

Abstract
INTRODUCTION
Hyperinsulinemia activates brain Akt and PKC-λ/ι and increases Aβ1–40/42 and phospho-tau in insulin-resistant animals.
METHODS
Here, we examined underlying mechanisms in mice, neuronal cells and mouse hippocampal slices.
RESULTS
Like Aβ1–40/42, β-secretase activity was increased in insulin-resistant mice and monkeys. In insulin-resistant mice, inhibition of hepatic PKC-λ/ι is sufficient to correct hepatic abnormalities and hyperinsulinemia simultaneously reversed increases in Akt, aPKC, β-secretase and Aβ1–40/42, and restored acute Akt activation by insulin; However, two aPKC inhibitors additionally blocked insulin’s ability to activate brain PKC-λ/ι and thereby increase β-secretase and Aβ1–40/42. Furthermore, direct blockade of brain aPKC simultaneously corrected an impairment in novel object recognition in high-fat-fed insulin-resistant mice. In neuronal cells and/or mouse hippocampal slices, PKC-ι/λ activation by insulin, metformin or expression of constitutive PKC-ι provoked increases in β-secretase, Aβ1–40/42 and phospho-thr-231-tau that were blocked by various PKC-λ/ι inhibitors, but not by an Akt, inhibitor.
CONCLUSIONS
PKC-λ/ι provokes increases in brain β-secretase, Aβ1–40/42 and phospho-thr-231-tau. Excessive signaling via PKC-λ/ι may link hyperinsulinemia and other PKC-λ/ι activators to development of pathological and functional abnormalities in Alzheimer’s disease.
Keywords: Alzheimer’s, Aβ, phospho-tau, beta-secretase, atypical PKC, PKC-iota/lambda, PKM-zeta, insulin, metformin, Akt
Graphical abstract
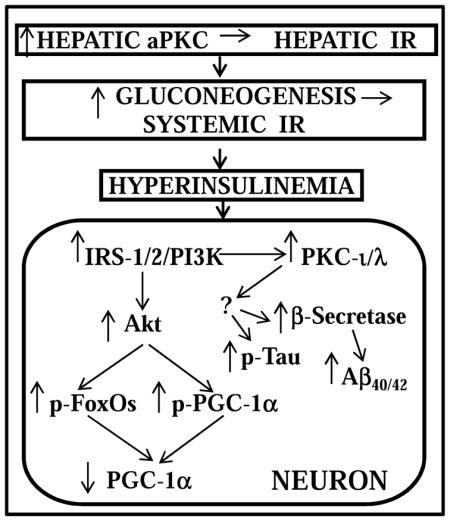
Schematic of pathogenesis of neuronal signaling abnormalities in insulin-resistant states that lead to production of factors that may abet development of Alzheimer’s disease. In this scheme, diet-induced increases in hepatic aPKC activity lead to impaired Akt activation by insulin, i.e., hepatic insulin resistance (IR), increases in hepatic gluconeogenesis, systemic IR, and hyperinsulinemia, which persistently hyperactivates brain Akt and aPKC. Increases in brain Akt activity lead to phosphorylation and thus diminished activities of all FoxOs (1/3a/4/6), and decreased activity and expression of PGC-1α (all needed for neuronal function and integrity). Increases in brain aPKC activity, either directly or indirectly, provoke increases in b-secretase activity, and levels of Aβ1-40/42 and phospho-thr-231-tau, and thus abet plaque and tangle development.
1. INTRODUCTION
Alzheimer’s disease (AD) is prevalent in insulin-resistant obesity and type 2 diabetes mellitus (T2DM). In the USA, obesity, the metabolic syndrome and T2DM now afflict approximately 50% of people over age 50, and this may explain why AD afflicts approximately 50% of people over age 85. In some series, T2DM or “pre-T2DM” is present in 80% of AD patients (Janson et al, 2004).
As obesity/T2DM generally predates AD, it is reasoned that systemic insulin resistance abets AD development, and some investigators postulate that the brain itself is insulin-resistant. However, in examining insulin signaling factors in brains of multiple insulin-resistant obesity/T2DM models, viz., high-fat-fed (HFF) mice, ob/ob mice, heterozygous muscle-specific PKC-λ knockout (Het-MλKO) mice [wherein impaired muscle glucose transport secondarily hyperactivates hepatic aPKC causing hyperexpression of gluconeogenic and lipogenic enzymes], and monkeys with long-standing diet-dependent insulin-resistant obesity/T2DM, we found that hyperinsulinemia in each model was accompanied by, and apparently responsible for, maximal or near-maximal increases in resting/”basal” activities of both phosphatidylinositol 3-kinase(PI3K)-dependent protein kinases that mediate most insulin effects, i.e., Akt and the atypical protein kinase C (aPKC) isoform, PKC-λ/ι (Sajan et al., 2016).
Further, with persistent activation of brain Akt in each obesity/T2DM model, brain FoxOs-1/3a/4/6 are maximally phosphorylated and therefore inhibited, and FoxO1-dependent PGC-1α levels were reduced (Sajan et al., 2016), thus diminishing availability of multiple transcriptional factors needed for memory function and neuronal integrity (Renault et al., 2009; Paik et al., 2009; Qin et al., 2009; Salih et al., 2012; Gong et al., 2013; Sweeney and Song, 2015). Additionally, tau phosphorylation was increased in more advanced insulin-resistant models (Sajan et al., 2016) by a mechanism independent of glycogen synthase kinase-3β (GSK3β), which was inhibited phosphorylated and thus inhibited by hyperactivated Akt. Most importantly, both acute 15-minute insulin treatment of normal mice, and hyperinsulinemia in each insulin-resistant model were accompanied by increases in Aβ1-40/42 levels. Moreover, with correction of hyperinsulinemia in the Het-MλKO mouse obesity/T2DM model that followed pharmacological reduction of excessive hepatic aPKC activity [elicited by liver-selective aPKC inhibitor, aurothiomalate (ATM), which does not inhibit brain aPKC], all CNS aberrations in Akt, PKC-λ/ι, FoxOs, GSK3β, mTOR and Aβ1-40/42 reverted to normal, and acute stimulatory effects of exogenous insulin on each of these factors were fully restored (Sajan et al., 2016). This reversal of hyperinsulinemia-induced CNS aberrations and restoration of normal brain insulin signaling suggested that signaling aberrations in brains of insulin-resistant animals that could reasonably abet development of AD pathologies, i.e., phospho-tau “tangles” and β-amyloid plaques, were provoked by hyperinsulinemia.
Here, we examined the role of insulin-sensitive protein kinases, in particular PKC-ι, for increasing levels of Aβ1-40/42 and phospho-tau, and we questioned whether β-secretase may underlie increases in Aβ1-40/42. We first revisited previous findings showing that (a) acute 15-min insulin treatment in normal mice and chronic hyperinsulinemia in Het-MλKO provoke comparable increases in Aβ1-40/42, and (b) selective inhibition of hepatic aPKC by aurothiomalate (ATM) in Het-MλKO mice reverses hyperinsulinemia, and thereby restores normal basal and insulin-stimulated levels of brain Aβ1-40/42 (Sajan et al., 2016). However, instead of using ATM, which does not alter brain aPKC activity, we used a PKC-λ/ι-specific inhibitor, [1H-imidazole-4-carboxamide,5-amino]-[2,3-dihydroxy-4-[(phosphono-oxy) methyl]cyclopentane-[1R-(1a,2b,3b,4a)] (ICAPP), in a dose that inhibits PKC-λ/ι in brain as well as liver. And, as previously reported, ICAPP, like ATM, effectively inhibited liver aPKC and thereby corrected hyperinsulinemia in Het-MλKO mice (Sajan et al, 2012b), and, as herein found, the correction of hyperinsulinemia reversed signaling aberrations mediated by both aPKC and Akt in brains of Het-MλKO mice. More interestingly, ICAPP, by directly inhibiting brain aPKC, blocked not only chronic hyperinsulinemia-induced increases, but also acute insulin-induced increases, in PKC-ι/λ (but not Akt) activity, β-secretase activity and Aβ1-40/42 production in both Het-MλKO mice and normal mice. We also used another aPKC inhibitor, 2-acetyl-cyclopentane-1,3-dione, (ACPD), which, like ICAPP, directly inhibited brain aPKC (but not Akt), and, in insulin-resistant HFF mice, ACPD diminished HF diet-induced increases in brain PKC-ι/λ activity, Aβ1-40/42 production and thr-231-tau phosphorylation. Of further note, ACPD simultaneously improved an impairment in acute memory function induced by HF feeding. Finally, we examined stimulatory effects of insulin and other aPKC activators, metformin and constitutive PKC-ι, on β-secretase activity, Aβ1-40/42 production and thr-231-tau phosphorylation in cultured neuronal cells, and insulin effects on Aβ1-40/ and thr- 231-tau in incubations of mouse hippocampal slices, and similarly found that PKC-λ/ι was required for these increases.
2. METHODS & MATERIALS
2.1 aPKC and Akt Inhibitors
1H-imidazole-4-carboxamide,5-amino]-[2,3-dihydroxy-4-[(phosphono-oxy) methyl]cyclopentane-[1R-(1a,2b,3b,4a)] (ICAPP) was identified as a potential aPKC inhibitor by high throughput screening (HTS) of a chemical library for virtual docking with the crystallographic structure of PKC-ι catalytic domain, custom-synthesized by the Southern Research (Birmingham, AL, USA), and found to preferentially inhibit recombinant PKC-ι (Pillai et al., 2011), and inhibit recombinant PKC-ζ only at 10–30-fold higher concentrations (MP Sajan and RV Farese unpublished findings). Because of cost and limited availability of ICAPP, in long-term in vivo studies, we more recently used 2-acetyl-cyclopentane-1,3,-dione (ACPD), which was similarly identified by the same HTS that identified ICAPP, purchased from Sigma/Allied Chemical Co. (St Louis, MO, USA), and found to inhibit recombinant PKC-ι and PKC-ζ with equal potency (Sajan et al., 2014; Sajan et al., 2015). Neither ICAPP (Sajan et al., 2012b) nor ACPD (Sajan et al., 2014; Sajan et al., 2015) inhibit conventional PKCs, novel PKCs, AMP-dependent protein kinase and Akt; and ACPD had no effect on a battery of protein kinases, including, Akt2, FGFR1/2/3/4, mTOR, GSK3β, IRAK1/4, JAK1/2, MEK1, ERK1/2, JNK1/2, PKA, Src, ROCK2, ROS1, or PI3Kα/α, as tested by Life Technologies (Madison WI, USA). Akt inhibitor Akti was purchased from Calbiochem/EMD Millipore (Billerica, MA, USA).
2.2 In Vivo Mouse Studies
Brain samples were obtained from normal and insulin-resistant mice and monkeys used previously [9], and includes mice originally used in studies of insulin signaling and resistance in liver: (a) 4–6 month old C57Bl/6 mice used in studies of high fat feeding (University of South Florida College of Medicine (USF-COM) Vivarium colony) (Sajan et al., 2014); 4–6 month-old male C57Bl/6 ob/ob and lean ob+ littermate control mice (Jackson Labs, Bar Harbor, Maine) (Sajan et al., 2015); and (c) 10–12 month-old C57Bl/6 Het-MλKO mice (USF Vivarium colony) (Sajan et al., 2012b). Males and females were comparably present in experimental groups, and sex didn’t appreciably alter combined findings. Hepatic alterations, clinical characteristics and ameliorating effects of liver-selective aPKC inhibitors were reported previously (Sajan et al., 2012b; Sajan et al., 2014; Sajan et al., 2015). Mice were housed in environmentally-controlled rooms and fed standard mouse chow supplying 10% of calories from fat, or diets supplying either 40% or 60% of calories from fat (Harlan Industries, Madison, Wisconsin) (results in mice consuming these HF diets were in most respects, comparable, and thus combined (Sajan et al., 2016)).
Where indicated, Het-MλKO were injected subcutaneously (SC) once daily for 8 days with saline or ICAPP (0.4mg/kg body weight) in saline, to inhibit hepatic aPKC, and thus diminish aPKC-dependent expression of hepatic gluconeogenic and lipogenic enzymes, and thereby reduce serum insulin levels to normal (Sajan et al., 2012b). Where indicated, normal mice were given a single SC injection of saline or saline containing 1.5mg/kg body weight ICAPP to follow time-related alterations in brain signaling factors. Where indicated, mice were treated intraperitoneally (IP) with insulin (1U/kg body weight) or vehicle 15-min before euthanitization by administration of Xylazine/Ketamine, followed by whole body perfusion with phosphate-buffered saline and rapid removal of brain and other tissues.
Note that, in accordance with an earlier report showing that insulin signaling responses in whole brain largely occur in neurons, rather than glial or endothelial cells, most notably, in the hippocampus and hypothalamus (Freude et al., 2005), we previously documented that activating effects of insulin were readily seen in neurons of the anterior cortex and hippocampus (Sajan et al., 2016), and measurements of total brain insulin signaling correlated well with alterations in these individual neurons, both in normal and insulin-resistant HFF and ob/ob mice (Sajan et al., 2016). Also note that the effects of insulin in vivo were measured at 15 min in these experiments, as this time is optimal for observing maximal insulin-induced increases in Akt and aPKC activities in classical insulin target tissues, i.e., liver, muscle, and adipose tissues, which were also under study in these experiments, and, fortuitously, changes in brain insulin signaling to Akt and aPKC also appeared to be maximal at this time. All experimental procedures involving mice were approved by the Institutional Animal Care and Use Committees (IACUCs) of the (USF-COM) or Roskamp Institute, and the James A. Haley Veterans Administration Research and Development Committee.
2.3 In Vivo Monkey Studies
As described (Sajan et al., 2016), brains were obtained immediately post-mortem from 16–30-year-old male and female lean non-diabetic and hyperinsulinemic obese/T2D Macaca mulatta rhesus monkeys; note that obesity and T2D were present for many years; see [9] further details. Euthanitization was initiated with ketamine-HCl (10–15mg/kg body weight) followed by intravenous Euthasol (0.22ml/kg body weight). Anterior cortical samples of monkey brains were taken and stored at −150°C. Experimental procedures involving monkeys, including euthanitization, were approved by USF-COM IACUC.
2.4 Tissue Preparations
As described (Sajan et al., 2016), mouse brains were hemisected sagittaly, and one-half was frozen in liquid N2, stored at −80°C, and samples thereof were homogen ized in buffer containing 0.25M sucrose, 20mM Tris/HCl (pH, 7.5), 2mmol/l EGTA, 2mM EDTA, 1mM phenyl-methyl-sulfonyl-fluoride, 20μg/ml leupeptin, 10μg/ml aprotinin, 2mM Na4P2O7, 2mM Na3VO4, 2mM NaF, and 1μM microcystin, and then supplemented with 1% TritonX-100, 0.6% Nonidet and 150mM NaCl.
2.5 Incubations of Mouse Hippocampal Slices
As described (Talbot et al., 2012), mouse hippocampal slices (400- micron thickness) were prepared with a McIlwain slicer and incubated at 37° in Krebs Ringer bicarbonate HEPES buffer under 95%O2/5%CO2.
2.6 β-Secretase (BACE1, beta-site APP-cleaving enzyme-1) Activity
β-Secretase was measured using a kit purchased from Thermo-Fisher Scientific. In this fluorescent resonance transfer (FRET) assay, a weakly fluorescent synthetic peptide becomes highly fluorescent upon proteolytic cleavage by β–secretase, and enzymatic activity is linearly related to the initial rate of proteolysis, which is measured spectrophotometrically by fluorescence at 545 nm excitation and 585 nm emission.
2.7 Western Analyses
As described (Sajan et al., 2016), Western analyses were conducted with rabbit polyclonal antisera or, where indicated mouse monoclonal antibodies (mMab) using targeting: aPKC, anti-glyceraldehyde phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnologies, Santa Cruz, CA, USA); phospho-threonine- 560/555-PKC-ζ/λ/ι (Invitrogen, Carlsbad, CA, USA); p-serine-256-FoxO1 and anti-FoxO1 (Abnova, Walnut, CA, USA); Akt (mMab), phospho-serine-473-Akt, phospho-serine-9-GSK3β, anti-GSK3β, phospho-serine-253-FoxO3a, FoxO3a, phospho-serine-256-FoxO1, phospho-serine-193-FoxO4; anti-FoxO1, -phospho-serine-2448-mTOR, - mTOR, 4kDa-Aβ1-40/42 and amyloid precursor protein (120kDa APP) (Cell Signaling Technologies, Danvers, MA, USA); and phospho-serine-202-tau and phospho-threonine-231-tau (GeneTex, Irvine, CA, USA). Samples from experimental groups were compared on the same blots, and routinely checked with loading controls. Note that: insulin-sensitive 70kDa aPKC is largely PKC-λ in mouse brain and orthologous PKC-ι (98% aa homology) in monkey and other primate brains; brain PKC-ζ exists as a 50kDa moiety that, lacking an auto-inhibitory regulatory domain, is constitutively-active and much less or unresponsive to insulin. In some cases, we also measured Aβ1-40 by an ELISA method (ThermoFischer; Rockford, Il, USA) and obtained results indistinguishable from Western analysis of 4kDa Aβ1- 40/42 Also note that insulin receptor activity was measured by Western analysis of the pY (mMab) content of the immunoprecipitated insulin receptor β-subunit (antiserum and mMab from Santa Cruz Biotechnologies, Santa Cruz, CA, USA), as described (Sajan et al., 2014b)
2.8 Novel Object Recognition Testing
This test is dependent on neuronal activity in cortical areas of the para-hippocampal region of the temporal lobe, and was conducted as described (Sajan et al., 2016) (Antunes and Biala, 2012), In brief, after acclimation to handling for three consecutive days, and then to daily 5-min placements in a chamber for another 3 days, the mice were then placed for 5-min in the same chamber containing two copies of an object to allow familiarization, and, 3 hours later, returned for 5-min to the same chamber containing one copy of the initial/familiar object and one copy of a new object. As mice are innately drawn to explore new versus familiar objects, the ratio of time spent exploring the novel object to time spent exploring the initial/familiar object (measured by camera and computer analysis) serves as an index of acute visual memory of the initial/familiar object.
2.9 Statistical Analyses
Data were expressed as mean ± SEM, and compared using one-way ANOVA and Tukey post hoc test for analysis of significance.
3. RESULTS
3.1 Alterations in Akt Activity in untreated and ICAPP-treated Het-MλKO mice
Brain Akt activity (Fig 1a) and phosphorylation of Akt substrates, mTOR (Fig 1b), FoxO3a (Fig 1c) and GSK3β (Fig 1d) were increased by acute 15- min insulin treatment in normal wild-type (WT) mice, and to similar levels in the resting/ “basal” state in Het-MλKO mice that were apparently maximal or near-maximal, as acute insulin treatment was without further effect. Interestingly, 8-day treatment of Het-MλKO mice with ICAPP diminished resting/basal Akt activity and phosphorylation of Akt substrates, and this was attended by restored ability of insulin to acutely activate Akt and increase phosphorylation of Akt substrates, mTOR (Fig 1b), FoxO3a (Fig 1c) and GSK3β (Fig 1d) (Fig 1a). These improvements in resting/basal and insulin-stimulated brain Akt signaling presumably reflected correction of hyperinsulinemia elicited by inhibitory effects of ICAPP on hepatic PKC-λ/ι and consequent decreases in expression of hepatic gluconeogenic and lipogenic enzymes (Sajan et al, 2012b).
Figure 1.

Hepatic aPKC inhibitor ICAPP reverses hyperinsulinemia-induced increases in basal/resting Akt activity (a) and phosphorylation of Akt substrates, ser-2448-mTOR (b), ser-253-FoxO3a (c), and ser-9-GSK3β (d) in brains of insulin-resistant Het-MλKO (KO) mice. Where indicated, PKC-λ/ι inhibitor, ICAPP (0.4mg/kg body weight), was administered subcutaneously once daily for 8 days, and insulin (1U/kg body weight) (shaded bars) was administered intraperitoneally 15-min before killing. Other features of these Het-MλKO (KO) and littermate wild type (WT) mice were reported previously (Sajan et al., 2012b; Sajan et al., 2016). Note that, by inhibiting hepatic aPKC and aPKC-dependent increases in hepatic gluconeogenic enzymes, ICAPP corrects hyperinsulinemia in Het-MλKO mice (Sajan et al., 2012b), which in turn reduces hyperinsulinemia-dependent increases in resting/basal brain Akt activity and Akt substrate phosphorylation ((Sajan et al., 2016); accordingly, acute effects of exogenous insulin treatment on these Akt-dependent parameters were restored by ICAPP treatment. Representative Western blots of indicated proteins are shown; loading controls are shown below phospho-proteins. Relative bar values are mean ± SEM of 6 mice. Asterisks: *, P<0.05; **, P<0.01; ***, P<0.001 (ANOVA).
3.2 Alterations in PKC-λ/ι activity and Aβ1-40/42 levels in untreated and ICAPP-treated Het-MλKO mice
As with brain Akt activity, insulin acutely increased activity of 70kDa PKC-λ/ι in brains of WT mice, and ICAPP diminished resting/basal increases in activity of PKC-λ/ι in brains of Het-MλKO mice (Fig 2a). However, in marked contrast to the restoration of the ability of insulin to activate Akt (Fig 1a), ICAPP blocked the ability of insulin to acutely activate PKC-λ/ι in Het-MλKO mice (Fig 2a), and this loss of PKC-λ/ι activation was accompanied by a comparable loss in the ability of insulin to acutely increase Aβ1-40/42 levels (Fig 2b).
Figure 2.

aPKC inhibitor ICAPP inhibits hyperinsulinemia-stimulated increases in basal PKC-λ/ι activity (a), Aβ1-40/42 peptide production (b), and β-secretase (BACE1) activity (c) in brains of Het-MλKO (KO) mice, and subsequent stimulatory effects of acute insulin treatment on these parameters. PKC-λ/ι inhibitor, ICAPP (0.4mg/kg body weight), was administered subcutaneously once daily for 8 days to Het-MλKO (KO) mice, and insulin (1U/kg body weight) (shaded bars) was administered intraperitoneally 15-min before killing. Other features of these mice are described in Fig 1 or were previously reported [8,9]. Representative Western blots of indicated proteins are shown; loading controls levels, which were not significantly altered, are not shown in lower panels. Relative bar values are mean ± SEM of 6 mice. Asterisks: *, P<0.05; **, P<0.01; ***, P<0.001 (ANOVA).
3.3 Alterations in β-secretase activity in untreated and ICAPP-treated Het-MλKO mice
We next focused on β-secretase, which initiates and is thought to be rate-limiting for proteolytic release of Aβ1-40/42 from β-amyloid precursor protein (β-APP). In conjunction with increases in brain Aβ1-40/42 levels in Het-MλKO mice (Fig 2b), chronic hyperinsulinemia provoked increases in β-secretase activity (BACE1) (Fig 2c). Further, in brains of Het-MλKO mice, in conjunction with inhibition of PKC-λ/ι, 8-day ICAPP treatment reduced resting/basal β-secretase activity to nearnormal levels, but, on the other hand, ICAPP fully blocked acute insulin-stimulated increases in β-secretase activity (Fig 2c).
3.4 Alterations in β-secretase activity in HFF and Ob/OB Mice and Obese/T2D monkeys
We previously reported (Sajan et al., 2016) that (a) brain Aβ1-40/42 levels are increased acutely by insulin in normal mice and by hyperinsulinemia in HFF and ob/ob mice, and obese/T2D monkeys; (b) β-APP levels are not altered in insulin-resistant mouse models; and (c) in monkeys with long-standing obesity and T2DM, in conjunction with increases in Aβ1-40/42 peptides, there are modest but significant decreases in β-APP. These findings suggested that increases in Aβ1-40/42 were occurring at the expense of β-APP, which, over protracted periods in monkeys, was measurably decreased by proteolysis. Coincident with this idea, acute insulin treatment in normal chow-fed (Fig 3a) and lean ob+ control mice (Figs 3b), and hyperinsulinemia in HFF mice (Fig 3a), ob/ob mice (Fig 3b) and obese/T2D monkeys (Fig 3c) provoked increases in resting/basal β-secretase activity.
Figure 3.

Activation of brain β-secretase activity (BACE1) by insulin (1U/kg body weight) administered intraperitoneally 15-min before killing (shaded bars) in control chow-fed (a) and lean (ob+) control (Con) mice (b), and activation of brain β-secretase (BACE1) by hyperinsulinemia in high fat-fed (HFF) mice (a), ob/ob mice (b), and obese/type 2 diabetic (T2DM) monkeys (c). Increases in fasting serum or plasma insulin levels in obese/diabetic versus lean/non-diabetic mice were 4.9 for HFF mice, 4.2 for ob/ob mice, and 2.2 for obese/T2D monkeys (actual data are given in Sajan et al., 2014, Sajan et al., 2015, and Sajan et al., 2016., respectively). Other features of these mice, including alterations in brain Aβ1-40/42, were reported previously [8,9]. Relative bar values are mean ± SEM of (N) mice or monkeys. Asterisks: *, P<0.05; **, P<0.01; ***, P<0.001 (ANOVA).
3.5 Alterations in PKC-λ/ι activity, β-secretase activity and Aβ1-40/42 levels in untreated and ICAPP-treated normal mice
In addition to findings suggesting PKC-λ/ι-dependence of β-secretase activation in Het-MλKO mice, we found in normal mice that administration of a single dose of PKC-λ/ι inhibitor ICAPP provoked time-related, well-correlated decreases in insulin-dependent PKC-λ/ι activity, β-secretase activity and Aβ1-40/42 peptide levels, without altering insulin-stimulated increases in Akt activity (Fig 4).
Figure 4.

ICAPP provokes time-related decreases in insulin-stimulated aPKC activity (blue), β-secretase (BACE1) activity (black) and Aβ1-40/42 peptide production (red), but spares insulin-stimulated Akt activation (green) in brains of normal mice. ICAPP (1.5mg/kg body weight) was administered subcutaneously as a single dose at zero time, and, at indicated times, insulin (1U/kg body weight) was administered intraperitoneally15-min before killing. Results in insulin-stimulated samples were compared to results in vehicle-injected control mice (see Figs 1–3 for comparison of control and insulin-stimulated values). Relative insulin-stimulated values plotted here are mean ± SEM of 3–4 mice. Asterisks: *, P<0.05; **, P<0.01; ***, P<0.001 (ANOVA). Representative blots of insulin-stimulated parameters are shown at top, Constant values of p-ser-273-Akt and glyceraldehydes-3-phosphate dehydrogenase (GAPDH) serve as loading controls.
3.6 Effects of Insulin and ICAPP on Akt, PKC-λ/ι and β-secretase activity and Aβ1-40/42 levels in cultured neuronal cells
In concert with findings in brains of normal mice, 24-hour insulin treatment elicited increases in: Akt and aPKC activities; phosphorylation of Akt substrates, GSK3β and mTOR; β-secretase activity; and levels of Aβ1-40/42 in cultured LA1-5s human neuroblastoma neuronal cells (Fig 5a). Further, in these cells, ICAPP provoked dose-dependent decreases in insulin-stimulated aPKC activity, β-secretase activity and Aβ1-40/42 levels, without altering stimulatory effects of insulin on Akt activity (Fig 5b) and phosphorylation of Akt substrates (not shown). Interestingly, dose-dependent inhibitory effects of ICAPP on insulin-stimulated aPKC activity in these neuronal cells were similar (IC50, approximately 1-10nM) to those previously seen in isolated human hepatocytes (Sajan et al., 2012a), and isolated mouse adipocytes and recombinant PKC-λ/ι (Sajan et al., 2012b).
Figure 5.

Effects of insulin and ICAPP on aPKC and Akt activities, phosphorylation of Akt substrates (GSK3β and mTOR), β-secretase activity (BACE1) and Aβ1-40/42 peptide levels in LA1-5s human neuroblastoma cells. Effects of 24- hour treatment ± 200nM insulin are shown at left (a), and dose-related effects of ICAPP on insulin-stimulated parameters are shown at right (b). Loading controls, which did not differ significantly; in panel a are shown below phospho proteins; and in panel b, unchanged glyceraldehyde phosphate dehydrogenase (GAPDH) served as a loading control. Relative bar values in panel a are mean ± SEM of 6 determinations. Values in panel b are means ± SEM of 2–4 determinations. Asterisks: *, P<0.05; **, P<0.01; ***, P<0.001 (ANOVA). Note that the incubation time of 24 hours was arbitrarily chosen in anticipation of conducting studies requiring time for expression studies as in Figure 7, wherein cells were incubated for 48 hours to allow time for virally-mediated expression of mutated enzymes.
3.7 Effects of inhibition of aPKC versus Akt on insulin-stimulated increases in Aβ1-40/42 and p-thr-231-tau levels in cultured neuronal cells and mouse hippocampal slices
In addition to increases in Aβ1-40/42, insulin provoked increases in phospho-thr-231-tau in both cultured neuronal cells and in slices of mouse hippocampus; moreover, insulin-induced increases in both Aβ1-40/42 and p-thr-231-tau were blocked by aPKC inhibitor ACPD, but not by Akt inhibitor, Akti (Fig 6).
Figure 6.

Effects of aPKC versus Akt inhibition on insulin-stimulated increases in Aβ1-40/42 and p-thr-231-tau in LA1-5s human neuroblastoma neuronal cells (A) and hippocampal slices (B). Neuroblastoma neuronal cells were treated for 24 hours without or with 200nM insulin, 1μM ACPD and 10μM Akti, as indicated. [Note that the dose of 200nM insulin was arbitrarily chosen to be certain that any losses of insulin owing to internalization and/or degradation by insulin-degrading enzyme would not limit maximal effects of insulin; the incubation time of 24 hours was arbitrarily chosen to allow attainment of maximal effects of a variety of agents, including insulin, metformin, metabolites, etc., on tissue levels of activated PKC-λ and increases in PKC-λ-dependent parameters.] Hippocampal slices were incubated for 1 hour without or with 100nM insulin, 1μM ACPD and 10μM Akti. Loading controls, which did not differ significantly in are shown below phospho proteins. The loading control for Aβ was 120kDa APP. Values are Mean ± SEM of 4 determinations. *, P<0.05 vs, unstimulated control mean value (clear bars); #, P<0.05, vs. insulin-stimulated mean value in absence of inhibitor treatment.
3.8 Effects of adenovirally-mediated expression of constitutively-active and kinase-inactive PKC-ι on basal and insulin-stimulated PKC-ι activity, p-thr-231-Tau, β-secretase activity (BACE1) and Ab1-40/42 levels in cultured neuronal cells
As insulin activates signaling factors other than aPKC, it was important to find that constitutively-active (CA) PKC-ι provoked insulin-like increases in aPKC activity (Fig 7a), β-secretase activity (Fig 7c), phospho-thr-231-tau (Fig 7b) and Aβ1-40/42 levels (Fig 7d), without altering basal levels and insulin-stimulated increases in Akt activity (Akt data not shown) in cultured neuronal cells. Further, as chemical inhibitors may target unintended factors, it was important to find that expression of kinase-inactive (KI) PKC-ι blocked the stimulatory effects of insulin on aPKC activity (Fig 7a), phospho-thr-231-tau levels (Fig 7b), β-secretase activity (Fig 7c) and Aβ1- 40/42 (Fig 7d) levels.
Figure 7.

Adenovirus (Adv) encoding constitutively active (CA) PKC-λ/ι (in blue) dose-relatedly phosphorylates/activates total aPKC (a) and increases phospho-tau levels (b), β-secretase activity (BACE1) (c) and Ab 1-40/42 levels (d) in LA1-5s human neuroblastoma cells. Adv encoding kinase-inactive (KI) PKC-λ/ι (in red) dose-relatedly blocks effects of 200nM insulin on these parameters. Cells were incubated for 48 hours with (red) and without (blue) 200nM insulin and indicated Adv multiplicity of infection (MOI), the total level of which kept constant at 100 MOI for all samples by adding non-coding Adv. Note that the explanation for inhibitory effects of kinase-inactive (KI) PKC-λ [mutagenized in the ATP-binding site of the catalytic domain (Kotani et al, 1998) on endogenous wild type (WT) PKC-λ during insulin action is that KI-PKC-λ competes with WT-PKC-λ for either the PKC-λ-activating lipid, PIP3, or for the substrate-recognition motif (including the auto(trans)phosphorylation site at thr-555), or both. Shown here are Mean ± SEM (N=4) values relative to the mean initial basal or insulin-stimulated level of indicated protein/peptide. Symbols: *, P<0.05; ¥, P<0.01; #, P<0.001 (ANOVA). Abbreviations: KI, kinase-inactive PKC-λ; LC, loading control, glyceraldehyde phosphate dehydrogenase; and CA, constitutively-active PKC-λ.
3.9 Effects of metformin and ICAPP on PKC-ι activity, β-secretase activity (BACE1), Ab1-40/42 levels and p-thr- 231-tau levels in cultured neuronal cells
Metformin reportedly activates β-secretase and increases Aβ1-40/42 in isolated neuronal cells and intact mouse brain (Chen et al., 2012). Metformin also promotes tau aggregation and exacerbates abnormal behavior in a mouse tauopathy model (Barini et al., 2016). Presently, in addition to insulin and constitutive PKC-ι, we found that metformin increased activity of aPKC (Fig 8a), but not Akt (Fig 8e), and simultaneously increased β-secretase activity (8b), and levels of Aβ1-40/42 (Fig 8c) and phospho-thr-231-tau (Fig 8d) in isolated neuronal cells; and, very importantly, aPKC inhibitor ICAPP dose-dependently inhibited each of these metformin effects.
Figure 8.

Metformin activates aPKC (a), and increases β-secretase activity (BACE1) (b), A-beta (Aβ1-40/42) levels (c), and phospho-thr-231-tau levels (d), but not Akt activity (e) in LA1-5s human neuroblastoma cells, and PKC-λ/ι inhibitor, ICAPP, dose-relatedly blocks metformin-induced increases in aPKC activity, β-secretase activity (BACE1), and levels of Aβ1-40/42 and phospho-thr-231-tau. As in Fig 6, cells were incubated for 24 hours with metformin and ICAPP. Shown here are Mean ± SEM (N=4) values relative to the mean initial basal or insulin-stimulated level of indicated immunoreactive protein/peptide. Asterisks: *, P<0.05; **, P<0.01; ***, P<0.001 (ANOVA). Abbreviation: GAPDH, glyceraldehyde phosphate dehydrogenase, was used for loading controls.
3.10 Effects of ACPD-mediated inhibition of brain aPKC on high-fat-feeding-induced (a) elevations of brain Aβ1-40/42 and phospho-thr-231-tau, and (b) impairment of novel object recognition memory function
Different from ATM- and ICAPP-treated Het-MλKO mice in which hyperinsulinemia was fully/sufficiently improved (Sajan et al., 2012b), the hyperinsulinemia in HFF and ob/ob mice treated with ACPD at a dosage of 10mg/kg body weight/day was only modestly improved (Sajan et al., 2014; Sajan et al., 2015), but not sufficiently to reverse resting/basal increases in brain 70kDa PKC-λ/ι and Akt activities (Sajan et al., 2016), and this dose of ACPD did not alter brain aPKC activity. Presently, we used at a higher dose of 20mg/kg, ACPD [administered every other day, 3 days per week, as inhibition of hepatic aPKC activity persists for 48 hours after subcutaneous ACPD injection (Sajan et al., 2014)]. Interestingly, this dose of ACPD was more (but not fully) effective in diminishing resting/ad lib-fed hyperinsulinemia, and was sufficient to substantially reduce resting PKC-λ/ι activity, and, moreover, fully blocked acute stimulatory effects of insulin on 70kDa PKC-λ/ι activity in these HFF mice (Fig 9a), without inhibiting HF diet-induced increases in resting/ad lib-fed activities of brain Akt (Fig 9b) and 50kDa PKM-ζ (Fig 9e). In conjunction with the blockade of insulin effects on 70kDa PKC-λ/ι activity in ACPD-treated HFF mice, the ability of insulin to increase levels of both Aβ1-40/42 [as measured by both Western analysis (Fig 9g) and ELISA (Fig 9c)] and thr-231-phospho-tau (Fig 9f), were similarly inhibited. Perhaps most remarkably, ACPD simultaneously reversed an impairment in novel object recognition induced by HF feeding (Fig 9h).
Figure 9.

Treatment of high-fat-fed mice with aPKC inhibitor, ACPD, reduces high fat diet-induced and acute insulin-stimulated increases in 70kDa PKC-ι/λ activity (a), Aβ1-40 levels (as per ELISA) (c), 4kDa Aβ1-40/42 levels (as per Western) (g), and phospho-thr-231-tau (f) levels, and simultaneously reverses/prevents a memory impairment in novel object recognition (NOR) (h), without affecting high fat (HF) diet-induced and acute insulin-stimulated increases in Akt activity (b) or PKM-ζ activity (e). Effects of hyperinsulinemia in HFF mice, and acute insulin treatment in regular chow (RC) fed and HFF mice, on insulin receptor (IR) activity (as per total pY content of immunoprecipitated IR β-subunit) are shown in panel (d). Mice were fed regular chow (RC) or a high fat (HF) diet, and treated ± ACPD (20mg/kg body weight, Monday, Wednesday and Friday of each week) over 10 weeks. Novel Object Recognition (NOR) testing was conducted at week 9. At 10 weeks, mice were treated acutely ± insulin (1U/kg body weight) or vehicle 15 min before killing. Values are mean ± SEM (N=4). Asterisks: *, P<0.05; **, P<0.01; ***, P<0.001 (ANOVA).
Finally, note that both acute insulin treatment in normal chow-fed mice, and chronic hyperinsulinemia (± acute insulin treatment) in HFF mice, provoked comparable, apparently maximal increases in activity of the insulin receptor, as measured by the total pY content of the insulin receptor β-subunit (Fig 9d).
4. DISCUSSION
The present findings showed that: (a) along with previously-reported increases in brain PKC-λ/ι activity and Aβ1-40/42 levels (Sajan et al., 2016), insulin treatment in vivo provoked rapid increases in β-secretase activity in mouse brain; (b) hyperinsulinemia in insulin-resistant HFF, ob/ob and Het-MλKO mice and obese/T2D monkeys was accompanied by increases in brain β-secretase activity, as well as previously-reported increases in PKC-λ/ι activity and Aβ1-40/42 production; (c) correction of hyperinsulinemia in insulin-resistant Het-MλKO mice by ICAPP, which improves systemic insulin resistance sufficiently by inhibiting hepatic aPKC in this model (Sajan et al., 2012b), simultaneously reduced the resting/basal hyperactivity of both Akt and aPKC to normal, and, very importantly, restored insulin effects on Akt activation, but, in contrast, blocked acute stimulatory effects of insulin on PKC-λ/ι activity, β-secretase activity, and Aβ1-40/42 production in brains of Het-MλKO mice; (d) PKC-λ/ι was required for acute stimulatory effects of insulin on β-secretase activity and Aβ1-40/42 production in brains of normal mice; (e) in cultured neuronal cells, aPKC activators, insulin, metformin and expression of CA-PKC-λ/ι, increased PKC-λ/ι activity, β-secretase activity, Aβ1-40/42 levels, and phospho-thr-231-tau levels, and aPKC inhibitor, ICAPP, and/or expression of KI-PKC-λ/ι, blocked effects of insulin, metformin and constitutive PKC-λ/ι on these parameters; and (f) in both cultured neuronal cells and incubations of mouse hippocampal slices, insulin -induced increases in Aβ1-40/42 levels and phospho-thr-231-tau were blocked by inhibition of aPKC, but not by inhibition of Akt. And, of particular interest, two specific PKC-λ/ι inhibitors, ICAPP and ACPD, were apparently able to pass the mouse blood brain barrier (BBB) and not only inhibit chronic hyperinsulinemia- and acute insulin-dependent increases in brain PKC-λ/ι activity, β-secretase activity, Aβ1-40/42 production, and thr-231-tau phosphorylation, but also prevented/repaired a HF-diet-induced impairment in memory function, i.e., acute visual novel object recognition. Together, these findings suggested that aPKC activation may play an important role in abetting development of AD pathology and cognitive dysfunction in hyperinsulinemic states and possibly in other conditions.
The finding that PKC-λ/ι was required for acute stimulatory effects of insulin on β-secretase activity and Aβ1-40/42 production in brains of intact mice and isolated neuronal cells and mouse hippocampal slices further suggested that alterations in β-secretase contributed importantly to the increases in Aβ1-40/42.levels seen in both normal mice after acute insulin treatment, and in response to persistent hyperinsulinemia in insulin-resistant mice and monkeys. Nevertheless, further studies are needed to see if factors other β-secretase contribute to increases in Aβ1-40/42, e.g., Aβ1-40/42 clearance by insulin-degrading enzyme (IDE) (aka, “amyloidase”) may also be altered by a competition between insulin and Aβ1-40/42 peptides for IDE binding. However, alterations in IDE cannot explain increases in brain β-secretase activity and Aβ1-40/42 production seen in mice treated with metformin (Chen et al., 2009) [note that we have corroborated these effects of metformin in mouse brain, and found them to be dependent on aPKC (MP Sajan and RV Farese, unpublished findings)].
In addition to the present findings showing that insulin acts through aPKC to activate β-secretase and thereby increase tissue levels of Aβ1-40/42, Gasparini et al., 2001, reported that insulin increases Aβ1-40/42 secretion by a mechanism involving mitogen-activated protein kinase-dependent trafficking of Aβ1-40/42-containing vesicles to the plasma membrane (PM) from the trans-Golgi network (TGN), where Aβ1-40/42 is generated from β-amyloid precursor protein (βAPP) by sequential actions of (a) β-secretase, which splits the C-terminal portion of βAPP from the Aβ1-40/42 sequence attached to the N-terminal remainder of βAPP, and (b) γ-secretase, which splits the Aβ1-40/42 sequence from the N-terminal remainder of βAPP. In this regard, note that vesicle-associated βAPP trafficking through the endocytic pathway to the TGN, rather than to the degradative lysosomal compartment, is phosphorylation-dependent, and the co-localization of vesicles containing βAPP and vesicles containing β-secretase in endosomes and/or TGN may increase Aβ1-40/42 production (Viera et al., 2010). Further note that conventional and novel PKCs may influence Aβ1- 40/42 metabolism, e.g., chronic phorbol ester-induced downregulation of PKC-α is accompanied by increases in PKC- ε, expression of βAPP, βAPP accumulation in the TGN, and Aβ1-40/42 generation (da Cruz e Silva et al., 2009). Needless to say, further studies are needed to see if aPKC regulates intracellular trafficking of vesicles containing β-secretase, βAPP and/or Aβ1-40/42, and/or increases actual enzymatic activity of β-secretase.
That insulin increased β-secretase activity, Aβ1-40/42 production and thr-231-tau phosphorylation in mouse brain, and may therefore abet development of AD pathology, is seemingly at odds with the fact that intra-nasal insulin treatment is currently being used in clinical trials for patients with AD or mild cognitive impairment (MCI) (Craft, 2007; Craft, 2012). The rationale for using insulin therapy in AD and MCI derives from findings indicating that levels and/or activities of the insulin receptor and/or post-receptor insulin signaling factors, are deficient in brains of AD humans (Craft, 2007, Craft, 2012; De la Monte, 2012; De Felice, 2013; Talbot et al., 2012). Indeed, AD has been called “type 3 diabetes”, and it has led to postulation that insulin action in brain is impaired in insulin-resistant states, and the assumption that brain insulin resistance per se predisposes to AD development. However, contrariwise, the present and previous findings (Sajan et al., 2016) suggest that prior to the development of significant AD pathology that may eventually lead to actual resistance to insulin, the brains of insulin-resistant obese and T2D subjects, presumably including those destined to develop AD, are, if anything, hyperinsulinized, and this hyperinsulinization, over time, may promote development of AD pathology by multiple mechanisms, as discussed here and previously (Sajan et al., 2016).
Our findings in isolated neuronal cells also suggested that increases in phospho-thr-231-tau levels seen previously [9] in brains of hyperinsulinemic ob/ob mice and obese/T2D monkeys, and seen presently in HFF mice consuming a diet supplying 60% of calories from fat, may also be due, in part, to increases in aPKC activity, and may contribute to the development of intraneuronal neurofibrillary “tangles” in AD and other dementias. However, for uncertain reason, increases in phospho-thr-231-tau were not seen in either HFF mice consuming a diet supplying 40% of calories from fat over a 10-week period, or in Het-MλKO mice (Sajan et al., 2016), and this may reflect lesser degrees of insulin resistance therein, or involvement of factors other than insulin.
It should be emphasized that, in addition to the hyperactivation of brain PKC-λ/ι and subsequent increases in β-secretase activity and Aβ1-40/42 levels, hyperactivation of brain Akt also occurs in insulin-resistant hyperinsulinemic states, and this leads to persistent Akt-dependent phosphorylation and thus inhibition, of the all four brain FoxOs3, 1/3a/4/6, and subsequent decreases in activity and levels of PGC-1α (Sajan et al., 2016). These alterations may be important in AD, as FoxOs and PGC-1α are needed to maintain brain memory function and neuronal integrity [reviewed in (Sajan et al., 2016)]. However, Akt may also have salutary effects in the brain, e..g., by diminishing apoptosis.
The fact that the aPKC inhibitors, ICAPP and ACPD, which can improve hyperinsulinemia, obesity, metabolic syndrome features and T2DM by inhibiting hepatic PKC-λ/ι (Sajan et al., 2014; Sajan et al., 2015; Sajan et al., 2012b) can, in higher doses, simultaneously inhibit insulin-stimulated PKC-λ/ι in brain, and thereby diminish insulin-dependent increases in β-secretase activity, Aβ1-40/42 levels and phospho-thr-231-tau levels, may have relevance, not only for hyperinsulinemia-dependent increases in brain aPKC activity, but also for increases provoked by other aPKC activators. In this regard, a variety of non-insulin PKC-λ/ι activators have been implicated in AD development, e.g., sphingomyelins, ceramide, hypoxia, hyperglycemia, proinflammatory cytokines, e.g., tumor necrosis factor-α, and various agonists, e.g., insulin-like growth factor-1 (as well as insulin) (Frolich et al., 1998; Gontier et al., 2014; Zemva and Schubert, 2014) and metformin (Chen et al., 2009; Wang et al., 2012), each of which reportedly increases β-secretase and/or Aβ1-40/42 levels. This “promiscuity” of aPKC raises the possibility that inhibition of CNS PKC-λ/ι, either directly or indirectly via improvement in aPKC-dependent hepatic aberrations, may be useful for preventing/diminishing β-amyloid plaque and phospho-tau accumulation in AD and pre-AD states.
Although the present findings are particularly relevant to situations wherein AD develops in response to, or in association with, hyperinsulinemic states of obesity, the metabolic syndrome and T2DM, it should be noted that (a) AD pathology may affect other brain areas, e.g., the hypothalamic appetite center (e.g., Kohjima et al., 2010), or distant organs altered by increases in circulating Aβ1-40/42, e.g., pancreatic islets (e.g., Vandal et al., 2017), that regulate energy and glucose metabolism, and therefore may alter systemic insulin sensitivity; and (b) there are unexplained increases in activities of both aPKC and Akt in hippocampal CA1 pyramidal neurons in hippocampi of non-diabetic AD humans (Talbot et al., 2012).
As to the first point, it is interesting that transgenic (Tg) mice that develop increases in β-secretase, Aβ1-40/42 and phospho-tau, and associated memory deficits, in response to mutations in βAPP, tau and α-secretase [α-secretase is protective for AD, as it preemptively neutralizes non-salutary β-secretase action by splitting the Aβ1-40/42 sequence in βAPP between the sites of cleavage by β-secretase and γ-secretase, thereby leading to production of more soluble, less harmful peptides], also develop unexplained, age- and sex-dependent forms of obesity, glucose intolerance, systemic insulin resistance and hyperinsulinemia, that accelerate AD development (Barron et al. 2013; Macklin et al., 2017; Vandal et al., 2017). Although some investigators postulate that, as in liver and muscle of insulin-resistant mice, the brain is insulin-resistant in these glucose-intolerant Tg-AD mice, the evidence for this conclusion usually hinges upon the finding that exogenous insulin has no or diminished effects on brain insulin signaling factors that are already strongly activated by hyperinsulinemia. Oppositely, our previous and present findings suggest that hyperinsulinemia, acting via brain insulin receptors in systemically insulin-resistant Tg- AD mice, underlies the exacerbation of Aβ1-40/42 production, tau phosphorylation and memory impairment.
As to the second point, from a detailed post-mortem study of brain insulin signaling factors, it is of considerable interest that CA1 hippocampal neurons of non-diabetic AD humans have elevations of phosphorylated/activated forms of Akt, aPKC, ERK2 and Akt substrate, mTOR (Talbot et al., 2012), comparable in magnitude to elevations produced by insulin and hyperinsulinemia in the mouse. But, militating against insulin as a cause for these elevations in AD brain, and, moreover, taken as evidence that the AD brain is insulin-resistant, basal activity (pY content) of the insulin receptor (IR) is normal, rather than elevated in AD post-mortem brains, and acute insulin-stimulated IR activity is modestly diminished by15–30% in hippocampal slices of AD humans. On the other hand, IRS-1 and IRS-2 activities (pY content) and the binding of both IRS-1 and IRS-2 to the p85α subunit of phosphatidylinositol 3-kinase (PI3K) (which activates Akt and aPKC) are very substantially increased in hippocampi of AD humans, and, with elevated baselines, in our opinion, the observed decreases in insulin-stimulated activities of these and other downstream PI3K-dependent signaling factors does not provide convincing evidence that insulin action is impaired. Moreover, activities of the IR and IRS-1 may be partially downregulated by negative feedback mechanisms owing to strong steady-state increases in activities of aPKC, Akt, ERK, and mTOR. In addition, IRs may be internalized and thus unavailable for activation in hyperinsulinemic states, and post-receptor signaling factors, such as aPKC and Akt may be maximally activated at sub-maximal levels of IR occupancy, owing to operation of “spare receptors”. In any case, regardless of whether it is insulin and/or other agonists that operate via IRS1/2 and PI3K that underlie increases in aPKC and Akt in hippocampi of AD humans, the present findings suggest that the these increases in aPKC may contribute importantly to increases in β-secretase activity, Aβ1-40/42 production and thr-231-tau phosphorylation. In any case, future studies are needed to identify the responsible agonist(s) in brains of AD humans.
It was important to find that the insulin receptor was maximally activated in brains of systemically insulin-resistant, hyperinsulinemic HFF mice, Thus, it may be surmised that (a) insulin- and hyperinsulinemia-induced increases in Akt, aPKC and aPKC-dependent factors truly reflect insulin receptor activation, and (b) unlike the marked insulin resistance seen in muscles, and, to a lesser extent, in livers, of HFF mice (Sajan et al., 2009; Sajan et al., 2014), the brain is fully responsive to the elevated insulin levels in HFF mice and presumably other insulin-resistant states. In this regard, it is interesting to note that type A IRs in brain differ structurally from type B IRs in peripheral tissues in that brain IR has shortened α-subunits; however, further studies are needed to explain differences in downregulation of brain and peripheral IRs.
Finally, in considering the use of aPKC inhibitors that act directly in brain, note that constitutively-active 50kDa PKMζ, which is abundant and present only in brain, is thought to play a key role in long-term potentiation (LTP) and spatial long-term memory formation (Sacktor, 2008). In this regard, note that, although brain-specific PKMζ knockout does not impair LTP and memory functions (Lee et al., 2013; Volk et al, 2013), it was recently found that hippocampal PKC-λ/ι can increase in amount and compensate for losses of hippocampal PKMζ in PKMζ knockout mice, and thereby maintain LTP and spatial memory functions (Tsokas et al, 2016). Further note that PKC-λ/ι may play a role in short-term and LTP memory functions (Ren et al, 2013: Wang et al., 2016). However, as seen here, despite inhibiting insulin- and hyperinsulinemia-related increases in activity of 70kDa PKC-ι/λ in HFF mice, high-dose ACPD treatment did not diminish activity of 50kDa PKMζ, and, moreover, corrected a HF diet-induced impairment in acute memory function, i.e., novel object recognition. Obviously, further studies are needed to clarify the roles of PKC- λ/ι and PKMζ in various memory functions, and to determine CNS effects of doses of PKC-λ/ι inhibitors that act only at the hepatic level (and thus indirectly affect the brain by improving hyperinsulinemia), vis-à-vis doses of PKC-λ/ι inhibitors that act at brain, as well as, hepatic, levels. In any case, the present findings suggest that the activation of brain aPKC may link aPKC activators to abetment of AD pathology, and control of this signaling pathway may provide a means to alter AD pathology
5. CONCLUSIONS
Alzheimer’s disease is characterized by increases in poorly-soluble Aβ peptides and phospho-tau that accumulate to form pathological intraneuronal plaques and interneuronal tangles, respectively, that impair memory functions and destroy neurons. Although specific mutations can underlie increases in Aβ peptides and phospho-tau, they are abetted by various agents, in particular, hyperinsulinemia in insulin-resistant states. However, the actual intracellular signaling factors that mediate increases in Aβ peptide and phospho-tau production are uncertain. Here, we found that insulin-induced increases in brain atypical PKC-ι/λ activity mediate increases in β-secretase, Aβ production and phospho-thr-231-tau. These findings therefore provide a novel signaling mechanism whereby hyperinsulinemia in insulin-resistant states of obesity and diabetes, and other aPKC activators, can promote plaque and tangle formation, and thus abet development of Alzheimer’s disease and other tauopathies.
Figure 10.
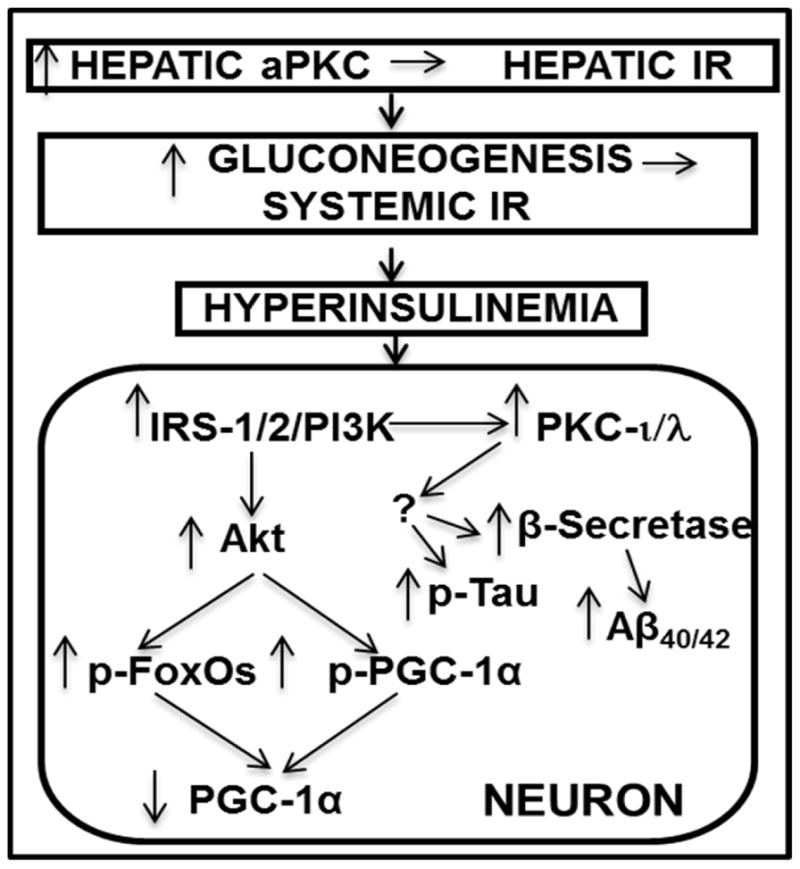
Schematic of pathogenesis of neuronal signaling abnormalities in insulin-resistant states that lead to production of factors that may abet development of Alzheimer’s disease. In this scheme, diet-induced increases in hepatic aPKC activity lead to impaired Akt activation by insulin, i.e., hepatic insulin resistance (IR), increases in hepatic gluconeogenesis, systemic IR, and hyperinsulinemia, which persistently hyperactivates brain Akt and aPKC. Increases in brain Akt activity lead to phosphorylation and thus diminished activities of all FoxOs (1/3a/4/6), and decreased activity and expression of PGC-1α (all needed for neuronal function and integrity). Increases in brain aPKC activity, either directly or indirectly, provoke increases in β-secretase activity, and levels of Aβ1-40/42 and phospho-thr- 231-tau, and thus abet plaque and tangle development.
Highlights.
Alzheimer’s disease is characterized by increases in Aβ peptides and phospho-tau
These increases are abetted by hyperinsulinemia in insulin-resistant states
CNS intracellular signaling factors that increase Aβ and phospho-tau are uncertain
In mice, and isolated neurons and hippocampal slices, aPKC increases β-secretase, Aβ and phospho-tau
Deleterious effects of hyperinsulinemia mice can be blocked by aPKC inhibitors
Acknowledgments
Supported by funds from the Department of Veterans Affairs Merit Review Program to R.V. Farese, and the National Institutes of Health Grants DK 065969-09 to R.V. Farese and DK300136 to C.R. Kahn, and the Deutsche Forschungsgemeinschaft Sta314/2-1 and KE246/7-2 to M. Leitges. Dr. Robert V. Farese is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. This work does not represent the views of the Department of Veteran Affairs or the United States government.
Abbreviations
- AD
Alzheimer’s Disease
- Aβ
amyloid-β peptide
- aPKC
atypical protein kinase C
- T2DM
type 2 diabetes mellitus
- βAPP
beta-amyloid precursor protein
- HFF
high-fat-fed
- ICAPP
1H-imidazole-4-carboxamide,5-amino]-[2,3-dihydroxy-4-[(phosphono-oxy) methyl] cyclopentane-[1R-(1a,2b,3b,4a)]
- ACPD
2-acetyl-cyclopentane- 1,3-dione
- ATM
aurothiomalate
- Het-MλKO
heterozygous muscle-specific PKC-λ knockout
- GSK3β
glycogen synthase kinase-3β
- mTOR
mammalian target of rapamycin
- IR
insulin receptor
- IRS-1
insulin receptor substrate-1
- PI3K
phosphatidylinositol 3-kinase
- ERK
extracellular-regulated kinase
- IDE
insulin degrading enzyme
- FoxO
forkhead box O-type receptor
- PGC-1α
Peroxisome Proliferator-Associated Receptor-γ Coactivtor-1-alpha
- WT
wild type
- pY
phospho-tyrosine
Footnotes
Conflicts
The authors report no conflicts of interest
There are no actual or potential conflicts of interest amongst the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antunes M, Biala G. The novel object recognition memory: neurobiology, test procedure and its modification. Cogn Process. 2012;13:93–110. doi: 10.1007/s10339-011-0430-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barini E, Antico O, Zhao Y, Asta F, Tucci V, Catelani T, Marotta R, Xu H, Gasparini L. Metformin promotes tau aggregation and exacerbates abnormal behavior in a mouse model of tauopathy. Molec Neurodegen. 2016;11:16–36. doi: 10.1186/s13024-016-0082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron AM, Rosario ER, Pike CJ. Sex-specific effects of high fat diet on indices of metabolic syndrome in 3xTg-AD mice: Implications for Alzheimer’s disease. LPOS ONE. 2013;8:1–8. doi: 10.1371/journal.pone.0078554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhou K, Wang R, Liu Y, Kwak Y-D, Ma T, Thompson RC, Zhao Y, Smith L, Gasparini L, Luo Z, Xu H, Liao F-F. Antidiabetic drug metformin (Glucophage) increases biogenesis of Alzheimer’s amyloid peptides via upregulating BACE1 transcription. Proc Natl Acad Sci USA. 2009;106:3907–3912. doi: 10.1073/pnas.0807991106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft S. Insulin resistance and Alzheimer’s disease pathogenesis: mechanisms and implications for treatment. Curr Alzheimer Res. 2007;4:147–152. doi: 10.2174/156720507780362137. [DOI] [PubMed] [Google Scholar]
- Craft S. Intranasal insulin therapy for Alzheimer’s disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69:29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cruz e Silve O-AB, Rebelo S, Viera SI, Gandy S, da Cruz e Silva EF, Greengard P. Enhanced generation of Alzheimer’s amyloid-β following chronic exposure to phorbol ester correlates with differential effects on alpha and epsilon isozymes of protein kinase C. J Neurochem. 2009;108:319–330. doi: 10.1111/j.1471-4159.2008.05770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG. Alzheimer’s disease and insulin resistance: translating basic science into clinical applications. J Clin Invest. 2013;123:531–539. doi: 10.1172/JCI64595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Monte SM. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr Alzheimer Res. 2012;9:35–66. doi: 10.2174/156720512799015037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolich P, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, Muschner D, Thalheimer A, Turk A, Hoyer S, Zochling R, Boissl KW, Jellinger K, Riederer P. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Transm. 1998;105:423–438. doi: 10.1007/s007020050068. [DOI] [PubMed] [Google Scholar]
- Freude S, Plum L, Schnitker J, Leeser U, Udelhoven M, Krone W, Bruning JC, Schubert M. Peripheral hyperinsulinemia promotes tau phosphorylation in vivo. Diabetes. 2005;54:3343–3348. doi: 10.2337/diabetes.54.12.3343. [DOI] [PubMed] [Google Scholar]
- Gasparini L, Gouras GK, Wang R, Gross RS, Flint Beal M, Greengard P, Xu H. Stimulation of β-amyloid precursor protein trafficking by insulin reduces intraneuronal β-amyloid and requires mitogen-activated protein kinase signaling. J Neurosci. 2001;21:2561–2570. doi: 10.1523/JNEUROSCI.21-08-02561.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Pan Y, Pasinetti GM. Nicotinamide riboside restores cognition through an upregulation of proliferatoractivated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol Aging. 2013;34:1581–1588. doi: 10.1016/j.neurobiolaging.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gontier G, George C, Chaker Z, Holzenberger M, Aid S. Blocking IGF signaling in adult neurons activates Alzheimer’s disease pathology through amyloid-β clearance. J Neurosci. 2015;35:11500–11513. doi: 10.1523/JNEUROSCI.0343-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janson J, Laedtke T, Parisi JE, O’Brien P, Peterson RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer’s disease. Diabetes. 2004;53:478–481. doi: 10.2337/diabetes.53.2.474. [DOI] [PubMed] [Google Scholar]
- Kohjima M, Sun Y, Chan L. Increased food intake leads to obesity and insulin resistance in the Tg2576 Alzheimer’s disease mouse model. Endocrinol. 2010;151:1532–1540. doi: 10.1210/en.2009-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotani K, Ogawa W, Matsumoto M, Kitamura T, Sakaue H, Hino Y, Miyaki K, Sano W, Akimoto K, Ohno S, Kasuga M. Requirement of atypical protein kinase Cλ for insulin stimulation of glucose uptake but not for Akt activation in 3T3/L1 adipocytes. Mol Cell Biol. 1998;18:6971–6982. doi: 10.1128/mcb.18.12.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AM, Kanter BR, Wang D, Lim JP, Zou ME, Qiu C, McMahon T, Dagdar J, Fischbach-Weiss SC, Messing RO. Prkcζ null mice show normal learning and memory. Nature. 2013;493:416–420. doi: 10.1038/nature11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macklin L, Griffith CM, Cai Y, Rose GM, Yan X-X, Patrylo PR. Glucose tolerance and insulin sensitivity are impaired in APP/PS1 transgenic mice prior to amyloid plaque pathogenesis and cognitive decline. Exp Gerontol. 2017;88:9–18. doi: 10.1016/j.exger.2016.12.019. [DOI] [PubMed] [Google Scholar]
- Paik J-H, Ding Z, Narukar R, Ramkissoon S, Muller F, Kamoun WS, Ghae S-S, Zheng H, Ying H, Mahoney J, hiller D, Jiang S, Protopopov A, Wong WH, Chin L, Logon KL, DePinho RA. FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell. 2009;5:540–553. doi: 10.1016/j.stem.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai P, Desai S, Patel R, Sajan M, Farese R, Ostrov D, Acevedo-Duncan M. A novel PKC-ι inhibitor abrogates cell proliferation and induces apoptosis in neuroblastoma. Int J Biochem Cell Biol. 2011;43:784–794. doi: 10.1016/j.biocel.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Qin W, Haroutunian V, Katsel P, Cardozo CP, Lo H, Buxbaum JD, Pasinetti GM. PGC-1α expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol. 2009;66:352–361. doi: 10.1001/archneurol.2008.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren S-Q, Yan J-Z, Zhang XY, Bu Y-F, Pan W-W, Yao W, Tian T, Lu W. PKCλ is critical in AMPA receptor phosphorylation and synaptic incorporation in LTP. The EMBO J. 2013;32:1365–1380. doi: 10.1038/emboj.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renault VM, Rafalski VA, Morgan A, Salih DA, Brett JO, Webb AE, Villeda S, Thekkat PU, Guillerey C, Denko NC, Palmer TD, Butte AJ, Brunet A. FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell. 2009;5:527–539. doi: 10.1016/j.stem.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajan MP, Acevedo-Duncan ME, Standaert ML, Ivey RA, III, Lee MC, Farese RV. Akt-dependent phosphorylation of hepatic FoxO1 is compartmentalized on a WD40/Propeller/FYVE scaffold and is selectively inhibited atypical PKC in early phases of diet-induced obesity. A mechanism for impairing gluconeogenic but not lipogenic enzyme expression. Diabetes. 2014;63:2690–2701. doi: 10.2337/db13-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajan MP, Farese RV. Insulin Signalling in Hepatocytes of Type 2 Diabetic Humans. Excessive Expression and Activity of PKC-ι and Dependent Processes and Reversal by PKC-ι Inhibitors. Diabetologia. 2012a;55:1446–1457. doi: 10.1007/s00125-012-2477-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajan MP, Ivey RA, III, Farese RV. Brain insulin signaling is increased in insulin-resistant states and decreases in FOXOs and PGC-1α and increases in Aβ1–40/42 and phospho-tau may abet Alzheimer’s development. Diabetes. 2016;65:1892–1903. doi: 10.2337/db15-1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajan MP, Ivey RA, III, Lee MC, Farese RV. Hepatic insulin resistance in ob/ob mice involves increases in ceramide, atypical PKC activity and selective impairment of Akt-dependent FoxO1 phosphorylation. J Lipid Res. 2015;56:70–80. doi: 10.1194/jlr.M052977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajan MP, Ivey RAIII, Lee M, Mastorides S, Jurczak M, Samuels VT, Shulman GI, Braun U, Leitges M, Farese RV. PKCλ haplo-insufficiency prevents diabetes by a mechanism involving alterations in hepatic enzymes. Molecular Endocrinology. 2014b;28:1097–1107. doi: 10.1210/me.2014-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajan MP, Nimal S, Mastorides S, Acevedo-Duncan M, Kahn CR, Fields AP, Braun U, Leitges M, Farese RV. Correction of Metabolic Abnormalities in a Rodent Model of Obesity, Metabolic Syndrome and Type 2 Diabetes by Inhibitors of Hepatic Protein Kinase C-iota. Metabolism. 2012b;61:459–469. doi: 10.1016/j.metabol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salih DAM, Rashid AJ, Colas D, de la Torre-Libieta L, Zhu RP, Morgan AA, Santo EE, Ucar D, Devarajan K, Cole CJ, Madison DV, Shamloo M, Butte AJ, Bonni A, Josselyn SA, Brunet A. FoxO6 regulates memory consolidation and synaptic function. Genes and Development. 2012;26:2780–2801. doi: 10.1101/gad.208926.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajan MP, Standaert ML, Nimal S, Varanasi U, Pastoor T, Mastorides S, Braun U, Leitges M, Farese RV. Critical role of atypical protein kinase C in activating hepatic SREBP-1c and NFkB in obesity. J Lipid Res. 2009;50:1133–1145. doi: 10.1194/jlr.M800520-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney G, Song J. The association between PGC-1α and Alzheimer’s disease. Anat Cell Biol. 2016;49:1–6. doi: 10.5115/acb.2016.49.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokas P, Hsieh C, Yao Y, Lesburgueres E, Wallace EJC, Tcherepanov A, Jothaniandan D, Hartley BR, Pan L, Rivard B, Farese RV, Sajan MP, Bergold PJ, Hernandez AI, Cottrell J, Harel Z, Shouval HZ, Fenton AA, Sacktor TC. Compensation for PKMζ in LTP and spatial long-term memory in mutant mice. eLife. 2016;5:e14846. doi: 10.7554/eLife.14846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandal M, White PJ, Chevrier G, Tremblay CF, St-Amour I, Planel E, Marette A, Calon F. Age-dependent impairment of glucose tolerance in the 3xTg-AD mouse model of Alzheimer’s disease. The FASEB J. 2017;29:4273–4284. doi: 10.1096/fj.14-268482. [DOI] [PubMed] [Google Scholar]
- Viera SI, Rebelo S, Esselman H, Wiltfang J, Lah J, Lane R, Small SA, Gandy S, da Cruz e Silva EF, da Cruz e Silva O-AB. Retreival of the Alzheimer’s amyloid precursor protein from the endosome to the TGN is S655 phosphorylation state-dependent and retromer-mediated. Molec Neurodegen. 2010;5:40–61. doi: 10.1186/1750-1326-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk LJ, Bachman JL, Johnson R, Yu Y, Huganir RL. PKM-ζ is not required for hippocampal synaptic plasticity, learning and memory. Nature. 2013;493:420–425. doi: 10.1038/nature11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Gallagher D, DeVito LM, Cancina GI, Tsui D, He L, Keller GM, Frankland PW, Kaplan DR, Miller FD. Metformin activates an atypical PKC-CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell. 2012;11:23–35. doi: 10.1016/j.stem.2012.03.016. [DOI] [PubMed] [Google Scholar]
- Wang S, Sheng T, Ren S, Tian T, Lu W. Distinct roles of PKCι/λ and PKMζ in the initiation and maintenance of hippocampal long-term potentiation and memory. Cell Reports. 2016;16:1954–1961. doi: 10.1016/j.celrep.2016.07.030. [DOI] [PubMed] [Google Scholar]
- Zemva JM, Schubert The role of neuronal insulin/insulin-like growth factor-1 signaling for the pathogenesis of Alzheimer’s disease: possible therapeutic implications. CNS Neurol Disord Drug Targets. 2014;13:322–337. doi: 10.2174/18715273113126660141. [DOI] [PubMed] [Google Scholar]