

Abstract
The evolution of virulence traits is central for the emergence or re-emergence of microbial pathogens and for their adaptation to a specific host1–5. Typhoid toxin is an essential virulence factor of the human-adapted bacterial pathogen Salmonella Typhi6,7, the cause of typhoid fever in humans8–12. Typhoid toxin has a unique A2B5 architecture with two covalently linked enzymatic “A” subunits, PltA and CdtB, associated to a homopentameric “B” subunit made up of PltB, which has binding specificity for N-acetylneuraminic acid (Neu5Ac) sialoglycans6,13 predominantly present in humans14. Here we examined the functional and structural relationship between typhoid toxin and ArtAB, an evolutionarily related AB5 toxin encoded by the broad-host Salmonella Typhimurium15. We found that ArtA and ArtB, homologs of PltA and PltB, can form a functional complex with the typhoid toxin CdtB subunit after substitution of a single amino acid in ArtA, while ArtB can form a functional complex with wild type PltA and CdtB. We also found that after addition of a single terminal Cys residue, a CdtB homolog from cytolethal distending toxin can form a functional complex with ArtA and ArtB. In line with the broad host specificity of S. Typhimurium, we found that ArtB binds human glycans, terminated in N-acetylneuraminic acid, as well as glycans terminated in N-glycolylneuraminic acid (Neu5Gc), which are expressed in most other mammals14. The atomic structure of ArtB bound to its receptor shows the presence of an additional glycan-binding site, which broadens its binding specificity. Despite equivalent toxicity in vitro, we found that the ArtB/PltA/CdtB chimeric toxin exhibits reduced lethality in an animal model, indicating that the host specialization of typhoid toxin has optimized its targeting mechanisms to the human host. This is a remarkable example of toxin evolution to broaden its enzymatic activities and adapt to a specific host.
Keywords: Salmonella Typhi, bacterial pathogenesis, evolution of virulence, glycobiology, sialylated glycans
Typhoid toxin appears to have evolved through the combination of two “A” subunits independently present in other bacterial toxins, into a single toxin7. PltA exhibits significant amino acid sequence similarity to ADP-ribosyl transferases that serve as A subunits in several AB5 exotoxins, while CdtB is a homolog of the A subunit of the cytolethal distending toxins (CDT) encoded by several bacterial pathogens16 (Fig. 1a and Supplementary Fig. 1). CdtB and PltA are linked together by a single disulfide bond coordinated by uniquely positioned cysteine residues (Fig. 1a and Supplementary Fig. 1)6. The crystal structure of typhoid toxin shows that the atomic interface between CdtB and PltA is rather limited and reduction of the disulfide bond results in the dissociation of the complex6. Despite the very high amino acid sequence similarity of CdtB and PltA with its homologs in other bacteria, the Cys residues involved in tethering them to form typhoid toxin are unique to these two subunits (Fig. 1a and Supplementary Fig. 1). These observations suggest that the evolution of this multi-subunit toxin is likely to be relatively recent. In support of this hypothesis, homologs of each of the components of typhoid toxin are encoded within various Salmonella enterica subspecies as components of other toxins17,18. For example, available genome sequencing data indicate the presence of an intact locus encoding all the CDT components including CdtB in Salmonella Arizonae and Diarizonae18. Furthermore, certain strains of S. Typhimurium encode a toxin ArtAB, which exhibits very significant amino acid sequence similarity to the PltA and PltB components of typhoid toxin (Fig. 1a and Supplementary Fig. 1 and 2)15. A degraded copy of the artAB locus is present in several Salmonella enterica serovars15. Since Salmonella can readily exchange genetic materials across different serovars and strains, the presence of the different components of typhoid toxin in different Salmonella lineages, sometimes within mobile genetic elements, provides a framework through which typhoid toxin could have emerged.
Figure 1.
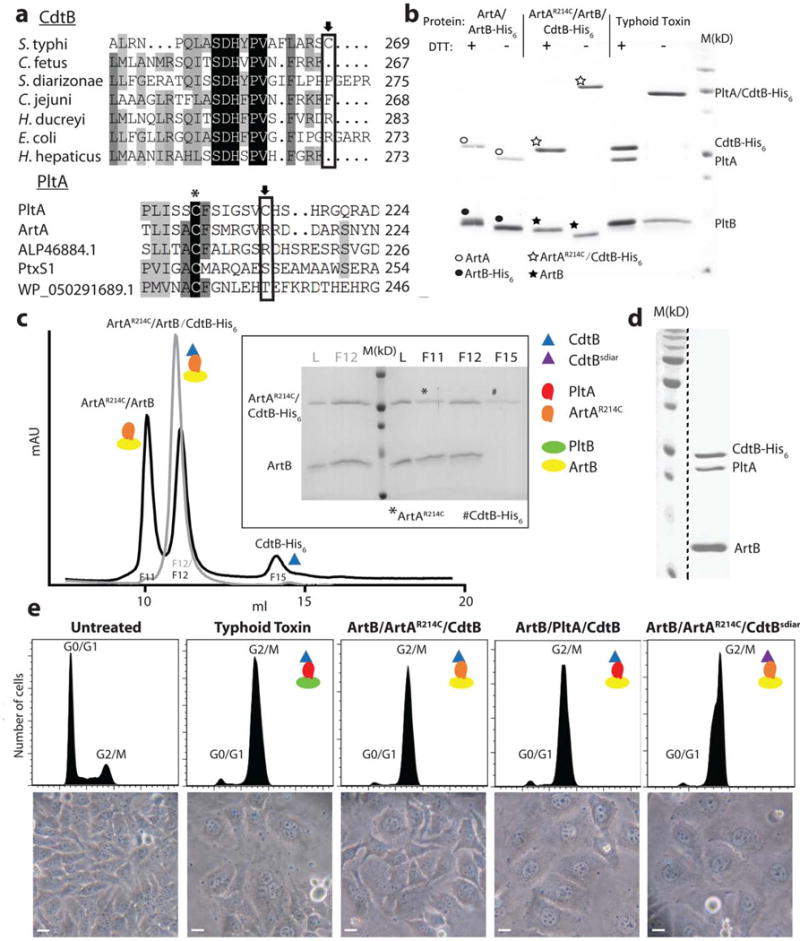
The ArtAB toxin components can form a functional complex with typhoid toxin subunits. a, Amino acid sequence comparison of CdtB and PltA homologs. Conserved and unique cysteines are indicated with an asterisk and an arrow, respectively. ALP46884.1 and WP_050291689.1 are PltA homologs from Escherichia coli and Yersinia kristensenii, respectively. PltxS1 is the A subunit from pertussis toxin. b, Purified ArtB/ArtA, ArtB/ArtAR214C/CdtB, or typhoid toxin (PltB/PltA/CdtB) protein complexes were analyzed by SDS-PAGE in the presence or absence of DTT to release CdtB, which is linked to the complex by a disulfide bond. The migration of ArtA and His6-tagged CdtB are very similar in SDS-PAGE so in the presence of DTT the two bands overlap. In the absence of DTT, ArtA and CdtB (like CdtB and PltA in the case of typhoid toxin, see right lanes) migrate as a single, slower moving band, an indication that these subunits are linked by a disulfide bond. ○: indicates ArtA; ●: indicates ArtB-His6; ☆: indicates ArtAR214C/CdtB-His6; ★: indicates ArtB. This experiment was carried out twice with equivalent results. c, The ArtAR214C/ArtB/CdtB chimeric toxin complex was analyzed by ion exchange chromatography before (gray) and after (black) treatment with DTT. L: loading control; M: molecular weight markers; F: chromatographic fraction. Inset shows SDS-PAGE analyzes of the indicated chromatographic fractions. *: ArtAR214C; #: CdtB-His6. This experiment was carried out once. d, ArtB can form a complex with wild-type PltA and CdtB. The ArtB/PltA/CdtB complex was purified by ion exchange and size exclusion chromatography and subsequently analyzed by SDS-PAGE and coomassie blue staining. This experiment was carried out three times with equivalent results. The dashed black line indicates that this panel is a composite image of two discontinuous lanes from the same gel. e, Toxicity of the chimeric toxin complexes. Cultured Henle-407 epithelial cells were treated with Typhoid Toxin (3.5 pM), ArtB/ArtAR214A/CdtB (15 pM), ArtB/PltA/CdtB (15 pM), or ArtB/ArtAR214A/CdtBsdiar, and the CdtB-mediated cell cycle arrest was assayed by flow cytometric analysis. CdtBsdiar: CdtB from S. diarizonae. Light microscopic images of mock or toxin treated cells are also shown. Scale bar: 50μm. This experiment was carried out three times with equivalent results.
We therefore investigated the potential evolutionary relatedness between ArtAB and typhoid toxin. We introduced a Cys residue in ArtA (ArtAR214C) at the equivalent position of the Cys residue in PltA (PltAC214) that forms a disulfide bond with CdtB, co expressed it with ArtB and CdtB, and examined their ability to form a complex. We found that ArtB, ArtAR214C, and CdtB formed a complex in the predicted stoichiometry of 5:1:1 (Fig. 1b and 1c). Importantly, addition of a reducing agent resulted in the dissociation of CdtB from ArtA (Fig. 1b and 1c), demonstrating that the assembled complex has an equivalent architecture to that of typhoid toxin. We then tested the ability of the “B” subunit ArtB to form a complex with PltA and CdtB, the two “A” subunits from typhoid toxin. We found that co-expression of all these components led to the assembly of a stable complex of a size consistent with that of an ArtB5-PltA-CdtB complex (Fig. 1d and Supplementary Fig. 3). We then examined the functionality of the resulting chimeric toxins by testing their CdtB-dependent toxicity when applied to cultured cells. We found that consistent with the formation of stable complexes (Supplementary Fig. 4), the CdtB-dependent toxic activity of the different chimeric toxins was similar to that of typhoid toxin (Fig. 1e). We also examined the ability of a homolog of CdtB from the S. diarizonae cytolethal distending toxin to form a complex with ArtA and ArtB (Fig. 1e). We found that after addition of single Cys residue as its last amino acid, CdtBCys276 was able to form a functional complex with ArtA and ArtB (Fig. 1e). Taken together, these results demonstrate a very close structural relationship among the components of these evolutionary related toxins and, more importantly, a remarkable plasticity to form functional chimeric complexes, which likely facilitates the evolution of this toxin family.
Typhoid toxin exhibits exquisite preference for human-specific Neu5Ac-terminated sialoglycans on surface glycoproteins that serve as its receptors13. The amino acid sequence similarity between PltB and ArtB (Supplementary Fig. 2) suggests that the ArtB may also bind sialoglycans. Consistent with this hypothesis, removal of surface sialic acids from cultured cells reduced ArtB binding (Fig. 2a and Supplementary Fig. 5). Since ArtAB is encoded by the broad host range S. Typhimurium, we hypothesized that the binding specificity of ArtB may differ from that of the human-adapted typhoid toxin. To test this hypothesis we probed the ability of fluorescently-labeled ArtB to bind a diverse group of glycans arrayed on a solid surface. We found that similar to typhoid toxin, ArtB was able to bind a diverse group of sialylated glycans with the consensus structure Neu5Acα2–3Galβ1–3/-4Glc/GlcNAc (Fig. 2b and Supplementary Table S1). ArtB was also able to bind an additional group of sialylated glycans with the consensus Neu5Acα2–6Gal/GalNAc, which typhoid toxin did not (Fig. 2b and Supplementary Table S1). Importantly, however, unlike typhoid toxin, ArtB was also able to bind Neu5Gc-terminated glycans (Fig. 2c and Supplementary Table S2). The broad binding specificity of ArtB is entirely consistent with the broad host range of the Salmonella eterica serovar that harbors it.
Figure. 2.
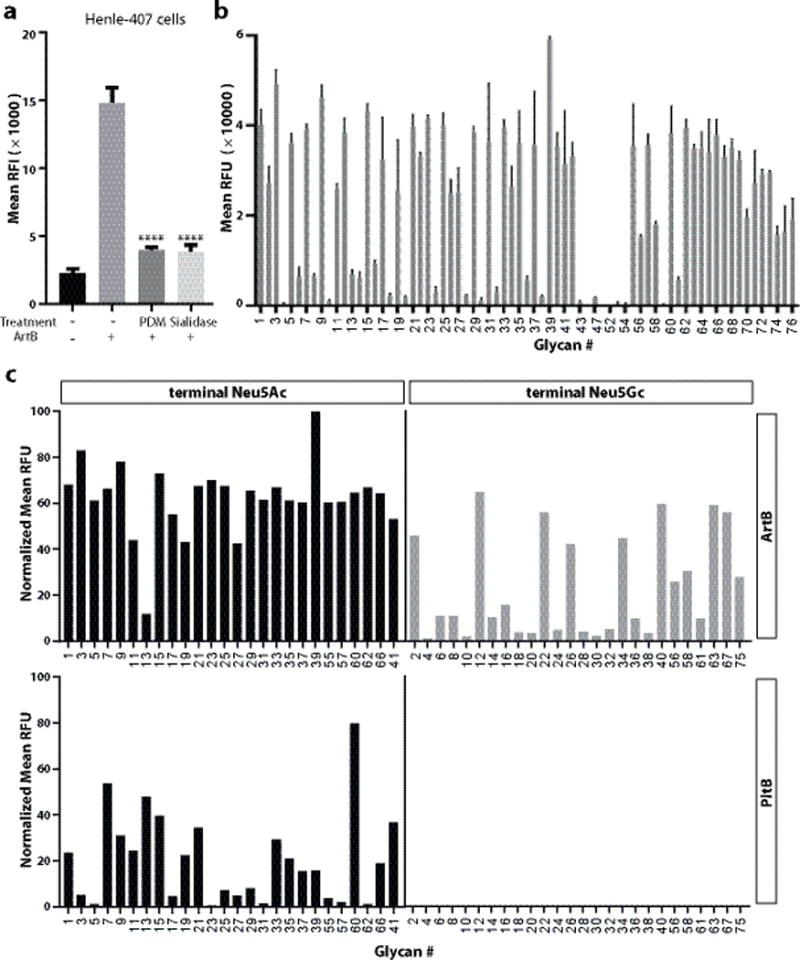
ArtB binds Neu5Ac- and Neu5Gc-terminated glycans. a, Removal of surface glycans reduces ArtB binding to cultured cells. Henle-407 cells were treated with a mixture of glycosidases (PDM, protein deglycosylation mix) or a sialidase (α2–3/6/8-neuraminidase) and the ability of treated and control cells to bind fluorescently labeled ArtB (2.5 μM) was evaluated by flow cytometry. The y-axis values represent the relative fluorescence intensity (RFI). Bars represent mean ± standard deviation of at least three independent measurements. Two-tailed Student’s t tests were performed to determine the statistical significance for two group comparisons. ****: P<0.0001, compare to the relative fluorescence intensity of ArtB-binding to the untreated cells. b, ArtB-binding to a customized glycan microarray. The y-axis values represent average and standard deviation of the relative fluorescence units (RFU) from four independent experiments, and the x-axis indicates the glycan numbers (see also Table S1). c, Comparison of ArtB-binding to paired Neu5Ac- and Neu5Gc- terminated glycans. The y-axis values represent the normalized average relative fluorescence units (RFU) from four independent experiments and the x-axis depicts the glycan numbers (see also Table S2).
We have previously shown that a single amino acid substitution in the glycan-binding site of PltB (PltBS35A) completely abolished the ability of typhoid toxin to bind its glycan receptors and to intoxicate human cells6. However, we found that in contrast to PltB, the equivalent mutation in ArtB (ArtBS31A) retained significant ability to bind cultured cells (Supplementary Fig. 6). These results suggested substantial differences between the mechanisms of glycan binding in ArtB and PltB. To gain insight into the structural bases for the binding specificity of ArtB, we determined its atomic structures alone (at 2.3 Å resolution, Fig. 3a) or bound to its glycan receptor Neu5Acα2–3Galβ1–4Glc (at 2.2 Å resolution, Fig. 3b)(Supplementary Table S3). For comparison, we also solved the structure of PltB bound to the same glycan (Fig. 3c). In the crystal, each asymmetric unit of ArtB contained 10 molecules organized into two homopentamers as predicted by its homology to other AB5 toxins. In each of the pentamers, the ArtB protomer shows a typical oligosaccharide-binding fold located on the side of the pentamer, which corresponds to the equivalent binding site observed in the PltB structure. Clear electron density corresponding to the Neu5Acα2–3Galβ was observed at this site in two of the five subunits of the ArtB and in three of the PltB pentamers (Fig. 3a–3c). The other sites were occupied by 2-(N-morpholino)ethanesulfonic acid (present in the crystallization buffer), which exhibits a similar structure to Neu5Ac. Close view of the ArtB canonical glycan-binding site shows that the Neu5Ac moiety interacts through multiple direct hydrogen bonds and water-mediated hydrogen bonds with Asn27, Tyr29, Gln30, Ser31, Arg59 and Asn65 in ArtB (Fig. 3d). In addition, the Neu5Ac ring makes a hydrophobic contact with the aromatic ring of Tyr29. Overall, the arrangement of Neu5Ac and the interactions between Neu5Ac and ArtB are very similar to what is seen in PltB, which makes contact with the glycan moiety with equivalent interaction involving Tyr33, Tyr34, Ser35, Lys59 and Thr65 (Supplementary Fig. 7). However, ArtB has a Tyr103 that is absent from PltB (Fig. 3d). The corresponding residue in the related toxin SubB has been shown to form a critical hydrogen bond with the extra hydroxyl group of Neu5Gc19. This structural feature is consistent with the observation that ArtB, like SubB, can also bind glycans terminated with Neu5Gc.
Figure. 3.
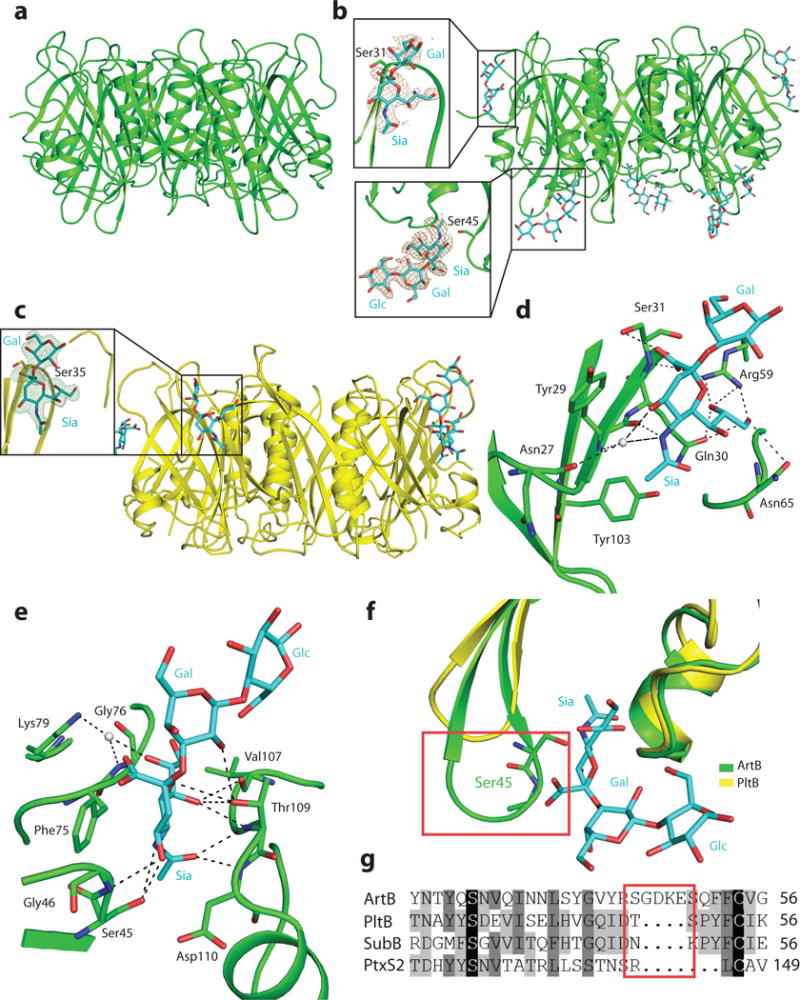
The atomic structure of ArtB bound to its receptor shows the presence of an additional glycan-binding site. a, Atomic structure of the ArtB pentamer shown as a ribbon cartoon. b, Atomic structure of the ArtB pentamer in complex with the Neu5Acα2–3Galβ1–4Glc oligosaccharide shown as a ribbon cartoon. Cyan, blue and red sticks represent carbon, nitrogen and oxygen atoms in the sugar backbone. Insets show the close-up views of Neu5Acα2–3Galβ1–4Glc and Neu5Acα2–3Gal. Brown mesh represents the sugar composite annealed omit difference density map contoured at 2.0σ. c, Atomic structure of the PltB pentamer in complex with the Neu5Acα2–3Gal oligosaccharide is shown as a ribbon cartoon. Cyan, blue and red sticks represent carbon, nitrogen and oxygen atoms in the sugar backbone. Insets show the close-up views of Neu5Acα2–3Galβ1–4Glc. Green mesh represents the sugar composite annealed omit difference density map contoured at 2.5σ. d and e, Interactions between ArtBSer31 (d) and ArtBSer45 (e) with Neu5Acα2–3Gal and Neu5Acα2–3Galβ1–4Glc, respectively. ArtB is shown as a green colored ribbon cartoon, the sugar and the amino acids interacting with the sugar are shown as sticks, the interactions are shown in black dashes and water is shown as gray balls. f, Structural comparison of ArtBSer45 sugar-binding site with the equivalent surface in PltB. Blue and red sticks in the sugar backbone represent nitrogen and oxygen atoms, respectively. g, Amino acid sequence alignment of ArtBSer45 glycan-binding site with the equivalent regions in PltB, SubB and PtxS2. The red boxes depicted in f and g highlight the insert sequence and the associated structural features that are uniquely present in ArtB.
Surprisingly, an additional unambiguous glycan density was detected away from the predicted glycan-binding site of the ArtB pentamer in 3 or 4 out of the five protomers in different pentamers (Fig. 3b). No glycan density was detected at an equivalent position of PltB. At this additional glycan-binding site, ArtB makes contact with both the Neu5Ac and Gal moieties in Neu5Acα2–3Galβ1–4Glc through multiple interactions. For example, Ser45, Gly46, Phe75, Gly76, Lys79, Val107, Thr109 and Asp110 interact with the Neu5Ac moiety through multiple direct hydrogen bonds and water-mediated hydrogen bonds. In addition, the aromatic ring of Phe75 makes hydrophobic contact with the ring structure of Neu5Ac and Thr109 makes contact with the Gal moiety of Neu5Acα2–3Galβ1–4Glc through hydrogen bonding (Fig. 3e). Comparison of this additional glycan-binding site in ArtB with the equivalent surface in PltB reveals the presence in ArtB of a spoon-like structure critical for ligand binding, which is absent in PltB (Fig. 3f). Amino acid sequence alignment of this region of ArtB with the equivalent region in PltB showed the presence of a unique 5 amino acid insertion in ArtB (Fig. 3g), which is responsible for the structural features associated with glycan binding. Comparison of the atomic structures of the receptor-bound and apo forms of ArtB indicates that, similar to PltB, binding to its glycan receptors does not result in detectable conformational changes (Supplementary Figs. 8 and 9). Taken together, these findings indicate that ArtB has an additional glycan-binding site that may contribute to its expanded glycan-binding repertoire and that may have been eliminated from typhoid toxin by introduction of a small deletion during the process of its adaptation to humans.
The residual glycan-binding activity of the ArtBS31A mutant coupled to the finding of a second glycan-binding site in ArtB prompted us to explore the potential specific contribution of each one of these sites to its overall glycan binding activity. We generated and purified ArtBS31A, and ArtBS45A single mutants as well as ArtBS31A/S45A double mutant proteins (Supplementary Fig. 10) and compared their ability to bind cultured epithelial cells. We found that, in comparison to wild type ArtB, the binding of ArtBS31A and ArtBS45A was reduced but not abrogated. In contrast, the binding of the ArtBS31A/S45A double mutant was undetectable (Supplementary Fig. 11). These observations suggest that both sites contribute to ArtB binding to cultured cells. We then examined the binding of the different ArtB mutants in a customized glycan array. We found that the ArtBS45A mutant exhibited less residual glycan-binding activity than ArtBS31A (Supplementary Fig. 12 and Supplementary Table S3), indicating that the ArtBSer45 site plays a more prominent role in glycan binding. Importantly, comparison of the binding profiles of the ArtBS31A and ArtBS45A mutants indicated that the ArtBSer45 site exhibits binding preference for glycans terminated in Neu5Ac-α2–6Gal/GalNAc (Supplementary Fig. 13 and Supplementary Table S4), a binding-specificity absent in PltB13. Furthermore, we found that both ArtB mutants retained the ability to bind Neu5Gc-terminated glycans indicating that both sites contribute to this expanded binding specificity (Supplementary Fig. 14 and Supplementary Table S5). To further explore the impact of delivering the typhoid toxin-associated toxic activities through the different ArtB glycan-binding sites, we assembled chimeric toxins consisting of wild type and different mutants of ArtB (ArtBS31A, ArtBS45A or ArtBS31A/S45A) in complex with PltA and CdtB (Supplementary Fig. 15) and tested their toxic and binding activities in culture human (displaying Neu5Ac-terminated glycans) and mouse (displaying Neu5Gc- and Neu5Ac-terminated glycans) cells. We found that relative to one another, PltB exhibited greater binding to human cells, while ArtB exhibited higher binding to mouse cells (Fig. 4a). Although the chimeric and typhoid toxins exhibited similar toxic activity to mouse cells (Fig. 4b and Supplementary Fig. 16), typhoid toxin showed more toxicity towards human cells (Fig. 4b). We also found that both ArtB glycan-binding sites played an important role in toxin delivery since each of the individual mutants exhibited a significant reduction in toxicity, and simultaneous introduction of mutations in both sites resulted in the complete loss of toxicity (Fig. 4c). Taken together, these results indicate that the presence of an additional glycan-binding site broadens the glycan binding specificity of ArtB relative to PltB.
Figure 4.
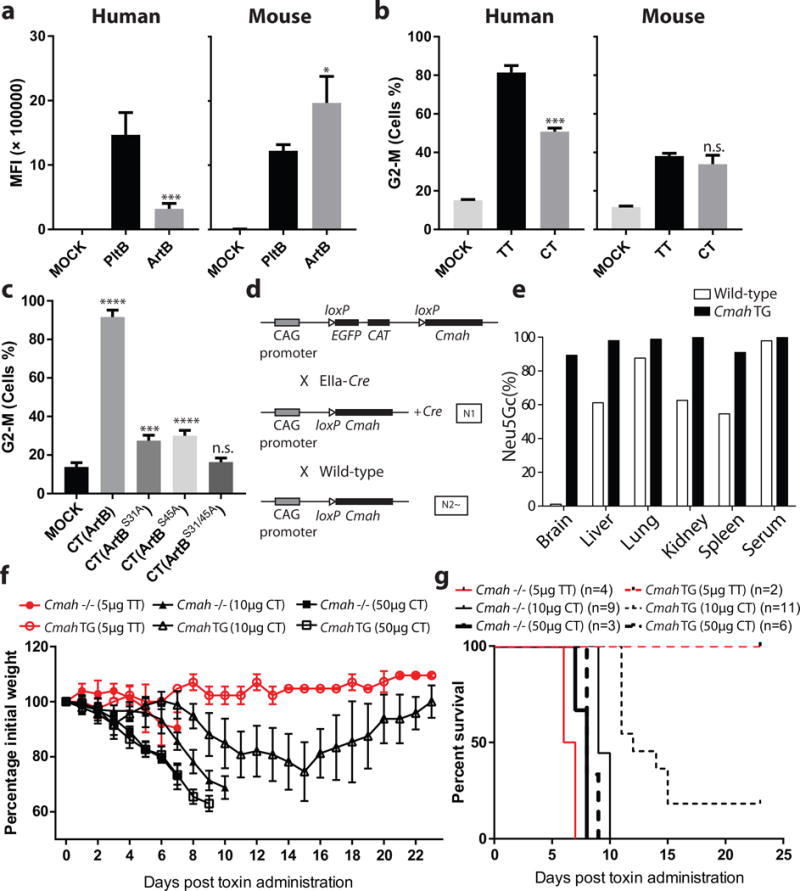
The ArtB/PltA/CdtB chimeric toxin exhibits reduced lethality in mice relative to typhoid toxin. a, ArtB and PltB (2. 5 μM each) binding to human (Henle-407) or mouse (embryo fibroblasts) cells. The binding of fluorescently-labeled PltB and ArtB was evaluated by flow cytometry. The y-axis values represent the mean fluorescence index (MFI) and are the mean ± SD of at least three independent measurements. *: p < 0.05; ***: p < 0.0005. b, Relative toxicity to human (Henle-407) or mouse (embryo fibroblasts) cells of purified typhoid toxin (TT) (3 pM) and ArtB/PltA/CdtB chimeric toxin (CT) (3 pM). The percentage of cells in G2/M, a measured of CdtB toxic activity, after application of the indicated toxins was determined by flow cytometry. Data are the mean ± SD of at least three independent determinations. ***: p < 0.0005, n.s. (no significance): p>0.05 compared to untreated cells. c, Relative contribution of the different ArtB glycan-biding sites to toxicity. Different ArtB/PltA/CdtB chimeric toxin (CT) preparations containing the indicated ArtB mutants were tested for their ability to intoxicate cultured cells. Equal amounts (250 pM) of chimeric toxin preparations were applied to the cultured cells and the percentage of cells in G2/M was determined by flow cytometry. Data are the mean ± SD of at least three independent determinations. ****: p < 0.0001, ***: p < 0.0005, n.s. (no significance): p>0.05 compared to untreated cells. In a-c two-tailed Student’s t tests were performed to determine the statistical significance for two group comparisons. d, Scheme of the strategy used to generate a Cmah TG mouse. Targeted Cre-inducible Cmah TG mice (transgene insertion in H11 locus, top) were crossed with EIIa-Cre mice that induce Cre expression at the preimplantation embryo stage and the resulting N1 generation mouse with the Cmah transgene and Cre was further mated with wild-type mice. Among the resulting N2 generation, the mice that had Cmah transgene but lacked Cre were selected as systemic Cmah TG mice and maintained by crossing with wild-type mice. e, Tissue homogenates obtained from 11-week-old male mice were hydrolyzed in 2M acetic acid to release sialic acids after treatment with 0.1M sodium hydroxide to remove O-acetylation of sialic acids. The percentage of Neu5Gc in total sialic acids was determined by HPLC using DMB-derivatization method. Each bar represents the average of samples from two mice per genotype. Cmah TG mice showed remarkably high Neu5Gc expression in all tissues tested. f and g, Mouse toxicity of the ArtB/PltA/CdtB chimeric toxin relative to typhoid toxin. Cmah −/− or Cmah transgenic (TG) mice were administered intraorbitally either typhoid toxin (TT) (5μg) or ArtB/PltA/CdtB chimeric toxin (CT) (10 or 50 μg) and their body weight (f) and survival (g) at the indicated times were recorded. Values in (f) are the mean and standard deviations. The difference in weight loss between groups (f) was analyzed by the Mann-Whitney test (TT vs CT in Cmah TG: p <0.0001, TT vs CT in Cmah −/−: p<0.05). The difference in survival (g) was analyzed by the Mantel Cox test. (TT vs CT in Cmah TG: p <0.05; TT (5 μg) vs CT (10 μg) in Cmah −/−: p <0.001.
We compared the toxicity of typhoid toxin with that of the ArtB/PltA/CdtB chimera in an animal model of intoxication. Despite equivalent in-vitro toxic activity (Fig. 4b and Supplementary Fig. 16), administration of the same amount of toxin to C57BL/6 mice resulted in a more rapid death in animals receiving typhoid toxin than those receiving the ArtB/PltA/CdtB chimera toxin (Supplementary Fig. 17). Since Cmah expression in C57BL/6 mice is variable, these mice display both Neu5Ac- and Neu5Gc-terminated glycans20. We therefore compared the toxicity of the different toxin preparations in Cmah −/− mice, which display Neu5Ac-terminated human-like sialoglycans20. In addition, we constructed a transgenic mouse that constitutively expresses Cmah under a strong promoter and therefore almost exclusively displays Neu5Gc-terminated glycans (Fig. 4d and 4e and Supplementary Fig. 18 and 19). We found that as predicted by its expanded glycan-binding specificity, the chimeric ArtB/PltA/CdtB toxin displayed toxicity to the Cmah transgenic mice while, as predicted by its binding-specificity13, typhoid toxin did not (Fig. 4f and 4g). Surprisingly, however, despite its expanded binding specificity, the chimeric toxin exhibited significantly less lethality than typhoid toxin for Cmah −/− mice. Furthermore, there were clear differences in the course of intoxication as the mice receiving the ArtB/PltA/CdtB chimeric toxin did not show signs of stupor, which were readily apparent in mice that received typhoid toxin. In contrast, leukopenia was apparent in all mice regardless whether they got typhoid toxin or the chimeric toxin (Supplementary Fig. 20). These results indicate that although typhoid toxin has narrowed its receptor-binding specificity, the process of host adaptation has resulted in heightened toxicity presumably by gaining efficiency in the targeting of specific tissues and/or in its escape from soluble sialylated decoy ligands21.
Taken together, this study indicates that typhoid toxin may have evolved from an ArtAB-like ancestor in at least two steps. One step involved the incorporation of an additional active subunit to an already existing AB5 toxin by evolving specifically positioned Cys residues to coordinate the linking of PltA to CdtB by a disulfide bond. The interface between PltA and CdtB buries only ~ 950 Å2 and CdtB is released from the toxin under reducing conditions indicating that the disulfide bond is all that maintains these two subunits tethered to one another. Indeed, simply adding a terminal Cys residue to a CdtB homolog from a S. diarizonae cytolethal distending toxin was sufficient to form a functional complex with ArtAB. The other step may have involved the narrowing and optimization of its glycan-binding site to maximize the toxin’s effectiveness in its human host. In fact, the locus that encodes a typhoid toxin homolog in S. diarizonae contains two pltB homologs, pltBSdiar-1, which exhibits very close amino acid and structural similarity to S. Typhi PltB, and pltBSdiar-2, a more divergent homolog, which is more related to ArtB (Supplementary Fig. 21–24). Notably, we found that, unlike typhoid toxin, chimeric toxins assembled with either of these homologs were able to intoxicate cells displaying either Neu5Ac or Neu5Gc-terminated glycans (Supplementary Fig. 25 and 26). However, a chimeric toxin assembled with pltBSdiari-1 exhibited more toxicity to cells displaying Neu5Ac-terminated glycans, while a chimeric toxin assembled with pltBSdiari-2 showed the opposite preference (Supplementary Fig. 25 and 26). It is therefore possible that PltBSdiari-1 and PltBSdiari-2 represent intermediate step in the evolution of S. Typhi’s PltB from an ArtB-like ancestor. In summary, these findings describe a remarkable example of virulence factor evolution in which relatively minor genetic changes coupled to horizontal gene exchange within the same bacterial species may have led to the emergence of a powerful toxin exquisitely adapted to the human host.
METHODS
Toxin Expression and Purification
Coding sequences for artA and artB were amplified from Salmonellla enterica serovar Typhimurium DT104 genomic DNA and cloned into the expression vector pET28b. Coding sequences for pltB homologs from Salmonella diarizonae were amplified directly from bacterial cell lysates and cloned into the expression vector pET28b. Expression and purification of ArtB, ArtB chimeric toxin, PltBsdiar chimeric toxin and typhoid toxin were carried out as previously described6. Plasmids expressing ArtB point mutations were constructed using standard recombinant DNA techniques.
Crystallization
Expression and purification of C-terminal His6-tagged ArtB and PltB used for crystallization have been described previously6. Five mg/ml of full-length ArtB protein was used to perform initial spare matrix crystal screening with a crystallization robot at the Yale University School of Medicine Structural Biology Core facility. After optimization trials, the ArtB crystals grew at room temperature using the hanging-drop vapour-diffusion method, mixing 1 μl protein with 1μl reservoir solution consisting of 44% (w/v) PEG200, 0.1 M calcium chloride and 0.1 M Mes, pH 5.5. Crystals appeared in ~3 days and matured in ~1 week. In order to obtain 3′-sialyllactose (3′SL)-bound ArtB crystals, purified ArtB protein were mixed 5 mM 3′SL before setting up crystallization trays. The crystals used to collect diffraction data grew in the presence of 35% (w/v) PEG200, 0.1 M calcium chloride and 0.1 M MES buffer, pH 5.5. To grow 3′SL-bound PltB crystals, purified protein was mixed 5 mM of 3′SL in the presence of 26% (w/v) PEG1500, 0.1 M sodium acetate, pH 4.6.
X-ray data collection and structure determination
X-ray data were collected at a wavelength of 1.5418 Å on a Rigaku Homelab system at the Yale University Chemical and Biophysical Instrumentation Center (http://cbic.yale.edu) or at the Yale University School of Medicine X-ray Crystallography Core Facility. Data were integrated and scaled using the HKL-200022 or Mosflm23 software packages. Further processing was performed with programs from the CCP4 suite. The apo and 3′SL bound ArtB and PltB structures were determined by molecular replacement using PHASER24 with the atomic coordinates of PltB (Protein Data Bank [PDB] ID 4RHR) as the initial search model. To complete the model, manual building was carried out in COOT. Figures were prepared using PyMol25. The structure refinement was done by PHENIX26. The data collection and refinement statistics are summarized in Table S6. Coordinates for the atomic structures have been deposited in the RCSB Protein Data Bank under PDB numbers XXX, XXX and XXX (pending) http://www.rcsb.org/pdb/explore.do?structureId=4RHS.
Alexa 555 and Oregon Green 488 Labelling
Purified wild-type PltB, ArtB and different ArtB mutants were fluorescently labeled with Alexa-555 or Oregon Green 488 (Invitrogen) according to the vendor’s instruction. Purified PltB, ArtB and its mutants (2 mg/ml in 500 μl of 20 mM Hepes buffer, pH 8.1) were incubated for 1 hr at room temperature with reactive dyes and applied to the desalt spin columns to separate dye-protein conjugates from the free dyes.
Glycan Microarray Analysis
Glycan microarray analysis was carried out as previously described13. Briefly, glycan microarrays were fabricated using epoxide-derivatized glass slides as previously described27. Printed glycan microarray slides were blocked by ethanolamine, washed and dried, and then fitted in a multiwell microarray hybridization cassette (ArrayIt) to divide into subarrays. The subarrays were blocked with Ovalbumin (1% w/v) in PBS (pH 7.4) for 1 hr at RT in a humid chamber with gentle shaking. Subsequently, the blocking solution was discarded, and the appropriate dilutions of the different Alexa Fluor 555-labeled protein samples (64 μg/ml) were added to each subarray. After incubation for 2 hs at RT with gentle shaking, the slides were extensively washed to remove nonspecifically bound proteins. The developed glycan microarray slides were then dried and subjected to scanning by a Genepix 4000B microarray scanner (Molecular Devices). Data analysis was carried using the Genepix Pro 7.0 analysis software (Molecular Devices).
Mammalian Cell Culture Conditions
Henle-407 human intestinal epithelial cells (Roy Curtiss laboratory collection), HEK293T, mouse-embryo fibroblasts (MEF), and NIH3T3 cells (from the American Type Culture Collection) were cultured in DMEM containing 10% fetal bovine serum (FBS) at 37°C in a cell culture incubator with 5% CO2. All cell lines were routinely tested for the presence of mycoplasma by a standard PCR method. The cells were frequently checked for their morphological features, growth characteristics, and functionalities, but were not authenticated by short tandem repeat (STR) profiling. Cells were split into 12-well plates at a cell density of 2.5 × 104 cells per well for the intoxication assays or at 2 × 105 cells per well for the toxin-binding assay. The next day the cells were used for the different assays as described below.
Metabolic incorporate of Neu5Ac or Neu5Gc
Metabolic incorporation of N-acetylneuraminic acid (Neu5Ac) or N-glycolylneuraminic acid (Neu5Gc) was carried out as previously described13. Briefly, a stock solution containing 50 mM Neu5Ac or Neu5Gc was freshly prepared in DMEM medium, pH 7.0. Henle-407 cells (1.5 × 105) were seeded onto 6-well culture plates in 3 ml media containing 10 mM Neu5Ac or Neu5Gc. During the feeding period, the cells were continuously monitored and maintained below 80% confluence. After 3 days of growth, the cells were harvested and seeded onto 12-well plates at a cell density of 2.5 × 104 cells per well in 1 ml media containing 10 mM Neu5Ac or Neu5Gc. The next day the cells were used for intoxication assays as described below.
Toxin Binding Assay
Cultured cells were seeded in 12-well plates and 16 hs later were treated with different preparations of OG488-labeled PltB, ArtB, or its mutants in HBSS for 30 minutes on ice. Cells were then washed three times with PBS, fixed with 2% paraformaldehyde and ~ 10,000 cells by FACS on a BD Accuri™ C6 Flow Cytometeranalyzed. The toxin binding profile was then analyzed using FlowJo cytometry software (Tree Star, Inc., Ashland, OR) (Supplementary Fig. 27). When indicated, cells were treated with 2 μl Protein deglycosylation mix (NEB #P6039S; contains PNGase F, O-glycosidase, neuraminidase, β1–4 galactosidase and β-N-acetylglucosaminidase) or 50 units of α2–3,6,8 neuraminidase (NEB#P0720S) and incubated for 2hs at 37°C before toxin-binding assay and flow cytometric analyses.
Mammalian Cell Intoxication Assay
Cell-cycle arrest after intoxication by the ArtB chimeric or typhoid toxins was examined by flow cytometry as previously described28. Briefly, cells were treated with the different toxin preparations and 66 hr (for Henle-407) or 48 hr (for NIH3T3) after treatment, cells were trypsinized, collected, washed, and fixed overnight in ~70% ethanol/PBS at −20°C. Fixed cells were washed with PBS and resuspended in 500 μl of PBS containing 50 μg/ml propidium iodide, 0.1 mg/ml RNase A, and 0.05% Triton X-100. After incubation for 30 min at 37°C, cells were washed with PBS, resuspended in 500 μl PBS, filtered, and analyzed (3,000–5,000 cells) by a flow cytometry on a BD Accuri™ C6 Flow Cytometer. The DNA content of treated cells was determined using Flowjo software (Supplementary Fig. 28).
Generation of a Neu5Gc-overexpressing Cmah transgenic mouse
A Cmah transgenic mouse overexpressing Neu5Gc was generated following standard approaches following the scheme depicted in Fig 4.
Mouse Intoxication Experiments
Mouse lines have been previously described13 and all animal experiments were conducted according to protocols approved by Yale University’s Institutional Animal Care and Use Committee. Seven to ten-week old C57BL/6, Cmah −/− or Cmah transgenic (TG) mice were anesthetized with 30% w/v isoflurane in propylene glycol and 100 μl of toxin solution was administered via the retro-orbital route. Changes in behavior, weight and survival of the toxin-injected mice were closely monitored for the duration of the experiment. Blood samples were collected by cardiac puncture 4 days after toxin administration in Microtainer tubes coated with EDTA, kept at room temperature and analyzed within 2 hs after blood collection using a HESKA Veterinary Hematology System. The number of animals used in each experiment were empirically determined using prior experience with equivalent type of experiments.
Structural modelling
Structural modelling was performer online by using SWISS-MODEL SERVER https://swissmodel.expasy.org) with the PltB structure (Protein Data Bank [PDB] ID 4RHR) as the template. The highest scored structure was selected for the structural comparisons.
Statistical Analysis
Mice were randomly allocated to different groups. Investigators were not blinded to the allocation during the experiments or to the outcome assessment. Two-tailed Student’s t tests were performed to determine the statistical significance for two group comparisons. Given the nature of the experiments and the types of samples, mouse weight loss was assessed with a non-parametric Mann–Whitney test and mouse survival was assessed with a Mantel Cox test (Log-rank test). A p value of < 0.05 was considered to be statistically significant. All analyses were performed by using GraphPad Prism (GraphPad software).
Data Availability
The data that support the findings of this study are available from the corresponding author upon request. Coordinates for the atomic structures have been deposited in the RCSB Protein Data Bank under PDB numbers 5WHV, 5WHU and 5WHT. All data that support the findings of this study are available from the corresponding author upon request.
Supplementary Material
Acknowledgments
We thank J. Wang for suggestions and providing help with structure refinement and the Galan laboratory for careful review of the manuscript. G.S. is supported in part by a Postdoctoral Fellowship from the EMBO, ALTF 172-2015. Crystal screening was conducted at the Yale Macromolecular X-ray Core Facility (1S10OD018007-01). This work was supported by National Institute of Health grants AI079022 (to J.E.G.) and GM32373 (to A.V).
Footnotes
Author contribution
X. G., G. S., L. D., A. V. & J. E. G. designed the research and analyzed data.
X. G., G. S. & L. D., performed the research.
Y. N.-M., H. Y. and X. C. provided critical reagents.
X. G. and J. E. G. wrote the manuscript with input from all the authors.
Competing interests
The authors declare no competing interests
References
- 1.Jackson R, Johnson L, Clarke S, Arnold D. Bacterial pathogen evolution: breaking news. Trends Genet. 2011;27:32–40. doi: 10.1016/j.tig.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Donnenberg MS, Whittam TS. Pathogenesis and evolution of virulence in enteropathogenic and enterohemorrhagic Escherichia coli. J Clin Invest. 2001;107:539–548. doi: 10.1172/JCI12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bentley S, Parkhill J. Genomic perspectives on the evolution and spread of bacterial pathogens. Proc Biol Sci. 2015;282:20150488. doi: 10.1098/rspb.2015.0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alizon S, Michalakis Y. Adaptive virulence evolution: the good old fitness-based approach. Trends Ecol Evol. 2015;30:248–254. doi: 10.1016/j.tree.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 5.Daugherty M, Malik H. Rules of engagement: molecular insights from host-virus arms races. Annu Rev Genet. 2012;46:677–700. doi: 10.1146/annurev-genet-110711-155522. [DOI] [PubMed] [Google Scholar]
- 6.Song J, Gao X, Galan JE. Structure and function of the Salmonella Typhi chimaeric A(2)B(5) typhoid toxin. Nature. 2013;499:350–354. doi: 10.1038/nature12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galán J. Typhoid toxin provides a window into typhoid fever and the biology of Salmonella Typhi. Proc Natl Acad Sci U S A. 2016;113:6338–6344. doi: 10.1073/pnas.1606335113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parry C, Hien TT, Dougan G, White N, Farrar J. Typhoid fever. N Engl J Med. 2002;347:1770–1782. doi: 10.1056/NEJMra020201. [DOI] [PubMed] [Google Scholar]
- 9.Crump J, Mintz E. Global trends in typhoid and paratyphoid Fever. Clin Infect Dis. 2010;50:241–246. doi: 10.1086/649541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raffatellu M, Wilson R, Winter S, Bäumler A. Clinical pathogenesis of typhoid fever. J Infect Dev Ctries. 2008;2:260–266. doi: 10.3855/jidc.219. [DOI] [PubMed] [Google Scholar]
- 11.Wain J, Hendriksen R, Mikoleit M, Keddy K, Ochiai R. Typhoid fever. Lancet. 2015;385:1136–1145. doi: 10.1016/S0140-6736(13)62708-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dougan G, Baker S. Salmonella enterica serovar Typhi and the pathogenesis of typhoid fever. Annu Rev Microbiol. 2014;68:317–336. doi: 10.1146/annurev-micro-091313-103739. [DOI] [PubMed] [Google Scholar]
- 13.Deng L, et al. Host adaptation of a bacterial toxin from the human pathogen salmonella typhi. Cell. 2014;159:1290–1299. doi: 10.1016/j.cell.2014.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varki N, Strobert E, Dick EJ, Benirschke K, Varki A. Biomedical differences between human and nonhuman hominids: potential roles for uniquely human aspects of sialic acid biology. Annu Rev Pathol. 2011;6:365–393. doi: 10.1146/annurev-pathol-011110-130315. [DOI] [PubMed] [Google Scholar]
- 15.Saitoh M, et al. The artAB genes encode a putative ADP-ribosyltransferase toxin homologue associated with Salmonella enterica serovar Typhimurium DT104. Microbiology. 2005;151:3089–3096. doi: 10.1099/mic.0.27933-0. [DOI] [PubMed] [Google Scholar]
- 16.Lara-Tejero M, Galan JE. Cytolethal distending toxin: limited damage as a strategy to modulate cellular functions. Trends Microbiol. 2002;10:147–152. doi: 10.1016/s0966-842x(02)02316-8. [DOI] [PubMed] [Google Scholar]
- 17.Desai P, et al. Evolutionary Genomics of Salmonella enterica Subspecies. MBio. 2013;4:e00579–00512. doi: 10.1128/mBio.00579-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang C, et al. Complete genome sequence of Salmonella enterica subspecies arizonae str. RKS2983. Stand Genomic Sci. 2015;10 doi: 10.1186/s40793-015-0015-z. Stand Genomic Sci. 2015 Jun 2013;2010:2030. doi: 2010.1186/s40793-40015-40015-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Byres E, et al. Incorporation of a non-human glycan mediates human susceptibility to a bacterial toxin. Nature. 2008;456:648–652. doi: 10.1038/nature07428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hedlund M, et al. N-glycolylneuraminic acid deficiency in mice: implications for human biology and evolution. Molecular and cellular biology. 2007;27:4340–4346. doi: 10.1128/MCB.00379-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beddoe T, Paton A, Le Nours J, Rossjohn J, Paton J. Structure, biological functions and applications of the AB5 toxins. Trends Biochem Sci. 2010;35:411–418. doi: 10.1016/j.tibs.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 23.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeLano WL. The PyMOL Molecular Graphics System. Schrödinger; 2002. http://www.pymol.org. [Google Scholar]
- 26.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Padler-Karavani V, et al. Rapid evolution of binding specificities and expression patterns of inhibitory CD33-related Siglecs in primates. FASEB J. 2014;28:1280–1293. doi: 10.1096/fj.13-241497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spano S, Ugalde JE, Galan JE. Delivery of a Salmonella Typhi exotoxin from a host intracellular compartment. Cell Host Microbe. 2008;3:30–38. doi: 10.1016/j.chom.2007.11.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request. Coordinates for the atomic structures have been deposited in the RCSB Protein Data Bank under PDB numbers 5WHV, 5WHU and 5WHT. All data that support the findings of this study are available from the corresponding author upon request.