

SUMMARY
Mutations in PARK6 (PINK1) and PARK2 (Parkin) are linked to rare familial cases of Parkinson’s disease (PD). Mutations in these genes result in pathological dysregulation of mitophagy, contributing to neurodegeneration. Here, we report that environmental factors causing a specific posttranslational modification on PINK1 can mimic these genetic mutations. We describe a molecular mechanism for impairment of mitophagy via formation of S-nitrosylated PINK1 (SNO-PINK1). Mitochondrial insults simulating age- or environmental-related stress lead to increased SNO-PINK1, inhibiting its kinase activity. SNO-PINK1 decreases Parkin translocation to mitochondrial membranes, disrupting mitophagy in cell lines and human iPSC-derived neurons. We find similar levels of SNO-PINK1 in brains of α-synuclein transgenic PD mice as in cell-based models, indicating the pathophysiological relevance of our findings. Importantly, SNO-PINK1-mediated deficits in mitophagy contribute to neuronal cell death. These results reveal a direct molecular link between nitrosative stress, SNO-PINK1 formation, and mitophagic dysfunction that contributes to the pathogenesis of PD.
Keywords: PARK6 (PINK1), PARK2 (Parkin), Parkinson’s disease, mitophagy, S-nitrosylation
eTOC Blurb
Nitrosative stress and mitochondrial dysfunction represent key pathological events in Parkinson’s disease. Oh et al. identify a molecular link between these events in which increased nitric oxide (NO)-related species S-nitrosylate a critical thiol group in PINK1, thus compromising its ability to eliminate damaged mitochondria via mitophagy.
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disorder and the most common affecting the motor system. It initially results from loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) in the mesencephalon of the brainstem (Lang and Lozano, 1998). Mutations in PTEN-induced kinase 1 (PINK1, PARK6) and Parkin (PARK2) have been linked to rare familial forms of autosomal recessive juvenile parkinsonism (AR-JP) (Kitada et al., 1998; Valente et al., 2004). The proteins encoded by these genes are known to be critical for mitochondrial quality control by regulating mitophagy, a selective form of macroautophagy that is specific for mitochondria (Pickrell and Youle, 2015).
Under basal conditions, PINK1 is integrated into the mitochondrial outer membrane, but is rapidly cleaved by mitochondrial proteases such as mitochondrial processing peptidase β and PARL, generating an N-terminally cleaved form of PINK1. The cleaved form of PINK1 retranslocates to the cytosol where it is degraded by the ubiquitin proteasome system, thus maintaining very low levels of PINK1 on healthy mitochondria (Greene et al., 2012; Jin et al., 2010; Muqit et al., 2006). In contrast, loss of mitochondrial membrane potential (ΔΨm), caused by mitochondrial toxin exposure, environmental pesticides or other forms of mitochondrial stress that in excess have been associated with PD pathogenesis, disrupts PINK1 turnover, leading to stabilization of full-length PINK1 on the damaged mitochondrial membrane (Lin and Kang, 2008). Full-length PINK1 then phosphorylates a number of proteins, including ubiquitin and Parkin. In turn, the PINK1-mediated phosphorylation of ubiquitin enhances Parkin’s mitochondrial translocation and ubiquitin E3 ligase activity (Kane et al., 2014; Koyano et al., 2014). Subsequently, activated Parkin polyubiquitinates mitochondrial outer membrane proteins to trigger mitophagy (de Vries and Przedborski, 2013; Kondapalli et al., 2012; Okatsu et al., 2012; Pickrell and Youle, 2015). When properly functioning, mitophagy ameliorates cytotoxicity by removing damaged or dysfunctional mitochondria.
Aging and environmental toxins are thought to contribute to a number of neurodegenerative disorders including PD, in part, by generating excessive reactive oxygen and nitrogen species (ROS/RNS) (Bazan, 2006; Lin and Beal, 2006). For example, recent evidence has shown that RNS/nitric oxide (NO)-induced protein S-nitrosylation triggers endoplasmic reticulum stress, protein misfolding, and excessive mitochondrial fragmentation, causing bioenergetic compromise with consequent synaptic loss (Hess et al., 2005; Nakamura et al., 2013; Raju et al., 2015). However, although NO has been reported to affect macroautophagy in general and mitophagy in particular (Han et al., 2015; Sarkar et al., 2011), further mechanistic insight into this process is needed. Here, we present evidence that excessive levels of NO, as seen in neurodegenerative disorders like PD, S-nitrosylate PINK1 to inhibit its kinase activity, thus inhibiting PINK1/Parkin-mediated mitophagy and contributing to dopaminergic neuronal cell death.
RESULTS
Cys568 is the Predominant Site of PINK1 S-Nitrosylation
Initially, we investigated whether endogenous PINK1 is S-nitrosylated in the SH-SY5Y dopaminergic human-derived neural cell line 20 min after exposure to the physiological NO donor S-nitrosocysteine (SNOC). When PINK1 degradation was prevented with a proteasome inhibitor or the protonophore/uncoupler carbonyl cyanide 3-chlorophenylhydrazone (CCCP), a mitochondrial depolarizing agent that triggers accumulation of full-length PINK1 on damaged mitochondria (Muqit et al., 2006; Lin and Kang, 2008), we observed formation of SNO-PINK1 (Figure 1A; Figure S1). Next, we asked if NO produced from neuronal NO synthase (nNOS) can S-nitrosylate PINK1. For this purpose, HEK-nNOS cells (which stably express nNOS) were transiently transfected with PINK1-Flag (C-terminally Flag-tagged PINK1), and exposed to the calcium ionophore A23187 to activate nNOS. SNO-PINK1 levels were augmented in cells incubated with A23187, but not in the presence of the NOS inhibitor Nω-nitro-L-arginine (L-NNA) (Figure 1B).
Figure 1. Endogenous PINK1 is S-Nitrosylated in Cultured Neural Cells and in a Transgenic PD Mouse Model.

(A) SH-SY5Y cells were pre-incubated with 2.5 μM MG132 for 4 hr prior to incubation for 20 min in 200 μM SNOC (or, as a control, ‘old’ SNOC from which NO had been dissipated). Cell lysates were subjected to biotin-switch assay to detect endogenous SNO-PINK1. Negative controls were performed in the absence of ascorbate, which selectively reduces SNO. SNO-PINK1 and total (Input)-PINK1 were detected by immunoblot with anti-PINK1 antibody.
(B) HEK-nNOS cells were transfected with wild-type (wt) PINK1-Flag. After 1 day, cells were incubated for 3 hr with 10 μM MG132 and A23187 in the presence or absence of 1 mM L-NNA. Cell lysates were then subjected to biotin-switch assay, immunoblotted, and probed with anti-Flag antibody.
(C) SH-SY5Y cells were transfected with wt PINK1-Flag or mutant PINK1(C568A)-Flag. After 1 day, cells were pre-treated with 10 μM MG132 for 4 hr and then incubated in 200 μM fresh or old SNOC for 20 min. Cell lysates were subjected to biotin-switch assay and immunoblotted with anti-Flag antibody.
(D) Ratio of SNO-PINK1/Input (total) PINK1 from wt or C568A mutant PINK1 transfected SH-SY5Y cells. Data are mean + SEM; ***p < 0.001; n = 3 experiments.
(E) Mass spectrometry identification of Cys568 on human PINK1 as the site of S-nitrosylation after labeling with biotin (see Method Details). Fragmentation of the amide bonds of the peptide resulted in formation of ‘b’ and ‘y’ ion series corresponding to the N-terminal and C-terminal fragments, respectively. All fragments have a charge of +1. The spectrum indicates that Cys568 indicated as C* possesses a 428.1916 mass shift, which corresponds to the addition of biotin. Manual validation of the spectra was performed to determine the source of the large peaks that were un-assigned by the software: The peak at 463(***) is the c4 ion with loss of the biotin modification; 429 peak (**) is the c4 ion with the loss of biotin as well as H2S from the side chain of cysteine; 714 peak (#) represents charge +2 of the y9 ion; the 1056 peak(@) is the b10 ion with biotin.
(F and G) Ratio of SNO-PINK1 to total PINK1 in a transgenic PD mouse model. Brain lysates from 4-month-old control or human α-synuclein—overexpressing mice were subjected to biotin-switch assay, followed by immunoblotting with anti-PINK1, anti-α-synuclein, and anti-Parkin antibodies (F). Ratio of SNO-PINK1/Input (total) PINK1 from mouse brains (G). Data are mean + SEM; *p < 0.05; n = 4 experiments.
(H) Schematic drawing showing PINK1 protein. MTS, mitochondria-targeting sequence; TM, transmembrane domain.
In order to determine the site of S-nitrosylation, we mutated candidate cysteine residues 92, 166, 564 and 568, all of which are surrounded by a partial SNO-motif (Stamler et al., 1997), and monitored their S-nitrosylation status (Figure S2). The PINK1(C568A) mutation decreased S-nitrosylation by ~70% compared to wild-type (wt) PINK1 (Figures 1C and 1D). Next, we performed Mass Spectrometry (MS), exposing recombinant PINK1 to SNOC, and showed that PINK1 can be S-nitrosylated at cysteine 568 (Figure 1E). Collectively, these data suggest that Cys568 is the predominant site of S-nitrosylation on PINK1.
S-Nitrosylation of PINK1 in the α-Synuclein Transgenic Mouse Model of PD
To assess the potential pathophysiological relevance of SNO-PINK1 formation to PD, we asked if SNO-PINK1 levels are elevated in the α-synuclein (α-syn) transgenic mouse that partially mimics sporadic PD with regard to increased expression of α-syn and abnormal mitochondrial morphology (Rockenstein et al., 2002; Nakamura et al., 2011). Consistent with the notion that SNO-PINK1 might contribute to PD-related pathology in vivo, we observed a significant increase in SNO-PINK1 expression in α-syn mouse brains early on in the disease process (Figures 1F and 1G). In contrast, although we and others have published that Parkin can also be S-nitrosylated in sporadic cases of advanced PD (Chung et al., 2004; Ozawa et al., 2013; Yao et al., 2004), we found that SNO-Parkin was undetectable in these early, pre-symptomatic α-syn transgenic mice, suggesting that PINK1 is even more sensitive to SNO modification and occurs earlier in the disease process than Parkin (Figure 1F).
S-Nitrosylation of PINK1 Downregulates Its Kinase Activity, and Non-Nitrosylatable PINK1 Mutant Mimics This Effect
The site of S-nitrosylation on PINK1, Cys568, is located in the C-terminus and is present in both full-length and cleaved forms (Figure 1H). Interestingly, PD-causing mutations encountered in the C-terminus of PINK1 abolish the enzyme’s kinase activity, supporting the notion that this region plays a critical role in enzyme function (Plun-Favreau et al., 2007; Sim et al., 2006). For this reason, we examined if S-nitrosylation of PINK1 affects its kinase activity. We exposed SH-SY5Y cells transfected with PINK1-Flag to SNOC in the presence or absence of CCCP. Consistent with prior reports, our phos-tag immunoblotting analysis, in which phosphorylated proteins appear as slower migrating bands (Kinoshita et al., 2006), showed that CCCP exposure increased auto-phosphorylation of PINK1 (Figure 2A) (Okatsu et al., 2012). In contrast, exposure to SNOC decreased the level of auto-phosphorylated PINK1 both in the presence and absence of CCCP (Figure 2A). To further verify the effect of SNOC on PINK1 kinase activity, we examined the phosphorylation status of Parkin, a substrate of PINK1 kinase during mitophagy. Similar to our observations on auto-phosphorylation, we found that CCCP elevated Parkin phosphorylation, while SNOC diminished this effect (Figures 2B and 2C; Figure S3) (Shiba-Fukushima et al., 2012). Hence, these results are consistent with the notion that S-nitrosylation inhibits PINK1 kinase activity. Next, we co-transfected wt or non-nitrosylatable mutant PINK1(C568A) plus GFP-Parkin into SH-SY5Y cells, and found, similar to the inhibitory effect of S-nitrosylation on PINK1 activity, that mutation of PINK1 at Cys568 significantly decreased its ability to phosphorylate Parkin (Figures 2D and 2E; Figure S3).
Figure 2. S-Nitrosylation of PINK1 Decreases Its Kinase Activity.

(A) SH-SY5Y cells were transfected with wt PINK1-Flag. After 1 day, cells were exposed to 10 μM CCCP in the presence or absence of 200 μM SNOC. Ninety minutes later, cell lysates were subjected to phos-tag SDS-PAGE to detect auto-phosphorylated PINK1 with anti-Flag antibody (upper panel). Open arrow indicates phosphorylated PINK1. Immunoblot of PINK1-Flag and GAPDH using standard SDS-PAGE (lower panels).
(B) SH-SY5Y cells were transfected with wt PINK1-Flag and GFP-Parkin. After 1 day, cells were exposed to 10 μM CCCP in the presence or absence of 200 μM SNOC. Ninety minutes later, cell lysates were subjected to phos-tag SDS-PAGE and immunoblotting with anti-Parkin or anti-Flag antibody. Open arrow indicates phosphorylated Parkin.
(C) Phosphorylated Parkin normalized to total PINK1 from immunoblots. Data are mean + SEM; **p < 0.01 by ANOVA; n = 3 experiments.
(D) SH-SY5Y cells were transfected with GFP-Parkin plus wt PINK1-Flag or mutant PINK1(C568A)-Flag. The amount of transfected plasmid DNA was adjusted to produce equal expression of wt and mutant PINK1. After 1day, cells were lysed and subjected to phos-tag SDS-PAGE and standard immunoblotting with anti-Parkin or anti-Flag antibody.
(E) Phosphorylation of Parkin normalized to total PINK1 from immunoblots. Data are mean + SEM; *p < 0.05; n = 3 experiments.
(F) SH-SY5Y cells were transfected with wt PINK1-Flag or mutant PINK1(C568A)-Flag. After 1 day, cells were exposed to 10 μM CCCP for 3 hr. Cell lysates were subjected to immunoblotting with anti-ubiquitin, p-ubiquitin, Flag, or GAPDH antibodies.
(G) Phosphorylation of ubiquitin normalized to total Ubiquitin from immunoblots. Data are mean + SEM; ***p < 0.001; n = 3 experiments.
Importantly, during the initiation steps of mitophagy, PINK1 phosphorylates ubiquitin to activate Parkin E3 ligase activity (Kane et al., 2014; Koyano et al., 2014). Hence, we tested whether non-nitrosylatable mutant PINK1(C568A) can decrease ubiquitin phosphorylation in SH-SY5Y cells after exposure to CCCP. Consistent with a reduction in PINK1 kinase activity, phosphorylated poly-ubiquitin chains were also decreased by non-nitrosylatable PINK1 (Figures 2F and 2G) (Fiesel et al., 2015). From these results, we reasoned that the PINK1(C568A) mutant mimics SNO-PINK1 in that it manifests decreased kinase activity, allowing us to exploit the use of this cysteine mutant in lieu of SNO-PINK1 in subsequent experiments. In these experiments, we monitor the effects of the PINK1(C568A) mutant compared to wt PINK1 to elucidate potential effects of SNO-PINK1 on Parkin translocation and mitophagy.
Interestingly and quite unexpectedly, we also found that exposure to SNOC decreased the level of full-length PINK1 in SH-SY5Y cells (Figure 2A; Figures S4A and S4B). While cleaved PINK1 appeared to transiently increase after SNOC exposure, this form was also found subsequently to decrease as well (see lane 3 of Figure S4A obtained 90 min after SNOC exposure vs. last lane of Figure S4B obtained 180 min after SNOC exposure); the proteasome inhibitor MG132 failed to prevent this downregulation (Figure S4C). This finding may account at least in part for the decrease in cleaved PINK1 that we observed in both postmortem human PD patient brain and in human induced pluripotent stem cell-derived dopaminergic neurons (hiPSC-DA) generated from PD patient fibroblasts (compared to isogenic, mutation-corrected controls) (Figures S4D–G; Table S1) (Ryan et al., 2013). Notably, non-nitrosylatable mutant PINK1(C568A) possessed similar stability compared to wt PINK1, consistent with the notion that the decreased expression of PINK1 after exposure to SNOC occurred independently of SNO-PINK1 formation (Figures S4H and S4I).
S-Nitrosylation of PINK1 Decreases Parkin Translocation to Mitochondria in Neural Cells
Since we found that S-nitrosylation of PINK1 decreases its kinase activity and that NO exposure decreases total PINK1 levels, we expected that both of these effects would summate to limit the ability of PINK1 to phosphorylate and recruit Parkin to damaged mitochondria, thus interfering with mitophagy. Since non-nitrosylatable PINK1(C568A) mimicked the effect of SNO-PINK1 in that it exhibited decreased kinase activity, we used PINK1(C568A) to simulate SNO-PINK1 in the absence of NO exposure; this approach eliminated the confounding effect of NO-induced decrease in PINK1 levels. Since exogenous expression of wt PINK1 is sufficient to recruit Parkin to mitochondria in the absence of mitochondrial damage (Figure 3A) (Vives-Bauza et al., 2010), we examined the ability of exogenous wt PINK1 vs. mutant PINK1(C568A) to effect Parkin translocation. For this experiment, GFP-Parkin was co-transfected with wt, C568A, or a kinase dead (KD) mutant of PINK1-Flag in SH-SY5Y cells. Expression of wt PINK1-FLAG significantly increased translocation of GFP-Parkin to mitochondria within 6 hours of transfection (Figure 3B; Figure S5A). In contrast, transfection with PINK1(C568A)-Flag displayed significantly delayed translocation of GFP-Parkin to the mitochondrial membrane (at both 6 and 24 hr). Additionally, as controls, transfection with GFP-Parkin alone or KD PINK1-Flag did not increase translocation of GFP-Parkin to mitochondria.
Figure 3. S-Nitrosylation of PINK1 Decreases PINK1/Parkin-Mediated Mitophagy.

(A) SH-SY5Y cells were transfected with GFP-Parkin and wt PINK1-tomato. After 1 day, cells were fixed with 4% PFA and immunostained with anti-Tom20 antibody (blue). Arrows indicate complete translocation, and arrowheads indicate partial translocation of GFP-Parkin to the mitochondrial membrane. Scale bar, 20 μm.
(B) SH-SY5Y cells were transfected with GFP-Parkin and wt-, non-nitrosylatable C568A, or kinase dead (KD) PINK1-Flag. After 6 or 24 hr, cells were scored for complete, partial, or no translocation of GFP-Parkin to mitochondria (See Figure S5A). Data are mean + SEM; ***p < 0.001; **p < 0.01 by ANOVA; n = 3 experiments.
(C) SH-SY5Y cells were transfected with GFP-Parkin and wt PINK1-Flag. After 6 hr, cells were exposed to 100 μM GSNO or GSH control, and then incubated an additional 24 hr. Cells with complete translocation of GFP-Parkin to the mitochondrial membrane were scored (See Figure S5D). Data are mean + SEM; **p < 0.01 by ANOVA; n = 3 experiments.
(D) SH-SY5Y cells were transfected with mt-Keima and wt PINK1-GFP or mutant PINK1(C568A)-GFP. After 1 day, cells were exposed to 20 μM CCCP for 16 hr. Cells were then imaged with 458 nm (measuring mitochondria with neutral pH) and 561 nm (measuring mitochondria with acidic pH) laser excitation for mt-Keima (emission at 610 nm) and at 488 nm for GFP (emission at 530 nm). mt-Keima can be used to differentially label mitochondria localized in the cytoplasmic (458 nm) and lysosomal (561 nm) compartments. Thus, a high ratio of mt-Keima-derived florescence (561 nm/458 nm), originating from low pH compartments, i.e., mitochondria within lysosomes, appears as red. Scale bar, 20 μm.
(E) Quantification of mitophagy index, derived as the proportion of high-ratio signal area (561 nm/458 nm) to total mitochondrial area (561 nm plus 458 nm) from three random fields in each experiment. Data are mean + SEM; ***p < 0.001 by ANOVA; n = 3 experiments.
To exclude the effects of endogenous PINK1 on these results, we repeated these experiments by co-transfecting GFP-Parkin and wt PINK1 or PINK1(C568A) into PINK1 null cells generated using CRISPR/Cas9 technology (Nezich et al., 2015). In PINK1 null HeLa cells, overexpressed GFP-Parkin was not translocated to the mitochondrial membrane even after exposure to CCCP, whereas co-transfection with wt PINK1-FLAG increased GFP-Parkin mitochondrial translocation. In contrast, co-transfection with GFP-Parkin and PINK1(C568A) resulted in significantly less GFP-Parkin translocation to mitochondria in both the presence and absence of CCCP (Figures S5B and S5C).
Moreover, since S-nitrosylation of PINK1 suppresses its kinase activity, we hypothesized that exposure to NO would decrease Parkin translocation to the mitochondrial membrane. To test this premise, we incubated SH-SY5Y cells expressing GFP-Parkin and wt PINK1-Flag with the physiological NO donor, S-nitrosoglutathione (GSNO, a longer-lasting donor than SNOC), and indeed found that GSNO decreased GFP-Parkin translocation compared to control (Figure 3C; Figure S5D). Taken together, these findings support the notion that S-nitrosylation of PINK1 may impair Parkin translocation to the mitochondrial membrane.
S-Nitrosylation of PINK1 Decreases Mitophagy
Next, since Parkin translocation to mitochondria is important for mitophagy, we wanted to assess the effect of SNO-PINK1 on the process of mitophagy in SH-SY5Y cells. To monitor mitophagy, we utilized mt-Keima (mitochondria targeted Keima), a ratiometric, pH-sensitive fluorescent protein that is resistant to lysosomal proteases. Moreover, the mt-Keima probe is targeted to the mitochondrial matrix. As Keima is excited by 561 nm light under acidic conditions and by 458 nm wavelength at neutral pH, a high ratio of fluorescence (561 nm/458 nm; red in Figure 3D) represents the presence of mitochondria in acidic lysosomes, e.g., mitochondria that are undergoing mitophagy. In contrast, a low ratio of fluorescence reflects normal mitochondria in the cytoplasm (Figure 3D). As a measure of mitochondrial delivery to the lysosome in PINK1-transfected cells, we then determined the ‘mitophagy index,’ the ratio of the area of red signal (indicating engulfment of mitochondria within lysosomes) to total mitochondrial area following previously described protocols (Bingol et al., 2014; Katayama et al., 2011). Because mt-Keima provides robust assessment of mitophagy flux (Bingol et al., 2014; Katayama et al., 2011; Lazarou et al., 2015; Sun et al., 2015), this fluorescence-based strategy was employed for this experiment (LC3 was also used in subsequent experiments described below). Here, in the mt-Keima assay, CCCP exposure significantly increased the mitophagy index in wt PINK1-GFP-overexpressing cells, whereas expression of mutant PINK1(C568A)-GFP caused a decrease in the mitophagy index (Figures 3D and 3E). Collectively, given that mutant PINK1(C568A) mimics the effect of SNO-PINK1, these results are consistent with the concept that S-nitrosylation of PINK1 impairs PINK1/Parkin-mediated mitophagy.
Endogenous NO Suppresses Mitochondrial Toxin-Induced Parkin Translocation to Mitochondria and Decreases Mitophagy in hiPSC-Derived Dopaminergic Neurons
Next, to more definitely determine the mechanistic role of endogenous NO on SNO-PINK1 formation in PD in a human context, we studied hiPSC-DA neurons. These human A9-type DA neurons were prepared as we have previously described (Ryan et al., 2013) and were found to be more comparable in terms of PINK1 expression to in vivo systems. For instance, in contrast to neural cell lines like SH-SY5Y and more reminiscent of the PINK1 expression pattern in human and mouse brain, we found that hiPSC-DA neurons contained a considerable amount of endogenous, cleaved PINK1 (Figure 4A). This fact allowed us to investigate the role of SNO-PINK1 in mitophagy using endogenous human PINK1.
Figure 4. Endogenous PINK1 Levels and Mitophagy in hiPSC-DA Neurons as a Model for Human PD.

(A) Lysates of SH-SY5Y cells, wt hiPSC-DA neurons, A53T mutant α-synuclein hiPSC-DA neurons, control human postmortem brain, and wt mouse brain were immunoblotted for PINK1, Parkin and GAPDH. The differentiation of hiPSCs into DA neurons was verified as previously described.(Ryan et al., 2013) Abbreviations: hPINK1, human PINK1; mPINK1, mouse PINK1.
(B–D) hiPSC-DA neurons were exposed to 1 μM valinomycin for 9 hr. Cell lysates were immunoblotted for PINK1, ubiquitin, phospho-ubiquitin and GAPDH. Phosphorylation of ubiquitin levels were normalized to total ubiquitin (D). Data are mean + SEM; ***p < 0.001; **p < 0.01; n = 3 experiments.
For this purpose, we exposed hiPSC-DA neurons to various mitochondrial insults known to trigger mitophagy and, at higher concentration, to induce parkinsonian-type neuronal damage, including rotenone (a pesticide that inhibits complex I of the electron transport chain), CCCP and FCCP (uncoupling agents), or valinomycin (a neutral ionophore that is selective for potassium ions and often used experimentally to induce mitophagy) (Okatsu et al., 2012; Vives-Bauza et al., 2010). Consistent with prior observations (Grenier et al., 2013), valinomycin led to stabilization of the levels of full-length PINK1 (Figure 4B). Moreover, we found that valinomycin increased ubiquitin phosphorylation in hiPSC-DA neurons (Figures 4C and 4D). Valinomycin is known to depolarize mitochondria, thus increasing the influx of L-arginine, the NOS substrate leading to NO formation (Grossini et al., 2009; Lowry et al., 1998; Signorello et al., 2003). Accordingly, we found that valinomycin exposure, like rotenone, induced production of endogenous NO in hiPSC-DA neurons (Figure 5A). Furthermore, incubation of hiPSC-DA neurons with valinomycin caused Parkin and LC3 translocation to mitochondria (Figures 5B–E). To test whether NO affects valinomycin-induced mitophagy, hiPSC-DA neurons were incubated with valinomycin plus the NOS inhibitor Nω-nitro-L-arginine methyl ester (L-NAME). Strikingly, we found that inhibition of NO production led to enhanced stability of full-length PINK1, translocation of Parkin and LC3 to mitochondria, and an increase in the mitophagy index assessed with mt-Keima (Figures 5B–H). These results indicate that high levels of endogenous NO suppress valinomycin-induced mitophagy.
Figure 5. Mitochondrial Insult Induces Mitophagy in hiPSC-DA Neurons.

(A) hiPSC-DA neurons were exposed for 9 hr to 1 μM valinomycin in the presence or absence of 1 mM L-NAME. To monitor NO, cells were incubated with 2.5 μM DAF-FM for 30 min. Data are mean + SEM from five random fields in each experiment; ***p < 0.001 by ANOVA; n = 3 experiments.
(B) hiPSC-DA neurons were exposed for 9 hr to 250 nM valinomycin in the presence or absence of 1 mM L-NAME, and immunostained for Parkin and Tom20. Scale bar, 20 μm.
(C) Parkin translocation to the mitochondrial membrane assessed by co-localization with Tom20. Data are mean + SEM from three random fields in each experiment; ***p < 0.001; **p < 0.01 by ANOVA; n = 3 experiments.
(D) hiPSC-DA neurons were exposed for 9 hr to 250 nM valinomycin in the presence or absence of 1 mM L-NAME, and then immunostained for LC3 and Tom20. Scale bar, 10 μm.
(E) LC3 translocation to the mitochondrial membrane assessed by co-localization with Tom20. Data are mean + SEM from three random fields in each experiment; ***p < 0.001; **p < 0.01 by ANOVA; n = 3 experiments.
(F) hiPSC-DA neurons were exposed for 9 hr to 1 μM valinomycin in the presence or absence of 1 mM L-NAME; lysates were then immunoblotted for PINK1 and GAPDH.
(G) hiPSC-DA neurons were electroporated with mt-Keima. Transfected cells were exposed to 250 nM valinomycin for 16 hr in the presence or absence of L-NAME, and imaged following excitation at 458 nm and 561 nm. After obtaining an initial set of images (top three rows), 50 mM NH4Cl was added to neutralize the pH in the lysosomal lumen before acquiring a second set of images (bottom row); this reversed the high-ratio (561 nm/458 nm) to low-ratio signal (Bingol et al., 2014). Scale bar, 10 μm.
(H) Quantification of mitophagy index in three random fields in each experiment. Data are mean + SEM; ***p < 0.001; *p < 0.05 by ANOVA; n = 3 experiments.
Mitochondrial Toxin-Induced SNO-PINK1 Formation Suppresses Mitophagy and Increases Cell Death in hiPSC-Derived Neurons
Having shown that NO negatively regulates valinomycin-induced mitophagy, we next asked if pathophysiogically-relevant levels of SNO-PINK1 play a role in this pathway. Using the biotin-switch assay, we found that exposure of hiPSC-DA neurons to valinomycin resulted in formation of endogenous SNO-PINK1 (Figures 6A and 6B). Importantly, when we compared the ratio of endogenous SNO-PINK1 (determined by biotin-switch assay) to total PINK1 (from immunoblots) in hiPSC-DA and in brains of the α-syn transgenic PD mouse model, we found that the degree of SNO-PINK1 formation was comparable (Figures 1F and 6B). These results indicate that pathophysiologically-relevant amounts of SNO-PINK1 are present in our hiPSC-DA model after exposure to valinomycin.
Figure 6. S-Nitrosylation of PINK1 Decreases Mitophagy in hiPSC-DA Neurons Exposed to Valinomycin.

(A) hiPSC-DA neurons were exposed to 250 nM valinomycin for 3 hr. Cell lysates were then subjected to biotin-switch assay to detect endogenous SNO-PINK1. Control was performed in the absence of ascorbate. SNO-PINK1 and Input-PINK1 were detected by immunoblotting with anti-PINK1 antibody.
(B) Ratio of SNO-PINK1/Input-PINK1 from hiPSC-DA neurons. Data are mean + SEM; **p < 0.01; n = 3 experiments.
(C) hiPSC-DA neurons were electroporated with mt-Keima and wt PINK1-GFP or mutant PINK1(C568A)-GFP. Subsequently, transfected cells were exposed to 250 nM valinomycin for 16 hr and imaged for mt-Keima and GFP. Scale bar, 20 μm.
(D) Mitophagy index determined from three random fields in each experiment. Data are mean + SEM; ***p < 0.001 by ANOVA; n = 3 experiments.
(E and F) hiPSC-DA neurons were exposed for 9 hr to 250 nM valinomycin in the presence or absence of 1 mM L-NAME. Cells were then fixed with 4% PFA and assayed for apoptotic neurons by TUNEL (total number of cells was assessed by Hoechst nuclear staining [E], see Figure S6A) and by the presence of condensed nuclei stained with Hoechst dye (F). Data are mean + SEM from three random fields in each experiment; ***p < 0.001 by ANOVA; n = 3 experiments.
Next, to determine if these levels of SNO-PINK1 suppress mitophagy and increase neuronal cell death in a disease-specific context, we used hiPSC-DA neurons since they produce similar levels of SNO-PINK1 as found in vivo during the pathogenesis of PD. For this experiment, we transfected hiPSC-DA neurons with wt PINK1-GFP or mutant PINK1(C568A)-GFP, which mimics the inhibition of kinase activity by SNO-PINK1, in conjunction with mt-Keima in order to assess mitophagy. The resulting increase in the mitophagy index revealed that exogenous expression of wt PINK1-GFP enhanced valinomycin-induced mitophagy, but mutant PINK1(C568A)-GFP failed to do so (Figures 6C and 6D). These results are consistent with the notion that S-nitrosylation of PINK1 abrogates its ability to mediate valinomycin-induced mitophagy. In general, through the removal of damaged mitochondria, mitophagy is thought to protect neurons. In contrast, in PD, defective mitophagy appears to contribute to neuronal cell death. Since we now report that S-nitrosylation of PINK1 inhibits the PINK1/Parkin-dependent mitophagy pathway, our findings suggest that formation of SNO-PINK1 could mediate this disruption in mitophagy, resulting in neuronal cell death and thus modeling disease pathogenesis in PD. Consistent with this premise, we found that inhibition of NO production by L-NAME decreased the neuronal cell death that occurred in response to valinomycin exposure, and, conversely, expression of mutant PINK1(C658A) increased cell death (Figures 6E and 6F; Figure S6).
DISCUSSION
Prior work had shown that agents that produce mitochondrial insult, such as valinomycin, rotenone and CCCP, produce dysfunctional mitochondria, thus activating the PINK1/Parkin pathway to enhance mitophagy. In contrast, high levels or more prolonged mitochondrial insults are known to induce PD-like neuronal damage and can thus serve as a useful tool for modeling defects in mitophagy that are relevant to PD. If mitophagy fails to remove sufficient numbers of damaged mitochondria, these mitochondrial toxins can then trigger neuronal cell death and onset of disease (Martinez-Vicente et al., 2010). Here, we demonstrate that the mitochondrial depolarizing agent valinomycin facilitates production of high levels of endogenous NO, resulting in formation of SNO-PINK1. S-Nitrosylation of PINK1 inhibits its kinase activity, thus repressing PINK1/Parkin-dependent mitophagy and increasing neuronal cell death (Figure 7). Moreover, we show in a human context using hiPSC-DA neurons that endogenous PINK1 participates in mitochondrial toxin-induced mitophagy, and, in opposition to this pathway, SNO-PINK1 functions as a negative regulator of mitophagy. Our work thus provides a molecular link between ‘nitrosative stress’ and ‘mitophagic defects’ observed in PD.
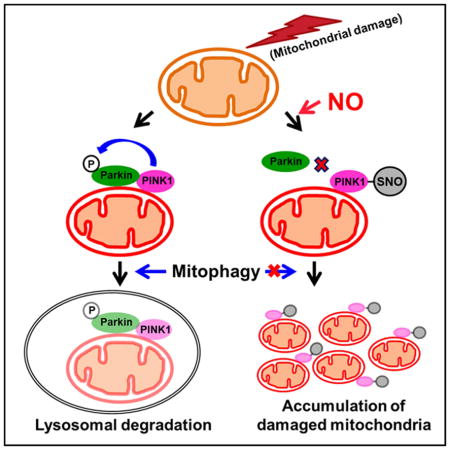
Figure 7. Proposed Model Illustrating SNO-PINK1-Mediated Inhibition of Mitophagy.

Dissipation of mitochondrial membrane potential stabilizes PINK1 on the mitochondrial membrane. The accumulated PINK1 phosphorylates Parkin and other substrates, facilitating lysosome-dependent degradation of damaged mitochondria (mitophagy). Low levels of NO can enhance mitophagy, possibly via SNO-Parkin-mediated pathways. In contrast, under conditions causing nitrosative stress, excessively produced NO can S-nitrosylate PINK1, inhibit its kinase activity, and decrease translocation of Parkin to mitochondria. This leads to accumulation of damaged mitochondria and increased cell death, contributing to the pathogenesis of neurodegenerative diseases such as PD.
To put these unique findings into a broader context, under physiological conditions, NO levels are maintained at low levels and mediate important signaling pathways for normal neuronal function, such as basal synaptic activity and mitochondrial bioenergetics. For instance, low levels of NO enhance Parkin translocation to mitochondria to increase mitophagy in a PINK1-independent manner (Han et al., 2015). In contrast, high levels of NO cause further mitochondrial impairment, thus contributing to the pathogenesis of neurodegenerative diseases (Nicotera et al., 1997). Along these lines, in the present study we found a significant increase in SNO-PINK1 formation in vivo early in the pathogenesis of PD in the α-syn mouse model (Figure 1E). In contrast, we could not find detectable levels of SNO-PINK in postmortem human PD brain (data not shown). Taken together, these results are consistent with the notion that SNO-PINK1 contributes to early stages of disease pathogenesis. Moreover, we establish that hiPSC-DA neurons exposed to excessive mitochondrial stress (e.g., in the form of valinomycin) mimic the α-syn PD mouse model with regard to increased SNO-PINK1 levels, thus inhibiting mitophagy and enhancing neuronal cell death (Figures 5 and 6A). We conclude that pathophysiologically relevant levels of SNO-PINK1 in hiPSC-DA neurons disrupt mitophagy and contribute to PD-related neurodegeneration. Furthermore, in our hiPSC-DA cell-based model, we demonstrate that the PINK1(C568A) mutant, which mimics S-nitrosylation, also disrupts PINK1/Parkin-dependent mitophagy (Figure 6). Taken together, these findings support the premise that formation of SNO-PINK1 represents a key mediator of defective mitophagy in PD.
Also, we and others have previously demonstrated that Parkin is S-nitrosylated in postmortem brain of sporadic cases of human PD and in transgenic mouse models of PD, but this nitrosylation events occurs relatively late in the progression of the disease (Chung et al., 2004; Yao et al., 2004) (Figures 1F and 1G); SNO-Parkin leads to dysregulation of its ubiquitin E3 ligase activity and compromises its neuroprotective function. In addition, another group recently reported that NO transiently promotes mitophagy via S-nitrosylation of Parkin, possibly representing a neuroprotective effect of NO. However, after this transient event, excessive oxidative/nitrosative stress leads to a decrease in the removal of damaged mitochondria (Ozawa et al., 2013). Along these lines, in the present study we show that SNO-PINK1 forms under pathophysiological conditions of high nitrosative stress, occurs early in the disease process in vivo, and inhibits mitophagy. These findings lead us to conclude that SNO-PINK1 formation may be, at least in part, responsible for dysfunctional mitophagy during PD pathogenesis.
Mutations in the PINK1 gene are known to result in loss of its kinase activity, leading to the development of a rare inherited form of PD. Analogous to these effects of genetic mutations, S-nitrosylation of PINK1 impairs its kinase activity and causes mitophagic deficits, suggesting that SNO-PINK1 may contribute to the development of sporadic PD attributed to environmental- and age-related nitrosative stress. Thus, the discovery reported here, namely that S-nitrosylation of PINK1 occurs early in the pathogenesis of PD and inhibits mitophagy, may suggest innovative therapeutic strategies for PD and other neurodegenerative diseases associated with aberrant mitophagy due to excessive nitrosative stress.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
SH-SY5Y neural cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM: Sigma) supplemented with 10% fetal bovine serum (FBS; HyClone), 2 mM L-glutamine (Gibco-Invitrogen), 50 IU/ml penicillin (Omega Scientific), and 50 μg/ml streptomycin (Omega Scientific) in a 5% CO2 incubator at 37 °C. HEK-nNOS cells (representing a stably expressing nNOS cell line, a gift from Drs. David Bredt and Solomon Snyder) were maintained in DMEM with FBS, 2 mM L-glutamine, 50 IU/ml penicillin, 50 μg/ml streptomycin, and 100 μg/ml geneticin. Transfections were performed in 6-well plates using 1 mg/ml polyethyleneimine solution (Sigma) with DMEM; after 3 hr, mixtures were replaced with fresh culture medium. After 2 days, cells were analyzed by immunoblotting. Transfection of PINK1 null HeLa cells (Nezich et al., 2015) was performed using FuGENE® HD Transfection Reagent (Promega) mixed with plasmids at an 8:2 ratio in Opti-MEM according to the manufacturer’s protocol. Empirically, we found that to obtain equivalent expression of wt PINK1 and mutant PINK1(C568A) in these experiments we had to transfect 20% more cDNA encoding the mutant.
PD Mouse Model and Human Brain Tissue
Transgenic mice overexpressing human wild type α-synuclein under the Thy1 promoter were used (Rockenstein et al., 2002). To detect SNO-PINK1 formation, brains were collected from 4-month-old control (C57BI/6) or α-synuclein transgenic mice (male; n = 4) and homogenized in HENTS buffer. Brain lysates were subjected to the biotin-switch assay. All animal experiments were approved by Sanford Burnham Prebys Institutional Animal Care and Use Committee, where the brains were collected. Patient brain tissues were obtained postmortem from subjects whose age, postmortem interval, and gender have been described in Table S1. Human brain samples were analyzed with institutional permission under the state of California and NIH guidelines. Informed consent was obtained according to procedures approved by Institutional Review Boards at the University of California, San Diego.
Biotin-Switch Assay for Detection of S-Nitrosylated Proteins
The biotin-switch method for detecting S-nitrosylated proteins was used as described previously (Haun et al., 2013; Uehara et al., 2006; Yao et al., 2004). In brief, cells were lysed with HENTS buffer (100 mM Hepes, pH 7.4, 1 mM EDTA, 0.1 mM neocuproine, 0.1% SDS, and 1% Triton X-100). Cell lysates (500 μg to 1 mg) were mixed with blocking buffer (2.5% SDS, 10 mM methyl methane thiosulfonate [MMTS] in HEN buffer [100 mM HEPES, pH 7.4, 1 mM EDTA, and 0.1 mM Neocuproine]) and incubated for 20 min at 50 °C with frequent vortexi ng to block free thiol groups. After removing excess MMTS by acetone precipitation, S-nitrosothiols were reduced to thiols with 20 mM ascorbate. Newly formed thiols were then linked with the sulfhydryl-specific biotinylating reagent N-[6-biotinamido]-hexyl]-l′-(2′pyridyldithio) propionamide (Biotin-HPDP). Unreacted Biotin-HPDP was removed by acetone precipitation, and the pellet was resuspended with HENS buffer (100 mM HEPES, pH 7.4, 1 mM EDTA, 0.1 mM neocuproine, 1% SDS), neutralized, and centrifuged to clear undissolved debris. Five percent of the supernatant was used as the input for the loading control. Biotinylated proteins were pulled down with Streptavidin-agarose beads and analyzed by immunoblotting for SNO-PINK1. In some experiments, exogenous NO donors were used to supply NO for S-nitrosothiol formation. The rapidly-decaying NO donor SNOC was employed for short-term additions of NO. SNOC was synthesized as described previously (Lei et al., 1992). The more stable NO donor GSNO was used for more prolonged NO addition and was prepared as previously described (Gaston et al., 1993).
Quantification and Statistical Analysis
All data represent at least three independent experiments, presented as mean + SEM. Unless otherwise stated, statistical significance between two samples were analyzed by a Student’s t-test (Excel), and for multiple samples by ANOVA with a Tukey’s post hoc test (GraphPad Prism). Our data met the assumptions of these tests.
Supplementary Material
Highlights.
Nitric oxide inhibits PINK1 kinase activity via S-nitrosylation of PINK1 at Cys568
S-Nitrosylation of PINK1 decreases translocation of Parkin to mitochondria
S-Nitrosylation of PINK1 thus impairs mitophagy in hiPSC-derived neurons
S-Nitrosylation of PINK1 exacerbates neuronal death in Parkinson’s disease models
Acknowledgments
We thank Dr. Eliezer Masliah (UC San Diego and NIA/NIH) for providing human biological materials, Zhuohua Zhang (Sanford Burnham Prebys Medical Discovery Institute) for the PINK1-Flag plasmid, Richard Youle (NINDS/NIH) for PINK1 null HeLa cells, and Scott R. McKercher (The Scripps Research Institute) for help with manuscript critical review and preparation. This work was supported in part by NIH grants R01 NS086890, P01 ES016738, DP1 DA041722, RF1 AG057409, R01 AG056259 and P30 NS076411 (to S.A.L.), by P41 GM103533 (to J.R.Y.), and by R37 HD045022, R37 CA084198 and R01 NS088538 (to R.J.), a Distinguished Investigator Award from the Brain & Behavior Research Foundation (to S.A.L.), and the Michael J. Fox Foundation (to S.A.L. and T.N.).
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and one table, and can be found with this article online at http://doi.org/xxxxxx.
AUTHOR CONTRIBUTIONS
C.O., T.N., and S.A.L. designed the research, C.O., A.S., J.P., N.D., F.S., D.B.M., J.K.D., and R.J. performed experiments or generated reagents, C.O., D.B.M., J.R.Y., R.A., T.N., and S.A.L. analyzed the data, and C.O., T.N. and S.A.L. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bazan NG. Cell survival matters: docosahexaenoic acid signaling, neuroprotection and photoreceptors. Trends Neurosci. 2006;29:263–271. doi: 10.1016/j.tins.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. The mitochondrial deubiquitinase USP30 opposes Parkin-mediated mitophagy. Nature. 2014;510:370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM. S-Nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- Cociorva D, DLT, Yates JR. Validation of tandem mass spectrometry database search results using DTASelect. Curr Protoc Bioinformatics. 2007;Chapter 13(Unit 13):14. doi: 10.1002/0471250953.bi1304s16. [DOI] [PubMed] [Google Scholar]
- de Vries RL, Przedborski S. Mitophagy and Parkinson’s disease: be eaten to stay healthy. Mol Cell Neurosci. 2013;55:37–43. doi: 10.1016/j.mcn.2012.07.008. [DOI] [PubMed] [Google Scholar]
- Fiesel FC, Ando M, Hudec R, Hill AR, Castanedes-Casey M, Caulfield TR, Moussaud-Lamodiere EL, Stankowski JN, Bauer PO, Lorenzo-Betancor O, et al. (Patho-)physiological relevance of PINK1-dependent ubiquitin phosphorylation. EMBO Rep. 2015;16:1114–1130. doi: 10.15252/embr.201540514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaston B, Reilly J, Drazen JM, Fackler J, Ramdev P, Arnelle D, Mullins ME, Sugarbaker DJ, Chee C, Singel DJ, et al. Endogenous nitrogen oxides and bronchodilator S-nitrosothiols in human airways. Proc Natl Acad Sci USA. 1993;90:10957–10961. doi: 10.1073/pnas.90.23.10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–385. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenier K, McLelland GL, Fon EA. Parkin- and PINK1-Dependent Mitophagy in Neurons: Will the Real Pathway Please Stand Up? Front Neurol. 2013;4:100. doi: 10.3389/fneur.2013.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossini E, Molinari C, Caimmi PP, Uberti F, Vacca G. Levosimendan induces NO production through p38 MAPK, ERK and Akt in porcine coronary endothelial cells: role for mitochondrial K(ATP) channel. Br J Pharmacol. 2009;156:250–261. doi: 10.1111/j.1476-5381.2008.00024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JY, Kang MJ, Kim KH, Han PL, Kim HS, Ha JY, Son JH. Nitric oxide induction of Parkin translocation in PTEN-induced putative kinase 1 (PINK1) deficiency: functional role of neuronal nitric oxide synthase during mitophagy. J Biol Chem. 2015;290:10325–10335. doi: 10.1074/jbc.M114.624767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haun F, Nakamura T, Shiu AD, Cho DH, Tsunemi T, Holland EA, La Spada AR, Lipton SA. S-nitrosylation of dynamin-related protein 1 mediates mutant huntingtin-induced mitochondrial fragmentation and neuronal injury in Huntington’s disease. Antioxid Redox Signal. 2013;19:1173–1184. doi: 10.1089/ars.2012.4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol. 2011;18:1042–1052. doi: 10.1016/j.chembiol.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics. 2006;5:749–757. doi: 10.1074/mcp.T500024-MCP200. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the Parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2:120080. doi: 10.1098/rsob.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, et al. Ubiquitin is phosphorylated by PINK1 to activate Parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- Lang AE, Lozano AM. Parkinson’s disease. N Engl J Med. 1998;339:1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei SZ, Pan ZH, Aggarwal SK, Chen HS, Hartman J, Sucher NJ, Lipton SA. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron. 1992;8:1087–1099. doi: 10.1016/0896-6273(92)90130-6. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Lin W, Kang UJ. Characterization of PINK1 processing, stability, and subcellular localization. J Neurochem. 2008;106:464–474. doi: 10.1111/j.1471-4159.2008.05398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry MA, Goldberg JI, Belosevic M. Induction of nitric oxide (NO) synthesis in murine macrophages requires potassium channel activity. Clin Exp Immunol. 1998;111:597–603. doi: 10.1046/j.1365-2249.1998.00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muqit MM, Abou-Sleiman PM, Saurin AT, Harvey K, Gandhi S, Deas E, Eaton S, Payne Smith MD, Venner K, Matilla A, et al. Altered cleavage and localization of PINK1 to aggresomes in the presence of proteasomal stress. J Neurochem. 2006;98:156–169. doi: 10.1111/j.1471-4159.2006.03845.x. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, Munishkina L, Zhang J, Gardner B, Wakabayashi J, et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein α-synuclein. J Biol Chem. 2011;286:20710–20726. doi: 10.1074/jbc.M110.213538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, Lipton SA. Aberrant protein S-nitrosylation in neurodegenerative diseases. Neuron. 2013;78:596–614. doi: 10.1016/j.neuron.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nezich CL, Wang C, Fogel AI, Youle RJ. MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J Cell Biol. 2015;210:435–450. doi: 10.1083/jcb.201501002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicotera P, Brune B, Bagetta G. Nitric oxide: inducer or suppressor of apoptosis? Trends Pharmacol Sci. 1997;18:189–190. [PubMed] [Google Scholar]
- Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M, et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun. 2012;3:1016. doi: 10.1038/ncomms2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa K, Komatsubara AT, Nishimura Y, Sawada T, Kawafune H, Tsumoto H, Tsuji Y, Zhao J, Kyotani Y, Tanaka T. S-Nitrosylation regulates mitochondrial quality control via activation of Parkin. Sci Rep. 2013;3:2022. doi: 10.1038/srep02202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res. 2003;2:43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ. The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald N, et al. The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat Cell Biol. 2007;9:1243–1252. doi: 10.1038/ncb1644. [DOI] [PubMed] [Google Scholar]
- Raju K, Doulias PT, Evans P, Krizman EN, Jackson JG, Horyn O, Daikhin Y, Nissim I, Yudkoff M, Nissim I, et al. Regulation of brain glutamate metabolism by nitric oxide and S-nitrosylation. Sci Signal. 2015;8:ra68. doi: 10.1126/scisignal.aaa4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing α-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J, Soldner F, Sunico CR, Nagar S, Talantova M, et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell. 2013;155:1351–1364. doi: 10.1016/j.cell.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Korolchuk VI, Renna M, Imarisio S, Fleming A, Williams A, Garcia-Arencibia M, Rose C, Luo S, Underwood BR, et al. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell. 2011;43:19–32. doi: 10.1016/j.molcel.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep. 2012;2:1002. doi: 10.1038/srep01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signorello MG, Pascale R, Leoncini G. Transport of L-arginine and nitric oxide formation in human platelets. Eur J Biochem. 2003;270:2005–2012. doi: 10.1046/j.1432-1033.2003.03572.x. [DOI] [PubMed] [Google Scholar]
- Sim CH, Lio DS, Mok SS, Masters CL, Hill AF, Culvenor JG, Cheng HC. C-terminal truncation and Parkinson’s disease-associated mutations down-regulate the protein serine/threonine kinase activity of PTEN-induced kinase-1. Hum Mol Genet. 2006;15:3251–3262. doi: 10.1093/hmg/ddl398. [DOI] [PubMed] [Google Scholar]
- Soldner F, Laganiere J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, Khurana V, Golbe LI, Myers RH, Lindquist S, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146:318–331. doi: 10.1016/j.cell.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Toone EJ, Lipton SA, Sucher NJ. (S)NO signals: translocation, regulation, and a consensus motif. Neuron. 1997;18:691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II, Holmstrom KM, Fergusson MM, Yoo YH, Combs CA, et al. Measuring in vivo mitophagy. Mol Cell. 2015;60:685–696. doi: 10.1016/j.molcel.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, et al. ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics. 2015;129:16–24. doi: 10.1016/j.jprot.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM, Zhang Z, Masliah E, et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci USA. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.