

Abstract
P1, P2-Diimidazolyl derivatives of pyrophosphate and halomethylene-bis-phosphonates have been synthesized and characterized, and the mechanism of their formation was studied. These reagents enable synthesis of dinucleoside tetraphosphates and tetraphosphonates conveniently and in high yields.
Introduction
Nucleoside polyphosphates play fundamental roles in living systems, and much effort has been focused in the development of their synthetic chemistry.1,2 The subclass of bis-nucleoside polyphosphates, initially thought of as mere artifacts or by-products in the synthesis of mononucleoside polyphosphates, has gained increased attention in recent years,3 as they have emerged as important intra- and extracellular signal messengers which take part in the regulation of many biological processes.4,5 For instance, they have been implicated in regulation of blood pressure,6 cellular stress response,7 insulin and glucose levels,8 platelet activation,9 and neurotransmission.10 The class has been subject to drug development efforts,11–14 which resulted in late-stage clinical trials of bis-uridine tetraphosphate (Up4U) and its analogs for treatment of cystic fibrosis15 and dry eye;16 and bis-adenosine tetraphosphate (Ap4A) for control of blood pressure during anesthesia.17
Bis-nucleoside tetraphosphates and their phosphonate analogs (Scheme 1, 3) are most often synthesized by extension of the method for synthesis of nucleoside triphosphates in which activated nucleoside mono-, or thiomonophosphates 1 are reacted with pyrophosphate, or methylene-bis-phosphonates 2. If excess of 2 is used the reaction leads to mono-nucleoside triphosph(on)ates. If a limiting amount of 2 is used, the reaction gives bis-nucleoside tetraphosph(on)ates 3, although in low yield rarely exceeding 30%, together with significant amount of byproducts, mainly mono-, or bis-nucleoside polyphosphates with variable chain length. For instance, reaction of 2 equivalents of adenosine monophosphate (AMP), activated as the phosphoromorpholidate (1, X = 1-morpholinyl, Y = O, B = adenine) with one equivalent of pyrophosphate (2a), gave Ap4A, 23%, together with bis-adenosine diphosphate (Ap2A), 8%, triphosphate (Ap3A), 18%, and pentaphosphate (Ap5A), 4%, along with adenosine 5′-triphosphate (ATP), 7%; adenosine 5′-tetraphosphate, 8%; and minor amounts of AMP and adenosine 5′-diphosphate (ADP).18 Activated nucleoside monophosphates 1 have included morpholidates,19 imidazolidates,20 and diphenylphosphoryl anhydrides.21
Scheme 1.
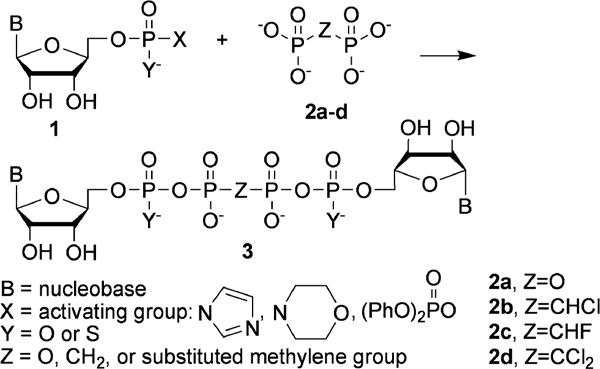
Synthesis of bis-nucleoside tetraphosph(on)ates from activated nucleoside monophosph(on)ates.
The need for activation of the nucleoside mono- or thiomonophsphates is a definite disadvantage of this approach, as it is often a source of side reactions.22
In a modification of this method the activated nucleoside monophosphate 1 is reacted with nucleoside triphosphates or analogs to prepare bis-nucleoside tetraphosphates. For instance, the P2, P3-dichloromethylene analog of Ap4A (3, Y = O, Z = CCl2) was prepared in 46% yield from the morpholidate of AMP (1, X = 1-molpholinyl, Y = O, B = adenine) and the P2, P3-dichloromethylene analog of ATP, AppCCl2p.19 In this method the higher yield is offset by the need to prepare the corresponding nucleoside triphosphates.
Another approach to the synthesis of bis-nucleoside tetraphosphates is based on the reaction of a P1, P3-cyclic nucleoside triphosph(on)ates (“nucleoside trimetaphosphates”, Scheme 2A, 4) with a nucleoside monophosphates or thiomonosphates (5). The nucleoside trimetaphosphates 4 can be prepared by treatment of the corresponding nucleoside triphosphates with carbodiimides14,23 or by phosphitylation of the 5′-hydroxy group of 2′,3′-protected nucleosides with the salicyl phosphoanhydride reagent 6 (Scheme 2B), followed by double displacement of the salicylate with pyrophosph(on)ates (2) and oxidation or sulfurization of the resulting cyclic phosphite to 2′,3′-protected 4.24 These methods require the availability or the synthesis of the corresponding nucleoside triphosph(on)ates or protected nucleosides.
Scheme 2.
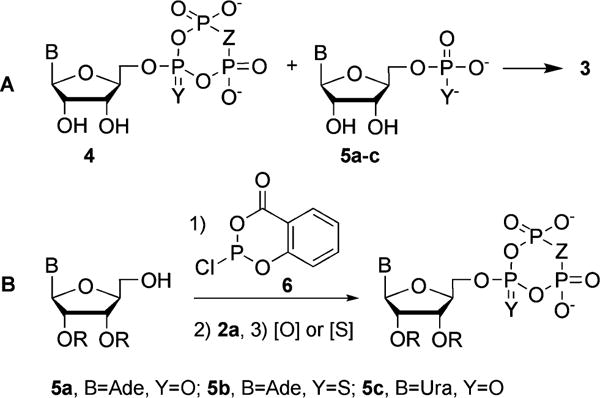
Synthesis of nucleoside bis-tetraphosph(on)ates from nucleoside P1, P3-cyclic triphosph(on)ates.
In yet another approach Tarussova et al.25 reacted ADP with carbonylditriazole or carbonyldibenzimidazole to prepare Ap4A in 16%, or 21% yield, respectively, presumably by formation of P2-imidazolide of ADP, and its condensation with unreacted ADP. The same activating agents, as well as carbonyldi-4(5)-bromoimidazole were used in the same work, and 1,1′-carbonyldiimidazole (CDI) was used in a previous work26 to activate methylene-bis-phosphonic acid, which was then reacted with AMP to afford the P2, P3-methylene analogue of Ap4A in 5 to 40% yield.
In our search for a more efficient and cost-effective way to prepare bis-nucleoside tetraphosph(on)ates 3, we found that organic salts of pyrophosphoric acid and its halomethylene-bis-phosphonate analogs 2 react with excess CDI to give stable, isolable diimidazolides (Scheme 3, 7), and that these diimidazolides react with nucleoside 5′-mono- or thiomonophosphates 5 to give bis-nucleoside tetraphosph(on)ates 3 directly and in high yields (Scheme 3). We present studies on the mechanism of formation of the novel diimidazolides 7, their properties and stability, and their utility in efficient, high yield synthesis of bis-nucleoside tetraphosph(on)ates.
Scheme 3.
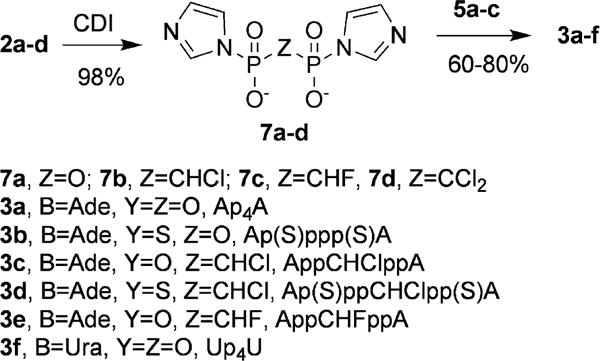
Synthesis of bis-imidazolides of pyrophosphoric and halomethylene-bis-phosphonic acids 7 and their use for synthesis of bis-nucleoside tetraphosph(on)ates 3.
Results and discussion
Synthesis of the diimidazolides of pyrophosphate and halomethylene-bis-phosphonates
If excess CDI is added to a solution of tetrabutylammonium pyrophosphate in DMF release of carbon dioxide is observed, and, at the end of this process, 31P NMR of the reaction mixture reveals that the pyrophosphate signal is replaced by a singlet at −21.26 ppm (Fig. 1). This reaction product was isolated by addition of 2 M NaClO4 in acetone to the reaction mixture followed by further dilution with acetone. This resulted in a fine precipitate which was collected by centrifugation and washed repeatedly with acetone. This product was shown by 1H, 31P, 13C NMR and ESI-MS to be the sodium salt of the P1, P2-diimidazolide of pyrophosphoric acid (7a). It was obtained in nearly quantitative yield and in high purity (>97% by 31P NMR).
Fig. 1.
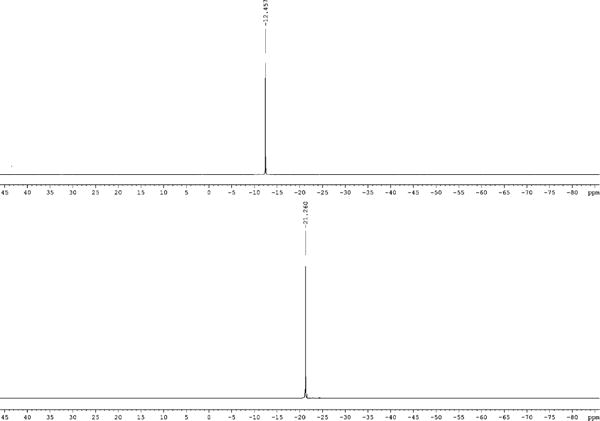
31P NMR of the reaction of tetrabutylammonium pyrophosphate with CDI in DMF. Top, before addition of CDI; bottom, after completion of the reaction.
Using the above procedure, we also prepared the diimidazolides of monochloromethylene-bis-phophonate (7b), monofluoromethylene-bis-phosphonate (7c), and dichloromethylene-bis-phosphonate (7d). As with 7a, these compounds were isolated in almost quantitative yields and high purity as the disodium salts and characterized by their 1H, 31P, 13C, and MS spectra.
It should be noted that the rate of the reaction is strongly dependent on the acidity/basicity of the reaction medium. For instance, it took 24 h for compound 2b as the the mono-pyridinium salt to be converted into compound 7b when reacted with 5 mol mol−1 CDI in DMF, and the final product was only of 94% purity (31P NMR), whereas, the same conversion took only 45 min when additionally 3 mol of triethylamine were present, and the purity of 7b was above 98%. Similarly, mono-tetrabutylammonium pyrophosphate reacted with a large excess of CDI very slowly, giving significant amounts of by-products resulting from cleavage of the pyrophosphate bond, whereas, after addition of 3 equivalents of triethylamine, the reaction with 3 equivalent of CDI was very fast, giving almost exclusively 7a (31P NMR).
Mechanism of formation of the diimidazolides
The high selectivity of the formation of the diimidazolides and the lack of any significant amount of by-products was quite surprising, having in mind that pyrophosphate and its analogs are bi-functional compounds. The formation of the diimidazolides is not instantaneous, and intermediate mono-imidazolides could self-condense, resulting in formation of polyphosphate by-products. To shed light on this unexpected high selectivity we studied the kinetics of this reaction by 31P NMR27 in DMF-d7. When CDI and tetrakis-tributylammonium pyrophosphate were reacted in the molar ratio 1.1: 1, the 31P NMR of the reaction mixture revealed a complex and dynamic mixture of pyrophosphate derivatives (Fig. 2, A). When the reaction was carried out with an excess of CDI (molar ratio 3: 1), significantly simpler spectra were obtained, in which two intermediates gradually converted into the final reaction product, the bis-imidazolide 7a, represented by the singlet at −20.8 ppm (Fig. 2, B)
Fig. 2.
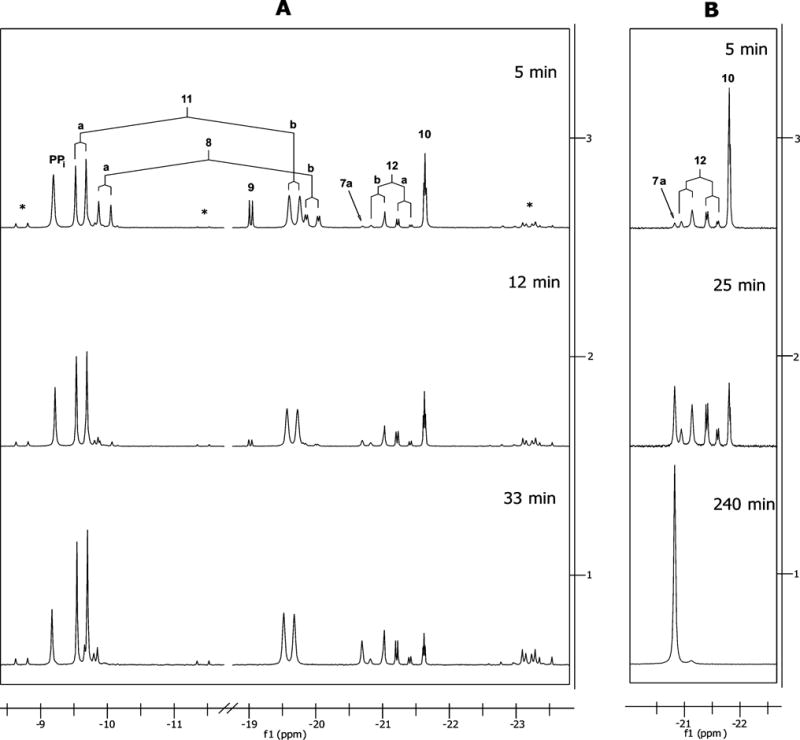
31P NMR spectra (proton-coupled) of the of the time course of reaction of tetrakis-tributylammonium pyrophosphate, 0.16 M, in DMF-d7 with 13C-labeled CDI. Panel A: 1.1 mol CDI per mol pyrophosphate; from top to bottom: 5, 12 and 33 min after CDI addition. Panel B: 3 mol CDI per mol pyrophosphate, from top to bottom: 5, 25 and 240 min after CDI addition. “*” Designates the condensation by-products (tetraphosphates).
To assign the structures of the intermediates we carried out the above reaction using CDI labeled with 13C on the carbonyl group (13C-CDI). We used the 2-bond 13C-31P coupling to assign the 31P resonances of intermediates that contained the –P–O–13C(O)– mixed anhydride groups. Additionally, the resonances arising from phosphorus directly connected to imidazole (P–Im groups) were assigned thanks to the rather small 3-bond 1H–31P coupling through the nitrogen, which resulted in broadening of those signals in the proton-coupled 31P spectra, when compared with the corresponding proton-decoupled spectra. Using this technique, as well as the strong 2-bond POP coupling in the non-symmetric structures and the information from the chemical shifts and the relative intensities, we were able to assign the structures of all intermediates observed in the 31P spectra. Those structures are shown in Scheme 4, and the assignments of their corresponding 31P resonances are given in Fig. 2A, spectrum 3 and listed in Table 1. The relative concentrations of each intermediate at different time points were determined by integration of the 31P spectra, and are plotted in Fig. 3 (panel A for pyrophosphate/CDI ratio 1: 1.1 and panel B for ratio 1: 3).
Scheme 4.
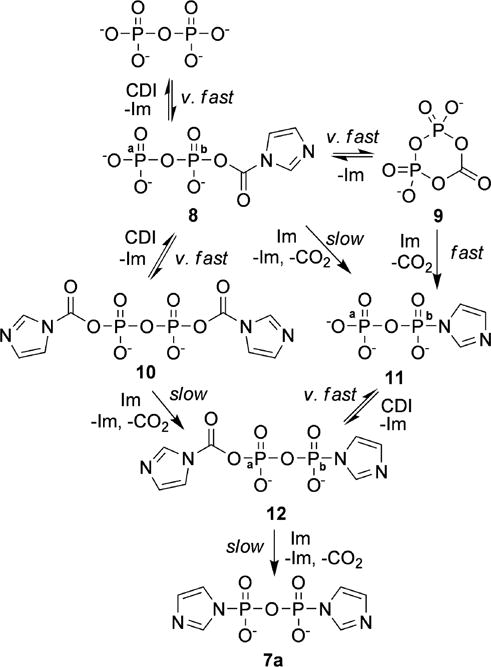
Mechanism of the reaction of CDI with pyrophosphate.
Table 1.
Phosphorus chemical shifts and coupling constants of the intermediates in Scheme 4
| Chemical shiftsa (ppm)
|
Coupling constants (Hz)
|
||||
|---|---|---|---|---|---|
| Cpd. No. | Pa | Pb |
|
JP–C | |
| 8 | −9.93 | −19.91 | 22.4 | 4.2 | |
| 9 | −18.99 | — | — | 5.9 | |
| 10 | −21.62 | — | — | 2.1 | |
| 11 | −9.56 | −19.64 | 19.4 | — | |
| 12 | −21.28 | −20.95 | 23.8 | 4.2 | |
For compound 12 from simulated fitting of an AB system. For all other compounds directly measured from spectra.
Fig. 3.
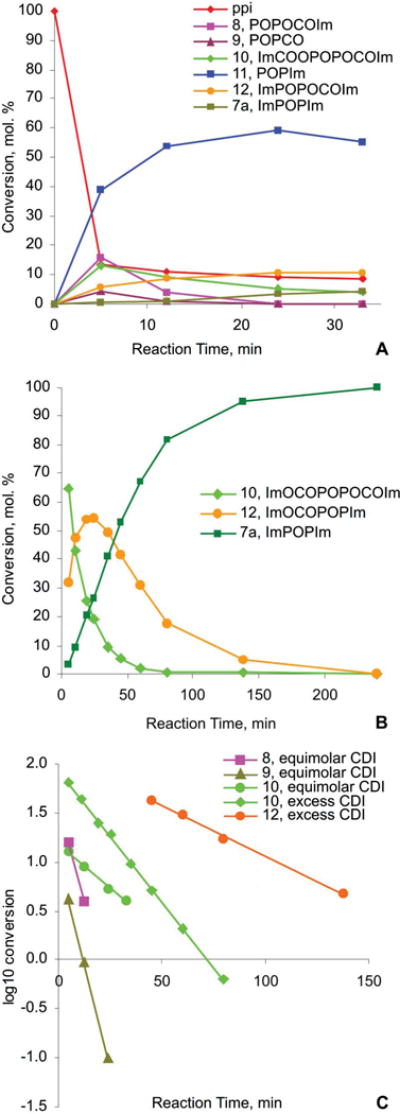
Time course of the reaction of tetrakis-tributylammonium pyrophosphate, 0.16 M, with CDI. A: 1.1 mol CDI per mol pyrophosphate; B: 3 mol CDI per mol pyrophosphate; C: log10(conversion)–time plot.
Based on the structural assignments and the rate of formation and consumption of the intermediates, a mechanism of the reaction of pyrophosphate with CDI, leading to 7a was proposed, and is presented in Scheme 4.
First, CDI reacts quickly with the primary phosphate groups of PPi to form the mixed anhydrides 8 and 10. Because of the high rate of this reaction, only intermediate 10 (together with a small amount of the products of its reaction with imidazole, 12 and 7a) and no intermediate 8 is observed after 5 min in the reaction of PPi with excess CDI (Fig. 2B, spectrum 3). This very fast conversion of the primary phosphate groups to mixed anhydrides explains why no by-products arising from self condensation of PPi are observed when excess of CDI is employed, while they are present and gradually increase with time when equimolar amount of CDI is used (indicated with “*” on Fig. 2A). Second, the mixed anhydrides 8, 10, and 12 react with imidazole to form the imidazolides 11, 12, and 7a, respectively. Here we observed significant difference in the rate of reaction of the mixed anhydride 8, which has a neighboring primary phosphate group, and the mixed anhydrides 10 and 12, in which the neighboring phosphate group is di-substituted. Analysis of the logarithmic plots of the decay portions of the reactions (Fig. 3, plot C) revealed that compound 8 is consumed much faster than compounds 10 and 12. The reactions of imidazolysis of the mixed anhydrides 8, 10, and 12 appear to be of first overall order, which is not surprising if we take into account that imidazole is both consumed and released in these reactions, resulting in zero net imidazole concentration change. In the preceding reactions of formation of the mixed anhydrides the net imidazole concentration increases, but they are much faster to influence the order of the following reactions of imidazolysis. The first order rate constant of imidazolysis of compound 8 in the experiment with 1: 1.1 molar ratios is 0.199 min−1 (t1/2 = 3.47 min), whereas the corresponding rate constant for compound 10 is 0.042 min−1 (t1/2 = 16.5 min). The decay portion of the concentration– time curve of compound 12 was not observed in this experiment, but was clearly observed in the experiment with excess of CDI, where it corresponds to a pseudo-first-order process with a rate constant of 0.023 min−1 (t1/2 = 30.1 min). In the same experiment the corresponding rate constant and half-life of compound 10 were 0.062 min−1, and 11.2 min, respectively. The higher reaction rate for this compound in the experiment with excess of CDI is a result of the higher overall imidazole concentration.
While the higher reactivity of 8 in comparison with 10 and 12 can be attributed to an intramolecular general base catalysis by the doubly charged primary phosphate group, interacting either with the attacking imidazole, or with the leaving group, the observation of the doublet centered at −18.99 ppm in the 31P spectrum (Fig. 2A, spectrum 3), assigned to the cyclic mixed anhydride 9 (3JC–P = 5.9 Hz) in Scheme 4, and the fact that the rate of decay of 9 equals that of compound 8 (k = 0.195 min−1 for compound 9, and 0.199 min−1 for compound 8) make the mechanism proposed in Scheme 3 more likely. According to this mechanism, compound 8 undergoes very fast intramolecular nucleophilic displacement at the carbonyl group to give 9. A nucleophilic attack by imidazole on the carbonyl group of compound 9 results in formation of compound 8, thus establishing very fast equilibrium between the two compounds with a equilibrium constant of 0.25, estimated from the 31P NMR data. Thus, compound 11 can be formed by a nucleophilic displacement by imidazole at the phosphorus atom of compound 9, followed by decarboxylation, or directly from compound 8. Based on the ratio of the pseudo-first-order rate constants of decay of compounds 10, 8 and 9, and the equilibrium constant for interconversion of 8 and 9, we estimated that compound 9 reacts with imidazole 20 times faster than compound 8. To our knowledge this is the first observation of the cyclic anhydride 9, and further studies of its reactivity/stability are under way.
Properties and stability of diimidazolides of diphosph(on)ates
The disodium salts of diimidazolides of pyrophosphoric and halomethylene-bis-phosphonic acids (7a–d) are white, non-hygroscopic powders, which when protected from atmospheric moisture are stable at room temperature for at least 8 months. They gradually (within a couple of days) decompose when exposed to moist air. They have excellent solubility in water, moderate solubility in methanol, low solubility in DMF, and are not soluble in acetone and other less polar organic solvents. As the bis-triethylammonium, -tributylammonium, or -tetrabutylammonium salts they are viscous oils, which are very soluble in anhydrous DMF and DMSO. The resulting solutions are quite stable – storage under Ar at room temperature for one week, and for months in the refrigerator, did not result in any signs of degradation (31P NMR).
In an attempt to prepare the internally protonated, zwitterion form of 7a, we subjected its triethylammonium salt to repeated evaporations from DMF and then from DMSO at high vacuum and room temperature. This resulted only in products of its self-condensation and degradation (31P NMR). On the other hand, the non-volatile bis-tetrabutylammonium salt was completely stable under the same treatment, which is an indication that the internally protonated, zwitterion form is highly reactive and unstable.
The diimidazolides are remarkably stable in water at high pH. For instance, compound 7a remained unchanged in 50 mM sodium carbonate/bicarbonate buffer at pH 9.8 for 48 h at room temperature (31P NMR). At pH 7.8 in 50 mM sodium bicarbonate buffer ca. 50% of it decomposed after 19 h (determined by integration of 31P NMR spectrum), resulting in a mixture of mono-imidazolide 11, 16.4%; PPi, 34.9%; and 7a, 48.7%. This result indicates that the diimidazolides are more stable hydrolytically than the mono-imidazolides, which could be attributed either to an intramolecular catalysis by the primary phosphate group of the mono-imidazolides; or by a catalytic effect of the bicarbonate anion, involving as reactive intermediate compound 9. At pH lower than 7 the decomposition was too fast to be kinetically observed by phosphorus NMR.
Use of the diimidazolides of diphosph(on)ates for synthesis of bis-nucleoside tetraphosph(on)ates
Compounds 7a–c, as the tri- or tetrabutylammonium salts, react with nucleoside mono- or thiomonophosphates 5a–c at room temperature in concentrated DMF solutions to give the corresponding bis-nucleoside tetraphosph(on)ates 3a–f. For instance the diimidazolide 7b reacted with the trioctylammonium salt of AMP for 30 h at room temperature in DMF to give 70% AppCHClppA (3c, isolated yield). It is important to keep the reaction concentration high: below 0.1 M the reaction rates become unacceptably low.
This reaction is catalyzed by 1H-tetrazole, pyridinium hydrochloride, and, most effectively, anhydrous zinc chloride.28 The latter catalyst not only shortened the reaction times from 24–48 h to 0.5–2 h, but also, when used in sufficiently large excess (5–10 mol per mol diimidazolide) rendered the sodium salts of the diimidazolides 7 soluble in anhydrous DMF. This was a significant improvement, because the sodium salts, being non-hygroscopic powders, are much easier to handle than the tributylammonium salts, which are hygroscopic oils or glassy residues. The zinc chloride catalyst also made possible the use of the sodium salts of the nucleoside mono- or thiomonophosphates instead of their tributylammonium salts.28 After completion of the reaction the zinc was sequestered by treatment with CHELEX® ion exchange resin in the sodium form or with EDTA, and the product was isolated by either ion-exchange chromatography on QEA-Sephadex® with a gradient of triethyammonium bicarbonate (TEAB) or reverse-phase chromatography with a gradient of acetonitrile in TEAB. After removal of the volatile buffer the products were converted into the sodium salts by either precipitation with sodium perchlorate/acetone, or by passing through strong cationite (in the sodium form) and lyophilization. Thus a series of bis-nucleoside teraphosph(on)ates 3a–f was prepared in yields in the range of 60–70% for the pyrophosphate analogs and 70–80% for the halomethylene-bis-phosphonate analogs, and were characterized by 1H, 31P NMR and UV spectra and LC–MS in the positive and negative ionization modes.
Experimental
General
Chloromethylene-bis-phosphonic acid, 2b,29 dichloromethylene-bis-phosphonic acid, 2d,30 fluoromethylene-bis-phosphonic acid, 2c,31 and 1,1′-carbonyldiimidazole labeled with 13C on the carbonyl group,32 were prepared by the corresponding literature methods. All transfers and manipulations at moisture-sensitive steps were done under argon, using syringe techniques and oven-dried and argon-filled glassware. The purity of the synthesized bis-nucleoside tetraphosph(on)ates was determined by analytical reverse-phase HPLC (column, XBridge Shield C18; mobile phases – A, 20 mM triethylammonium acetate in water pH 6.8; B, acetonitrile; linear gradient from 0 to 30% B in A for 30 min; UV detection at 260 nm) and was above 95%.
Solutions of tributylammonium pyrophosphate and tetrabutylammonium pyrophosphate in DMF
Tetrasodium pyrophosphate decahydrate (10.00 g, 22.4 mmol) was dissolved in 100 ml water. This solution was passed through a column (20 × 6.3 cm) of Dowex® 50X4-100 in the H+ form, and eluted with ice-cold water until the eluate pH increased to 5. The eluate was collected into a cooled (ice bath) and stirred solution of tri-n-butylamine (8.31 g, 10.7 ml, 44.8 mmol) in 200 ml of 2-propanol. The resulting solution was concentrated under vacuum (bath temperature 35 °C), and then re-evaporated under high vacuum (less then 1 torr) twice from 150 ml DMF. The resulting oil was transferred quantitatively with 20 ml of dry (SureSeal®) DMF into a pre-tared flask, evaporated under high vacuum, dissolved in 20 ml dry DMF, and partially evaporated to give 27.93 g tributylammonium pyrophosphate solution in DMF with concentration of 0.80 mmol g−1 solution.
To prepare tetrabutylammonium pyrophosphate a 55% w/w tetrabutylammonium hydroxide solution in water (21.8 ml, 21.15 g, 44.8 mmol) was added with stirring and cooling to the ice-cooled eluate of pyrophosphoric acid from the ion-exchange column, and the resulting solution was evaporated and rendered anhydrous as described for the tributylammonium salt to obtain 46.5 g of DMF solution at a concentration 0.482 mmol g−1. Both the tributylammonium and the tetrabutylammonium salt solutions were sealed under argon and stored at −5 to −10 °C. After 1 year, 31P NMR of the tributylammonium salt solution showed less than 1 mol% disproportionation to ortho- and tripolyphosphate.
Monitoring the reaction of tributylammonium pyrophosphate with 1,1′-13C-carbonyldiimidazole by 31P-NMR
Tributylammonium pyrophosphate (0.175 g of 0.80 mmol g−1 solution, 0.144 mmol, measured by means of a pre-tared syringe) was mixed with 0.6 ml DMF-d7, and evaporated on a speedvac to 0.2 ml. After breaking the vacuum with argon the residue was mixed with a solution of 13C-CDI (25 mg, 0.154 mmol) and tributylamine (71 μl, 52 mg, 0.28 mmol) in 0.6 ml DMF-d7. The mixture was transferred into a 5 mm NMR tube, and 31P spectra (32 scans each) were recorded at 5, 12, 24, and 33 min at 25 °C. To improve the integration accuracy, long (5 s) acquisition times without proton decoupling were used.
In a separate experiment 75 mg (0.46 mmol) of 13C-CDI was used, and the spectra were recorded at 5, 11, 19, 25, 35, 45, 60, 80, 138, and 240 min.
P1, P2-Di(1-imidazolyl)pyrophosphate, 7a
N, N′-Carbonyldiimidazole (2.43 g, 15.0 mmol) was suspended in dry DMF (5 ml) under argon. The flask was sealed with a septum, connected with a mineral oil bubbler, and tributylammonium pyrophosphate (6.25 g of 0.80 mmol g−1 solution in DMF, 5.0 mmol) was added by means of pre-tared syringe with stirring, followed by tributylamine (0.927 g, 1.19 ml, 5.0 mmol). The mixture was stirred overnight at r.t., during which time carbon dioxide evolution ceased. Water (200 μl) was added and the mixture was stirred for 5 min, and then concentrated under 0.5 mm Hg vacuum at 32 °C to an oil. This oil was taken up in 50 ml dry DMF and concentrated partially as above to give 15.4 g of DMF solution of 7a as the tributylammonium salt. This solution contains 0.324 mmol g−1 7a and 1.30 mmol g−1 imidazole, and can be used for synthesis of bis-nucleoside tetraphosph(on)ates, as in the example for synthesis of 3a below without farther purification or imidazole removal.
Pure disodium salt of 7a was isolated as follows: The DMF solution from above was evaporated under vacuum (0.5 mm Hg, 32 °C) to oil. This oil was added with vigorous stirring to 250 ml of 2 M solution of sodium perchlorate in acetone, followed by 250 ml acetone. The mixture was stirred vigorously for 30 min, during which time the initially formed sticky residue converted into a white fine powder. This powder was separated by centrifugation and washed twice by resuspension in 100 ml acetone, centrifugation and decanting. After initial drying under a stream of nitrogen the product was dried under high vacuum for 16 h. Yield, 1.58 g, 98%. 1H NMR (300 MHz, D2O) δ 7.64 (s, 1H), 7.06 (s, 1H), 6.94 (s, 1H); 31P NMR (D2O) −20.87 (s); 13C NMR (D2O) 140.28, 129.36 (t, 3JPC = 5.1 Hz), 120.6; MS (ESI in the negative ionization mode): calcd. for C6H7N4O5P2− (M – H) 276.99 (100%), 277.99 (8.2%), 278.99 (1.1%); found 276.97 (100%), 277.97 (7.4%), 278.96 (1.0%); positive ionization mode: calcd. for C6H8N4NaO5P2+ (M + Na) 300.99 (100%), 301.99 (6.8%), 301.98 (1.5%); found 300.86 (100%), 301.89 (7.9%), 302.88 (1.3%).
Disodium chloromethylene-bis(1-imidazolyl)phosphonate, 7b
Monochloromethylene-bis-phosphonic acid mono-pyridinium salt (500 mg, 1.173 mmol), CDI (840 mg, 5.18 mmol), and dry DMF were mixed under argon in a dry vial equipped with a stirring bar. The vial was sealed, equipped with a bubbler, and triethylamine (725 μl, 524 mg, 5.18 mmol) was added via a syringe with stirring. After a few min the mixture became homogenous. After 4 h sodium perchlorate (5 ml of 2 M solution in acetone) was added with vigorous stirring, followed by acetone (15 ml). The mixture was stirred vigorously for 2 h, and then centrifuged. The supernatant was discarded, and the solid was suspended in anhydrous acetone (30 ml). The suspension was centrifuged, the supernatant discarded, and the acetone washing was repeated two more times. After the final wash the solid was dried under a stream of dry nitrogen and then under high vacuum at r.t. for 24 h to give 610 mg (99.6%) of white powder. 1H NMR (300 MHz, DMSO-d6) δ 7.66 (2H, bs, H-2), 7.15 (2H, bs, H-4), 6.80 (2H, bs, H-5), 3.69 (1H, t, CH–Cl, 2JP–H = 15.48 Hz); 31P NMR (121 MHz, DMSO-d6), ppm: 0.63 (1H decoupled, s; 1H coupled, d, 2JP–H = 14.92 Hz); MS (ESI in the negative ionization mode): Calcd. for C7H8ClN4O4P2− (M – H), 309.0 (100%), 310.0 (9.0%), 311.0 (32%); Observed, 309.1 (100%), 310.0 (6.5%), 311.1 (35%)
Tetrasodium P1, P4-bis(adenosine-5′)-P2, P3-(chloromethylene)-tetraphosphate, 3c (uncatalyzed reaction)
Adenosine 5′-monophosphate, free acid, monohydrate (1.09 g, 2.98 mmol), tri-octylamine (1.055 g, 2.98 mmol, 1.31 ml), and methanol (50 ml) were stirred until a clear solution was obtained. The solution was evaporated (30 °C, 10–15 mm Hg vacuum). The residue was dissolved in 100 ml dry DMF, and the solution was evaporated (30 °C, 0.5–1 mm Hg vacuum). The residue was re-dissolved in another 100 ml of dry DMF, and the evaporation was repeated under the same conditions.
Chloromethylene-bis-phosphonic acid, mono-pyridinium salt (216 mg, 0.746 mmol) and tributylamine (277 mg, 1.49 mmol, 0.355 ml) were stirred with 15 ml methanol until a clear solution was obtained (ca. 15 min.). This solution was evaporated (30 °C, 10–15 mm Hg vacuum) to a glassy residue. This residue was dissolved in 50 ml dry DMF, and evaporated (30 °C, 0.5–1 mm Hg vacuum) to an oily residue. The residue was re-dissolved in another 50 ml of dry DMF, and the evaporation was repeated under the same conditions. The resulting chloromethylene-bis-phosphonate, bis-tributylammonium salt, was dissolved in 5 ml dry DMF under argon, and CDI (0.605 g, 3.73 mmol) was added in one portion with stirring. The flask was sealed and connected to a bubbler filled with paraffin oil. The rate and the advance of the reaction was monitor by the rate of release of CO2. (After 12 h a small portion of the reaction mixture was diluted with DMF-d7 and checked by 31P NMR. No starting material was present, and only one phosphorus signal, corresponding to the diimidazolide was observed).
To decompose excess CDI, water (100 μl) was added. After 5 min the reaction mixture was concentrated to 2/3 of its volume (30 °C, 0.5–1 mm) and mixed with the solution of AMP, trioctylammonium salt, prepared above. The mixture was concentrated again (30 °C, 0.5–1 mm Hg vacuum) to a light oil. The vacuum was broken with argon, and the reaction mixture was stirred under argon for 36 h at r.t. Water (50 ml) was added, and the mixture was extracted twice with 150 ml diethyl ether containing 1% triethylamine. The aqueous layer was evaporated under vacuum to half of its volume, and then was loaded on a Toyopearl DEAE-650M column (5 × 45 cm) which was pre-equilibrated with 0.2 M triethylammonium bicarbonate (TEAB) buffer, pH 8, containing 10% v/v acetonitrile. The elution was carried out with a gradient from the equilibration buffer to 1 M TEAB/acetonitrile, 9: 1 v/v, for 300 min at a flow rate of 20 ml min−1. The fractions containing the product were pooled and evaporated (35 °C, 10–15 mm Hg vacuum) with periodic addition of 1-butanol to prevent foaming. The residue was evaporated 3 times from methanol (100 ml each) under vacuum and then dried for 4 h at 0.2 – 1 mm Hg vacuum in order to remove any residual TEAB, and then dissolved in methanol (5 ml). A solution of sodium perchlorate (1 g) in methanol (5 ml) was added dropwise with vigorous stirring. Acetone (50 ml) was added with stirring, and after 30 min the mixture was centrifuged, and the clear supernatant was decanted. The white solid was re-suspended in 10 ml acetone, and the suspension was filtered through a 0.45 μm glass fiber filter. The solid was washed with three portions of acetone (5 ml each), and with 5 ml diethyl ether, and dried under a stream of nitrogen, and then under high vacuum to give 502 mg (70%) of 3c as a fine white powder. 1H NMR (300 MHz, D2O) δ 8.32, 8.30 (2H, s, H-8), 8.01 (2H, bs, H-2), 5.86 (2H, d, H-1′, 3J1′–2′ = 7.48 Hz), 4.46 (2H, m, H-2′), 4.37 (2H, m, H-3′), 4.30 (2H, m, H-4′), 4.25 (1H, t, CHCl, 2JP–H = 17.1 Hz), 4.09–4.24 (4H, m, H-5′,5″); 31P NMR (121 MHz, D2O), ppm: 5.03 (m, P2 + P3, Hz), −8.02 (m, P1 + P4, Hz, Hz); MS (ESI in the negative ionization mode): Calcd. for C21H28ClN10O18P4− (M – H), 867.0 (100%), 868.0 (27.4%), 869.0 (39.3%), 870.0 (10.1%), 871.0 (2.6%); Observed, 867.1 (100%), 868.1 (27.6%), 869.0 (35.2%), 870.0 (9.4%), 871.0 (2.3%).
Tetrasodium P1, P4-bis(adenosine-5′)-P2, P3-(fluoromethylene)tetraphosphate, 3e (uncatalyzed reaction)
Trimethylbromosilane, 0.887 g, 0.767 ml, 5.8 mmol, was added dropwise under nitrogen and with stirring, to an ice cooled solution of tetraethyl (fluoromethylene)-bis-phosphonate28 (306 mg, 1 mmol) in 1.5 ml dichloromethane. After 24 h at r.t. the mixture was evaporated under vacuum (45 °C, 10–15 mm Hg), re-evaporated 3 times from 30 ml methanol, and once from 10 ml dry DMF (33 °C, 0.5–1 mm Hg vacuum). The resulting oil was dissolved in a mixture of 10 ml dry DMF and 0.524 ml (2.2 mmol) tributylamine and evaporated again as above to give the bis-tributylammonium salt of 2c. Following the procedure for compound 3c, this salt was converted into the diimidazolide 7c by reaction with CDI (0.811 g, 5 mmol) in 6.7 ml dry DMF. After decomposition of the excess of CDI with 130 μl water, 7c was condensed with 4 mmol AMP, trioctylammonium salt, to give, after work up 941 mg (68%) of tetrasodium P1, P4-bis-(5′-adenosine)-P2, P3-(monofluoromethylene)tetraphosphate, 3e. 1H NMR (300 MHz, D2O) δ 8.27 (2H, s, H-8), 8.02 (2H, s, H-2), 5.92 (2H, d, H-1′), 5.07 (1H, dt, CHF, 2JF–H = 45.6 Hz 2JP–H = 14.1 Hz), 4.63 (2H, m, H-2′), 4.44 (2H, m, H-3′), 4.25 (2H, m, H-4′), 4.11 (4H, m, H-5′, 5″); 31P NMR (121 MHz, D2O), ppm: 0.59–1.83 (m, P2 + P3, 2JP–F = 75.0 Hz), −9.61 – −10.30 (m, P1 + P4); MS (ESI in the negative ionization mode): Calcd. for C21H28FN10O18P4− (M – H), 851.1 (100%), 852.1 (27.4%), 853.1 (7.3%), 854.1 (1.3%); Observed, 851.1 (100%), 852.1 (21.5%), 853.0 (4.5%), 854.2 (0.2%).
Tetrasodium P1, P4-bis(adenosine-5′)-tetraphosphate, 3a (tetrazole catalysis)
Adenosine 5′-monophosphate, mono-tetrabutylammonium salt (3.81 g of DMF solution with concentration of 0.131 mmol g−1,33 0.500 mmol) and DMF solution of the tributylammonium salt of 7a, containing 0.324 mmol 7a and 1.30 mmol imidazole per gram (386 mg solution, 0.125 mmol 7a) were mixed and concentrated under vacuum (33 °C, 0.5–1 mm Hg) to a volume of ca. 2 ml. A solution of 1H-tetrazole in acetonitrile (0.45 M, 1 ml, 0.45 mmol) was added with stirring under argon, which resulted in formation of a bulky white precipitate. This mixture was concentrated under vacuum (r.t., 0.5–1 mm Hg) to a final volume of ca. 1 ml. During this evaporation the precipitate dissolved, resulting in a clear oil. This oil was stirred for 16 h at r.t. under argon, and then diluted with 100 ml water. The solution was loaded on a 5 × 45 cm Toyopearl DEAE-650M column. The column elution and the product isolation and conversion to the tetra-sodium salt were carried out as described above for compound 3c to give 85.0 mg (74%) of 3a as a fine white powder. 1H NMR (300 MHz, D2O) δ 8.32 (2H, s, H-8), 8.09 (2H, s, H-2), 5.97 (2H, d, H-1′, 3J1′–2′ = 5.97 Hz), 4.70–4.75 (1H, m, H-2′), 4.47–4.51 (2H, m, H-3′), 4.26–4.32 (2H, m, H-4′), 4.17–4.22 (4H, m, H-5′,5″); 31P NMR (121 MHz, D2O), ppm: −11.28 (m, P1 + P4, Hz, Hz, Hz, Hz34), −23.11 (m, P2 + P3); MS (ESI in the negative ionization mode): Calcd. for C20H27N10O19P4− (M – H), 835.0 (100.0%), 836.0 (26.4%), 837.0 (7.2%), 838.0 (1.1%); Observed, 835.1 (100%), 836.1 (24.0%), 837.1 (6.7%), 838.1 (0.9%).
Tetrasodium P1, P4-bis(uridine-5′)-tetraphosphate, 3f (zinc chloride catalysis)
Uridine 5′-monophosphate, disodium salt, hydrate (26% w/w water content, 274 mg, 0.576 mmol) was disolved in 3 ml of water. This solution was cooled in ice and passed through a pre-cooled column (12 × 2 cm) of Dowex R 50X4-100 in the H+ form, and eluted with ice-cold water until pH of the eluate was above 5. The eluate was collected into a stirred mixture of triethylamine (0.25 ml), isopropanol (10 ml) and water (10 ml). The resulting solution was evaporated under vacuum (35 °C, 10–15 mm Hg), and the glassy residue was dissolved in anhydrous DMF (20 ml) and evaporated under vacuum (35 °C, 0.5–1 mm Hg). This triethylammonium salt was dissolved in 1.5 ml of dry DMF under argon. Disodium P1, P2-di(1-imidazolyl)pyrophosphate, 7a (67.1 mg, 0.208 mmol) followed by anhydrous zinc chloride (138 mg, 1.015 mmol) were added with rigorous stirring under argon. After 15 min a clear solution was obtained. After 3 h a cooled suspension of Chelex® resin in the sodium form (5.5 ml resin in 11 ml water) was added. The mixture was shaken for 15–20 min until a clear supernatant was obtained. The resin was filtered and washed with water until the washings, when spotted on a TLC plate with fluorescent indicator, did not show a dark spot under UV light. The combined filtrate and washings were loaded on a column of DEAE Sephadex® (25 × 2.5 cm), which was equilibrated with 20 mM TEAB buffer, pH 8, containing 10% v/v acetonitrile. The elution was carried out with a linear gradient from the equilibration buffer to 1.5 M TEAB, pH 8, containing 10% acetonitrile for 800 min at 3 ml min−1 flow rate. The fractions containing the product were pooled, evaporated, and converted into the sodium salt as described for compound 3c to give 112 mg (61%) of 3f as a fine white powder. 1H NMR (300 MHz, D2O) δ 7.75 (2H, d, H-6, 3JH6–H5 = 7.91 Hz), 5.90 (2H, d, H-1′, 3J1′–2′ = 5.28 Hz), 5.80 (2H, d, H-5, 3JH5–H6 = 7.72 Hz), 4.21–4.31 (4H, m, H-2′+H-3′), 4.09–4.17 (6H, m, H-4′+H5′,5″); 31P NMR (121 MHz, D2O), ppm: −10.90 (m, P1 + P4, Hz, Hz, Hz), −22.34 (m, P2+P3, 2JP2–P3 = 15.8 Hz34); MS (ESI in the negative ionization mode): Calcd. for C18H25N4O23P− 4 (M – H), 789.0 (100%), 790.0 (22.1%), 791.0 (7.1%), 792.0 (1.2%); Observed, 789.1, (100%), 790.0 (20.4%), 791.1 (6.5%), 792.1 (0.9%).
Tetrasodium P1, P4-bis(adenosine-5′)-P1, P4-dithiotetraphosphate, 3b (zinc chloride catalysis)
Adenosine 5′-thiomonophophate bis-triethylammonium salt35 (80 mg, 0.141 mmol) was evaporated twice from 1 ml dry DMF under vacuum (30 °C, 0.5–1 mm Hg). Disodium P1, P2-di-(1-imidazolyl)pyrophosphate, 7a (11.0 mg, 0.035 mmol) and 0.4 ml dry DMF were added under argon. After 1 min sonification the mixture was evaporated as above. The vacuum was broken with argon, and the flask was sealed with a septum under argon. Anhydrous zinc chloride (96 mg, 0.707 mmol) was dissolved in 1 ml dry DMF. The solution was evaporated under vacuum (30 °C, 0.5–1 mm Hg), and re-dissolved under argon in 0.3 ml dry DMF. This solution was added by a syringe to the sealed flask with stirring at r.t. A clear solution quickly formed. After 2.5 h the reaction mixture was worked up with Chelex® resin, followed by chromatography on DEAE Sephadex® as described for compound 3f. After conversion to the sodium salt as described for compound 3c, using 1 ml of 2 M sodium perchlorate in acetone and 4 ml acetone, 3 washings with 5 ml acetone each, and drying under high vacuum, 22.5 mg of 3b (67.5%) were obtained as a fine white powder. Because of the stereogenic nature of the phosphorothioate modifications at P1 and P4 this compound is a mixture of 3 diastereomers with configurations RP, RP, SP, SP and RP, SP ≡ SP, RP (because of the molecular symmetry).36,37 These diastereomers were easily separable by analytical reverse-phase HPLC (see general methods) used to determine the product purity. The corresponding H-8 and H-2 resonances in the 1H NMR spectra are resolved, while the phosphorus resonances in the 31P NMR spectrum are only partially resolved. 1H NMR (300 MHz, D2O) δ 8.460, 8.447, 8.351 (2H, s, three diastereomers H-8 in ratios 0.275: 0.175: 0.555); 8.039, 8.033, 8.029 (2H, three diastereomers H-2); 5.946 (2H, d, H-1′, 3J1′–2′ = 5.39 Hz); 4.735–4.627 (2H, m, H-2′); 4.534–4.475 (2H, m, H-3′); 4.305–4.205 (6H, m, H-4′+H5′, 5″); 31P NMR (121 MHz, D2O), ppm: 41.487–40.852 (overlapping AA′–XX′ and AB–XY multiplets, P1 + P4); −26.016 – −26.451 (overlapping AA′–XX′ and AB–XY multiplets, P2 + P3); MS (ESI in the negative ionization mode): Calcd. for C20H27N10O17P4S2− (M – H), 867.0 (100.0%), 868.0 (27.9%), 869.0 (16.3%), 870.0 (3.7%), 871.0 (1.1%); Observed, 867.2 (100%), 868.1 (25.9%), 869.1 (13.6%), 870.0 (2.8%), 871.0 (0.7%).
Tetrasodium P1, P4-bis(adenosine-5′)-P2, P3-(chloromethylene)-P1, P4-dithiotetraphosphate, 3d (zinc chloride catalysis)
Compound 3d was prepared using the procedure for compound 3b but with 7b instead of 7a. The product was isolated in 72% yield. This analog also exists as a diastereomeric mixture (as for 3b). The stereogenic chloromethyl group causes four diastereomers to exist, however.21 In fact, four peaks were observed under analytical reverse-phase HPLC (see general methods), and 7 peaks were observed for H-8 in the 1H NMR spectrum. 1H NMR (300 MHz, D2O) δ 8.46–8.285 (2H, multiple singlets, H-8), 8.04–793 (2H, multiple singlets, H-2), 5.995–5.915 (2H, overlapping d, H-1′), 4.89–4.42 (1H, multiple overlapping triplets, CHCl),4.72–4.59 (2H, m, H-2′), 4.53–4.46 (2H, m, H-3′), 4.365–4.28 (2H, m, H-4′), 4.28–4.115 (4H, m, H-5′, 5″); 31P NMR (121 MHz, D2O), ppm: 44.61–43.71 (overlapping AA′, XX′ multiplets, P1 + P4), 3.45–2.90 (overlapping AA′, XX′ and AB, XY multiplets, P2 + P3); MS (ESI in the negative ionization mode): Calcd. for C21H28ClN10O16P4S2− (M – H), 899.0 (100.0%), 900.0 (28.9%), 901.0 (48.3%), 902.0 (13.1%), 903.0 (6.3%), 904.0 (1.4%); Observed, 899.0 (100%), 900.0 (28.7%), 900.9 (48.9%), 901.9 (11.6%), 903.0 (4.8%), 904.1 (0.5%).
Conclusions
Carbonyldiimidazole reacts with pyrophosphate and halomethylene-bis-phosphonates in DMF to form the corresponding P1, P2-bis-imidazolides. The mechanism of this reaction involves fast formation of mixed anhydrides with imidazolyl-1-carbonic acid, which react slowly with imidazole to give the imidazolides. It appears that the cyclic anhydride between pyrophosphoric and carbonic acid is involved as an intermediate as well. The resulting bis-imidazolides when protected from moisture are stable, and can be isolated in excellent yields as the disodium salts which can be stored at room temperature. They hydrolyze very slowly at high pH, and are highly reactive and quickly decompose at low pH. These compounds, both as the pure sodium salts and as the salts formed in situ with organic amines or tertiary ammonium salts, are excellent reagents for the synthesis of bis-nucleoside tetraphosphates, and their P1, P4-dithio-, and P2, P3-halomethylene phosphonate analogs.
Supplementary Material
Acknowledgments
The authors are grateful to Dr Wei-Chu Xu for the synthesis of tetraethyl (fluoromethylene)-bis-phosphonate, a precursor for the synthesis of 2c. This work was supported in part by SBIR grants HL081992 and HL088828 (to IBY) from the National Heart, Lung and Blood Institute.
Footnotes
Electronic supplementary information (ESI) available: Spectroscopic and chromatographic data for the synthesized compounds. See DOI: 10.1039/c0ob00542h
References and notes
- 1.Vaghefi M, editor. Nucleoside triphosphates and their analogs: chemistry, biotechnology, and biological applications. Taylor & Francis; Boca Raton: 2005. [Google Scholar]
- 2.Burgess K, Cook D. Chem Rev. 2000;100(6):2047–2060. doi: 10.1021/cr990045m. [DOI] [PubMed] [Google Scholar]
- 3.McLennan AG, editor. Ap4A and Other Dinucleoside Polyphosphates. CRC Press; Boca Raton: 1992. [Google Scholar]
- 4.Hoyle CHV, Hilderman RH, Pintor JJ, Schlüter H, King BF. Drug Dev Res. 2001;52:260–273. [Google Scholar]
- 5.McLennan AG. Pharmacol Ther. 2000;87(2–3):73–89. doi: 10.1016/s0163-7258(00)00041-3. [DOI] [PubMed] [Google Scholar]
- 6.Schlüter H, Offers E, Brüggemann G, von der Giet M, Tepel M, Nordhoff E, Karas M, Spieker C, Witzel H, Zidek W. Nature. 1994;367:186–188. doi: 10.1038/367186a0. [DOI] [PubMed] [Google Scholar]
- 7.Palfi Z, Suranyi G, Borbely G. Biochem J. 1991;276:487–491. doi: 10.1042/bj2760487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eugen J, Verspohl G, Blackburn MHN, Hagemann J, Lempka M. J Med Chem. 2003;46:1554–1562. doi: 10.1021/jm011070z. [DOI] [PubMed] [Google Scholar]
- 9.Chan SW, Gallo SJ, Kim BK, Guo MJ, Blackburn GM, Zamecnik PC. Proc Natl Acad Sci U S A. 1997;94:4034–4039. doi: 10.1073/pnas.94.8.4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pintor J, Diaz-Hernandez M, Gualix J, Gomez-Villafuertes R, Hernando F, Miras-Portugal MT. Pharmacol Ther. 2000;87(2–3):103–115. doi: 10.1016/s0163-7258(00)00049-8. [DOI] [PubMed] [Google Scholar]
- 11.Walkowiak B, Baraniak J, Cierniewski CS, Stec W. Bioorg Med Chem Lett. 2002;12(15):1959–1962. doi: 10.1016/s0960-894x(02)00318-9. [DOI] [PubMed] [Google Scholar]
- 12.Pendergast W, Yerxa BR, Douglass JG, 3rd, Shaver SR, Dougherty RW, Redick CC, Sims IF, Rideout JL. Bioorg Med Chem Lett. 2001;11(2):157–160. doi: 10.1016/s0960-894x(00)00612-0. [DOI] [PubMed] [Google Scholar]
- 13.Shirokova EA, Khandazhinskaya AL, Skoblov YS, Goryunova LY, Beabealashvilli RS, Krayevsky AA. Nucleosides, Nucleotides Nucleic Acids. 2001;20(4):1033–1036. doi: 10.1081/NCN-100002485. [DOI] [PubMed] [Google Scholar]
- 14.Douglass JG, Patel RI, Yerxa BR, Shaver SR, Watson PS, Bednarski K, Plourde R, Redick CC, Brubaker K, Jones AC, Boyer JL. J Med Chem. 2008;51(4):1007–1025. doi: 10.1021/jm701348d. [DOI] [PubMed] [Google Scholar]
- 15.Kellerman D, Mospan AR, Engels J, Schaberg A, Gorden J, Smiley L. Pulm Pharmacol Ther. 2008;21(4):600–607. doi: 10.1016/j.pupt.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 16.Tauber J, Davitt WF, Bokosky MD, Nichols KK, Yerxa BR, Schaberg AE, LaVange LM, Mills-Wilson MC, Kellerman DJ. Cornea. 2004;23(8):784–792. doi: 10.1097/01.ico.0000133993.14768.a9. [DOI] [PubMed] [Google Scholar]
- 17.Kikuta Y, Ohiwa E, Okada K, Watanabe A, Haruki S. Acta Anaesthesiol Scand. 1999;43(1):82–86. doi: 10.1034/j.1399-6576.1999.430117.x. [DOI] [PubMed] [Google Scholar]
- 18.Verheyden DLM, Wehrl WE, Moffatt JG. J Org Chem. 1965;30:3381–3385. doi: 10.1021/jo01021a029. [DOI] [PubMed] [Google Scholar]
- 19.Blackburn GM, Guo MJ, McLennan AG. In: Ap4A and Other Dinucleoside Polyphosphates. McLennan AG, editor. CRC Press; Boca Raton: 1992. pp. 305–342. [Google Scholar]
- 20.Tarussova NB, Osipova TI, Biriukov AI, Pokrovskaya MJ, Meshkov CV, Gnuchev NV. Nucl Acids Res Symp Ser. 1984;14:287–288. [Google Scholar]
- 21.Blackburn GM, Guo MJ. Tetrahedron Lett. 1990;31:4371–4374. [Google Scholar]
- 22.Maeda M, Patel AD, Hampton A. Nucleic Acids Res. 1977;4(8):2843–2853. doi: 10.1093/nar/4.8.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ko HJ, Carter RL, Cosyn L, Petrelli R, de Castro S, Besada P, Zhou YX, Cappellacci L, Franchetti P, Grifantini M, Van Calenbergh S, Harden TK, Jacobson KA. Bioorg Med Chem. 2008;16(12):6319–6332. doi: 10.1016/j.bmc.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han Q, Gaffney BL, Jones RA. Org Lett. 2006;8:2075–2077. doi: 10.1021/ol060491d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tarussova NB, Osipova TI, Purygin PP, Yakimova IA. Bioorg Khim. 1986;12:404–407. [Google Scholar]
- 26.Tarussova NB, Shumiyanzeva VV, Krylov AC, Karpeisky MY, Khomutov RM. Bioorg Khim. 1983;9:838–843. [Google Scholar]
- 27.Caesar JC, Griffiths DV, Tebby JC. J Chem Soc, Perkin Trans. 1988;1:175–178. [Google Scholar]
- 28.Kadokura M, Wada T, Urashima C, Sekine M. Tetrahedron Lett. 1997;38(48):8359–8362. [Google Scholar]
- 29.McKenna CE, Khawli LA, Ahmad WY, Pham P, Bongartz JP. Phosphorus Sulfur Relat Elem. 1988;37:1. [Google Scholar]
- 30.Vepsäläinen J, Nupponen H, Pohjala E, Ahlgren M, Vainiotalo P. J Chem Soc, Perkin Trans. 1992;2:835–842. [Google Scholar]
- 31.Xu Y, Qian L, Prestwich GD. Org Lett. 2003;5:2267–2270. doi: 10.1021/ol034597+. [DOI] [PubMed] [Google Scholar]
- 32.Nelson VC. J Labelled Compd Radiopharm. 1996;38:713–723. [Google Scholar]
- 33.This solution was prepared by neutralization of DMF solution of adenosine 5′-monophosphate, free acid, monohydrate with one equivalent of 55% (w/w) tetrabutylammonium hydroxide solution in water, and repeated evaporation under vacuum of the resulting solution from anhydrous DMF.
- 34.The coupling constants of this symmetrical spin system could not be measured directly from the NMR spectrum, and were determined by fitting of an AA′XX′ system to the experimental data (see ESI†) using the WinDNMR program (Hans J. Reich, Department of Chemistry, University of Wisconsin).
- 35.Eckstein F, Goumet M. Nucleic Acid Chem. 1978;2:861–864. [Google Scholar]
- 36.Dixon R, Lowe G. J Biol Chem. 1989;264:2069–2074. [PubMed] [Google Scholar]
- 37.Blackburn GM, Taylor GE, Thatcher GRJ, Prescott M, McLennan A. Nucleic Acids Res. 1987;15:6991–7004. doi: 10.1093/nar/15.17.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.