model for distinguishing copy number variants from cancer-related alterations. BMC Bioinformatics. 2010;11:297. doi: 10.1186/1471-2105-11-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiaromonte F, Yap VB, Miller W. Scoring pairwise genomic sequence alignments. Pac Symp Biocomput. 2002;2002:115–126. doi: 10.1142/9789812799623_0012. [DOI] [PubMed] [Google Scholar]
- 30.Bailey JA, et al. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 31.Jurka J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 2000;16:418–420. doi: 10.1016/s0168-9525(00)02093-x. [DOI] [PubMed] [Google Scholar]
- 32.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koboldt DC, et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 36.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saunders CT, et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 40.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Case DA, et al. AMBER 10 User Manual. University of California, San Francisco; San Francisco, California: 2008. [Google Scholar]
- 42.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684. [Google Scholar]
- 43.Darden T, York D, Pedersen L. Particle mesh Ewald: an N-logN method for Ewald sums in large systems. J Chem Phys. 1993;98:10089. [Google Scholar]
- 44.Li W, Zhu T, Guan KL. Transformation potential of Ras isoforms correlates with activation of phosphatidylinositol 3-kinase but not ERK. J Biol Chem. 2004;279:37398–37406. doi: 10.1074/jbc.M405730200. [DOI] [PubMed] [Google Scholar]
- 45.Chen M, et al. Enhanced growth inhibition by combined DNA methylation/HDAC inhibitors in lung tumor cells with silenced CDKN2A. Int J Oncol. 2010;37:963–971. doi: 10.3892/ijo_00000747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petrini I, et al. Copy number aberrations of genes regulating normal thymus development in thymic epithelial tumors. Clin Cancer Res. 2013;19:1960–1971. doi: 10.1158/1078-0432.CCR-12-3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zucali PA, et al. Insulin-like growth factor-1 receptor and phosphorylated AKT-serine 473 expression in 132 resected thymomas and thymic carcinomas. Cancer. 2010;116:4686–4695. doi: 10.1002/cncr.25367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.