

Abstract
Parkinson’s disease (PD) is characterized by accumulations of toxic α-synuclein aggregates in vulnerable neuronal populations in the brainstem, midbrain and cerebral cortex. Recent findings suggest that α-synuclein pathology can be propagated transneuronally, but the underlying molecular mechanisms are unknown. Advances in the genetics of rare early-onset familial PD indicate that increased production and/or reduced autophagic clearance of α-synuclein can cause PD. The cause of the most common late-onset PD is unclear, but may involve metabolic compromise and oxidative stress upstream of α-synuclein accumulation. As evidence, the lipid peroxidation product 4-hydroxynonenal (HNE) is elevated in the brain during normal aging and moreso in brain regions afflicted with α-synuclein pathology. Here we report that HNE increases aggregation of endogenous α-synuclein in primary neurons, and triggers the secretion of extracellular vesicles (EVs) containing cytotoxic oligomeric α-synuclein species. EVs released from HNE-treated neurons are internalized by healthy neurons which as a consequence degenerate. Levels of endogenously generated HNE are elevated in cultured cells overexpressing human α-synuclein, and EVs released from those cells are toxic to neurons. The EV-associated α-synuclein is located both inside the vesicles, and on their surface where it plays a role in EV internalization by neurons. Upon internalization, EVs harboring pathogenic α-synuclein are transported both anterogradely and retrogradely within axons. Focal injection of EVs containing α-synuclein into the striatum of wild type mice results in spread of synuclein pathology to anatomically connected brain regions. Our findings suggest a scenario for late-onset PD in which lipid peroxidation promotes intracellular accumulation and then extrusion of EVs containing toxic α-synuclein species; the EVs are then internalized by adjacent neurons, so propagating the neurodegenerative process.
Introduction
Parkinson’s disease (PD), dementia with Lewy bodies and multiple system atrophy are age-related neurodegenerative disorders characterized by accumulations of α-synuclein in the soma (Lewy bodies) and neurites (Baba et al., 1998; Jellinger, 2011; Spillantini et al., 1998a; Spillantini et al., 1998b; Spillantini et al., 1997; Wakabayashi et al., 1998). α-synuclein is a highly conserved 140 amino acid presynaptic protein which associates with membranes, and is believed to play a role in the regulation of neurotransmitter release (George et al., 1995; Iwai et al., 1995; Withers et al., 1997; Abeliovich et al., 2000; Burre et al., 2010; Vargas et al., 2014). Mutations in the gene encoding α-synuclein cause dominantly inherited PD (Polymeropoulos et al., 1997; (Kruger et al., 1998; Zarranz et al., 2004; Lesage et al., 2013), and duplication or triplication of the α-synuclein gene (Chartier-Harlin et al., 2004; Farrer et al., 2004; Kara et al., 2014; Singleton et al., 2003) indicating that overproduction of α-synuclein is sufficient to trigger PD during aging. When mutated or overexpressed, α-synuclein monomers tend to assemble into helically folded tetramers that resist aggregation in the cytosol (Bartels et al., 2011).
Normally, α-synuclein is degraded in proteasomes and by autophagy in lysosomes (Cuervo et al., 2004; Mak et al., 2010; Snyder et al., 2003; Webb et al., 2003; Xilouri et al., 2013). Knockdown of the autophagy pathway proteins Atg5 or Atg7 results in the accumulation of α-synuclein aggregates and neurological deficits similar to PD (Hara et al., 2006; Komatsu et al., 2006), while overexpression of wild type α-synuclein or its mutants inhibits autophagy (Chen et al., 2015; Chew et al., 2011; Choubey et al., 2011; Huang et al., 2012; Koch et al., 2015; Winslow et al., 2010). In addition to ubiquitination (Lee et al., 2008b; Nonaka et al., 2005), α-synuclein can be post-translationally modified by phosphorylation (Anderson et al., 2006; Hasegawa et al., 2002; Okochi et al., 2000) and acetylation (Trexler and Rhoades, 2012), which may influence its aggregation and degradation. One age-related factor that may contribute to the aggregation and cytotoxicity of α-synuclein is oxidative stress. In this regard the lipid peroxidation product 4-hydroxynonenal (HNE) is implicated in PD because of its accumulation in Lewy bodies (Castellani et al., 2002; Yoritaka et al., 1996), and because it can covalently modify α-synuclein (Qin et al., 2007; Nasstrom et al., 2011) and can impair lysosome function in neurons (Zhang et al., 2017). However, if and how HNE might cause intracellular accumulation and transcellular propagation of neurotoxic α-synuclein species is unknown.
Studies of PD patients at various stages of the disease process suggested that α-synuclein pathology spreads transcellularly, and may occur in peripheral neurons before spreading rostrally to the brainstem, and thence to the midbrain and cerebral cortex (Braak et al., 2006; Braak et al., 2003). Recent studies have shown that when preformed α-synuclein fibrils are introduced into the olfactory bulb (Rey et al., 2016) or striatum (Luk et al., 2012), the α-synuclein pathology spreads in a retrograde manner. In addition, when Lewy body extracts from PD patient brains were injected into the brains of monkeys, they induced a spreading α-synuclein pathology and neurodegeneration (Recasens et al., 2014). The mechanism by which α-synuclein pathology is transmitted between neurons is unclear, but may involve a prion-like seeding process (Goedert, 2015). It has also been reported that neurons can secrete pathogenic forms of amyloid β-peptide or α-synuclein in extracellular vesicles (EVs; also known as exosomes) in experimental models relevant to AD and PD, respectively (Danzer et al., 2012; Eitan et al., 2016). However, the possible involvement of membrane lipid peroxidation in the extrusion of α-synuclein in EVs and their transcellular pathogenicity is unknown. Here we report that HNE modifies α-synuclein which triggers intracellular accumulation and subsequent extrusion of toxic α-synuclein species in and on the surface of EVs. The EVs with HNE-modified α-synuclein are then internalized by and transported within adjacent neurons, thus propagating α-synuclein pathology and causing neuronal degeneration.
Materials and Methods
Reagents
HNE was from Cayman Chemical Company (Ann Arbor, MI). Anti- α-synuclein antibody (C20) was from Santa Cruz (Dallas, TX; catalog # sc-7011), anti- human α-synuclein antibody (Syn211) was from Thermo Fisher Scientific (Waltham, MA, catalog # MS-1572). Anti- S129 phosphorylated α-synuclein antibody was from Wako (Richmond, VA, catalog # 015-25191). Anti-actin antibody was from Sigma (St. Louis, MO; catalog #A2066). Anti-HNE antibody was from Alpha Diagnostic (San Antonio, TX; catalog #HNE13-M). Anti- flotillin-1 antibody (catalog # ab133497), anti- calnexin antibody (catalog # ab22595), anti α-synuclein filament antibody (FILA-1, catalog # ab209538) were from Abcam (Cambridge, MA). The MTS assay kit was from Promega Life Sciences (Madison, WI) and the LDH assay kit was from Roche Diagnostics (Indianapolis, IN). Microfluidic chambers were from Xona Microfluidics (Temecula, CA, catalog # SND450). The Elite ABC Kit (catalog # PK-6100) and DAB Peroxidase Substrate Kit (catalog # SK-4100) were from Vector Labs (Burlingame, CA). Blasticidin S HCl (catalog # A1113903), the ViraPower™ II Lentiviral Gateway® Expression System (catalog # K36720), antibiotic-antimycotic (catalog # 15240062), exosome-depleted FBS (catalog # A2720801) and the BCA protein assay kit (catalog # 23225) were from Thermo Fisher Scientific (Waltham, MA). α-synuclein CRISPR/Cas9 KO plasmid (catalog # sc-417273) and puromycin dihydrochloride (catalog # sc-108071) were from Santa Cruz (Dallas, TX). Ultra-Clear™ Beckman Coulter centrifuge tubes (344059), polypropylene centrifuge tubes (catalog #s 326823 and 326819) were from Beckman Coulter (Brea, CA). All other reagents were obtained from Sigma (St. Louis, MO).
Animals
Male B6.Cg-Tg(Thy-1-SNCA*A53T) M53Sud/J (SNCA) mice (Chandra et al., 2005) (original breeding pairs were purchased from Jackson Laboratories, Bar Harbor, ME) and wild type (WT) littermates were housed under a 12 hour light/12 hour dark cycle. SNCA mice express the A53T mutant human α-synuclein under the control of the murine Thy-1 promoter and exhibit α-synuclein accumulation in the midbrain and striatum, and age-dependent motor impairment. For biochemical studies, tissues were flash frozen after removal and stored at −80°C until used. Timed pregnant female Sprague-Dawley rats were purchased from Charles River Laboratories, Raleigh, NC). All procedures were approved by the Institutional Animal Care and Use Committee of the National Institute on Aging, and complied with NIH guidelines.
Stereotaxic injections
Wild type mice at 3 months of age or 24 months of age were anesthetized with isoflurane and EVs isolated from the culture medium of α-synuclein overexpressing HEK cells were injected stereotaxically (5 μg EV protein equivalents in 3 μl saline injected during a 5 minute period) using a 10 μl Hamilton syringe into the right dorsal striatum (coordinates: +0.2 mm relative to Bregma; +2.0 mm from the midline; 2.6 mm beneath the dura); control animals received EVs isolated from the culture medium of HEK SNCA knockout cells. Mice were monitored regularly following recovery from surgery, and euthanized at 5 days after EV injection by overdose with isoflurane. For histological studies the brain was removed after transcardial perfusion with PBS followed by 4% paraformaldehyde in PBS (pH 7.4). The brains were then post-fixed overnight in 4% paraformaldehyde in PBS before being immersed in 30% sucrose in PBS to cryopreserve the brains in preparation for cryostat sectioning.
Immunohistochemistry
Immunohistochemistry was performed on 40 μm thick free-floating coronal sections. After incubated with 3% hydrogen peroxide to exhaust endogenous peroxidases, brain sections were incubated with anti-human α-synuclein antibody (C20, 1:1000) at 4°C overnight, then processed with 3′-diaminobenzidine (Vector Laboratories) as a chromogen. Sections were subsequently stained with cresyl violet using the Nissl staining method. Images were captured with a Qimaging Retiga 2000R digital camera connected to a Nikon Eclipse E600 microscope (Melville, NY).
Primary neuronal culture
Cultures of primary cortical neurons were prepared from embryonic day 18 (E18) Sprague-Dawley rats. Cells were dissociated by trypsinization and trituration in Ca2+- and Mg2+-free Hank’s Balanced Saline Solution the presence of DNAase to prevent cell clumping. Dissociated cells were plated onto polyethyleneimine-coated plastic dishes or glass coverslips in MEM medium (Invitrogen Inc., Carlsbad, CA) supplemented with 10% Hyclone III (Thermo Fisher Scientific) at a density of 3.0 ×105 cells/cm2 for biochemistry and 1.0 × 104 cells/cm2 for imaging analyses. After cells attached to the surface (4 hours), the medium was changed to Neurobasal medium containing B27 supplements (Invitrogen). Every 7 days 50% of the medium volume was removed and replaced with fresh medium. Immediately prior to experimental treatment, the culture medium was replaced with fresh Neurobasal medium. HNE was prepared as a 2000X stock in ethanol; control cultures were exposed to ethanol in the same amount as the cultures exposed to HNE.
HEK cell cultures
The stable HEK-wt-SNCA cell line was generated with ViraPower™ lentiviral expression system from Invitrogen according to manufacturer’s instructions. The human wt-SNCA expressing plasmid was a generous gift from Mark R. Cookson at the National Institute on Aging. A stable HEK SNCA knockout cell line was generated using an α-synuclein CRISPR/Cas9 KO plasmid from Santa Cruz. All HEK cell lines were cultured in DMEM medium containing 10% Hyclone III, penicillin (100 U/ml), streptomycin (100 μg/ml) and amphotericin B (0.25 μg/mL). Cells overexpressing WT α-synuclein were maintained in the presence of 10 μg/ml blasticidin. SNCA-KO cells were maintained in the presence of 5 μg/ml puromycin dihydrochloride. For EV isolation, cells were grown in DMEM medium with exosome-depleted FBS (A2720801) for 48 hours. The supernatants were then collected and stored at −80°C for EV isolation and purification.
Isolation and purification of EVs
Isolation of EVs was performed as described previously (Vlassov et al., 2012; Eitan et al., 2016) with a few modifications. Briefly, 35 milliliters of culture medium was first centrifuged at 2000 g for 30 min at 4°C to remove dead cells and debris, and the supernatant was further centrifuged at 10,000 g for 40 min at 4°C. The resulting supernatant was collected and centrifuged at 120,000 g for 2 hours. The pellets were washed once in PBS and centrifuged at 120,000 g for 2 hours. The pellets were resuspended in 100 μl PBS for further experiments, subjected to PKH staining (PKH67-green; PKH26-red) with one more ultracentrifugation at 120,000 g for immunofluorescence imaging analyses, or reconstituted in RIPA lysis buffer for immunoblot analysis. To confirm the presence of α-synuclein in EVs, some isolated EVs were further purified using an OptiPrep™ density gradient. Briefly, a discontinuous iodixanol gradient was prepared by diluting a stock solution of OptiPrep™ (60% w/v) with 0.25 M sucrose/10 mM Tris, pH 7.5 to generate 40%, 30%, 25%, 20%, and 10% w/v iodixanol solutions. The discontinuous iodixanol gradient was generated by sequentially layering 2 mL each of 40, 30, 25, 20 and 10% (w/v) iodixanol solutions in 14×89 mm Ultra-Clear™ Beckman Coulter centrifuge tubes. A 2 ml volume of EVs was overlaid on the discontinuous iodixanol gradient and centrifuged in a SW 40 Ti rotor for 18 hours at 100,000 g at 4°C. Six 2 ml fractions were collected from the top of the gradient. Each fraction was diluted to 30 ml in PBS and centrifuged at 120,000 g for 2 hours at 4°C in a SW 28 Ti rotor. The resulting pellets were resuspended in 100 μl of 4X LDS sample buffer (Thermo Fisher Scientific).
q-Nano analysis of EVs
Isolated EVs were loaded on a 200 nm nanopore membrane on the qNano platform and their sizes and numbers were quantified with the q-Nano system (Izon Science, Cambridge, MA) according to the manufacturer’s instructions.
Electron microscopy
Isolated exosomes were allowed to adsorb to freshly ionized 300 mesh formvar/carbon-coated grids, washed briefly through 5–7 puddles of ddH2O; and negatively stained in 2% aqueous uranyl acetate. Images were acquired using a transmission electron microscope (FEI Tecnai G2 Spirit) with TWIN Lens operating at 100 kV with an Olympus Soft Imaging System Megaview III digital CCD.
Cell viability assays
The MTS assay (Promega Corporation, Madison, WI) and LDH assay (Roche Life Science, Indianapolis, IN) were performed according to the manufacturer’s instructions. Neurons were grown in 96-well plates at a density of 10,000 cells/well for 7 days. Different treatments were then added to the culture medium for designated time periods. The culture medium was removed for the LDH assay and cells were left in the wells for the MTS assay. For both assays, the absorbance (optical density) value was read at a wavelength of 492 nm with a microplate reader (Biotek, Winooski, VT).
Immunoblot assays for denatured proteins
For preparation of nonionic detergent-soluble and detergent-insoluble fractions, neurons were homogenized in ice-cold 1% Triton X-100 lysis buffer (50 mM Tris·HCl, pH 7.4, 150 mM NaCl, 0.1%SDS, 1% Triton X-100, 1% sodium deoxycholate, supplemented with protease inhibitor cocktail), followed by sonication. Homogenates were centrifuged at 15,600 × g for 20 min at 4°C to separate supernatants (Triton X-100-soluble fractions). The resulting pellets were further lysed by sonication in a buffer containing 2% SDS and a protease inhibitor cocktail, followed by centrifugation at 15,600 × g for 10 min at room temperature to separate the new supernatants (Triton X-100–insoluble fractions). For other immunoblot assays, cells, EVs or brain tissue were lysed in radioimmunoprecipitation assay buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 0.1% SDS, 1% NP-40, 2 mM EDTA, 1 mM DTT, 1 mM PMSF, 200 M Na3VO4, 50 mM NaF and protease inhibitors (Roche) on ice for 20 min with brief sonication, and centrifuged at 15,600 × g for 15 minutes. The supernatants were collected for immunoblot analysis. The protein concentration of samples was determined using a BCA protein assay kit (Thermo Fisher Scientific). Denatured samples were heated to 70°C in NuPAGE LDS sample buffer (Invitrogen) and loaded onto NuPAGE Bis-Tris gels (Invitrogen) for protein separation. Following electrophoresis, proteins were transferred from the gel to a nitrocellulose sheet by electrophoresis and the sheet was stained with Ponceau S solution (Sigma) to assess the efficiency of protein transfer. Then the sheet was washed with deionized-water and incubated overnight at 4°C with a primary antibody (see antibody sources in the first paragraph of the Methods section above) which included: anti-actin (1:500 dilution), anti-HNE (1:2000), anti- α-synuclein antibody C20 (1:1000), anti- human α-synuclein antibody Syn211(1:1000), anti- S129 phosphorylated α-synuclein antibody (1:1000), anti-flotillin-1 antibody (1:5000), and anti- calnexin antibody (1:1000). The sheet was then incubated in the presence of HRP-conjugated secondary antibodies (Cell Signaling Technology) and the immunolabeled proteins were visualized using an enhanced chemiluminescence kit (Thermo Fisher Scientific). Analysis of immunolabeled band intensity was performed using Image J software (NIH) and values were normalized to the intensity of the actin band for the same protein sample.
Native gel immunoblot analysis
For extraction of proteins for native gel immunobloting of α-synuclein, the cells or brain tissue were harvested, washed twice with ice-cold PBS, briefly sonicated in PBS, and centrifuged at 1,000 × g for 15 minutes at 4°C. The supernatants were analyzed using the NativePAGE™ Bis-Tris Gel system. After transfer of protein to a PVDF membrane, the membrane was incubated in 20 ml of 8% acetic acid for 15 minutes to fix the proteins. The membrane was then rinsed with deionized water and processed for immunodetection of designated proteins using the protocol for denatured proteins described above.
Dot Blot Assay
EV lysates were spotted directly on the nitrocellulose membrane, forced go through membrane by applying a vacuum, and let it dry completely. Membrane was subsequently blocked for 1 hour with 5% bovine serum albumin in TBST and incubated with FILA-1 at 4°C overnight. The following steps were the same as regular Western Blot assay. Immunoreactivity was visualized using chemiluminescence detection (Pierce) after incubations with anti-rabbit horseradish peroxidase-conjugated secondary antibody, using x-ray films. The immunoreactive dot intensities were determined by Image J software.
Proteinase K digestion
The lysates of EVs were treated with or without 50 μg/ml proteinase K (PK) for 20 min on ice. The reaction was stopped by addiction of 2 mm (final concentration) phenylmethyl sulfonyl fluoride.
Rat α-synuclein oligomer ELISA assay
Conditioned culture medium was harvested from rat primary cortical neuron cultures after 24 hours’ treatment of vehicle control or HNE. The medium was centrifuged at 2000 g for 30 min at 4°C to remove dead cells and debris, and the supernatant was further centrifuged at 10,000 g for 40 min at 4°C to remove large microvesicles. The supernatants were then used for α-synuclein oligomer detection using a rat α-synuclein oligomer ELISA kit from Mybiosource (San Diego, CA, catalog # MBS733196) according to the manufacturer’s protocol. To detect EV-associated α-synuclein oligomers, we lysed EVs in the supernatant in 1% Trition X-100 lysis buffer at a dilution of 1:2. Samples (100 μl) were assayed in duplicate. Optical density values were at 450 nm were determined using a microplate reader.
Trypsin treatment of EVs
EVs were incubated with trypsin (1:20, trypsin:protein ratio) with or without 0.1% saponin in PBS for 2 hours at 37°C. The digestion was halted with trypsin inhibitor (1:1, trypsin:trypsin inhibitor ratio). The resulting EVs were lysed directly in 4X LDS sample buffer for subsequent immunoblot analysis.
Immunofluorescence and confocal microscopy
Cortical neurons were grown on glass coverslips with or without specific treatments for designated time periods before immunostaining. Neurons were washed in PBS twice and then fixed in PBS containing 4% paraformaldehyde for 20 min at 23°C. Neurons were then incubated in blocking solution (PBS containing 3% bovine serum albumin and 0.2% Triton X-100) for 30 minutes, and then incubated with a primary antibody diluted in blocking solution overnight at 4°C. Primary antibodies included: anti- α-synuclein antibody C20 (1:1000), anti- human α-synuclein antibody Syn211 (1:400). Alexa Fluor 488, 568 and 633 (Invitrogen) were used as secondary antibodies. Cell nuclei were stained with Hoechst 33258 dye. Coverslips were mounted in Mountant Permafluor medium (Thermo Fisher Scientific) and visualized using a Zeiss LSM410 confocal microscope with a 40X objective lens. Images were analyzed with Image J software.
Statistics
All data are presented as mean ± SEM. Comparisons between control and treatment groups were performed by using Student’s unpaired t-test or analysis of variance where appropriate. Values of p<0.05 were considered statistically significant.
Results
Overexpression of human α-synuclein results in the accumulation of α-synuclein aggregates and HNE-modified proteins
To establish a cellular system for the production and release in EVs of potentially pathogenic forms of α-synuclein, we overexpressed wild type human α-synuclein in human embryonic kidney (HEK) cells (HEK-wt-SNCA cells), and then performed immunoblot analyses of cell lysates prepared in RIPA buffer. Lysates with the same amount of protein were loaded and separated in NuPAGE Bis-Tris gels. The analysis demonstrated that levels of monomeric α-synuclein (approximately 17 kDa) were more than 12-fold greater in cells overexpressing α-synuclein compared to non-transfected cells (Figure 1A and B). To determine if cells that accumulate α-synuclein aggregates exhibit increased oxidative stress, we performed immunoblot analysis of proteins from control and HEK-wt-SNCA cells using an antibody against HNE, a lipid peroxidation product that covalently modifies proteins on lysine residues (Mattson, 2009). We found a clear increase of HNE-modified proteins of a wide range of molecular weights in HEK-wt-SNCA cells compared to control cells (Figure 1C). To further measure the aggregation status of α-synuclein in control and HEK-wt-SNCA cells, we performed immunoblot analysis of non-denatured proteins using an α-synuclein antibody (Santa Cruz C20, recognizes α-synuclein from all species), and used proteins from cerebral cortex of 1 year old wild type and Thy1-A53T-SNCA human α-synuclein mutant transgenic mice as negative and positive controls, respectively. Large amounts of presumptive α-synculein aggregates with molecular weights ranging from approximately 30 – 70 kDa were present in samples from HEK-wt-SNCA cells but not in samples from control HEK cells (Figure 1D). As expected, samples from the brains of α-synuclein mutant transgenic mice exhibit large amounts of α-synuclein aggregates, ranging from approximately 30 kDa to over 500 kDa in size, whereas no α-synuclein aggregates were detected in samples from wild type mice (Figure 1D).
Figure 1. Cultured cells overexpressing wild type human α-synuclein accumulate oligomeric aggregates of α-synuclein.
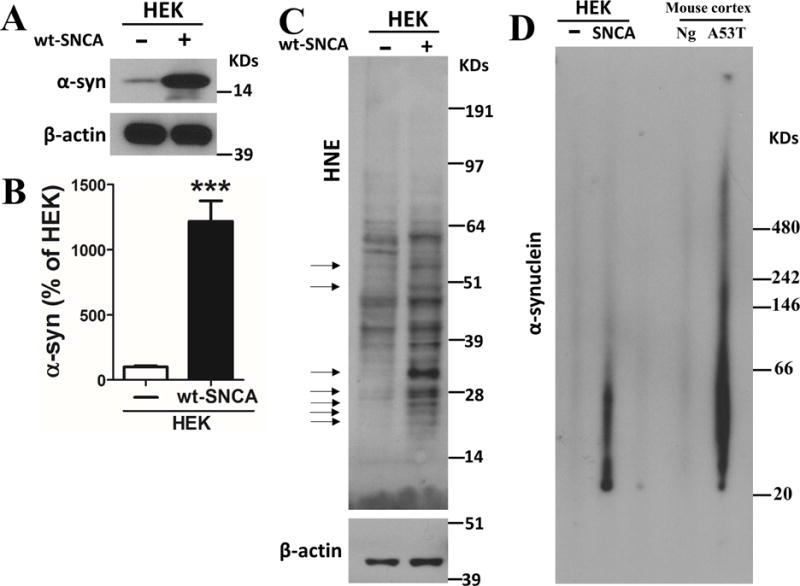
(A) Immunoblot showing relative levels of α-synuclein and β-actin as an internal control in HEK cells with or without human wildtype α-synuclein (wt-SNCA) overexpression. (B) Results of densitometric analysis of α-synuclein levels in HEK cells with or without human wildtype α-synuclein overexpression. Values are the mean and SEM of determinations made in 9 independent experiments. ***p<0.001. (C) Immunoblot showing relative levels of HNE-protein adducts and β-actin in HEK cells with or without human wild type α-synuclein overexpression. Arrows point to protein bands that exhibit much higher levels of 4-hydroxynonenal (HNE) immunoreactivity in samples from the cells overexpressing wild type α-synuclein. (D) Lysates prepared from HEK cells with or without α-synuclein overexpression, and cerebral cortex from nontransgenic (Ng) and Thy1-A53T-SNCA transgenic mice were run in NativePAGE™ Bis-Tris gels. The immunoblot reveals high amounts of α-synulcein aggregates in samples of HEK cells overexpressing α-synuclein and cortical tissue from Thy1-A53T-SNCA transgenic mice, compared to respective control HEK cells and wild type mice.
HNE induces α-synuclein aggregation in rat primary cortical neurons
Aging is the major risk factor for PD, and the accumulation of oxidatively modification of proteins, including covalent modification by HNE, occurs during normal aging, and is greatly exacerbated in vulnerable brain regions of PD patients and animal models of PD (Dexter et al., 1989; Castellani et al., 2002; Droge and Schipper, 2007; Di Domenico et al., 2016; Plotegher and Bubacco, 2016). We therefore determined whether exposure of neurons to HNE is sufficient to trigger accumulation of α-synuclein in neurons. Cultured primary cortical neurons were treated with vehicle control (ethanol) or 5 μM HNE, and 24 hours later the neurons were harvested and lysed in buffer containing 1% Triton X-100 buffer. After centrifugation, supernatants were collected and labeled as Triton X-100 soluble fraction (S). The pellets were resuspended in 2% SDS lysis buffer and designated as the insoluble fraction (IS). There was no difference in the amount of monomeric α-synuclein in control and HNE-treated neurons (Figure 2A and B). In contrast, there was an approximately 7-fold greater abundance of higher molecular weight α-synuclein aggregates in both the of S and IS fractions of HNE-treated neurons compared to control neurons (Figure 2A and B). In vehicle-treated control neurons, α-synuclein immunoreactivity was distributed diffusely throughout the cell body and neurites; in contrast, in neurons exposed to HNE the α-synuclein immunoreactivity was concentrated in aggregates of various sizes throughout the neurites and cell body (Figure 2C). Lewy bodies in PD patients are enriched in α-synuclein phosphorylated on serine residue 129 (Hasegawa et al., 2002). We therefore used an antibody that only binds pS129 α-synuclein to determine whether α-synuclein induced by HNE exhibit a Lewy body-like molecular signature. Immunoblot analysis of S and IS fractions of cell lysates from neurons that had been exposed to vehicle or HNE for 4 days revealed that α-synuclein aggregates in both fractions were immunoreactive with the pS129 antibody, with the relative intensities of bands in samples from HNE-treated neurons being greater than in samples from control neurons (Figure 2D and E).
Figure 2. HNE induces endogenous α-synuclein aggregation in rat primary cortical neurons.
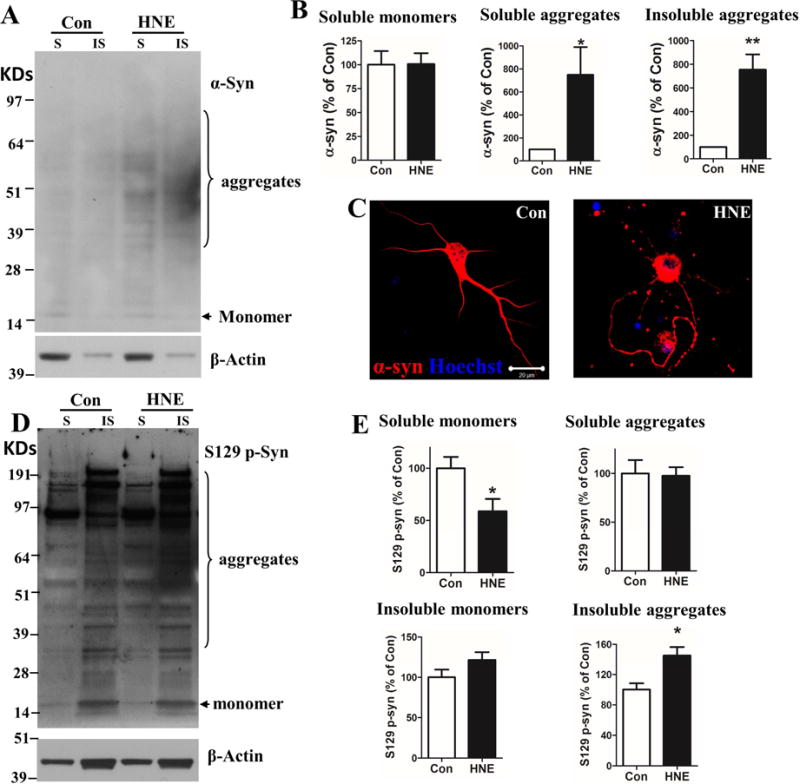
(A) Primary cortical neurons were treated with vehicle (Con) or 5 μM 4-hydroxynonenal (HNE) for 24 hours, lysed, and soluble (S) and insoluble (IS) fractions were isolated (see Methods). The immunoblot shown was probed with α-synuclein and actin antibodies. Note relatively greater amounts of higher molecular weight α-synuclein species in both S and IS fractions of HNE-treated neurons compared to control neurons. (B) Results of densitometric analysis of α-synuclein (monomer and aggregates) levels (normalized to the actin level). Values are the mean and SEM of determinations made in 3 independent experiments. *p<0.05, **p<0.01 compared to control value. (C) Cortical neurons were treated with vehicle (Con) or 5 μM HNE (HNE) for 24 hours, and the neurons were then fixed and immunostained with of α-synuclein antibody (red) and counterstained with Hoechst 33258 to label cell nuclei (blue). Scale bar = 20 μm. (D) Primary cortical neurons were treated with vehicle (Con) or 5 μM HNE (HNE) for 4 days, lysed, and soluble (S) and insoluble (IS) fractions were isolated. The immunoblot was probed with serine 129 phosphorylated α-synuclein (S129 p-Syn) and actin antibodies. (E) Results of densitometric analysis of S129 p-Syn (monomer and aggregates) levels (normalized to the actin level). Values are the mean and SEM of determinations made in 4 independent experiments. *p<0.05 compared to control value.
Cortical neurons secrete extracellular vesicles (EVs) containing aggregated and S129-phosphorylated α-synuclein
Pathogenic accumulations of α-synuclein in neurons impairs protein degradation systems, particularly the autophagy – lysosome pathway (Chen et al., 2015; Choubey et al., 2011; Huang et al., 2012). We and others previously showed that HNE can impair lysosome and proteasome degradation systems (Hyun et al., 2002; Ferrington and Kapphahn, 2004; Krohne et al., 2010a; Krohne et al., 2010b; Zhang et al., 2017). Inhibition of lysosome function can trigger the secretion of EVs containing undegraded proteins (Danzer et al., 2012; Eitan et al., 2016; Poehler et al., 2014). Here we asked if HNE causes neurons to release EVs containing pathogenic α-synuclein species. We isolated EVs from the medium bathing cultured primary cortical neurons using an ultracentrifugation protocol (Vlassov et al., 2012). To confirm the presence of exosomes in the isolated fraction we performed electron microscopy analysis, and we also measured the diameter of exosomes using q-Nano technology. Both approaches revealed membranous vesicles of a size range (30 – 100 nm) characteristic of exosomes (Figure 3A and B). We next performed immunoblot analysis of presumptive EVs isolated from the culture medium of HNE-treated and control cortical neurons. Flotillin-1, an EV marker protein, was present in similar amounts in vesicle-containing preparations isolated from medium of HNE-treated and control neurons (Figure 3C). Greater amounts of α-synuclein oligomers and pS129 α-synuclein monomer were present in EVs released from HNE-treated neurons compared to control neurons (Figure 3C). To further test the existence of pathological α-synuclein in EVs, we did dot blot assay in order to detect toxic α-synuclein oligomers/fibrils with anti α-synuclein filament antibody (FILA-1). EV lysates with or without Proteinase K (PK) digestion was spotted onto nitrocellulose membrane directly without protein denaturing steps. To our expectation, the amounts of alpha-synuclein oligomers/fibrils increased 2.5 folds in EVs from HNE-treated neurons than EVs from control neurons (Figure 3D, E). Interestingly, most of alpha-synuclein aggregates are not PK resistant (Figure 3D), suggesting that most of alpha-synuclein species in HNE-EVs are oligomers and not fibrils. To determine the amounts of oligomeric α-synuclein in the EV-free supernatant and in EVs we performed an ELISA assay with conditioned medium supernatants from primary rat neuron cultures that had been treated for 24 hours with vehicle or HNE. We found that the amount of α-synuclein oligomers was increased in the supernatant of HNE-treated neurons (117.1 ± 8.4 pg/ml) compared with control neurons (73.8 ± 15.7 pg/ml) (Figure 3F). When supernatants were treated with Triton X-100 to lyse EVs, the levels of oligomeric α-synuclein were increased by approximately 3-fold and 5-fold in samples from control (327.0 ± 82.2 pg/ml) and HNE-treated (565.3 ± 60.7 pg/ml) neurons (Figure 3F). These data provide evidence that elevated levels of HNE cause neurons to release increased amounts of EVs containing potentially pathogenic oligomeric α-synuclein species.
Figure 3. Cortical neurons secrete extracellular vesicles (EVs) containing aggregated α-synuclein and S129 phosphorylated α-synuclein.
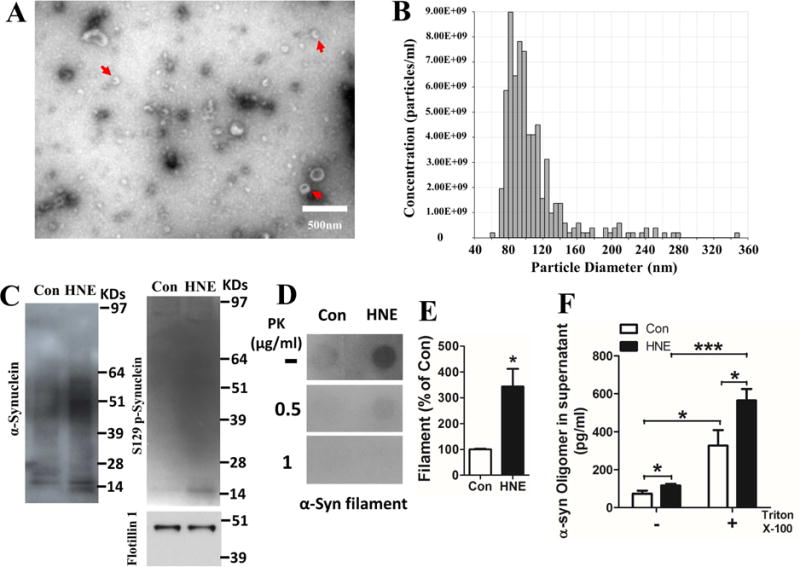
EVs were isolated from the culture medium of primary rat cerebral cortical neurons and the EV preparations were evaluated by electron microscopy, nanoparticle tracking and immunoblot analysis. (A) Electron micrograph showing EVs (arrows). Scale bar = 500 nm. (B) The size and number of EVs was analyzed using a qNano instrument. Histogram shows numbers of EVs of the indicated diameters. (C) Cortical neurons were treated with vehicle (Con) or 5 μM HNE (HNE) for 24 hours, and then EVs were isolated from the culture medium and lysed in RIPA buffer. EV proteins were separated on a NuPAGE Bis-Tris gel and immunoblotted using antibodies against total α-synuclein and S129 phospho-α-synuclein. An antibody against the EV marker protein flotillin-1 was used as an internal control. (D) Dot blot of EV samples from control and HNE-treated neurons using an anti-α-synuclein filament antibody. Samples had been incubated without or with the indicated concentrations of proteinase K (PK). (E) Result of densitometric of α-synuclein dot blots (without PK digestion). Values are the mean and SEM (n = 3 independent experiments; *p<0.05). (F) The concentration of oligomeric a-synuclein in conditioned medium supernatants were measured with an ELISA kit. Triton X-100 was used to permeabilize EVs in the supernatant. Values are the mean and SEM (n = 3 independent experiments; *p<0.05, ***p<0.001).
EVs secreted by HNE-treated neurons are internalized by and toxic to previously healthy neurons
After being released from one cell, EVs can be internalized by neighboring cells (Alvarez-Erviti et al., 2011; Danzer et al., 2012; Emmanouilidou et al., 2010). To determine whether EVs released from cortical neurons can be internalized by other cortical neurons, we isolated EVs from the medium of cultured control or HNE-treated neurons, and labeled the EVs with a lipophilic fluorescent probe (PKH67 green), and then added the labeled EVs to the culture medium (20 μg exosome protein/ml medium) of healthy cortical neurons. We then fixed neurons at 2, 6 or 24 hours after exposure to EVs, immunostained them with α-synuclein antibody, and then simultaneously imaged the PKH67 green fluorescence and the α-synuclein immunoreactivity. EVs from both control and HNE-treated neurons were internalized by recipient neurons in a time-dependent manner with significantly more EVs from HNE-treated neurons being internalized compared with EVs from control neurons (Figure 4A and B). Enlarged Z-stack images showed that α-synuclein immunoreactivity is co-localized with many of the internalized EVs, especially with HNE-EVs (Figure 4A). To determine whether internalized EVs affected neuronal viability we exposed cortical neurons to EVs isolated from the medium of control and HNE-treated neurons for 48 hours and then performed MTS and LDH assays. EVs from HNE-treated neurons exhibited concentration-dependent neurotoxicity on recipient neurons, as indicated by lower levels of MTS signal in the neurons and increased levels of LDH released into the culture medium (Figure 4C). To determine if exosomes from HNE-treated neurons modify the vulnerability of neurons to another insult relevant to PD, we treated cortical neurons with a low concentration of the mitochondrial complex I inhibitor rotenone (1 nM) in combination with EVs released from control or 5 M HNE-treated neurons (20 μg protein/ml) for 48 hours. Rotenone at this concentration did not cause a reduction in neuronal viability, but did significantly exacerbate the toxicity of EVs from HNE-treated neurons (Figure 4D). These results suggest that α-synuclein oligomer-containing exosomes released from neurons are readily internalized by healthy neurons and can increase their vulnerability to mitochondrial stress.
Figure 4. EVs secreted by HNE-treated neurons are readily internalized by and toxic to healthy neurons.
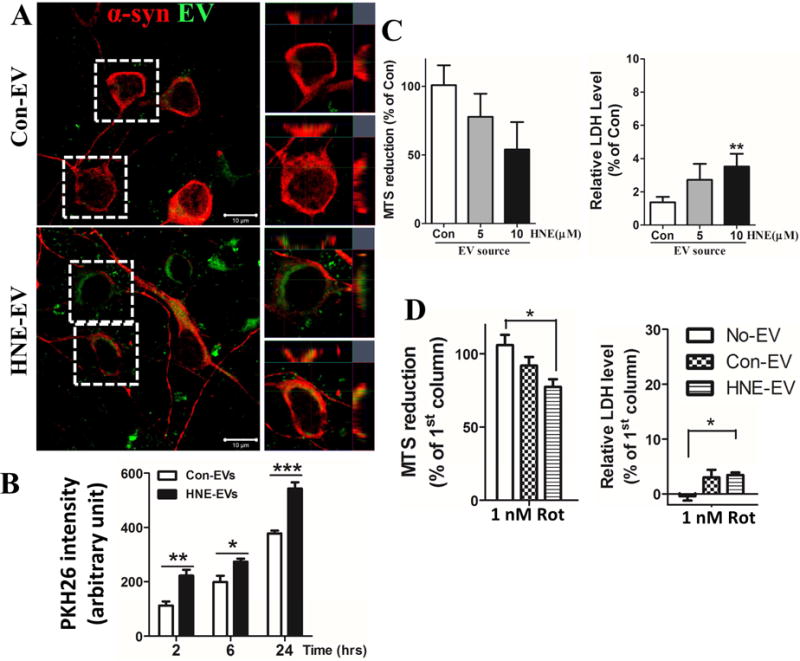
(A) EVs secreted by control or HNE-treated neurons (24 hour exposure to 5 μM HNE) were labeled with lipophilic fluorescent dye PKH67 (green) and were then added at a concentration of 20 μg EV protein/ml medium to cultured primary cortical neurons for 4 hours. Neurons were then fixed and immunostained with anti-α-synuclein antibody (magenta). Confocal images show that labeled EVs were internalized by recipient neurons. Scale bar = 20μm. (B) Neurons were grown in 96-well plates for 7 days and exposed for 2, 6 or 24 hours to PKH26-labeled EVs isolated from the medium of cultured vehicle- (Con) or HNE-treated neurons. Cultures were then washed to remove EVs in the medium and the fluorescence intensity of cell-associated PKH26 fluorescence corresponding to internalized EVs was quantified in a plate reader. Values are the mean and SEM of determinations made in 4 independent experiments. *p<0.05, *p<0.01, *p<0.001. (C) Naïve cortical neurons were treated for 48 hours with EVs (20 μg/ml) released from neurons that had been treated with vehicle (Con) or the indicated concentrations of HNE, and neuron viability was measured with MTS and LDH assays. Values are the mean and SEM of determinations made in 6 independent experiments. **p<0.01. (D) Cortical neurons were exposed for 48 hours to vehicle or 1 nM rotenone, alone or in combination with EVs released from vehicle-treated control neurons (Con EVs) or EVs released from HNE-treated neurons (HNE EVs). Neuronal viability was quantified using MTS and LDH assays. Values are the mean and SEM of determinations made in 3 independent experiments. * p<0.05.
To better understand the subcellular fate of the α-synuclein entering neurons in EVs, we isolated EVs from the medium of HEK-wt-SNCA cells. We confirmed that the EV preparation was indeed enriched in EVs as demonstrated by the presence of the EV marker flotillin-1 and the absence of the endosome marker calnexin (Figure 5A). Human α-synuclein was present in the isolated EVs (Figure 5A). The quality of ultracentrifugation protocol on EV isolation was confirmed by Opti-prep gradient ultracentrifugation (Figure S1A). EV pellets from ultracentrifugation were further evaluated by discontinuous gradient ultracentrifugation; 6 fractions were collected. Both α-synuclein and flotillin-1 were enriched in fractions 2 and 3, but not in the other fractions (Figure S1B). We then treated cultured cortical neurons with EVs (20 μg protein/ml medium) derived from HEK-wt-SNCA cells. After 2 hours the neurons were fixed and double-label immunostained with an antibody that recognizes both human and rat α-synuclein (C20) and an antibody that is specific for human α-synuclein (Syn211). The specificity of Syn211 antibody was confirmed by immunoblot and immunostaining analyses (Figure S2). No human α-synuclein immunoreactivity was detected in rat primary neurons with antibody Syn211, while rat α-synuclein was detectable with antibody C20 (Figure S2A, B). In addition, human α-synuclein was recognized by antibody Syn211 in brain lysates from 1 year old Thy1-A53T-SNCA mice, but not in brain lysates from age-matched wild type mice (Figure S2C). We found that the majority of internalized human α-synuclein was located in the axon and dendrites of recipient neurons, with some but not all of the human α-synuclein colocalizing with rat α-synuclein (Figure 5B). Z-stack image analyses confirmed that EV-derived human α-synulcein was internalized by neurons rather than attached to the cell surface membrane (Figure 5B).
Figure 5. EVs containing human α-synuclein are internalized by primary cerebral cortical neurons.
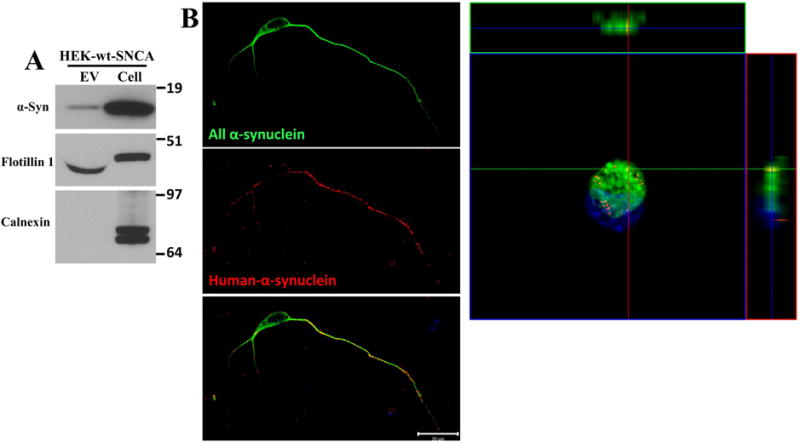
(A) EVs were isolated from the culture medium of HEK cells overexpressing wild type α-synuclein, and proteins in the EVs and the cells that released the EVs were separated in a NuPAGE Bis-Tris gel and immunoblotted using antibodies against human α-synuclein, flotillin-1 (EV marker protein) and calnexin (a protein not known to be in EVs). (B) Cerebral cortical neurons were treated with HEK-wt-SNCA EVs at a concentration of 20 μg/ml for 2 hours. Neurons were then fixed and immunostained with an α-synuclein antibody that recognizes both human and rat α-synuclein (C20 antibody, green) and an antibody specific for human α-synuclein (Syn211 antibody, red). Confocal images reveal EV-derived human α-synuclein immunoreactivity within the axon, and to a lesser extent in the cell body and dendrites of a recipient neuron. The Z-stack image shows the distribution of EV-derived human, and endogenous rat α-synuclein, in the cell body of the same neuron shown at the left. These images are representative of 20 neurons examined in 3 independent experiments. Scale bar, 20 μm.
α-synuclein is essential for EV-mediated neurotoxicity
To determine the role of α-synuclen in EV-induced neurotoxicity, we knocked out the Snca gene in HEK cells with CRISPR-cas9 technology. Immunoblot analysis confirmed that HEK Snca KO cells and EVs secreted from those cells lacked α-synuclein protein (Figure 6A). We measured viability of cortical neurons that were treated with EVs from HEK, HEK-wt-SNCA, or HEK-SNCA-KO cells. After 24 hours, only neurons treated with HEK-wt-SNCA EVs exhibited a significant decrease in MTS reduction (Figure 6B), and neurons treated with either HEK-EVs or HEK-wt-SNCA EVs exhibited significantly greater LDH release compared to neurons treated with EVs lacking α-synuclein, or no EV treatment (Con) (Figure 6B).
Figure 6. α-synuclein is essential for EV mediated neurotoxicity.
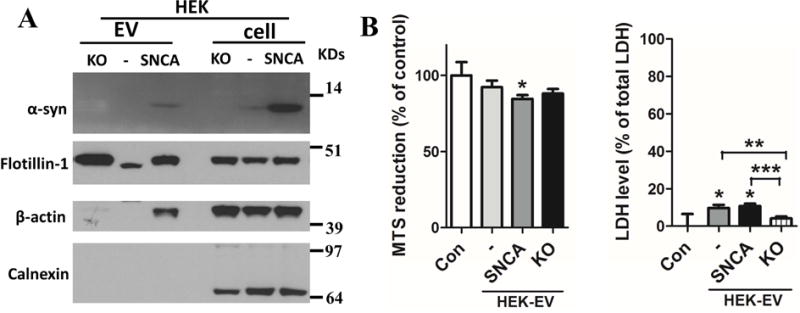
(A) EVs were isolated from the medium of cultured control HEK cells (−), HEK cells overexpressing wildtype human α-synuclein (+), and α-synuclein knockout HEK cells (KO). Proteins in the EVs and the cells that released the EVs were separated in a NuPAGE Bis-Tris gel and immunoblotted using antibodies against α-synuclein, flotillin-1, calnexin and actin. (B) Primary cortical neurons were incubated for 24 hours in the presence of EVs (20 μg protein/ml) isolated from the medium of control HEK cells (−), HEK cells overexpressing wildtype human α-synuclein (+), and α-synuclein knockout HEK cells (KO). Neuronal viability was assessed using MTS and LDH assays. Values are the mean and SEM of determinations made in 3 independent experiments. * p<0.05, ** p<0.01, ***p<0.001.
EV membrane surface-associated α-synuclein is required for EV internalization by neurons
To determine the location of α-synuclein in EVs, we incubated EVs with trypsin alone or in combination with 0.1% saponin (membrane permeabilizer). HEK-wt-SNCA EVs were incubated with trypsin (1:20, trypsin:total EV protein) at 37°C for 2 hours. EVs without trypsin or saponin treatment, and brain lysates from Thy1-A53T-SNCA mice were used as controls. After combined treatment with trypsin and saponin no α-synuclein was detected in EV samples (Figure 7A). EVs treated with trypsin alone exhibited reduced amounts of α-synuclein compared to untreated EVs, suggesting the association of α-synuclein with the membrane surface (Figure 7A). The EV marker flotillin-1 was present in EVs from each treatment group.
Figure 7. Evidence that α-synuclein is present within and on the surface of EVs, and that EV surface-associated α-synuclein enhances EV internalization by recipient neurons.
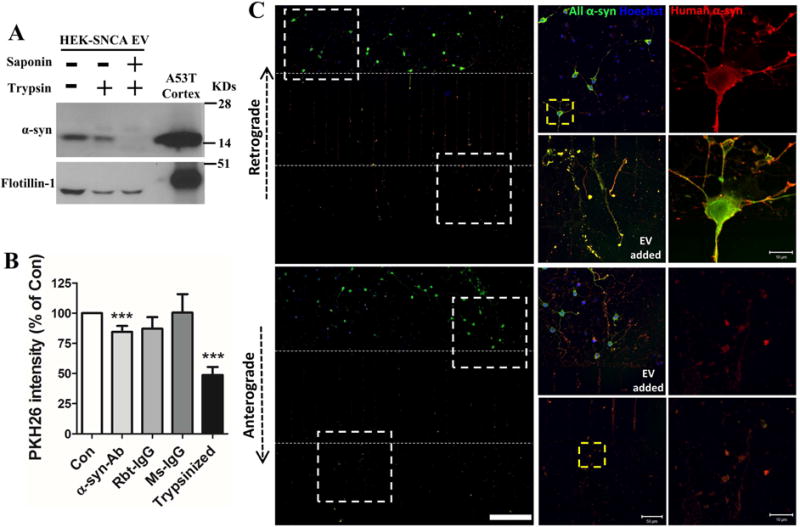
(A) EVs isolated from HEK-wt-SNCA cells were subjected to trypsin digestion without or with EV membrane permeabilization (see Methods). Proteins in the EVs and mouse cortical tissue were separated in a NuPAGE Bis-Tris gel and immunoblotted with α-synuclein and flotillin-1 antibodies. (B) Control and trypsin digested EVs from HEK-wt-SNCA cells were labeled with fluorescent PKH26 dye. EVs were preincubated with or without human α-synuclein antibody, rabbit IgG, or mouse IgG for 30 minutes before being added to the medium of cortical neurons cultured in 96 well plates. Neurons were incubated with the different EV preparations for 2 hours and the fluorescence intensity of PKH26 inside neurons was quantified in a plate reader. Values are the mean and SEM of determinations made in 3–5 independent experiments. ***p<0.001 compared to the control value. (C) Rat primary cortical neurons were grown in microfluidic chambers for 10 days. EVs released from HEK-wt-SNCA cells were then added to the medium of either the axonal compartment (upper panels) or the somatodendritic compartment (lower panels), and 4 hours later neurons were fixed and immunostained with anti-pan α-synuclein (green) and anti-human α-synuclein (red) antibodies. Confocal images show EV-derived human human α-synuclein immunoreactivity in axons, and the cell body and dendrites. The region of the image demarcated by the dashed white squares in left low magnification images are enlarged in the middle panels. The region of the image demarcated by the dashed yellow square in the middle panels are enlarged in right panels. Scale bar = 200 μm in panels at left, 50 μm in panels in middle, and 10 μm in the panels at the right.
To determine whether proteins on the outer membrane of HEK-wt-SNCA EVs play a role in α-synuclein internalization in neuron, we measured the internalization efficiency of EVs that either had or had not been treated with trypsin. We also pre-incubated other EVs with human α-synuclein antibody (Syn211) to block α-synuclein to determine if α-synuclein is required for EV internalization. All EVs were labeled with the fluorescent PKH26 lipophilic dye before adding them to the culture medium of primary neurons. After 2 hours, the cells were washed in fresh medium and the fluorescence intensity of the neuronal EV fluorescence signal was measured with a plate reader. We found a large decrease of EV internalization with trypsin-digested EVs, and with EVs that had been pre-incubated with α-synuclein antibody, compared to untreated EVs and EVs pre-incubated with either rabbit or mouse IgG (Figure 7B).
To determine whether EVs containing α-synuclein are preferentially internalized by axons, and to determine their subcellular fate after internalization, we grew neurons in microfluidic chambers in which the axons of neurons plated in one chamber grow through microgrooves into an adjacent chamber, thereby enabling selective application of EVs to the axons or to the cell body and dendrites. To confirm that EVs cannot move freely between compartments, we grew neurons in one chamber for only 2 days, a time period insufficient to allow axons to grow through the microgrooves into the second chamber. We then added HEK-wt-SNCA EVs to either the chamber with the neurons or to the empty chamber, waited 24 hours, and immunostained the neurons with the pan α-synuclein antibody and the human α-synuclein specific antibody. Human (i.e., EV-derived) α-synuclein was only detected in the neurons when the EVs were added to the neuronal compartment, and not when added to the cell-free compartment, demonstrating that the EVs do not pass through the microgrooves (Figure S3). We next maintained neurons in the microfluidic chamber cultures for 10 days, during which time numerous axons grew through the micrographs into the second chamber (Figure S4). We added HEK-wt-SNCA EVs into either the axon chamber or the somato-dendritic chamber for 4 hours. The neurons were then fixed and immunostained with antibodies that recognize only human α-synuclein or both rat and human α-synuclein. The EV-associated human α-synuclein was internalized by axons, and then retrogradely transported to the soma and dendrites (Figure 7C, upper panels). Interestingly, we also found that α-synuclein from EVs can be internalized by soma/dendrites, and then be transported anterogradely into the axon (Figure 7C, lower panels). These results suggest that EVs harboring α-synuclein can be internalized by both axons and dendrites, and transported either retrogradely or anterogradely.
Human α-synuclein in EVs placed in the striatum spreads to other brain regions
To determine whether EV-associated α-synuclein can move from one brain region to another in vivo, we injected EVs (5 μg total protein in 3 μl volume) from HEK-wt-SNCA or HEK-SNCA-KO cells into the dorsal striatum (unilaterally) of young (3 months) and old (24 months) wild type mice. Mice were euthanized 5 days after injection, and their brains processed for immunohistochemistry using the anti-human α-synuclein antibody Syn211, with cresyl violet counterstaining to label cell nuclei. In the hemisphere into which the EVs from HEK-wt-SNCA EVs were injected, human α-synuclein immunoreactivity was present not only in the dorsal striatum, but also in the cerebral cortex, substantia nigra and hippocampus (Figure 8A). Interestingly, we also observed human α-synuclein immunoreactivity in the cerebral cortex and hippocampus of the contralateral hemisphere, although the staining intensity was much lower than in the ipsilateral hemisphere (Figure 8A). Mice injected with EVs from HEK-SNCA-KO cells exhibited no human α-synuclein immunoreactivity (Figure 8B). When viewed at higher magnification, the human α-synuclein immunoreactivity was present in both neurites and cell bodies of neurons (Figure 9). Intense human α-synuclein immunoreactivity was present in occasional Lewy neurite-like structures and neuronal cell bodies in the ipsilateral striata of both young and old mice, and to a much lesser extent in the contralateral striatum (Figure 9A–H). Human α-synuclein immunoreactivity was also present in the cell bodies and neurites of some neurons in the ipsilateral, and to a lesser extent contralateral cerebral cortex of mice injected with HEK-wt-SNCA EVs (Figure 9I–P). Some neurons in the ipsilateral dentate gyrus of young and old mice exhibited human α-synuclein immunoreactivity in their soma (Figure 9Q–T). Neurons in both ipsilateral and contralateral substantia nigra pars compacta of both young and old mice exhibited EV-derived human α-synuclein immunoreactivity in their cell bodies and neurites. (Figure 9U–X).
Figure 8. When EVs harboring human α-synuclein are placed in the dorsal striatum of mice, the human α-synuclein is distributed to several different brain regions.
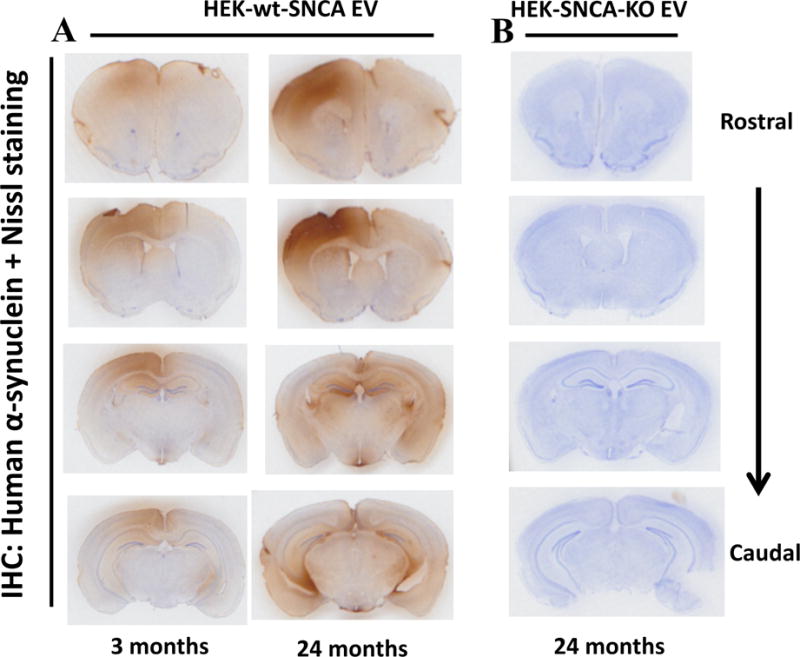
EVs released from HEK cells overexpressing human α-synuclein (panel A) or HEK-SNCA-KO cells (panel B) were injected into right dorsal striatum in wild type male mice at 3 months or 24 months of age. Mice were euthanized 5 days after EV injection and brains were fixed and coronal sections were immunostained with an antibody specific for human α-synuclein, and counterstained with cresyl violet to stain all neuronal cell bodies. Representative sections at four different rostral to caudal levels are shown. Similar patterns of immunoreactivity were observed in brain sections from 3 different mice.
Figure 9. Human α-synuclein derived from EVs placed in the dorsal striatum accumulates within neurons in substantia nigra, cortex and hippocampus in mice.
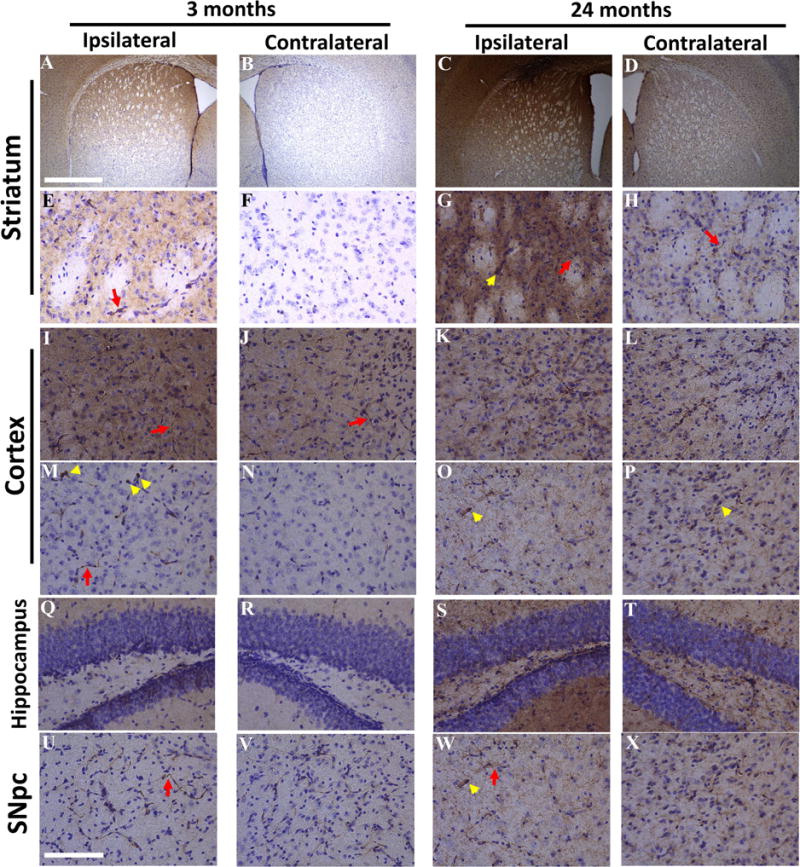
EVs released from HEK cells overexpressing human α-synuclein were injected into right dorsal striatum in wild type male mice at 3 months or 24 months of age. Mice were euthanized 5 days after EV injection and brains were fixed and coronal sections were immunostained with an antibody specific for human α-synuclein, and counterstained with cresyl violet. Images of human α-synuclein immunoreacitivty in different regions of brain are shown. (A–D) Lower magnification of ipsilateral and contralateral striatum. (E–H) Higher magnification images of ipsilateral and contralateral striatum. (I–L) Images of frontal cortex. (M–P) Images of posterior cortex. (Q–T) Images of dentate gyrus of hippocampus. (U–X) Images of substantia nigra pars compacta (SNpc). Scale bars = 1 mm in A–D, and 100 μm in E–X.
Discussion
Our findings demonstrate that: 1) EVs containing α-synuclein oligomers can be released from cultured primary neurons and a cell line overexpressing human α-synuclein; 2) EVs containing α-synuclein can be internalized by axons of healthy primary neurons, wherein they are transported retrogradely to the cell body and dendrites; 3) α-synuclein is present within EVs and on their surface, and the surface-associated α-synuclein plays a role in the internalization of the EVs by neurons; 4) Exposure of primary neurons to the lipid peroxidation product HNE enhances the accumulation of α-synuclein aggregates within the neurons and increases the release of EVs containing toxic forms of α-synuclein; 5) When EVs harboring human α-synuclein are placed in the dorsal striatum of mice, that α-synuclein spreads from the site of injection to the substantia nigra, cerebral cortex and hippocampus. In the light of previous findings showing that HNE accumulates in neurons in PD and that HNE can covalently modify α-synuclein (Yoritaka et al., 1996; Castellani et al., 2002; Qin et al., 2007; Nasstrom et al., 2011), our findings suggest a scenario in which membrane-associated oxidative stress promotes α-synuclein aggregation inside of neurons and consequent release of toxic α-synuclein species in EVs. The EVs are then internalized by and toxic to previously healthy neurons adjacent to those first affected by the accumulation of HNE-modified α-synuclein.
Two, not mutually exclusive, mechanisms for the spread of α-synuclein pathology between neurons and across regions of the nervous system are suggested from the results of previous studies. One mechanism is similar to prion proteins; pathogenic α-synuclein species ‘seed’ the acquisition of previously non-pathogenic α-synuclein, thereby propagating a molecular ‘chain reaction’. The latter mechanism is supported by data showing that extracts of PD patient brain, and pathogenic α-synuclein species isolated from the extracts, can trigger the accumulation of endogenous α-synuclein aggregates in the rodent brain (Guo et al., 2013; Mao et al., 2016; Volpicelli-Daley et al., 2014; Volpicelli-Daley et al., 2011; Luk et al., 2012). Moreover, mouse α-synuclein preformed fibrils are able to induce accumulation of LB-like intracellular inclusions and nigrostriatal degeneration in rats (Paumier et al., 2015)). The second mechanism, which is supported by our findings, is that neurons first affected by the accumulation of pathogenic α-synuclein species extrude those potentially cytotoxic α-synuclein species, which are then internalized by and transported within adjacent (synaptically connected) neurons. Similar to a recent study (Emmanouilidou et al., 2011; Danzer et al., 2012), we found that a cell line overexpressing human α-synuclein releases EVs that contain α-synuclein aggregates, and we further found that primary cortical neurons exposed to HNE release EVs harboring α-synuclein. Importantly, we found that EVs released in response to α-synuclein overexpression or exposure of cells to HNE can be internalized by neurons in cell culture and in the brain. Taking advantage of an antibody that binds only human and not rodent α-synuclein, we found that neurotoxic EV-associated α-synuclein species are transferred trans-neuronally.
A large literature has accumulated that supports pathogenic roles for HNE in several prominent age-related diseases including cardiovascular disease, diabetes and Alzheimer’s disease (for review see Mattson, 2009). For example, studies relevant to Alzheimer’s disease have shown that pathogenic forms of amyloid β-peptide can cause membrane lipid peroxidation and consequent destabilization of neuronal ion homeostasis and energy metabolism (Mark et al., 1995; Mark et al., 1997; Keller et al., 1997). Conversely, HNE promotes amyloidogenic processing of the β-amyloid precursor protein and aggregation of amyloid β-peptide. We found that HNE can cause neurons to release EVs harboring neurotoxic α-synuclein species. Although we did not establish the specific molecular mechanism by which HNE triggers release of EVs harboring pathogenic α-synuclein, previous findings suggest HNE may impair intracellular degradation of α-synuclein in proteasomes or via the autophagy – lysosome pathway. HNE can inhibit the proteasome (Keller et al., 2000; Farrington and Kapphahan, 2004), and HNE-modified protein aggregates may be refractory to proteasomal degradation (Shringarpure et al., 2000). In addition, exposure of cerebral cortical neurons to HNE impairs lysosome function resulting in the accumulation of polyubiquitinated proteins (Zhang et al. 2017). Moreover, it was recently reported that lysosome dysfunction triggers the release of EVs harboring pathogenic species of amyloid β-peptide, and EVs with similar pathogenic amyloid β-peptide species are present in the cerebrospinal fluid of AD patients (Eitan et al., 2016). It remains to be established if oxidative stress and impaired autophagy are pivotal for generation of EVs harboring pathogenic α-synuclein species in PD, and whether such EVs are critical for propagation of the disease process between different brain regions.
Compared to other neuronal populations, dopaminergic neurons in the SN may prone to very high levels of oxidative stress for several reasons. SN dopaminergic neurons have very high levels of iron which, when present in the free ionic Fe2+ form, is a potent inducer of membrane lipid peroxidation and HNE production (Jenner, 2003). Dopamine and its metabolites can themselves generate reactive oxygen species and cause lipid peroxidation (Napolitano et al., 2011). In addition, dopaminergic neurons experience high levels of excitability, and the consequent Ca2+ influx and uptake by mitochondria generates reactive oxygen species (Mattson, 2012; Surmeier et al., 2017). Recent findings suggest that α-synuclein pathology is propagated from peripheral neurons to the brainstem via the vagus nerve, thence to midbrain dopaminergic neurons and then to cerebral cortical neurons (Klingelhoefer et al., 2015). Because aggregating α-synuclein associates with membranes and aggregating α-synuclein can induce membrane lipid peroxidation, a cascade of events can be envisioned in which age-related factors (for example, impaired mitochondrial and lysosome function, inflammation) promote lipid peroxidation, and modification of α-synuclein by HNE resulting in α-synuclein aggregation and release from dendrites in EVs. The EVs harboring pathogenic α-synuclein species are then internalized by axon terminals of neurons innervating the affected neuron. The pathogenic α-synuclein then triggers oxidative stress and aggregation of endogenous α-synuclein in the previously unaffected neuron, thereby propagating the neurodegenerative cascade in a retrograde manner. Consistent with this retrograde propagation scenario, we found that when HNE-modified α-synuclein is introduced into the striatum where axons of SN dopaminergic neurons are located, α-synuclein pathology later appears in SN dopaminergic neuron cell bodies.
Our findings demonstrate that HNE induces the generation and release of EVs harboring neurotoxic species of human α-synuclein, and suggest that such EV-derived α-synuclein can be internalized by axons and distributed within and between neurons. We found that α-synuclein is present on the outer surface of EVs released from human cells that accumulate α-synuclein aggregates, and that a α-synuclein antibody inhibits the internalization of EVs harboring such pathogenic α-synuclein species, suggests that membrane-associated α-synuclein may play a critical role in trans-cellular propagation of α-synuclein pathology. Indeed, we found that such α-synuclein-laden EVs are neurotoxic and increase the vulnerability of neurons to a low concentration of the mitochondrial complex I inhibitor rotenone. Considerable evidence suggests that impaired complex I function contributes to neuronal vulnerability in PD including the fact that rodents, monkeys and humans exposed to complex I inhibitors (MPP+, 6-hydroxydopamine and rotenone) exhibit selective degeneration of dopaminergic neurons and clinical motor signs diagnostic of PD (Betarbet et al., 2002; Langston, 2017). Moreover, mutations in multiple genes that cause early-onset inherited PD (α-synuclein, Parkin, PINK1, DJ-1 and LRRK2) result, either directly or indirectly, in mitochondrial dysfunction (Pickrell and Youle, 2015; Ryan et al., 2015; Bose and Beal, 2016). Aging, the major risk factor for late-onset PD, involves accumulation of HNE-modified proteins in the brain (Di Domenico et al., 2016). Because HNE can impair mitochondrial function and autophagy (Keller et al., 1997; Zhang et al., 2017), and because impaired autophagy can trigger release of EVs containing pathogenic self-aggregating proteins (Eitan et al., 2016), our findings suggest a therapeutic potential for interventions that reduce the generation of EVs harboring α-synuclein or that inhibit the internalization of such potentially pathogenic EVs by other neurons.
Supplementary Material
The lipid peroxidation product HNE increases aggregation of endogenous α-synuclein in primary neurons.
HNE triggers the secretion of extracellular vesicles (EVs) containing cytotoxic oligomeric α-synuclein species.
EVs released from HNE-treated neurons are internalized by healthy neurons which as a consequence degenerate.
EV-associated α-synuclein is located both inside the vesicles, and on their surface where it plays a role in EV internalization by neurons.
Upon internalization, EVs harboring pathogenic α-synuclein are transported both anterogradely and retrogradely within axons.
Focal injection of EVs containing α-synuclein into the striatum of wild type mice results in spread of synuclein pathology to anatomically connected brain regions.
Acknowledgments
We thank Dr. Ruiqian Wan for technical assistance with stereotaxic injections. This work was supported by Intramural Research Program of National Institute on Aging.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, Cooper JM. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis. 2011;42:360–367. doi: 10.1016/j.nbd.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281:29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- Bartels T, Choi JG, Selkoe DJ. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, Greenamyre JT. Animal models of Parkinson’s disease. Bioessays. 2002;24:308–318. doi: 10.1002/bies.10067. [DOI] [PubMed] [Google Scholar]
- Bose A, Beal MF. Mitochondrial dysfunction in Parkinson’s disease. J Neurochem. 2016;139:S216–S231. doi: 10.1111/jnc.13731. [DOI] [PubMed] [Google Scholar]
- Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett. 2006;396:67–72. doi: 10.1016/j.neulet.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani RJ, Perry G, Siedlak SL, Nunomura A, Shimohama S, Zhang J, Montine T, Sayre LM, Smith MA. Hydroxynonenal adducts indicate a role for lipid peroxidation in neocortical and brainstem Lewy bodies in humans. Neurosci Lett. 2002;319:25–28. doi: 10.1016/s0304-3940(01)02514-9. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Chen L, Xie Z, Turkson S, Zhuang X. A53T human alpha-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J Neurosci. 2015;35:890–905. doi: 10.1523/JNEUROSCI.0089-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew KC, Ang ET, Tai YK, Tsang F, Lo SQ, Ong E, Ong WY, Shen HM, Lim KL, Dawson VL, et al. Enhanced autophagy from chronic toxicity of iron and mutant A53T alpha-synuclein: implications for neuronal cell death in Parkinson disease. J Biol Chem. 2011;286:33380–33389. doi: 10.1074/jbc.M111.268409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M, Zharkovsky A, Kaasik A. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem. 2011;286:10814–10824. doi: 10.1074/jbc.M110.132514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener. 2012;7:42. doi: 10.1186/1750-1326-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domenico F, Tramutola A, Butterfield DA. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic Biol Med. 2016 Oct 24; doi: 10.1016/j.freeradbiomed.2016.10.490. 2016. pii: S0891-5849(16)30980-7. [DOI] [PubMed] [Google Scholar]
- Dexter DT, Carter CJ, Wells FR, Javoy-Agid F, Agid Y, Lees A, Jenner P, Marsden CD. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J Neurochem. 1989;52:381–389. doi: 10.1111/j.1471-4159.1989.tb09133.x. [DOI] [PubMed] [Google Scholar]
- Dröge W, Schipper HM. Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell. 2007;6:361–370. doi: 10.1111/j.1474-9726.2007.00294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eitan E, Suire C, Zhang S, Mattson MP. Impact of lysosome status on extracellular vesicle content and release. Ageing Res Rev. 2016;32:65–74. doi: 10.1016/j.arr.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanouilidou E, Elenis D, Papasilekas T, Stranjalis G, Gerozissis K, Ioannou PC, Vekrellis K. Assessment of alpha-synuclein secretion in mouse and human brain parenchyma. PLoS One. 2011;6:e22225. doi: 10.1371/journal.pone.0022225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- Ferrington DA, Kapphahn RJ. Catalytic site-specific inhibition of the 20S proteasome by 4-hydroxynonenal. FEBS Lett. 2004;578:217–223. doi: 10.1016/j.febslet.2004.11.003. [DOI] [PubMed] [Google Scholar]
- George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- Goedert M. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science. 2015;349(6248):1255555. doi: 10.1126/science.1255555. [DOI] [PubMed] [Google Scholar]
- Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, et al. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154:103–117. doi: 10.1016/j.cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Fujiwara H, Nonaka T, Wakabayashi K, Takahashi H, Lee VM, Trojanowski JQ, Mann D, Iwatsubo T. Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J Biol Chem. 2002;277:49071–49076. doi: 10.1074/jbc.M208046200. [DOI] [PubMed] [Google Scholar]
- Huang Y, Chegini F, Chua G, Murphy K, Gai W, Halliday GM. Macroautophagy in sporadic and the genetic form of Parkinson’s disease with the A53T alpha-synuclein mutation. Transl Neurodegener. 2012;1:2. doi: 10.1186/2047-9158-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun DH, Lee MH, Halliwell B, Jenner P. Proteasomal dysfunction induced by 4-hydroxy-2,3-trans-nonenal, an end-product of lipid peroxidation: a mechanism contributing to neurodegeneration? J Neurochem. 2002;83:360–370. doi: 10.1046/j.1471-4159.2002.01125.x. [DOI] [PubMed] [Google Scholar]
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Synuclein deposition and non-motor symptoms in Parkinson disease. J Neurol Sci. 2011;310:107–111. doi: 10.1016/j.jns.2011.04.012. [DOI] [PubMed] [Google Scholar]
- Jenner P. Oxidative stress in Parkinson’s disease. Ann Neurol. 2003;53(Suppl 3):S26–36. doi: 10.1002/ana.10483. [DOI] [PubMed] [Google Scholar]
- Kara E, Kiely AP, Proukakis C, Giffin N, Love S, Hehir J, Rantell K, Pandraud A, Hernandez DG, Nacheva E, et al. A 6.4 Mb duplication of the alpha-synuclein locus causing frontotemporal dementia and Parkinsonism: phenotype-genotype correlations. JAMA Neurol. 2014;71:1162–1171. doi: 10.1001/jamaneurol.2014.994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, Uchida K, Waeg G, Mattson MP. 4-Hydroxynonenal, an aldehydic product of membrane lipid peroxidation, impairs glutamate transport and mitochondrial function in synaptosomes. Neuroscience. 1997;80:685–696. doi: 10.1016/s0306-4522(97)00065-1. [DOI] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Markesbery WR. Possible involvement of proteasome inhibition in aging: implications for oxidative stress. Mech Ageing Dev. 2000;113:61–70. doi: 10.1016/s0047-6374(99)00101-3. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Krohne TU, Kaemmerer E, Holz FG, Kopitz J. Lipid peroxidation products reduce lysosomal protease activities in human retinal pigment epithelial cells via two different mechanisms of action. Exp Eye Res. 2010a;90:261–266. doi: 10.1016/j.exer.2009.10.014. [DOI] [PubMed] [Google Scholar]
- Krohne TU, Stratmann NK, Kopitz J, Holz FG. Effects of lipid peroxidation products on lipofuscinogenesis and autophagy in human retinal pigment epithelial cells. Exp Eye Res. 2010b;90:465–471. doi: 10.1016/j.exer.2009.12.011. [DOI] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Langston JW. The MPTP Story. J Parkinsons Dis. 2017;7:S11–S22. doi: 10.3233/JPD-179006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT, Wheeler TC, Li L, Chin LS. Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum Mol Genet. 2008b;17:906–917. doi: 10.1093/hmg/ddm363. [DOI] [PubMed] [Google Scholar]
- Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N, Pieri L, Madiona K, Durr A, Melki R, et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol. 2013;73:459–471. doi: 10.1002/ana.23894. [DOI] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, Di Monte DA. Lysosomal degradation of alpha-synuclein in vivo. J Biol Chem. 2010;285:13621–13629. doi: 10.1074/jbc.M109.074617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin JH, et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science. 2016;353 doi: 10.1126/science.aah3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark RJ, Hensley K, Butterfield DA, Mattson MP. Amyloid beta-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci. 1995;15:6239–6249. doi: 10.1523/JNEUROSCI.15-09-06239.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP. Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J Neurosci. 1997;17:1046–1054. doi: 10.1523/JNEUROSCI.17-03-01046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp Gerontol. 2009;44:625–633. doi: 10.1016/j.exger.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Parkinson’s disease: don’t mess with calcium. J Clin Invest. 2012;122:1195–1198. doi: 10.1172/JCI62835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano A, Manini P, d’Ischia M. Oxidation chemistry of catecholamines and neuronal degeneration: an update. Curr Med Chem. 2011;18:1832–1845. doi: 10.2174/092986711795496863. [DOI] [PubMed] [Google Scholar]
- Näsström T, Fagerqvist T, Barbu M, Karlsson M, Nikolajeff F, Kasrayan A, Ekberg M, Lannfelt L, Ingelsson M, Bergström J. The lipid peroxidation products 4-oxo-2-nonenal and 4-hydroxy-2-nonenal promote the formation of α-synuclein oligomers with distinct biochemical, morphological, and functional properties. Free Radic Biol Med. 2011;50:428–437. doi: 10.1016/j.freeradbiomed.2010.11.027. [DOI] [PubMed] [Google Scholar]
- Nonaka T, Iwatsubo T, Hasegawa M. Ubiquitination of alpha-synuclein. Biochemistry. 2005;44:361–368. doi: 10.1021/bi0485528. [DOI] [PubMed] [Google Scholar]
- Okochi M, Walter J, Koyama A, Nakajo S, Baba M, Iwatsubo T, Meijer L, Kahle PJ, Haass C. Constitutive phosphorylation of the Parkinson’s disease associated alpha-synuclein. J Biol Chem. 2000;275:390–397. doi: 10.1074/jbc.275.1.390. [DOI] [PubMed] [Google Scholar]
- Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ, Steece-Collier K, Kemp CJ, Celano S, Schulz E, et al. Intrastriatal injection of preformed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis. 2015;82:185–199. doi: 10.1016/j.nbd.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotegher N, Bubacco L. Lysines, Achilles’ heel in alpha-synuclein conversion to a deadly neuronal endotoxin. Ageing Res Rev. 2016;26:62–71. doi: 10.1016/j.arr.2015.12.002. [DOI] [PubMed] [Google Scholar]
- Poehler AM, Xiang W, Spitzer P, May VE, Meixner H, Rockenstein E, Chutna O, Outeiro TF, Winkler J, Masliah E, et al. Autophagy modulates SNCA/alpha-synuclein release, thereby generating a hostile microenvironment. Autophagy. 2014;10:2171–2192. doi: 10.4161/auto.36436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Qin Z, Hu D, Han S, Reaney SH, Di Monte DA, Fink AL. Effect of 4-hydroxy-2-nonenal modification on alpha-synuclein aggregation. J Biol Chem. 2007;282:5862–5870. doi: 10.1074/jbc.M608126200. [DOI] [PubMed] [Google Scholar]
- Recasens A, Dehay B, Bove J, Carballo-Carbajal I, Dovero S, Perez-Villalba A, Fernagut PO, Blesa J, Parent A, Perier C, et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014;75:351–362. doi: 10.1002/ana.24066. [DOI] [PubMed] [Google Scholar]
- Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ, Lee VM, Brundin P. Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med. 2016;213:1759–1778. doi: 10.1084/jem.20160368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci. 2015;40:200–210. doi: 10.1016/j.tibs.2015.02.003. [DOI] [PubMed] [Google Scholar]
- Shringarpure R, Grune T, Sitte N, Davies KJ. 4-Hydroxynonenal-modified amyloid-beta peptide inhibits the proteasome: possible importance in Alzheimer’s disease. Cell Mol Life Sci. 2000;57:1802–1809. doi: 10.1007/PL00000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B. Aggregated and monomeric alpha-synuclein bind to the S6’ proteasomal protein and inhibit proteasomal function. J Biol Chem. 2003;278:11753–11759. doi: 10.1074/jbc.M208641200. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett. 1998a;251:205–208. doi: 10.1016/s0304-3940(98)00504-7. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A. 1998b;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Schumacker PT, Guzman JD, Ilijic E, Yang B, Zampese E. Calcium and Parkinson’s disease. Biochem Biophys Res Commun. 2017;483:1013–1019. doi: 10.1016/j.bbrc.2016.08.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trexler AJ, Rhoades E. N-Terminal acetylation is critical for forming alpha-helical oligomer of alpha-synuclein. Protein Sci. 2012;21:601–605. doi: 10.1002/pro.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE, Chandra SS. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci. 2014;34:9364–9376. doi: 10.1523/JNEUROSCI.4787-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlassov AV, Magdaleno S, Setterquist R, Conrad R. Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim Biophys Acta. 2012;1820:940–948. doi: 10.1016/j.bbagen.2012.03.017. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Lee VM. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc. 2014;9:2135–2146. doi: 10.1038/nprot.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, Hayashi S, Kakita A, Yamada M, Toyoshima Y, Yoshimoto M, Takahashi H. Accumulation of alpha-synuclein/NACP is a cytopathological feature common to Lewy body disease and multiple system atrophy. Acta Neuropathol. 1998;96:445–452. doi: 10.1007/s004010050918. [DOI] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, et al. alpha-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol. 2010;190:1023–1037. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers GS, George JM, Banker GA, Clayton DF. Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res Dev Brain Res. 1997;99:87–94. doi: 10.1016/s0165-3806(96)00210-6. [DOI] [PubMed] [Google Scholar]
- Xilouri M, Brekk OR, Stefanis L. alpha-Synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol. 2013;47:537–551. doi: 10.1007/s12035-012-8341-2. [DOI] [PubMed] [Google Scholar]
- Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci U S A. 1996;93:2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Zhang S, Eitan E, Mattson MP. Early involvement of lysosome dysfunction in the degeneration of cerebral cortical neurons caused by the lipid peroxidation product 4-hydroxynonenal. J Neurochem. 2017;140:941–954. doi: 10.1111/jnc.13957. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.