

Abstract
Purpose
Smoking is an established risk factor for a human papillomavirus (HPV) infection advancing to cervical precancer and cancer, but its role earlier in the natural history is less clear. Smoking is inversely associated with possessing HPV antibodies from a past infection suggesting that smoking may influence acquiring subsequent infections.
Methods
In a cohort of 1,976 U.S. women, we evaluate whether reduced antibodies to HPV-16 is a mechanism for smoking's role on acquiring a subsequent HPV-16 infection, through the analytic technique of causal mediation analysis. We posit a causal model and estimate two counterfactually-defined effects: a smoking impaired antibody-mediated indirect effect, and a non-mediated direct effect representing all other potential mechanisms of smoking.
Results
Compared to never smokers, current smokers had increased odds of HPV-16 infection by the antibody-mediated indirect effect (odds ratio=1.29, 95% confidence interval: 1.11, 1.73); the estimated direct effect was very imprecise (OR=0.57, 95% CI: 0.26, 1.13). We observed a stronger estimated indirect effect among women who smoked at least half a pack of cigarettes daily (OR=1.61, 95% CI: 1.27, 2.15) than among women who smoked less than that threshold (OR = 1.09; 95% CI: 0.94, 1.44).
Conclusions
This is the first study to directly test the mechanism underlying smoking as an HPV cofactor. The results support current smoking as a risk factor earlier in the natural history of HPV, and are consistent with the hypothesis that smoking increases the risk of a subsequent infection by reducing immunity.
Keywords: Human Papillomavirus, HPV, Smoking, Antibodies, Mediation, Mechanism, Indirect Effect
Introduction
Human papillomavirus (HPV) infections are commonly acquired shortly after sexual initiation and are the necessary cause of cervical carcinogenesis (1). Most infections are cleared within 1-2 years, but some persist. Women with a persistent carcinogenic HPV infection are at risk of developing precancerous lesions that may progress to cervical cancer (1, 2). Some infected women will develop antibodies, which could protect against a subsequent infection by that HPV type (3-6).
Risk factors are associated with acquiring antibodies from an HPV infection. Greater sexual activity increases a woman's exposure to HPV and thus her opportunity to develop antibodies (7, 8). Other factors, such as smoking, may impair a woman's antibody response (9-11). For smoking, a diminished antibody response could be a consequence of an impaired immune system. Smoking affects both the cellular and humoral immune response; it can reduce cytokines, natural killer cells, and immunoglobulins (12). Impairing the cellular response could lead to a persistent infection by inadequate clearance of HPV; while impairing the humoral response could lead to a subsequent infection by inadequate antibody protection. Despite smoking being associated with a lower natural antibody response, it is unclear whether it results in an increased risk of subsequent HPV infection. To directly test that causal mechanistic research question in human subjects requires an analytic technique called mediation analysis.
In a secondary analysis within a large clinical trial, we sought to evaluate the role of smoking and naturally acquired antibodies in subsequent HPV-16 infections. We posit a causal model of smoking, HPV antibodies, and HPV subsequent infection to estimate two causal pathways using mediation analysis: 1) a smoking impaired antibody-mediated pathway; and 2) an alternative non-mediated pathway representing all other potential mechanisms of smoking.
Methods
Study population
The Atypical Squamous Cells of Undetermined Significance (ASCUS) Low-Grade Squamous Intraepithelial Lesion (LSIL) Triage Study (ALTS) was a multicenter randomized controlled trial conducted by the National Cancer Institute from 1996 to 1998 – before the development of the HPV vaccine – comparing clinical management strategies for women with ASCUS or LSIL cytology results (13). The study enrolled 3,488 women with ASCUS and 1,572 with LSIL at four clinical centers in the United States and followed them for two years, during which the centers collected cervical specimens at six month intervals to test for HPV. In addition to completing a baseline questionnaire, 2,736 women also donated baseline blood samples allowing for the serological testing required for this analysis. We also required a population that was at risk for a subsequent HPV-16 infection during follow-up, meaning that each subject had been previously exposed to HPV-16. Given the ALTS inclusion criteria of ASCUS or LSIL, we assumed all subjects had a past exposure to HPV-16, thus had the opportunity to develop antibodies; we address this assumption by a sensitivity analysis described in the statistical analyses. To create our at risk population from those who donated blood, we excluded women who were not HPV-16 negative at baseline (n = 271); our results do not change if we include baseline HPV-16 positive women in the analytical study population after a negative HPV test during follow-up. We additionally excluded women with cervical intraepithelial neoplasia grade 2 or more severe because they are further along the natural history (n = 487). Two subjects were missing smoking information and were excluded, leaving the final analytical cohort with 1,976 women of whom 131 tested positive for HPV-16 during follow-up. Our exposure, smoking status (never [referent], former, current) and our mediator, HPV antibody status (positive, negative) were defined from the baseline questionnaire asking “Do you currently smoke” (and how much), and blood draw respectively. We defined our outcome, subsequent HPV-16 infection (yes, no), if any of a woman's follow-up cervical specimens tested positive for HPV-16.
HPV DNA genotyping and HPV serology
Cervical specimens were genotyped for HPV DNA using the Line Blot Assay (Roche Molecular Systems, Pleasanton, CA) (14), the predecessor to Linear Array. Extracted DNA was amplified using a PGMY09/11 L1 consensus primer, and the amplicon was tested using reverse line blot hybridization for the presence of 27 HPV genotypes, including type 16. Testing was done at baseline and up to four follow-up specimens (6, 12, 18, and 24 months).
A Luminex-based multiplex serology assay was used to test the baseline serum samples for HPV antibodies. Glutathione S-transferase (GST) fusion proteins targeted to the HPV L1 major capsid antigen measured seroreactivity for eight types, including type 16 (15). The proteins are bound to fluorescence-labeled polystyrene beads (Luminex, Austin, TX) and analyzed with a Luminex 100 that quantifies the fluorescence intensity associated with the antibodies to the viral antigens. The antigen types were loaded onto different colored bead sets, then loaded into a 96-well plate. Diluted serum specimens were incubated with a bead mixture, and antibodies that bound to beads were stained using biotinylated anti-human-Ig and streptavidin-R-phycoerythrin, and then analyzed. The assay reports median fluorescence intensity (MFI) of a minimum of 100 beads analyzed per bead set/antigen. The background fluorescence level was set using the MFI of empty GST-tag-loaded beads. The seroreactivity for a given HPV protein was its MFI level subtracting the background level. We defined the MFI antibody positive threshold as 3 standard deviations above the mean from a prior study of 125 South Korean women who reported being virgins (16), a threshold we have used previously (5, 10).
Statistical analyses
We conducted a formal mediation analysis to decompose the overall effect of cigarette smoking on subsequent HPV-16 infection into two separate causal effects: 1) a “total natural indirect effect” representing an antibody-mediated mechanism, and 2) a “pure natural direct effect” representing all other potential mechanisms. We used recognized counterfactually-defined direct and indirect effects to specify our effects of interest (17-20) and statistical estimation procedures based on those effects to obtain our results (21). The counterfactual natural direct and indirect effects are commonly written as an expected value of a difference between two potential outcomes: E(YX,M(X) – YX*,M(X*)); where YX,M(X) stands for what a woman's subsequent HPV infection outcome (Y) would be given two conditions: 1) an assigned smoking status (X: 1 for exposed, 0 for unexposed); and 2) her “natural” antibody status under either the same, or possibly different, assigned smoking status (M(X)) (see appendix for more details including how the potential outcome difference corresponds to a logistic regression analysis). In short, the pure natural direct effect aims to estimate the effect of smoking vs never smoking on subsequent HPV infection if smoking could not impair HPV antibodies [E(Y1,M(0) – Y0,M(0))], while the total natural indirect effect aims to estimate the effect of smoking vs never smoking on subsequent HPV infection because smoking impairs HPV antibodies [E(Y1,M(1) – Y1,M(0))]. We can estimate these effects by specifying our causal model, fitting statistical equations to that model, then applying the above counterfactual formulas (see appendix) (21, 22). For the estimates to be unbiased the specified causal model must be accurate and include all confounders of the effects for smoking-antibodies, smoking-infection, and antibodies-infection. Our model is as follows: smoking can impair a woman's antibody response following a past HPV exposure, and lowered antibodies can increase a woman's risk for a subsequent HPV infection; smoking may also affect subsequent infection through unmeasured alternative mechanisms; sexual behavior can confound the estimated effects (Figure 1). The statistical model representing the causal graph in Figure 1 has two logistic equations – one for the HPV infection outcome and one for the HPV antibody mediator; the HPV infection equation includes a multiplicative interaction term between smoking and HPV antibodies. We chose, a priori, to adjust each equation for age, age at sexual initiation, and lifetime number of sexual partners. We further adjusted for whether a subject reported a new sexual partner during follow-up and whether a subject had a previous sexually transmitted infection and found that including those variables did not change our results thus they were left out of our final models. We estimate odds ratios for the direct and indirect effects, and confidence intervals with parametric bootstrapping of 1,000 draws (23).
Figure 1.

Our proposed causal model between smoking, HPV antibodies, confounding factors, and subsequent HPV infection. We propose that smoking impairs the HPV-16 antibody response that in turn increases the risk of subsequent infection by that type (natural indirect effect). We also propose that smoking may have alternative mechanisms, represented here by the natural direct effect. Not depicted is the modeled interaction between smoking and HPV antibodies which could be a consequence of a more complex causal pathway involving smoking, sexual behavior, and immunity.
Our primary results are the natural direct and indirect effects of smoking status (never [referent], former, current) on subsequent HPV-16 infection from the method described above, which we will refer to as the causal method. Because this method is relatively new, we also report results obtained from a method more commonly used in epidemiology, which we will refer to as the traditional method. We include the traditional method not to support the results of the causal method, but rather to demonstrate how the results and conclusions from the traditional method may differ from the causal method. Though it may be more familiar to many readers, the traditional method is limited because it has historically not used counterfactuals to explicitly define the direct and indirect effects of interest. Thus, evaluating mediation with the traditional method can fail to have a causal interpretation even under the no bias assumptions required of all mediation analyses. The traditional method assesses mediation by observing the degree of attenuation between two estimated associations (in our case logistic regressions): 1) smoking's overall association with HPV-16 infection; and 2) the same association after further adjusting for the HPV-16 antibody mediator (24). Greater attenuation means greater explanation by the mediator, suggesting more of an indirect effect; while less attenuation means less explanation, suggesting more of a direct effect. We also report the two component associations in Figure 1 that comprise the indirect effect: 1) smoking's association with antibodies; and 2) antibodies' association with infection stratified by smoking status.
In a priori sub-analyses, we subcategorized smoking status by intensity (< ½ pack per day, ½ to < 1 pack per day, ≥ 1 pack per day), by median duration (≤ 7 years, > 7 years), and by a woman's smoking initiation relative to her sexual initiation (smoking at the same age or earlier than sexual initiation, after sexual initiation); never smokers were the referent for each subcategory. We also stratified by race to examine the mediation among non-Hispanic whites and African Americans. In a sensitivity analysis, we restricted to women with five or more sexual partners in their lifetime (median value). By doing so, we increase the probability that the women were previously exposed to HPV-16, testing our initial assumption. We also restricted our population to older women (24+ years) and those reporting a new sexual partner during follow-up based on results from the smoking-antibody interaction. Mediation analyses were conducted in Mplus version 7.2 (22), all other analyses were performed in SAS version 9.3 (25). We deemed statistical significance at the P < 0.05 two-sided level or, for bootstrapped effects, if the 95% confidence interval excluded the null value.
Results
Selected baseline characteristics of the study population are displayed in Table 1. Approximately 33% of the women were current smokers, 55% were never smokers. Current smokers were on average slightly younger and less educated than never smokers. Despite having a greater number of sexual partners and initiating sex at a younger age, fewer current smokers tested positive for HPV antibodies relative to never smokers (20% vs. 26%).
Table 1. Selected baseline characteristics of the ALTS population in the present analysis.
| Smoking | ||||||
|---|---|---|---|---|---|---|
| Never (n = 1,095) | Former (n = 244) | Current (n = 637) | ||||
|
| ||||||
| n | % | n | % | n | % | |
| HPV-16 infection during follow-up | ||||||
| positive | 73 | 7.0 | 13 | 5.7 | 45 | 7.9 |
| negative | 967 | 93.0 | 217 | 94.3 | 526 | 92.1 |
| HPV-16 antibodies | ||||||
| positive | 281 | 25.7 | 53 | 21.7 | 124 | 19.5 |
| negative | 814 | 74.3 | 191 | 78.3 | 513 | 80.5 |
| Age (mean, sd) | 28.9 | (9.6) | 35 | (12.0) | 27.5 | (8.4) |
| Race/Ethnicity | ||||||
| White | 546 | 50.0 | 198 | 81.1 | 439 | 69.4 |
| Black | 449 | 41.1 | 29 | 11.9 | 139 | 22.0 |
| Other | 98 | 8.9 | 17 | 7.0 | 55 | 8.6 |
| Education | ||||||
| < HS | 130 | 11.9 | 22 | 9.0 | 168 | 26.4 |
| Completed HS | 339 | 31.0 | 69 | 28.3 | 195 | 30.7 |
| Some College+ | 626 | 57.2 | 153 | 62.7 | 273 | 42.9 |
| Lifetime number of sex partners | ||||||
| 1-2 | 287 | 26.4 | 40 | 16.5 | 54 | 8.5 |
| 3-4 | 271 | 25.0 | 46 | 19.0 | 112 | 17.7 |
| 5-6 | 227 | 20.9 | 54 | 22.3 | 131 | 20.7 |
| 7+ | 301 | 27.7 | 102 | 42.1 | 336 | 53.1 |
| Age sexual debut | ||||||
| <16 | 283 | 25.9 | 74 | 30.3 | 300 | 47.2 |
| 16-17 | 388 | 35.5 | 81 | 33.2 | 233 | 36.6 |
| 18-19 | 251 | 23.0 | 60 | 24.6 | 78 | 12.3 |
| 20+ | 170 | 15.6 | 29 | 11.9 | 25 | 3.9 |
| History of an STI | ||||||
| Yes | 459 | 41.9 | 83 | 34.0 | 300 | 47.1 |
| No | 636 | 58.1 | 161 | 66.0 | 337 | 52.9 |
numbers not adding to total are due to missing data
Abbreviations: ALTS = Atypical Squamous Cells of Undetermined Significance/Low-Grade Squamous Intraepithelial Lesion Triage Study; HPV = human papillomavirus, HS = high school; sd = standard deviation; STI = sexually transmitted infection
We estimated counterfactually-defined natural direct and indirect effects for smoking on HPV infection (Table 2). For current compared to never smokers, we observed a statistically significant antibody-mediated indirect effect on HPV-16 infection (odds ratio [OR] = 1.29; 95% confidence interval [CI]: 1.11, 1.73); the direct effect was inverse but very imprecise (OR = 0.57; 95% CI: 0.26, 1.13). For former smokers, neither effects deviated from the null value (Table 2). In estimating these risks, there was a significant interaction between smoking status and antibody status (χ² = 8.38, df = 2, P value = 0.015). If we overlooked the interaction, the mediation analysis for current smokers would have produced an indirect effect OR = 1.08 (95% CI: 1.01, 1.18), and a direct effect OR = 1.03 (95% CI: 0.67, 1.47).
Table 2. Mediation analysis results of HPV-16 infection for current and former smokers compared to never smokers.
| Current Smokera | Former Smokera | |||
|---|---|---|---|---|
| OR | 95% CI | OR | 95% CI | |
| Overall associationb | 1.08 | (0.71, 1.62) | 1.02 | (0.54, 1.91) |
| Mediator adjusted associationb,c | 1.03 | (0.68, 1.55) | 1.01 | (0.54, 1.89) |
| Causal mediation analysis | ||||
| Total natural indirect effect | 1.29 | (1.11, 1.73) | 1.10 | (0.98, 1.53) |
| Pure natural direct effect | 0.57 | (0.26, 1.13) | 0.61 | (0.20, 1.30) |
| Causal mediation analysis without smoking-antibody interaction term | ||||
| Total natural indirect effect | 1.08 | (1.01, 1.18) | 1.03 | (1.00, 1.13) |
| Pure natural direct effect | 1.03 | (0.67, 1.47) | 1.00 | (0.48, 1.66) |
all models adjusted for model covariates: age, age at sexual debut, and lifetime number of sexual partners
never smokers are the referent category
the traditional method assesses the degree of attenuation between the overall association and the mediator adjusted association; higher attenuation suggests more of an indirect effect, less attenuation suggests more of a direct effect
additionally adjusted for the mediator, HPV-16 antibody status
To demonstrate how the two mediation methods can differ, we provided the results for a more traditional method for examining mediation (Table 2). The overall association between current smoking and HPV-16 infection was an OR = 1.08 (95% CI: 0.71, 1.62). Although that estimate attenuated after further adjusting for the antibody mediator (OR = 1.03; 95% CI: 0.68, 1.55), the attenuation does not suggest a substantial indirect effect because both results were weak and imprecise. Additionally, we estimated the component associations in Figure 1 that comprise the indirect effect: smoking with HPV-16 antibodies; and antibodies with infection (Table 3). Current smokers were less likely to test positive for HPV antibodies relative to never smokers (OR = 0.52; 95% CI: 0.40, 0.68). Testing positive for HPV antibodies had an inverse association with HPV infection, but this was only observed among smokers (Table 3), leading to significant interaction on the additive and multiplicative scales.
Table 3. Odds ratios for the component associations that comprise the antibody-mediated pathway: 1) HPV-16 antibody status with smoking status, and 2) HPV-16 antibody status with HPV-16 infection status stratified by never, former, and current smokers; first among the full population, then after restricting to five or more lifetime sexual partners.
| Never | Former | Current | ||||
|---|---|---|---|---|---|---|
| OR | 95% CI | OR | 95% CI | OR | 95% CI | |
| Full study population | ||||||
| 1) Positive antibody status with smoking statusa | 1.0 | ref | 0.76 | (0.54, 1.08) | 0.52 | (0.40, 0.68) |
| 2) Antibody status with infection, stratified by smokingb | ||||||
| negative | 1.0 | ref | 1.0 | ref | 1.0 | ref |
| positive | 1.03 | (0.60, 1.78) | 0.22 | (0.03, 1.79) | 0.18 | (0.04, 0.75) |
| Among women with five or more lifetime sexual partners | ||||||
| 1) Positive antibody status with smoking statusa | 1.0 | ref | 0.72 | (0.48, 1.10) | 0.52 | (0.39, 0.71) |
| 2) Antibody status with infection, stratified by smokingb | ||||||
| negative | 1.0 | ref | 1.0 | ref | 1.0 | ref |
| positive | 0.81 | (0.36, 1.80) | 0.32 | (0.04, 2.68) | 0.10 | (0.01, 0.72) |
all models adjusted for model covariates: age, age at sexual debut, and lifetime number of sexual partners
the stratification by smoking status is due to a significant interaction term between smoking and antibodies (P = 0.015);
negative antibody status is the referent group for each smoking category
When we restricted the study population to women with five or more lifetime sexual partners, the overall association between current smoking and HPV infection increased to an OR = 1.45 (95% CI: 0.88, 2.40) (Table 4). Further adjusting for the antibody mediator only slightly attenuated the overall association (OR = 1.34; 95% CI: 0.81, 2.23). For the causal mediation analysis, however, we observed results similar to our non-restricted study population: an OR = 1.29 (95% CI: 1.10, 1.81) for the indirect effect; and an OR = 0.93 (95% CI: 0.31, 1.97) for the direct effect (Table 4). The component associations of the indirect effect were similar though antibodies among never smokers were suggestive of an inverse association with infection (Table 3). We observed similar results when we restricted on age (24+ years), or reporting a new sexual partner during follow-up (Supplemental Table 1).
Table 4.
Odds ratios of current smoking relative to never on HPV-16 infection among women who have had five or more lifetime sexual partners.
| OR | 95% CI | |
|---|---|---|
| Overall associationa | 1.45 | (0.88, 2.40) |
| Mediator-adjusted associationb | 1.34 | (0.81, 2.23) |
| Causal mediation analysisa | ||
| Total natural indirect effect | 1.29 | (1.10, 1.81) |
| Pure natural direct effect | 0.93 | (0.31, 1.97) |
all models adjusted for model covariates: age, age at sexual debut, lifetime number of sexual partners
the traditional method assesses the degree of attenuation between the overall association and the mediator adjusted association; higher attenuation suggests more of an indirect effect, less attenuation suggests more of a direct effect
additionally adjusted for HPV-16 antibody status
As shown in Figure 2a-c, we performed the causal mediation analysis for 3 different subcategories of current smoking: intensity, duration, and initiation relative to sexual initiation. In all subcategories the estimated direct effects did not differ from the null value thus were not shown. For intensity, we did not observe a significant antibody-mediated effect for women who smoked less than half a pack of cigarettes per day compared to never smokers (OR = 1.09; 95% CI: 0.94, 1.44). But we did for women who smoked more: an OR = 1.59 (95% CI: 1.27, 2.27) for women smoking half a pack but less than a full pack; and an OR = 1.64 (95% CI: 1.27, 2.34) for women smoking at least a full pack per day. For duration, current smokers who had smoked for up to seven years prior had an OR = 1.30 (95% CI: 1.08, 1.82), while those who had smoked for more than seven years had an OR = 1.25 (95%: 0.99, 1.85). In the analyses relative to sexual initiation, current smokers who initiated smoking before sexual debut had an OR = 1.47 (95% CI: 1.18, 1.86), while those who initiated smoking after sexual debut had an OR = 1.14 (95% CI: 1.01, 1.52). Our results did not differ by race (Figure 2d).
Figure 2.
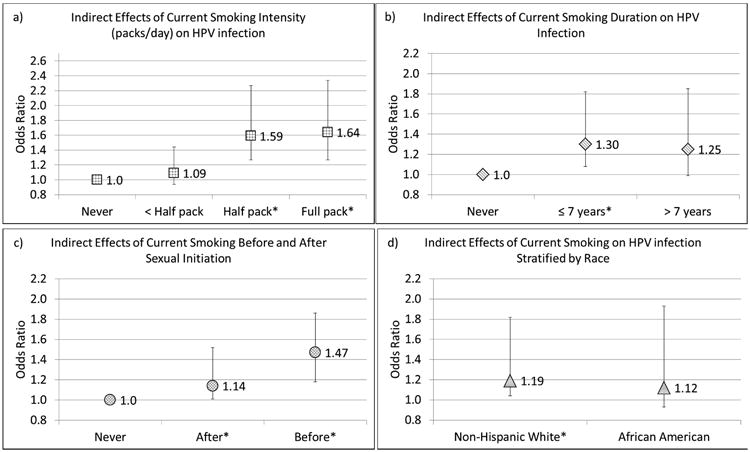
Odds ratios and confidence interval bars of indirect effects of current smoking on subsequent HPV-16 infection. a) by smoking intensity (packs per day); b) by smoking duration; c) by smoking initiation relative to sexual initiation; d) indirect effects among the non-hispanic white and African American populations with never smokers in each race category as the referent group. The * by the category name indicates statistical significance at P<0.05 two-sided level.
Discussion
We present a mediation analysis suggesting that current smoking increases the risk of acquiring a subsequent HPV-16 infection by lowering a woman's antibody titer. Though some past research may suggest this as a possible mechanism (9-11), this is the first study to test this hypothesis and estimate an increased risk. Although smoking is an established risk factor for an HPV infection advancing to cervical precancer (26), its role earlier in the natural history is less clear (27). The results of this study implicate smoking earlier in the natural history of HPV – increasing the risk of a subsequent infection, presumably due to impaired immunity.
Animal and epidemiologic evidence support that cigarette smoke impairs cellular and humoral immune responses. The tar and nicotine in cigarettes have immunosuppressive properties that can last weeks (12). Furthermore, cigarette smoking has been implicated in respiratory, HPV, and human immune-deficiency virus infections (28). Regarding the immune system and HPV infection, impairing the cellular response may be a mechanism for HPV persistence (29); and smokers may have longer persistence (30, 31). Impairing the humoral response and weakening immunity may be a mechanism for a heightened risk for subsequent infection (32) – which we observed in our study.
Past studies report an inverse association between current smoking and natural HPV antibody titers (9-11); and higher levels of antibodies may protect against subsequent infections (3-6). Combined, these results imply that smokers may have an increased risk of subsequent HPV infection through an immunity-based mechanism – by reducing antibodies, or preventing the development of antibodies that, if not impaired, may protect a woman from a subsequent infection. The results from our mediation analysis support this theory and estimate a statistically significant 29% increased odds of infection by that mechanism. Interestingly, that is stronger than the 8% increase we observed for the overall association between current smoking and HPV infection. Because of this discrepancy, our estimated direct effect was inverse, though very imprecise. The 8% overall association may be artificially low due to bias if not all subjects were previously exposed to HPV-16. When we tried to reduce this potential bias by restricting our study population to women reporting five or more sexual partners in their lifetime, the overall association grew from 8% higher odds to 45% but notably, the antibody-mediated indirect effect held at 29%. The resulting direct effect was null, thus suggesting that smoking has no direct effect. Among our mediation results, the antibody-mediated indirect effect was our only statistically significant finding, but that test may have the most power (33). Though our study is the first to attempt a mediation analysis to address the question of smoking's mechanism with HPV, our estimates are similar to the current literature, in which studies report weak to modest associations between smoking and HPV infection (34-39).
The antibody-mediated indirect effect for current smokers appears to be dose-dependent by the number of cigarettes smoked per day. We observed a 61% increased odds of infection for women who smoked at least half a pack of cigarettes compared to a 9% increase for those women who smoked less. This difference is in contrast to what we observed with smoking duration which showed no difference in the estimates between women who have smoked for 7 or fewer years or women who have smoked for more than 7 years. Given that we observed null results for former smokers, this suggests it is the current smoking behavior that increases a woman's risk, and not the cumulative effect of her past behavior. If true, reducing subsequent HPV risk is an added benefit to smoking cessation.
Our analysis focused on the biologic mechanisms related to subsequent HPV infection. Therefore, a previously exposed population is needed. Women were enrolled into the ALTS trial with an abnormal cytology result, enriching the population with subjects previously exposed to HPV. We assumed everyone had a past exposure to HPV-16, but we cannot be certain because other HPV types can also cause abnormal cytology. Testing positive for HPV-16 antibodies signifies past exposure, but to study HPV antibodies as a mechanism we cannot restrict on this factor. Restricting on greater sexual behavior will presumably increase the probability that subjects were previously exposed to HPV-16. When we restricted on five or more lifetime sexual partners the overall association between smoking and HPV infection got stronger, suggesting some possible bias in the overall association of 8% increased odds. We further investigated the issues of past infection and future risk by restricting on older age, and women reporting a new sexual partner during follow-up; we found similar results. Most importantly, the antibody-mediated results did not dramatically change in any of the populations, leaving us with the same conclusion – current smoking increases the risk of subsequent HPV infection by impairing immunity.
The results between the two mediation methods differed. The causal mediation analysis estimated a statistically significant antibody-mediated indirect effect with no convincing evidence of a direct effect. In contrast, the traditional analysis estimated imprecise overall associations that slightly attenuated when further adjusted for the mediator – suggesting more of a direct effect and less of an indirect effect. Though non-collapsibility of the odds ratio makes comparing the two methods more difficult (40), the difference between the two methods cannot be ignored. The discrepancy is due to the logistic equations for HPV antibodies and HPV infection, plus the exposure-mediator interaction we observed with smoking and HPV antibodies. Though we cannot be certain the statistical interaction represents a causal relationship, it could be the consequence of a more complex smoking, sexual behavior, immunologic causal pathway that we were unable to model. In the presence of interactions and nonlinear equations the results of the traditional method do not correspond to the mediating counterfactuals previously defined (19-21). Though the goal in employing the traditional method is to assess mediation, the method is limited by not guiding the appropriate estimation procedures with explicit mediation causal effects of interest – the causal method is not limited in this way. All mediation analyses, however, rely on the no confounding assumptions for unbiased estimation.
Some limitations of the current study should be noted. Importantly, the mediation analysis requires us to assume a causal model to estimate effects based off of that model. If our model is flawed – by perhaps omitting a confounder of the exposure-mediator effect or mediator-outcome effect – then our results will be biased. Using biological knowledge and past research, we built our model to the best of our ability. Though we acknowledge the possibility of unmeasured and residual confounding by sexual behavior, it would have to be independent of the sexual behavior variables that were available to us including reporting a new sexual partner during follow-up. We also recognize that some of the data may not fit our causal assumptions. For instance, HPV antibodies and smoking status we both measured at baseline, yet our model assumes a causal relationship. Past smoking status, however, may be causally relevant to HPV antibodies, and current smoking status is likely a good proxy (41). If we assume that past smoking behavior causes HPV infection, conditioning on infection status at baseline to obtain our at risk population opens the possibility of selection bias through an unmeasured risk factor for HPV infection (42, 43). This bias, however, is expected to be weak unless the unmeasured risk factor is particularly strong, and our model included all relevant risk factors available to us. Another potential source of bias is measurement error in the mediator, HPV antibodies. The GST-ELISA does not exclusively measure neutralizing antibodies – the antibodies that prevent internalization of the virus – thus we cannot rule out that our mediator may also be representing a mix of immune system mechanisms. The subsequent HPV infection outcome may, in some women, represent reactivation of a latent HPV infection. In either case, our findings of an impaired immune system mechanism would still be relevant. We were unable to extend this analysis to a neoplasia outcome because of the short follow-up time. Other datasets may be able to investigate the mechanisms of cervical neoplasia. Finally, our study population was collected before the implementation of the HPV vaccine thus our analysis focused on antibodies from natural infections. It does not address any role smoking may have in vaccine-initiated antibody titers, which are significantly greater than those induced by natural immunity.
By studying the underlying mechanism between smoking and subsequent HPV infection, we provide greater insight into the role of smoking in the natural history of HPV. We observed that current smoking increases the risk of re-acquiring HPV-16 by lowering a woman's antibody titer. This analysis implicates smoking earlier in the natural history of HPV and suggests an immune-related mechanism. To estimate the causal mechanism effects between smoking and HPV infection we used causal mediation analysis, and future studies can use this technique to investigate how smoking increases the risk for neoplasia, or how other cofactors are involved in the natural history of HPV.
Supplementary Material
Supplemental Table 1. Odds ratios and confidence intervals of our counterfactually-defined mediation analysis, and the component associations that comprise the indirect effect when restricting the population on having a new sexual partner during follow-up and on being 24+ years of age
Acknowledgments
This work was supported by the Intramural Research Program of the National Cancer Institute (Z01 CP010124) and the Cancer Prevention Fellowship Program of the National Cancer Institute
Abbreviations
- ALTS
Atypical Squamous Cells of Undetermined Significance Low-Grade Squamous Intraepithelial Lesion Triage Study
- ASCUS
Atypical Squamous Cells of Undetermined Significance
- CI
confidence interval
- DNA
deoxyribose-nucleic acid
- GST
glutathione S-transferase
- HPV
human papillomavirus
- LSIL
Low-Grade Squamous Intraepithelial Lesion
- MFI
median fluorescence intensity
- OR
odds ratio
Footnotes
Conflicts of Interest: The authors declare no potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. The Lancet. 370(9590):890–907. doi: 10.1016/S0140-6736(07)61416-0. [DOI] [PubMed] [Google Scholar]
- 2.Koshiol J, Lindsay L, Pimenta JM, Poole C, Jenkins D, Smith JS. Persistent Human Papillomavirus Infection and Cervical Neoplasia: A Systematic Review and Meta-Analysis. American Journal of Epidemiology. 2008;168(2):123–137. doi: 10.1093/aje/kwn036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ho GYF, Studentsov Y, Hall CB, Bierman R, Beardsley L, Lempa M, et al. Risk Factors for Subsequent Cervicovaginal Human Papillomavirus (HPV) Infection and the Protective Role of Antibodies to HPV-16 Virus-Like Particles. Journal of Infectious Diseases. 2002;186(6):737–742. doi: 10.1086/342972. [DOI] [PubMed] [Google Scholar]
- 4.Safaeian M, Porras C, Schiffman M, Rodriguez AC, Wacholder S, Gonzalez P, et al. Epidemiological Study of Anti-HPV16/18 Seropositivity and Subsequent Risk of HPV16 and -18 Infections. Journal of the National Cancer Institute. 2010 doi: 10.1093/jnci/djq384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson L, Pawlita M, Castle PE, Waterboer T, Sahasrabuddhe V, Gravitt PE, et al. Seroprevalence of 8 oncogenic human papillomavirus genotypes and acquired immunity against reinfection. J Infect Dis. 2014;210(3):448–55. doi: 10.1093/infdis/jiu104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wentzensen N, Rodriguez AC, Viscidi R, Herrero R, Hildesheim A, Ghosh A, et al. A Competitive Serological Assay Shows Naturally Acquired Immunity to Human Papillomavirus Infections in the Guanacaste Natural History Study. Journal of Infectious Diseases. 2011;204(1):94–102. doi: 10.1093/infdis/jir209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang SS, Schiffman M, Shields TS, Herrero R, Hildesheim A, Bratti MC, et al. Seroprevalence of human papillomavirus-16, -18, -31, and -45 in a population-based cohort of 10000 women in Costa Rica. Br J Cancer. 2003;89:1248–1254. doi: 10.1038/sj.bjc.6601272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porras C, Bennett C, Safaeian M, Coseo S, Rodriguez AC, Gonzalez P, et al. Determinants of seropositivity among HPV-16/18 DNA positive young women. BMC Infectious Diseases. 2010;10(1):238. doi: 10.1186/1471-2334-10-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiley DJ, Wiesmeier E, Masongsong E, Gylys KH, Koutsky LA, Ferris DG, et al. Smokers at higher risk for undetected antibody for oncogenic human papillomavirus type 16 infection. Cancer Epidemiology Biomarkers & Prevention. 2006;15(5):915–20. doi: 10.1158/1055-9965.EPI-05-0963. [DOI] [PubMed] [Google Scholar]
- 10.Wilson LE, Pawlita M, Castle PE, Waterboer T, Sahasrabuddhe V, Gravitt PE, et al. Natural immune responses against eight oncogenic human papillomaviruses in the ASCUS-LSIL Triage Study. Int J Cancer. 2013;133(9):2172–81. doi: 10.1002/ijc.28215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simen-Kapeu A, Kataja V, Yliskoski M, Syrjänen K, Dillner J, Koskela P, et al. Smoking impairs human papillomavirus (HPV) type 16 and 18 capsids antibody response following natural HPV infection. Scandinavian Journal of Infectious Diseases. 2008;40(9):745–751. doi: 10.1080/00365540801995360. [DOI] [PubMed] [Google Scholar]
- 12.Sopori M. Effects of cigarette smoke on the immune system. Nat Rev Immunol. 2002;2(5):372–377. doi: 10.1038/nri803. [DOI] [PubMed] [Google Scholar]
- 13.Schiffman M, Adrianza E. ASCUS-LSIL Triage Study. Acta Cytologica. 2000;44(5):726–742. doi: 10.1159/000328554. [DOI] [PubMed] [Google Scholar]
- 14.Gravitt PE, Peyton CL, Apple RJ, Wheeler CM. Genotyping of 27 human papillomavirus types by using L1 consensus PCR products by a single-hybridization, reverse line blot detection method. J Clin Microbiol. 1998;36(10):3020–7. doi: 10.1128/jcm.36.10.3020-3027.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waterboer T, Sehr P, Michael KM, Franceschi S, Nieland JD, Joos TO, et al. Multiplex Human Papillomavirus Serology Based on In Situ–Purified Glutathione S-Transferase Fusion Proteins. Clinical Chemistry. 2005;51(10):1845–1853. doi: 10.1373/clinchem.2005.052381. [DOI] [PubMed] [Google Scholar]
- 16.Clifford GM, Shin HR, Oh JK, Waterboer T, Ju YH, Vaccarella S, et al. Serologic Response to Oncogenic Human Papillomavirus Types in Male and Female University Students in Busan, South Korea. Cancer Epidemiology Biomarkers & Prevention. 2007;16(9):1874–1879. doi: 10.1158/1055-9965.EPI-07-0349. [DOI] [PubMed] [Google Scholar]
- 17.Robins JM, Greenland S. Identifiability and exchangeability for direct and indirect effects. Epidemiology. 1992;3(2):143–55. doi: 10.1097/00001648-199203000-00013. [DOI] [PubMed] [Google Scholar]
- 18.Pearl J. Direct and indirect effects In: Proceedings of the Seventeenth conference on Uncertainty in artificial intelligence. Seattle, Washington: Morgan Kaufmann Publishers Inc; 2001. pp. 411–420. [Google Scholar]
- 19.Vanderweele T, Vansteelandt S. Conceptual Issues Concerning Mediation, Interventions and Composition. Statistics and Its Interface. 2009;2:457–468. [Google Scholar]
- 20.Pearl J. The causal mediation formula--a guide to the assessment of pathways and mechanisms. Prev Sci. 2012;13(4):426–36. doi: 10.1007/s11121-011-0270-1. [DOI] [PubMed] [Google Scholar]
- 21.Muthén B, Asparouhov T. Causal Effects in Mediation Modeling: An Introduction With Applications to Latent Variables. Structural Equation Modeling: A Multidisciplinary Journal. 2014;22(1):12–23. [Google Scholar]
- 22.Muthén LK, Muthén BO. Mplus User's Guide. Seventh. Los Angeles, CA: Muthén & Muthén; 1998-2012. [Google Scholar]
- 23.MacKinnon DP, Lockwood CM, Williams J. Confidence Limits for the Indirect Effect: Distribution of the Product and Resampling Methods. Multivariate Behavioral Research. 2004;39(1):99–128. doi: 10.1207/s15327906mbr3901_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hafeman DM. “Proportion Explained”: A Causal Interpretation for Standard Measures of Indirect Effect? American Journal of Epidemiology. 2009;170(11):1443–1448. doi: 10.1093/aje/kwp283. [DOI] [PubMed] [Google Scholar]
- 25.SAS, Institute, Inc. Base SAS 9.3 Procedures Guide. Second. Cary, NC: SAS Institute Inc; 2012. [Google Scholar]
- 26.Castellsague X, Munoz N. Chapter 3: Cofactors in human papillomavirus carcinogenesis--role of parity, oral contraceptives, and tobacco smoking. J Natl Cancer Inst Monogr. 2003;(31):20–8. [PubMed] [Google Scholar]
- 27.Franco EL, Spence AR. Commentary: Smoking and human papillomavirus infection: the pursuit of credibility for an epidemiologic association. International Journal of Epidemiology. 2008;37(3):547–548. doi: 10.1093/ije/dyn057. [DOI] [PubMed] [Google Scholar]
- 28.Arcavi L, Benowitz NL. Cigarette smoking and infection. Archives of Internal Medicine. 2004;164(20):2206–2216. doi: 10.1001/archinte.164.20.2206. [DOI] [PubMed] [Google Scholar]
- 29.Welters MJP, de Jong A, van den Eeden SJF, van der Hulst JM, Kwappenberg KMC, Hassane S, et al. Frequent Display of Human Papillomavirus Type 16 E6-specific Memory T-Helper Cells in the Healthy Population as Witness of Previous Viral Encounter. Cancer Research. 2003;63(3):636–641. [PubMed] [Google Scholar]
- 30.Richardson H, Abrahamowicz M, Tellier PP, Kelsall G, du Berger R, Ferenczy A, et al. Modifiable risk factors associated with clearance of type-specific cervical human papillomavirus infections in a cohort of university students. Cancer Epidemiology Biomarkers & Prevention. 2005;14(5):1149–56. doi: 10.1158/1055-9965.EPI-04-0230. [DOI] [PubMed] [Google Scholar]
- 31.Koshiol J, Schroeder J, Jamieson DJ, Marshall SW, Duerr A, Heilig CM, et al. Smoking and Time to Clearance of Human Papillomavirus Infection in HIV-Seropositive and HIV-Seronegative Women. American Journal of Epidemiology. 2006;164(2):176–183. doi: 10.1093/aje/kwj165. [DOI] [PubMed] [Google Scholar]
- 32.Roden RB, Armstrong A, Haderer P, Christensen ND, Hubbert NL, Lowy DR, et al. Characterization of a human papillomavirus type 16 variant-dependent neutralizing epitope. J Virol. 1997;71(8):6247–52. doi: 10.1128/jvi.71.8.6247-6252.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Rourke HP, MacKinnon DP. When the test of mediation is more powerful than the test of the total effect. Behav Res Methods. 2015;47(2):424–42. doi: 10.3758/s13428-014-0481-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sellors JW, Karwalajtys TL, Kaczorowski J, Mahony JB, Lytwyn A, Chong S, et al. Incidence, clearance and predictors of human papillomavirus infection in women. Canadian Medical Association Journal. 2003;168(4):421–425. [PMC free article] [PubMed] [Google Scholar]
- 35.Muñoz N, Méndez F, Posso H, Molano M, van den Brule AJC, Ronderos M, et al. Incidence, Duration, and Determinants of Cervical Human Papillomavirus Infection in a Cohort of Colombian Women with Normal Cytological Results. Journal of Infectious Diseases. 2004;190(12):2077–2087. doi: 10.1086/425907. [DOI] [PubMed] [Google Scholar]
- 36.Collins S, Rollason TP, Young LS, Woodman CB. Cigarette smoking is an independent risk factor for cervical intraepithelial neoplasia in young women: a longitudinal study. Eur J Cancer. 2010;46(2):405–11. doi: 10.1016/j.ejca.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moscicki A, Hills N, Shiboski S, et al. Risks for incident human papillomavirus infection and low-grade squamous intraepithelial lesion development in young females. JAMA. 2001;285(23):2995–3002. doi: 10.1001/jama.285.23.2995. [DOI] [PubMed] [Google Scholar]
- 38.Winer RL, Lee SK, Hughes JP, Adam DE, Kiviat NB, Koutsky LA. Genital Human Papillomavirus Infection: Incidence and Risk Factors in a Cohort of Female University Students. American Journal of Epidemiology. 2003;157(3):218–226. doi: 10.1093/aje/kwf180. [DOI] [PubMed] [Google Scholar]
- 39.Schabath MB, Villa LL, Lin HY, Fulp WJ, Lazcano-Ponce E, Salmer ón J, et al. A prospective analysis of smoking and human papillomavirus (HPV) infection among men in The HPV in Men (HIM) Study. International journal of cancer Journal international du cancer. 2014;134(10):2448–2457. doi: 10.1002/ijc.28567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greenland S, Robins J, Pearl J. Confounding and collapsibility in causal inference. Stat Sci. 1999;14(1):29–46. [Google Scholar]
- 41.Soulakova JN, Hartman AM, Liu B, Willis GB, Augustine S. Reliability of adult self-reported smoking history: data from the tobacco use supplement to the current population survey 2002-2003 cohort. Nicotine Tob Res. 2012;14(8):952–60. doi: 10.1093/ntr/ntr313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greenland S. Quantifying biases in causal models: classical confounding vs collider-stratification bias. Epidemiology. 2003;14(3):300–6. [PubMed] [Google Scholar]
- 43.Flanders WD, Eldridge RC, McClellan WA. Nearly Unavoidable Mechanism for Collider Bias with Index-Event Studies. Epidemiology. 2014;25(5) doi: 10.1097/EDE.0000000000000131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Odds ratios and confidence intervals of our counterfactually-defined mediation analysis, and the component associations that comprise the indirect effect when restricting the population on having a new sexual partner during follow-up and on being 24+ years of age