

Abstract
Objective:
To determine whether baseline clinical and MRI features predict rate of clinical decline in patients with progressive apraxia of speech (AOS).
Methods:
Thirty-four patients with progressive AOS, with AOS either in isolation or in the presence of agrammatic aphasia, were followed up longitudinally for up to 4 visits, with clinical testing and MRI at each visit. Linear mixed-effects regression models including all visits (n = 94) were used to assess baseline clinical and MRI variables that predict rate of worsening of aphasia, motor speech, parkinsonism, and behavior. Clinical predictors included baseline severity and AOS type. MRI predictors included baseline frontal, premotor, motor, and striatal gray matter volumes.
Results:
More severe parkinsonism at baseline was associated with faster rate of decline in parkinsonism. Patients with predominant sound distortions (AOS type 1) showed faster rates of decline in aphasia and motor speech, while patients with segmented speech (AOS type 2) showed faster rates of decline in parkinsonism. On MRI, we observed trends for fastest rates of decline in aphasia in patients with relatively small left, but preserved right, Broca area and precentral cortex. Bilateral reductions in lateral premotor cortex were associated with faster rates of decline of behavior. No associations were observed between volumes and decline in motor speech or parkinsonism.
Conclusions:
Rate of decline of each of the 4 clinical features assessed was associated with different baseline clinical and regional MRI predictors. Our findings could help improve prognostic estimates for these patients.
Progressive apraxia of speech (AOS) is a neurodegenerative disorder characterized by slow speaking rate, abnormal prosody, distorted sound substitutions, additions, repetitions and prolongations, and syllable segmentation.1 Patients either can present with AOS in isolation or can also have agrammatic aphasia, which is characterized by agrammatic, telegraphic, or truncated spoken language. Patients who present with AOS and agrammatic aphasia are typically diagnosed as having agrammatic primary progressive aphasia (agPPA),2,3 whereas those who present with only progressive AOS, in the absence of agrammatic aphasia, are diagnosed with primary progressive AOS (PPAOS).4 Many individuals with PPAOS may later develop agrammatic aphasia.5 Patients with agPPA and PPAOS typically have frontotemporal lobar degeneration pathology at autopsy, including corticobasal degeneration, progressive supranuclear palsy (PSP), or TAR DNA-binding protein of 43-kDa pathology.6,7 Longitudinal studies have shown that disease progression is variable in these patients, with some patients rapidly developing features of PSP5,7–9 or corticobasal syndrome (CBS),7,8,10 others showing a decline in behavioral function,8,9,11,12 and some showing a more indolent rate of clinical progression.5 Therefore, prognosis is difficult in these patients, and biomarkers are urgently needed that can help predict future clinical course.
We aimed to determine whether baseline clinical and neuroimaging features could help predict rate of clinical progression in patients who presented with progressive AOS, either with or without aphasia, who had been followed up over time. We focused on predicting rate of decline in the 4 predominant clinical areas: aphasia, motor speech, parkinsonism, and behavioral dyscontrol.
METHODS
Patients.
Thirty-four patients with progressive AOS meeting diagnostic criteria for agPPA2 (n = 14) or PPAOS4 (n = 20) were recruited and followed up longitudinally between July 2010 and September 2015. All patients underwent a detailed speech/language battery4 at all visits that included tests of language severity, motor speech severity, repetition, single word comprehension, naming, object knowledge, and word knowledge. None of the patients met clinical criteria for PSP,13 CBS,14 or the semantic or logopenic variants of PPA2 at baseline.
AOS characteristics were assessed to subclassify AOS type.15 AOS type 1 subclassification was made if distorted sound substitutions or additions were judged to clearly dominate the speech pattern,15 while AOS type 2 subclassification was made if syllable segmentation within multisyllabic words or across words in phrases and lengthened intersegment durations between syllables, words, or phrases were judged to clearly dominate the speech pattern.15 If there was no clear predominance of type 1 or type 2 features, a designation of AOS not otherwise specified was made. These classifications were made by consensus between 2 speech-language pathologists, with excellent agreement as previously published.15 At a consensus meeting, the 2 speech-language pathologists viewed video of each patient’s speech-language evaluation and made a clinical judgment on the prominence of phonetic (AOS type 1) vs prosodic (AOS type 2) errors based on verbal output from spontaneous speech, the picture description task from the Western Aphasia Battery (WAB), and repetition of multisyllabic and single words. Videos providing patient examples of AOS types 1 and 2 and illustrating the important features of the 2 types have been previously published.15
Standard protocol approvals, registrations, and patient consents.
The study was approved by the Mayo Clinic Institutional Review Board. All patients consented to enrollment in the study.
Clinical outcome measures.
For this study, we selected 1 test each to reflect the severity of aphasia, motor speech, parkinsonism, and behavioral abnormalities. The WAB Aphasia Quotient (WAB-AQ)16 was used to measure global aphasia severity. The Motor Speech Disorders (MSD) scale (adapted from Yorkson et al.17) was used as a measure of motor speech severity. The MSD scale rates the severity of functional speech impairment (ranging from normal to effortful and rarely attempted vocalization) and has the advantage that it can be scored in every patient regardless of the degree of speech output. The Movement Disorders Society–sponsored revision of the Unified Parkinson's Disease Rating Scale part III (MDS-UPDRS III) 18 was used to assess parkinsonism. The Frontal Behavioral Inventory (FBI)19 was used to assess the severity of behavioral impairment. To reduce confounds with speech impairments, we calculated a total for the FBI removing items 10 (logopenia) and 11 (verbal apraxia) (modified FBI total = 66).
Neuroimaging.
All patients had a 3-dimensional magnetization-prepared rapid acquisition gradient echo (MPRAGE)4 performed at 3T at baseline. All MPRAGE images underwent preprocessing correction for gradient nonlinearity20 and intensity nonuniformity.21 Regional gray matter volumes were calculated with atlas-based parcellation22 in SPM5 and the automated anatomic labeling atlas.23 Regions of interest included the superior frontal cortex, precentral cortex, supplementary motor area, Broca area (pars triangularis + pars opercularis), and striatum (caudate nucleus + putamen), selected because they are atrophic in patients with agPPA and PPAOS.4,15 To assess lateral premotor cortex, which is associated with these syndromes4,15 and is not represented in the automated anatomic labeling atlas, a spherical region of interest (radius 10 mm) was placed in this region on the template with the use of Marsbar24 (left coordinates: x = −24, y = −5, z = 52. right coordinates: x = 32, y = −6, z = 52).4 The region of interest was applied to the MPRAGE scans in custom space. Total intracranial volume (TIV) was also measured.
Statistics.
Our analysis assessed the degree to which baseline clinical and MRI variables were associated with rate of decline in the 4 clinical outcome measures. For each patient and each clinical outcome, we calculated a rate of change measure by fitting a least-squares line to the data points. Rates of decline and baseline values were not associated with disease duration except for baseline MSD, so disease duration was not included in our models. Only baseline MDS-UPDRS III score was associated with age (p = 0.04). We calculated the Spearman rank correlation between patients' baseline values and their rates of change. To assess the associations between rate of decline and AOS type and MRI variables, we used all available data to fit linear mixed-effects regression models with models including patient-specific random intercepts and random slopes. For models comparing AOS type, we included an interaction between time from baseline and AOS type. For models with gray matter volume as a predictor, we included TIV as a covariate and included interactions between time and left volume and between time and the right volume. These 2 interactions allow the rate of change in the response variable to independently vary by left vs right volume after adjustment for TIV. For a given brain region, we summarized the association between volume and clinical change by displaying heat maps showing how rates of clinical decline vary for different combinations of left and right volume. Volume is modeled on a continuum, and estimates are available for any combination of left and right volumes, but to simplify the presentation, we chose to provide estimates for a 5 × 5 grid of volumes. In this grid, both the left and right volumes range from 15% below the cohort median for that region to 15% above the cohort median for that region. We report p values summarizing a likelihood ratio test of whether volume is predictive of clinical change. Because for each clinical outcome we examine 6 regions of interest, we also report false discovery rate–corrected p values for the 6 likelihood ratio tests.
For the dependent variables to be approximately conditionally normal, we used the following transformations: the square root of 100 minus WAB-AQ, 10 minus MSD, the square root of MDS-UPDRS III, and the square root of modified FBI. Models were fitted on the transformed scale, and then estimates were back-transformed with parametric bootstrap simulations.20 This allows us to report estimates and 95% confidence intervals on the measured (untransformed) scale based on the percentiles of the bootstrap simulations. Using the equivalence between a 95% confidence interval that does not include zero and a p < 0.05, we report p values based on “inverting” the widest confidence interval for bootstrap simulations that does not include zero. Mixed models were fitted in R (http://www-R-project.org) version 3.1.2 using version1.0-2 of the blme package, written to extend the standard lme4 package to fit mixed models with more stable variance component estimates.6
RESULTS
Demographic and baseline clinical features for the cohort are shown in the table. The median follow-up period was 2.4 years (range 0.9–5.0 years), and total number of visits available for analysis was 94. There was no difference in the number of visits between patients with agPPA and those with PPAOS (p = 0.44). Plots showing change over time in the 4 clinical outcome measures for each participant are shown in figure 1. Median rates of change (points per year) (interquartile range) were −3 (−7 to −1) for WAB-AQ, −1.0 (−1.4 to −0.6) for MSD, −7 (−14 to −2) for MDS-UPDRS III, and −1.9 (−6.6 to 0.0) for modified FBI.
Table.
Demographic and baseline clinical description of the cohort of 34 patients
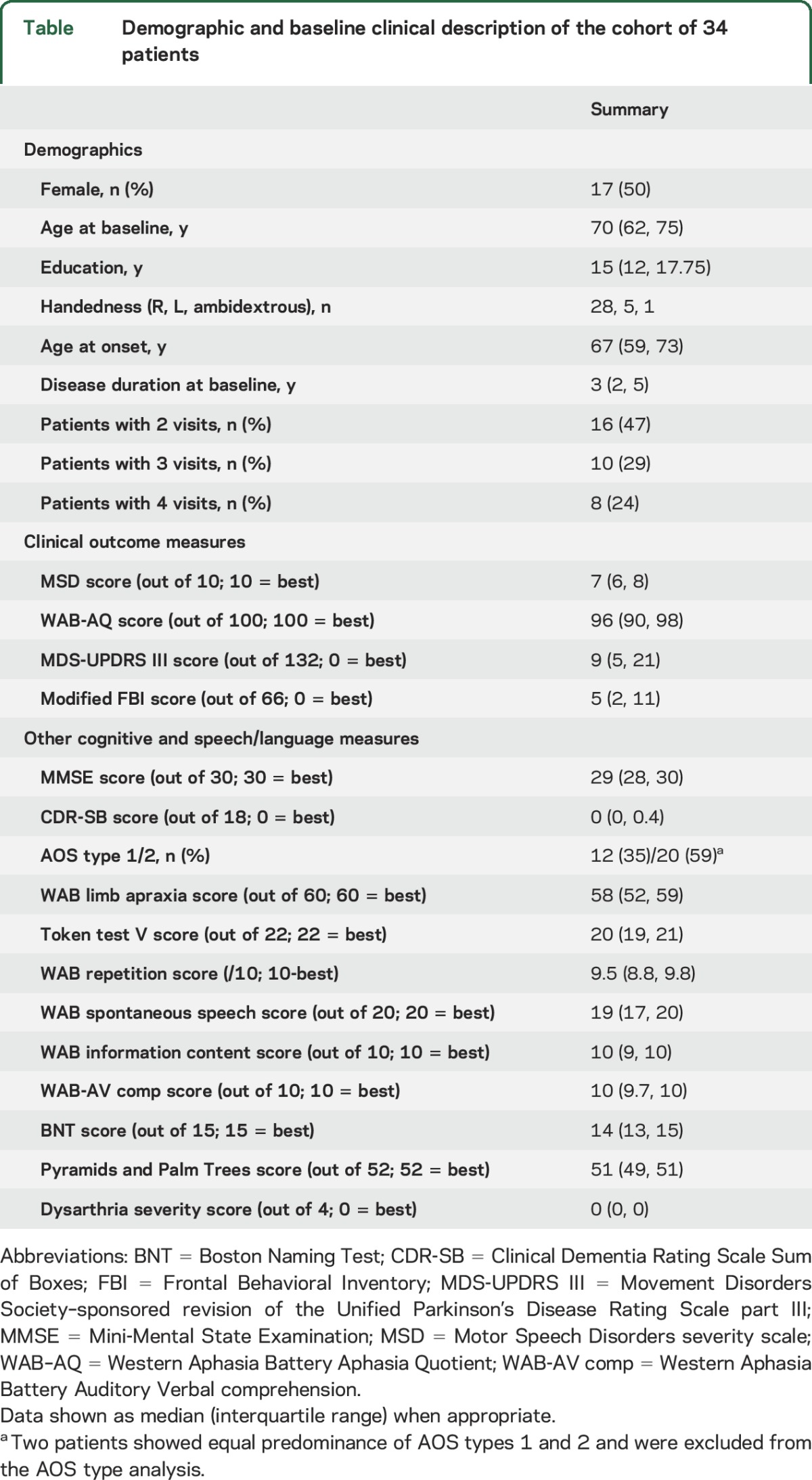
Figure 1. Performance of individual patients over time.

(A) WAB-AQ, (B) MSD, (C) MDS-UPDRS III, and (D) modified FBI. Blue-shaded points and lines were used for AOS type 1; orange-shaded points and lines were used for AOS type 2; and green-shaded points and lines were used for AOS not otherwise specified. The MDS-UPDRS III and modified FBI scores have been multiplied by −1 so that a worsening on all 4 measures corresponds to a declining score over time. Three patients had a baseline value only for WAB-AQ but contributed to the mixed model fit and thus were retained in the analyses. One patient had only a baseline FBI but was also retained in the analyses. AOS = apraxia of speech; FBI = Frontal Behavioral Inventory; MDS-UPDRS III = Movement Disorders Society–sponsored revision of the Unified Parkinson's Disease Rating Scale part III; MSD = Motor Speech Disorders; WAB-AQ = Western Aphasia Battery Aphasia Quotient.
Worse baseline MDS-UPDRS III was associated with faster rate of decline in MDS-UPDRS III (rank correlation r = 0.48, p = 0.004) (figure 2). No associations were identified for MSD, WAB-AQ, and modified FBI. Participants with AOS type 1 had a 6-point faster median rate of decline on WAB-AQ (p < 0.001) and a half-point faster rate of decline on MSD (p = 0.04) compared with participants with AOS type 2, while participants with AOS type 2 had a 7-point faster decline on MDS-UPDRS III compared to participants with AOS type 1 (p = 0.03) (figure 2). Participant-level clinical trajectories by AOS type are shown in figure 1. Participants with AOS type 2 also showed a trend for worse performance on MDS-UPDRS III at baseline compared to AOS type 1 (p = 0.08), with no difference observed in the other measures.
Figure 2. Relationship between clinical predictors and rate of decline.

(A) Relationship between baseline score and annual rate of decline for each of the 4 clinical outcome measures: (A.a) WAB-AQ, (A.b) MSD, (A.c) MDS-UPDRS III, and (A.d) modified FBI. The MDS-UPDRS III and modified FBI scores have been multiplied by −1 so that for all 4 scores higher is better and a worsening on all 4 measures corresponds to a declining score over time. Data on the vertical axis are slope estimates based on fitting a least-squares regression line to each patient. The rank correlation and corresponding p value between patients' slopes and baseline values are shown in each plot. (B) Box plots of the relationship between AOS type and rate of decline in the 4 clinical outcome measures: (B.a) WAB-AQ, (B.b) MSD, (B.c) MDS-UPDRS III, and (B.d) modified FBI. Box plots summarize the distribution of patient-specific rates of change based on fitting a least-squares regression line to each patient. AOS = apraxia of speech; FBI = Frontal Behavioral Inventory; MDS-UPDRS III = Movement Disorders Society–sponsored revision of the Unified Parkinson's Disease Rating Scale part III; MSD = Motor Speech Disorders; WAB-AQ = Western Aphasia Battery Aphasia Quotient.
We observed a trend for baseline volumes of Broca area and precentral gyrus to be associated with WAB-AQ, with fastest rates of decline in WAB-AQ observed in patients with relatively small left volumes but preserved right volumes (figure 3). The effect size for these associations is clinically significant with appreciably different rates of decline depending on volumes in this region. However, these trends did not survive correction for multiple comparisons. No significant associations were observed between the MSD (figure 3) or MDS-UPDRS III (figure 4) score and regional volumes. Bilateral volume reductions in lateral premotor cortex were associated with faster rates of decline in modified FBI, and this association survived correction for multiple comparisons (figure 4).
Figure 3. Heat maps of the relationship between baseline neuroimaging volumes and annual rate of decline for (A) WAB-AQ and (B) MSD.

Each subplot indicates the rate of annual decline for a given left and right baseline volume. The combinations shown represent volumes ranging from 15% below the cohort median to 15% above the cohort median. Colors are comparable across subplots only for the same outcome measure because of differences in scale and rate of decline. FDR = false discovery rate; MSD = Motor Speech Disorders; SMA = supplementary motor area; WAB-AQ = Western Aphasia Battery Aphasia Quotient.
Figure 4. Heat maps of the relationship between baseline neuroimaging volumes and annual rate of decline for (A) MDS-UPDRS III and (B) modified FBI.

Each subplot indicates the rate of annual decline for a given left and right baseline volume. The combinations shown represent volumes ranging from 15% below the cohort median to 15% above the cohort median. Colors are comparable across subplots only for the same outcome measure because of differences in scale and rate of decline. FBI = Frontal Behavioral Inventory; FDR = false discovery rate; MDS-UPDRS III = Movement Disorders Society–sponsored revision of the Unified Parkinson's Disease Rating Scale part III; SMA = supplementary motor area.
DISCUSSION
This study identified clinical and neuroimaging features at baseline that could help predict future rate of clinical decline in patients presenting with progressive AOS. In particular, we found that rate of decline of each of the 4 clinical features was associated with different baseline clinical and regional MRI predictors. Our findings are useful to help provide more informed prognostic estimates for individual patients.
Baseline clinical severity was clearly associated with future decline only for the MDS-UPDRS III, a measure of parkinsonism. Patients scored poorly on this test because of slowness on alternating motion rates and problems with balance and gait, consistent with the development of features of CBS and PSP.5 Our data show that the more parkinsonism a patient has at presentation, the faster the parkinsonism will decline over time, reflecting the fact that those patients who developed severe parkinsonism already displayed signs of parkinsonism at presentation. In fact, a subset (n = 9) did not develop features of parkinsonism, even after a disease duration of >8 years in some patients. Similar relationships were not present for WAB-AQ, MSD, or FBI, so other biomarkers are needed to help determine future rate of decline in these features. The reason for the lack of a relationship with WAB-AQ may be that many patients with PPAOS in the cohort did not show evidence of agrammatic aphasia at baseline but subsequently developed this feature over time, as we have previously shown.25
A clinical feature that was associated with rate of decline of WAB-AQ, MSD, and MDS-UPDRS III was AOS type.15 An AOS speech pattern dominated by distorted sound substitutions or additions, i.e., AOS type 1, was predictive of fast rates of decline in motor speech and agrammatic aphasia. In contrast, patients with AOS type 2 who show dominant syllable segmentation had worse baseline scores and faster rates of decline on the MDS-UPDRS III, suggesting that these patients are more likely to develop a parkinsonian disorder. This raises the possibility that the different AOS types may represent different diseases and may even show different pathologic correlates, although autopsy studies are needed to investigate this issue. Regardless of these implications, our data show that the assessment of AOS type could be particularly important for prognosis, especially because the baseline severity of aphasia and motor speech are not helpful to predict future rate of progression.
We observed some associations between baseline neuroimaging measures and future rate of clinical decline. The WAB-AQ tended to show faster rates of decline in patients with small left Broca area but relatively preserved right Broca area, i.e., in patients with left-sided asymmetric involvement of the Broca area. Patients with little atrophy of the left Broca area or patients with bilateral involvement of the Broca area showed lower rates of decline on the WAB-AQ. This conforms to the proposed central role of the Broca area in agrammatism26,27 and involvement of this region in agrammatic aphasia in both stroke and neurodegenerative disease.24,26–29 It also concurs with our previous findings that patients with PPAOS who develop aphasia have greater atrophy in the Broca area at baseline compared to patients with PPAOS who do not develop aphasia.25 However, atrophy of the left Broca area was not sufficient to result in fast rates of decline in agrammatism; the relative preservation of the right hemisphere was also important. This is consistent with the fact that patients with agrammatic aphasia consistently show left-sided asymmetry on MRI and the fact that patients with syndromes such as behavioral variant frontotemporal dementia that can involve bilateral damage to the inferior frontal lobes do not tend to display agrammatism.22,30,31 It is possible that an isolated breakdown of connectivity in the language network of the left hemisphere centered on the Broca area is necessary for the development of agrammatism. However, while this association is biologically reasonable, it did not survive correction for multiple comparisons. Larger studies are therefore needed to confirm the findings. We also observed the same trends in the precentral cortex, although resulting in a smaller change of WAB-AQ rates, likely showing that the participants who declined the fastest on the WAB-AQ also showed left-sided involvement of other regions in the frontal lobe.
Baseline neuroimaging measures were also associated with rates of change in the modified FBI, reflecting behavioral and personality change. In general, a trend was observed for faster rates of decline in patients with bilateral atrophy of frontal and striatal regions at baseline, although the most striking relationship was observed with lateral premotor cortex. Little neuroimaging work has been done on behavioral abnormalities in agPPA or PPAOS, although involvement of these structures is consistent with work linking behavioral abnormalities with a breakdown of frontal-basal ganglia circuits.32–34 The behavioral features observed in our cohort such as apathy and irritability overlap with those observed in behavioral-variant frontotemporal dementia, which is also most commonly associated with bilateral frontal atrophy.35
No regions showed significant associations with rate of decline on the MSD or MDS-UPDRS III, possibly because these clinical tests lack anatomic specificity. Alternatively, we may have lacked power to detect associations. Other cortical and subcortical brain regions that we did not assess may have been more strongly associated with these clinical measures, although we suspect this is unlikely given that we focused our analyses on the 6 imaging regions of interest that show the most involvement in these patients.
A strength of our study is that our models used all available time points, allowing us to sample a longer period of follow-up in some patients than if we had assessed only change over 1 set interval. We also included all patients in the study who had a progressive AOS, regardless of whether they had accompanying agrammatic aphasia. While this meant that our cohort included some patients who did not present with both features and may possibly show a different disease course, we feel that this unrestricted population will most closely mirror how these patients are typically classified by physicians, and hence, our findings should generalize across centers. All of the patients we studied are likely classified as having either agPPA2 or nonfluent variants of FTD21 at other centers. A potential limitation was that our model did not include disease duration, although we did not find evidence that disease duration was related to rate of decline. Our results were also limited by the inherent nature of each clinical test. For example, scores on the WAB-AQ were relatively truncated, with most scoring between 90 and 100. Assessment of AOS type in our study was qualitative in nature. Given that the AOS distinctions have only recently been described, it may take some time for the clinical description of AOS types to be reliably integrated into behavioral and movement disorder practices. Our study had missing data or attrition due to participants being too severe to undergo further longitudinal testing. We performed a sensitivity analysis to evaluate this possible bias by fitting 4 joint longitudinal and time-to-event models36 using the joineRML package in R. In the joint models, the longitudinal process was clinical decline on the WAB-AQ, MSD, MDS-UPDRS III, or modified FBI, and the time-to-event process defined an event as missing data as a result of severity (including death), with individuals who remained in the study censored at their last visit. We found that rates of clinical decline were very similar in the joint model: only 6% faster for WAB-AQ, 7% faster for MSD, 3% faster for MDS-UPDRS III, and 8% faster for modified FBI. These findings suggest that missing data and dropouts are not appreciably affecting our results.
This study highlights the value of careful assessment of both the AOS characteristics and MRI appearance when patients with progressive AOS present clinically. Although our study used a sophisticated atlas-based approach to assess atrophy, a simple visual assessment of asymmetry and atrophy of a few regions, particularly the Broca area and precentral and premotor cortex, may also be useful to help determine how an individual is likely to progress over the coming years. Any such clue could be useful to help educate patients and families and allow them to better plan for the future.
GLOSSARY
- agPPA
agrammatic primary progressive aphasia
- AOS
apraxia of speech
- CBS
corticobasal syndrome
- FBI
Frontal Behavioral Inventory
- MDS-UPDRS III
Movement Disorders Society–sponsored revision of the Unified Parkinson's Disease Rating Scale part III
- MPRAGE
magnetization-prepared rapid acquisition gradient echo
- MSD
Motor Speech Disorders
- PPAOS
primary progressive apraxia of speech
- PSP
progressive supranuclear palsy
- TIV
total intracranial volume
- WAB
Western Aphasia Battery
- WAB-AQ
Western Aphasia Battery Aphasia Quotient
AUTHOR CONTRIBUTIONS
Dr. Jennifer Whitwell: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, acquisition of data, study supervision or coordination, obtaining funding. Stephen Weigand: drafting/revising the manuscript, statistical analysis. Dr. Joseph Duffy, Dr. Heather Clark, Dr. Edythe Strand, Dr. Mary Machulda, and Anthony Spychalla: drafting/revising the manuscript, acquisition of data. Matthew Senjem: drafting/revising the manuscript, analysis or interpretation of data. Dr. Clifford Jack: drafting/revising the manuscript, acquisition of data. Dr. Keith Josephs: drafting/revising the manuscript, study concept or design, study supervision or coordination, obtaining funding.
STUDY FUNDING
Supported by the NIH (R01-DC12519 and R01-DC010367).
DISCLOSURE
J. Whitwell receives funding from NIH grants R01-DC12519, R01-NS089757, R01-AG050603, R01-AG037491, and R21-NS094684. S. Weigand reports no disclosures relevant to the manuscript. J. Duffy receives funding from NIH grants R01-DC12519 and R21-NS094684. H. Clark receives funding from NIH grants R01-DC12519 and R01-NS089757. E. Strand reports no disclosures relevant to the manuscript. M. Machulda receives funding from NIH grants R01-DC12519 and R01-AG050603. A. Spychalla and M. Senjem report no disclosures relevant to the manuscript. C. Jack serves as a consultant for Eli Lily and receives research funding from the NIH (R01-AG011378, RO1-AG041851, RO1-AG037551, U01-HL096917, U01-AG032438, U01-AG024904) and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Foundation. K. Josephs receives funding from NIH grants R01-AG037491, R01-NS089757, R01-DC12519, R01-AG050603, and R21-NS094684. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Duffy JR. Motor Speech Disorders: Substrates, Differential Diagnosis, and Management. 2nd ed. St. Louis: Mosby; 2005. [Google Scholar]
- 2.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grossman M. The non-fluent/agrammatic variant of primary progressive aphasia. Lancet Neurol 2012;11:545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Josephs KA, Duffy JR, Strand EA, et al. Characterizing a neurodegenerative syndrome: primary progressive apraxia of speech. Brain 2012;135:1522–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Josephs KA, Duffy JR, Strand EA, et al. The evolution of primary progressive apraxia of speech. Brain 2014;137:2783–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deramecourt V, Lebert F, Debachy B, et al. Prediction of pathology in primary progressive language and speech disorders. Neurology 2010;74:42–49. [DOI] [PubMed] [Google Scholar]
- 7.Josephs KA, Duffy JR, Strand EA, et al. Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain 2006;129:1385–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain 2005;128:1996–2005. [DOI] [PubMed] [Google Scholar]
- 9.Mochizuki A, Ueda Y, Komatsuzaki Y, Tsuchiya K, Arai T, Shoji S. Progressive supranuclear palsy presenting with primary progressive aphasia: clinicopathological report of an autopsy case. Acta Neuropathol 2003;105:610–614. [DOI] [PubMed] [Google Scholar]
- 10.Gorno-Tempini ML, Murray RC, Rankin KP, Weiner MW, Miller BL. Clinical, cognitive and anatomical evolution from nonfluent progressive aphasia to corticobasal syndrome: a case report. Neurocase 2004;10:426–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marczinski CA, Davidson W, Kertesz A. A longitudinal study of behavior in frontotemporal dementia and primary progressive aphasia. Cogn Behav Neurol 2004;17:185–190. [PubMed] [Google Scholar]
- 12.Van Langenhove T, Leyton CE, Piguet O, Hodges JR. Comparing longitudinal behavior changes in the primary progressive aphasias. J Alzheimers Dis 2016;53:1033–1042. [DOI] [PubMed] [Google Scholar]
- 13.Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 1996;47:1–9. [DOI] [PubMed] [Google Scholar]
- 14.Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Josephs KA, Duffy JR, Strand EA, et al. Syndromes dominated by apraxia of speech show distinct characteristics from agrammatic PPA. Neurology 2013;81:337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kertesz A. Western Aphasia Battery (Revised). San Antonio: PsychCorp; 2007. [Google Scholar]
- 17.Yorkson K, Strand EA, Miller R, Hillel A, Smith K. Speech deterioration in amyotrophic lateral sclerosis: implications for the timing of intervention. J Med Speech-Language Pathol 1993;1:35–46. [Google Scholar]
- 18.Goetz CG, Tilley BC, Shaftman SR, et al. Movement Disorder Society-sponsored revision of the Unified Parkinson's Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord 2008;23:2129–2170. [DOI] [PubMed] [Google Scholar]
- 19.Kertesz A, Davidson W, Fox H. Frontal behavioral inventory: diagnostic criteria for frontal lobe dementia. Can J Neurol Sci 1997;24:29–36. [DOI] [PubMed] [Google Scholar]
- 20.Jovicich J, Czanner S, Greve D, et al. Reliability in multi-site structural MRI studies: effects of gradient non-linearity correction on phantom and human data. NeuroImage 2006;30:436–443. [DOI] [PubMed] [Google Scholar]
- 21.Sled JG, Zijdenbos AP, Evans AC. A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans Med Imaging 1998;17:87–97. [DOI] [PubMed] [Google Scholar]
- 22.Whitwell JL, Przybelski SA, Weigand SD, et al. Distinct anatomical subtypes of the behavioural variant of frontotemporal dementia: a cluster analysis study. Brain 2009;132:2932–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tzourio-Mazoyer N, Landeau B, Papathanassiou D, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. NeuroImage 2002;15:273–289. [DOI] [PubMed] [Google Scholar]
- 24.Whitwell JL, Duffy JR, Strand EA, et al. Distinct regional anatomic and functional correlates of neurodegenerative apraxia of speech and aphasia: an MRI and FDG-PET study. Brain Lang 2013;125:245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitwell JL, Duffy JR, Machulda MM, et al. Tracking the development of agrammatic aphasia: a tensor-based morphometry study. Cortex 2016;90:138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Broca P. Sur le siege de la facultle due language articule. Bull Soc Anthropol 1865;6:337–393. [Google Scholar]
- 27.Geschwind N. The organization of language and the brain. Science 1970;170:940–944. [DOI] [PubMed] [Google Scholar]
- 28.Amici S, Ogar J, Brambati SM, et al. Performance in specific language tasks correlates with regional volume changes in progressive aphasia. Cogn Behav Neurol 2007;20:203–211. [DOI] [PubMed] [Google Scholar]
- 29.Grossman M, Powers J, Ash S, et al. Disruption of large-scale neural networks in non-fluent/agrammatic variant primary progressive aphasia associated with frontotemporal degeneration pathology. Brain Lang 2013;127:106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mandelli ML, Vitali P, Santos M, et al. Two insular regions are differentially involved in behavioral variant FTD and nonfluent/agrammatic variant PPA. Cortex 2016;74:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Couto B, Manes F, Montanes P, et al. Structural neuroimaging of social cognition in progressive non-fluent aphasia and behavioral variant of frontotemporal dementia. Front Hum Neurosci 2013;7:467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eslinger PJ, Moore P, Antani S, Anderson C, Grossman M. Apathy in frontotemporal dementia: behavioral and neuroimaging correlates. Behav Neurol 2012;25:127–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Josephs KA, Whitwell JL, Eggers SD, Senjem ML, Jack CR Jr. Gray matter correlates of behavioral severity in progressive supranuclear palsy. Mov Disord 2011;26:493–498. [DOI] [PubMed] [Google Scholar]
- 34.D'Anna L, Mesulam MM, Thiebaut de Schotten M, et al. Frontotemporal networks and behavioral symptoms in primary progressive aphasia. Neurology 2016;86:1393–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whitwell JL, Xu J, Mandrekar J, et al. Frontal asymmetry in behavioral variant frontotemporal dementia: clinicoimaging and pathogenetic correlates. Neurobiol Aging 2013;34:636–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Henderson R, Diggle P, Dobson A. Joint modelling of longitudinal measurements and event time data. Biostatistics 2000;1:465–480. [DOI] [PubMed] [Google Scholar]