

Abstract
AIRE is a well-established master regulator of central tolerance. It plays an essential role in driving expression of tissue-specific antigens in the thymus and shaping the development of positively selected T-cells. Humans and mice with compromised or absent AIRE function have markedly variable phenotypes that include a range of autoimmune manifestations. Recent evidence suggests that this variability stems from cooperation of autoimmune susceptibilities involving both central and peripheral tolerance checkpoints. Here we discuss the broadening understanding of the factors that influence Aire expression, modify AIRE function, and the impact and intersection of AIRE with peripheral immunity. This rapidly expanding body of knowledge will force a reexamination of the definition and clinical management of APS-1 patients as well as provide a foundation for the development of immunomodulatory strategies targeting central tolerance.
Introduction
The adaptive immune system is charged with the staggering task of maintaining a maximally diverse T-cell pool, capable of defending against constantly changing pathogenic and neoplastic challenges, while stringently preventing immune responses against healthy tissues. The latter process is broadly categorized as immune tolerance and when the mechanisms that promote immune tolerance fail, autoimmunity ensues [1].
T-cell development begins in the thymus where somatic rearrangement of T-cell receptors (TCRs) generates extraordinary diversity in peptide binding specificities [2,3]. Developing T-cells possessing TCRs with high affinity for self-peptides are eliminated in the medulla of the thymus through negative selection or become thymically derived T regulatory cells (tTregs) [2,3]. Therefore, medullary screening of TCR binding affinity provides the foundation for centrally mediated T-cell tolerance. This screening is achieved through promiscuous gene expression (PGE) of tissue-specific antigens (TSAs) by a specialized subset of medullary thymic epithelial cells (mTECs) [3,4]. Self-antigens are presented to thymocytes primarily by mTECs and medullary dendritic cells (DCs) as well as by a small subset of thymic B-cells [4,5]. Recent evidence has shown that this process exposes maturing thymocytes to a near complete representation of the protein coding genome, thereby ensuring the comprehensive projection of self [4].
The Autoimmune Regulator (Aire) protein is a transcriptional regulator that is highly expressed in a subset of mTECs where it plays a critical and non-redundant role in promoting PGE of a wide array of TSAs [6]. Over the last fifteen years, we and others have contributed to a large body of work delineating the crucial role of Aire in establishing and maintaining central tolerance. Underscoring the clinical significance of AIRE, loss of function in humans leads to the rare autosomal recessive disorder, Autoimmune Polyglandular Syndrome Type-I (APS-1/APECED), characterized by multiorgan autoimmunity as well as susceptibility to a “signature” infectious disease, chronic mucocutaneous candidiasis (CMC) [7]. Classically, APS-1 presents with the triad of hypoparathyroidism, adrenal insufficiency, and CMC (associated with neutralizing anti-IL-17F and IL-22 autoantibodies), and often involves multiple endocrine and non-endocrine organs and ectodermal dystrophies (described below) [7]. In the mouse, Aire deficiency has proven indispensable in dissecting Aire function as Aire−/− animals exhibit a multiorgan autoimmunity that closely resembles human disease [8]. Remarkably, several APS-1 human disease manifestations have been directly correlated to an absence or reduction in thymic expression of Aire-regulated TSAs in Aire−/− mice and escape of autoreactive T cells specific for those TSAs [9–12]. In such studies, epitope mapping and tetramer reagents in the murine model have been used to characterize autoreactive T-cell populations with striking granularity.
While numerous AIRE mutations have now been described and have recently been the subject of review elsewhere, the largest and most complete cohort of APS-1 patients harbor mutations in both AIRE alleles leading to a complete loss of AIRE function [13]. Curiously, age of disease onset, severity, and the spectrum of affected organs is highly variable in APS-1 patients and among Aire−/− mouse strains [7,13]. This long-standing observation suggests the possibility that Aire may cooperate with other central or peripheral immune tolerance checkpoints such that variable penetrance and pathology result from the confluence of these interactions as well as epigenetic, microbiomic, and environmental factors. The nature of such checkpoints, how they cooperate with Aire, and their role in regulating tolerance to specific TSAs is largely unknown.
Here we review recent insights from patients and mouse models that shed light onto the modifiers of Aire-dependent autoimmunity and discuss potential avenues for modulating Aire levels in vivo.
Aire as an autoimmune susceptibility gene
Although classical APS-1 is an autosomal recessive disease, more examples of families with autosomal dominant APS-1 have been recently identified [13]. Disease within these families is generally characterized by milder pathology, later onset, and fewer affected tissues. Aire functions as a homodimer or tetramer and is known to act as a binding partner for numerous nuclear factors [6,14]. Accordingly, mutations in a single Aire allele may act in a dominant negative or haploinsuffcicient manner and lead to reduced Aire function, which may be sufficient to significantly alter expression of some TSAs and promote escape of self-reactive T-cells [15]. Thus, the organ-specific pattern in patients harboring dominant negative mutations may be reflective of loss of tolerance to TSAs that are especially sensitive to specific perturbations in AIRE protein-protein interactions.
Recently, key insights into the role of Aire as an autoimmune susceptibility gene came from elegant work by Oftedal and colleagues describing a cohort of patients with dominant negative mutations localized to the PHD-1 zinc finger domain of AIRE [16••]. As might be predicted, disease was milder, with later onset. Interestingly, as opposed to pronounced endocrinopathies, disease manifested primarily as pernicious anemia and vitiligo. Indeed, examination of publicly available genetic databases has identified that the overall incidence of AIRE mutations in the general population may approach 1 in 1000 [16••]. Such a high incidence of AIRE mutations allows for the intriguing possibility that partial AIRE dysfunction could be significantly more prevalent than previously appreciated. Taken together, we suggest that mild AIRE dysfunction may represent one susceptibility factor in complex, multifactorial autoimmune diseases that have previously not been associated with AIRE or a central tolerance defect.
To address whether partial defects in AIRE can coordinate with a defined peripheral defect in immune regulation, we recently utilized a mouse model harboring a hypomorphic Aire mutation. The G228W mutation (AireGW) in the DNA-binding SAND domain of Aire was first identified in an Italian family with a late-onset autosomal dominant form of APS-1 which manifested primarily as autoimmune thyroiditis [17]. A mouse knock-in of this mutation on the autoimmune-resistant C57BL/6 background was observed to have extremely limited autoimmunity, with mild and variable involvement of the lacrimal and salivary glands [18]. However, when introduced onto the autoimmune-prone NOD background, a far broader subset of organs was affected compared to C57BL/6 and these organs were distinct from those affected in Aire−/− C57BL/6 mice [18]. These discrepancies pointed to the involvement of genetic modifiers and we hypothesized that they might be found amongst peripheral tolerance checkpoints. To test this possibility, we created a digenic model of autoimmune susceptibility by crossing AireGW mice to mice deficient for the Src-family kinase Lyn (Lyn−/−) [19••]. LYN is a negative regulator of innate immune cells and LYN-deficient mice develop a generalized, lupus-like inflammatory disease as they age [20].
Unexpectedly, almost half of AireGW.Lyn−/− mice developed bilateral autoimmune uveitis by 5–6 weeks [19••]. Importantly, uveitis was not observed in either of the monogenic AireGW or Lyn−/− parent strains. By crossing the lineage restricted CD11c-Cre to the AireGW.Lynfl/fl background, we showed that disease was driven by the rare escaped T-cells specific for an AIRE-dependent retinal antigen primed by hyperactive LYN-deficient DCs in eye-draining lymph nodes. This work provides direct evidence for a paradigm in which partial defects in thymic AIRE function can synergize with defects in DC tolerance to drive organ-specific autoimmunity. It is tantalizing to consider whether this mechanism might contribute to organ-specific manifestations observed in systemic autoimmune syndromes. Of note, uveitis and thyroiditits are common amongst patients with systemic autoimmune diseases such as lupus, rheumatoid arthritis and ankylosing spondylitis [21,22]
In an earlier report, Aire deficiency was also shown to synergize with a peripheral T-cell tolerance checkpoint controlled by the E3 ubiquitin ligase Cbl-b, which regulates responses to CD28 costimulation and T-cell anergy [23]. Mice deficient in both Aire and Cbl-b developed lethal exocrine pancreatitits as early as three weeks of age [23]. Significantly, in the same study no cooperation was found between Aire and several other peripheral T-cell checkpoints, including those controlling activation-induced cell death (Fasgld/gld) or inhibition of Tfh-cells (Rc3h1san/san), suggesting that co-stimulation by APCs leading to induction of anergy in autoreactive T-cells may be a major tolerance checkpoint for AIRE-mediated autoimmunity. It remains to be determined why broad defects in peripheral tolerance, such as Lyn or Cbl-b deficiency, predispose to activation and infiltration of T-cells specific for a particular organ while sparing the others, commonly affected in more penetrant Aire-dependent autoimmunity.
Taken together, these findings suggest that despite escape of self-reactive T-cells from the AIRE-dysfunctional thymus, and thus a break in central tolerance, autoimmunity may often require a failure of one or several peripheral tolerance checkpoints (Figure 1). Furthermore, these latter defects may interface with self-reactive T-cells in an antigen- or tissue-specific manner. Conceptually, this is well-aligned with an emerging consensus around a multi-hit hypothesis for autoimmune disease [24]. Current findings point to a model in which distinct peripheral regulatory mechanisms might exist for different AIRE-regulated TSAs. As a consequence, different patient populations will likely show differential susceptibility to specific autoimmune symptomology depending on their background frequency of specific peripheral tolerance defects and exposure to immunogenic environmental factors.
Figure 1. Defects in Aire cooperate with defects in peripheral tolerance to promote autoimmunity.
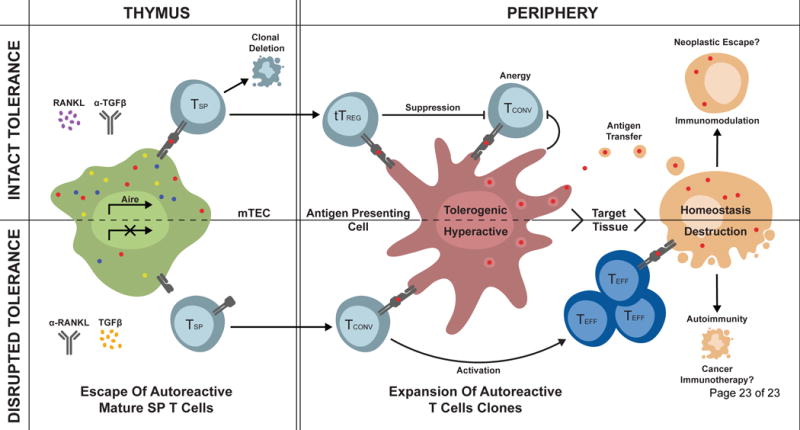
Aire induces expression of TSAs in thymic mTECs, which are presented to developing single positive (SP) T cells. Self-reactive T cells that bind peptide-MHC complexes with high affinity are either deleted by negative selection or become thymically derived natural tTregs. Mutations in the Aire gene leading to reduced or absent AIRE function result in decreased TSA presentation and survival and thymic escape of autoreactive T-cell clones as well as a loss of tTreg induction. Autoreactive T-cells can then encounter cognate self-antigen presented by APCs in secondary lymphoid organs or peripheral tissues. When peripheral tolerance checkpoints are intact, autoreactive T cells are suppressed by inhibitory signals from Tregs as well as resting APCs. Failure of peripheral tolerance leads to their activation and autoimmune attack. RANKL and TGFβ are two factors known to influence mTEC function, and thereby, AIRE and TSAs expression. Therapies directly targeting these molecules or their downstream pathways may provide therapeutic opportunities for immune modulation in the context of hyperactivity or release of tolerance in the context of neoplastic transformation and cancer immunotherapy.
Recent clinical, diagnostic, immunological and genetic insights from APS-1 patients
Cohorts of APS-1 patients had been primarily reported from Scandinavian and other European countries, with endocrinopathies being the predominant clinical manifestations [25,26]. Consecutive enrollment at the NIH of a large cohort of >80 APS-1 patients (American and, less so, European) has recently allowed patient evaluation by a multidisciplinary team of specialists in a prospective observational natural history study [27••]. This work has uncovered a far-broader clinical spectrum of the syndrome. Over 30 distinct manifestations can occur with >25 of them targeting non-endocrine tissues. This greater breadth of the phenotypic expression in APS-1 patients corroborates the broad-spectrum of endocrine and non-endocrine autoimmune disease encountered in Aire−/− mice, in which pulmonary, gastric, salivary and lacrimal gland, and ocular inflammation are common, but were thought to be infrequent or absent in the human disease [8,25,26]. These clinical observations, in addition to the susceptibility of Aire−/− mice to candidiasis (Lionakis lab, unpublished), strongly support the translational utility of the mouse model. These clinical findings have two important implications for patients: (1) expanded diagnostic criteria incorporating manifestations targeting the small intestine, enamel, and skin (which occur frequently and early in the course of the disease) may allow recognition of the syndrome before the development of life-threatening end-organ complications [27••] and (2) earlier patient diagnosis will provide a greater window of opportunity for successful lymphocyte-targeted immunomodulation and/or thymic manipulation, both of which are likely to be more efficacious in preventing and treating autoimmune pathology early in the course of APS-1.
Beyond the broader APS-1 clinical scope, patient cohort studies have also uncovered an impressive variation of phenotypic expression as it relates to AIRE genetics. As mentioned above, although most APS-1 patients carry biallelic AIRE mutations, the severity and spectrum of organ-specific clinical disease varies greatly among patients (including siblings) with identical AIRE mutations. On one end of the genotype-phenotype spectrum are patients with biallelic disease-causing AIRE mutations who suffer isolated APS-1 disease manifestations. While this has been described chiefly in the case of isolated hypoparathyroidism [28], it is likely applicable to other manifestations, and perhaps to individuals without any apparent clinical disease. Hence, the number of individuals with biallelic AIRE mutations who are not recognized as APS-1 patients due to single-disease or non-classical presentations may be larger than appreciated. On the other end of the spectrum are patients with completely penetrant clinical APS-1 but without biallelic AIRE mutations; these individuals typically carry heterozygous AIRE mutations/deletions that do not fall under the dominant-negative AIRE mutations in the SAND or PHD1 domains [29]. This spectrum underscores the existence of disease-modulating variables, including organ-specific disease-promoting and/or ameliorating AIRE-independent genetic elements that may modulate AIRE expression, function, or may affect immune tolerance via peripheral, AIRE-independent pathways.
Such coding and non-coding loci have recently been uncovered in mice and represent plausible candidates to explain the genetic etiology of the breadth of APS-1 clinical manifestations. For example, the recent characterization of the first cis-regulatory element of Aire represents a major conceptual advance [30,31]. Coding elements that affect mouse Aire expression include Jmjd6, Sirt1, Hipk2, Dgcr8, and Fbxo3 (Table 2) [32–36]. Importantly, the recent identification of Fezf2 as a key regulator of Aire-independent TSA expression in mTECs underscores that TSA representation in the thymus is likely to involve yet to be discovered factors that cooperate with AIRE and may help explain organ-specific phenotypic expressions of APS-1 or, conceivably, in patients with monogenic or multigenic autoimmune diseases [37].
Table 2.
Coding and non-coding genetic elements that affect AIRE expression or function.
| Gene | Mechanism | Reference |
|---|---|---|
| CNS1 | NF-kB responsive cis-regulatory element required for Aire expression in mTECs | [30,31] |
| JMJD6 | Splicing of intron 2 of Aire, required for mature Aire protein in mTECs | [32] |
| HIPK2 | Affects Aire phosphorylation and suppresses coactivator activity of Aire, modest impairment of Aire-dependent promiscuous gene expression in mTECs in vivo | [33] |
| SIRT1 | Affects Aire acetylation, required for expression of Aire-dependent TSA-encoding genes | [34] |
| DGCR8 | Required for accumulation of Aire+ mTECs in thymus | [35] |
| FBXO3 | Ubiquitinates Aire and promotes transcriptional activity of AIre | [36] |
Recent evidence describing the cell-specific contributions of different lymphoid effectors in mediating organ-specific autoimmune manifestations in Aire−/− mice has provided important insights with potential implications for the management of APS-1 patients. At present, successful management in APS-1 patients relies on azathioprine or mycophenolate monotherapy for those with autoimmune hepatitis or on azathioprine combined with rituximab for those with autoimmune pneumonitis (Lionakis, unpublished). Although it is well-established that self-reactive effector CD4+ T-cells drive multisystem autoimmunity in Aire−/− mice, it is now recognized that other lymphoid cells may also exert organ-specific modulating effects. For example, a defect in the neonatal output of tTregs in Aire−/− mice was recently shown to drive autoimmunity [38]. However, patients with immune dysregulation-polyendocrinopathy-enteropathy-X-linked (IPEX) syndrome, caused by FOXP3 mutations, lack Tregs and develop early-onset life-threatening autoimmunity with non-overlapping features relative to APS-1 patients [39]. Thus, while tTreg selection and function may play a role, it would appear that the contribution of impaired negative selection predominates in accounting for autoimmunity in APS-1 patients relative to a skewed tTreg repertoire. Additionally, Aire−/− mice have also been shown to have a thymically derived defect in V 6+V 1+ T-cells and B-cells that is a driver of organ-specific autoimmunity in the retina and lungs, respectively [40–42]. Specifically, B-cells appear to contribute to early T-cell priming and expansion as antigen-presenting cells. Although autoantibodies have not been proven directly pathogenic using mouse serum transfers, the role of autoantibodies in APS-1 disease remains an active area of investigation [41]. While Type 1 interferon autoantibodies are virtually diagnostic for APS-1, it is unclear whether they have a pathogenic role. Recently, investigators with the APECED patient collaborative described a broad spectrum of autoantibodies targeting >40% of the proteome in a cohort of 81 patients with notable prevalence of high-affinity, neutralizing anti-cytokine antibodies [43]. Based on the inverse correlation of type-I interferon autoantibodies with Type 1 diabetes, such antibodies may protect from diabetes in APS-1 patients [43]. In contrast, another recent study demonstrated that only a very limited portion of the proteome becomes targeted in APS-1 patients [44]. This difference in autoantibody repertoire contrasts with the broad defect of thymic presentation associated with AIRE deficiency and raises several questions with regard to other factors that may be needed for tolerance breakdown, some of which have been addressed above. The mechanisms by which Aire deficiency impairs B-cell central and/or peripheral tolerance checkpoints remain elusive, and the extent of overlap between the T- and B-cell tolerance defects in APS-1 patients are also largely unknown. These questions are highlighted by the recent demonstration that patients with thymomas or RAG mutations, who have decreased thymic AIRE expression, exhibit variable correlation between thymic AIRE expression, AIRE-dependent TSA expression levels, autoantibody presence and titers, and corresponding clinical autoimmune manifestations [45,46].
Aire as a therapeutic target
Immunomodulatory therapies are rapidly transforming the treatment of cancer, autoimmune, and inflammatory diseases. In recent years, cancer immunotherapy has been revolutionized by the development of biologics successfully targeting peripheral T-cell tolerance checkpoints. The most successful of these therapies modulate peripheral effector T-cell activation states and Treg function [47]. However, therapeutic strategies directed at central tolerance are currently lacking. The reasons for this are largely twofold. First, our understanding of the molecular underpinnings of thymic function are rapidly evolving. Second, manipulation of central tolerance is likely to have a durable and broad impact on the T cell repertoire in a way that targeted peripheral therapies may not. Despite this latter conceptual concern, we propose that the current pace of discovery will allow manipulation of thymic output to emerge as an attractive therapeutic target in T-cell mediated autoimmune diseases and cancer immunotherapy. In the former, augmentation of Aire and mTEC function might limit the escape of self-reactive T cells from the thymus as well as promote the generation of autoantigen-specific tTregs. In the latter, Aire and mTEC disruption can release autoreactive T-cells into the periphery where they might effectively target tumor-associated antigens. If medulla of the thymus can be thought of as a net preventing the passage of autoreactive T-cells, then these opposing approaches amount to a tightening or loosening of the webbing. Indeed, there is accumulating evidence that such manipulation is possible.
Medullary function can be modified through changes in overall cellularity, thereby decreasing the absolute number Aire-expressing mTECs, or by targeting pathways that directly affect Aire function, while maintaining mTEC cellularity. Aire expression is largely restricted to a subset of mTECs characterized by high levels of MHCII and CD80 (mTEChi) [4]. Among the best characterized positive regulators of the mTEC development and maintenance are members of the tumor necrosis factor superfamily (TNF), including receptor activator of NF-kB ligand (RANKL), CD40 ligand, and Lymphotoxin (Lt), which bind their cognate receptors to activate NF-kB signaling [4]. Of note, RANKL produced by developing thymocytes is especially important for maintenance of the mTEChi (Airehi) population [4]. In vivo blockade of RANKL with a neutralizing antibody not only greatly reduced mTEC cellularity but specifically reduced the mTEChi Aire-expressing subset [48]. This, in turn, led to the escape of autoreactive T-cells specific for Aire-regulated TSAs. It is now appreciated that numerous neoplasms, including melanoma, sarcoma, and prostate cancer express high levels of Aire-regulated TSAs and that the recognition of these proteins as self contributes to immune evasion [6]. Notably, we have shown that anti-RANKL treatment increased the pool of melanoma-specific T cells and enhanced the antitumor response and overall survival in a mouse model [48]. Importantly, no autoimmunity was observed in mice subjected to RANKL blockade (Anderson lab, unpublished) and the mTEC compartment was completely restored upon cessation of treatment, suggesting that transient interruption of central tolerance via RANKL blockade may allow robust anti-tumor responses while sparing healthy tissues [48]. It remains to be seen whether the anti-tumor effects of treatments diminishing Aire activity have the potential for combination therapy with peripheral checkpoint inhibitors, but it presents an exciting possibility.
TGF has potent effects on cellular growth and differentiation and has recently gained prominence as a negative regulator of mTEC development and function [49]. Blocking TEC TGF RII expression or systemic administration of TGF RI inhibitor resulted in increased generation of mTECs and greater expression of TSAs without considerable effects on cTECs or positive selection [50]. Significantly, blocking TGF was protective against autoimmunity induced by peripheral Treg depletion suggesting that thymic tolerance was enhanced. Given that TGF has a tolerogenic role in the periphery, selective blockade of this pathway in the thymus may be challenging and requires further research and careful consideration [49].
Conclusions and future directions
Maintenance of immune tolerance involves multiple central and peripheral checkpoints to ensure robust and redundant mechanisms to control autoimmunity. The work discussed in this review presents evidence for an an emerging rubric in which AIRE mutations that disrupt or diminish AIRE function may be a common feature of complex autoimmune diseases. Furthermore, heterogeneity of AIRE-dependent autoimmune manifestations should be viewed in the context of interaction between multiple tolerance checkpoints. It may prove particularly interesting to investigate peripheral tolerance defects in APS-1 patients and whether any such defects are shared amongst similar patterns of disease. Conversely, organ-specific manifestations in systemic autoimmunity may expose an otherwise obscured defect in thymic central tolerance and a search for AIRE mutations in this context could provide a new opportunity for understanding complex and heterogeneous autoimmune phenotypes.
Table 1.
Conditions that affect AIRE expression or function.
| Effect on thymic Aire | Mechanism | Reference | |
|---|---|---|---|
| Classical APS-1 | Absent | Autosomal recessive homozygous or compound heterozygous AIRE mutations | [7] |
| Non-classical APS | Reduced to absent | Autosomal dominant Aire mutations | [13] |
| Thymomas | Reduced | Reduced Aire transcripts in neoplastic TEC cells | [51] |
| Graft-versus-host disease | Reduced | Destruction of recipient mTEC hi cells by donor T cells | [52] |
| Sexual Dimorphism | Reduced by estrogen and progesterone Increased by androgens | Sex hormones affect Aire mRNA levels or TSA expression | [53,54] |
| Rag Mutations | Reduced | Reduced numbers of developing thymocytes lead to loss of Aire-enhancing signals | [55] |
Highlights.
APS-1/APECED patients exhibit marked phenotypic variability despite genotypic similarities
Defects in AIRE synergize with defects in peripheral tolerance to promote autoimmunity
Manipulation of AIRE-dependent thymic tolerance is an emerging therapeutic target
Acknowledgments
We would like thank Mickie Cheng for helpful discussions, constructive suggestions, and critical reading of this manuscript. This work was supported by the Division of Intramural Research (DIR), NIAID/NIH (to MSL); NIH-NIDDK (DK107383, R01 DK101622 to MSA); NIH-NIAID (AI 118688, AI1097457, AI35297 to MSA); and Larry Hillblom Foundation (2017-D-012-FEL to IP).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Theofilopoulos AN, Kono DH, Baccala R. The multiple pathways to autoimmunity. Nat Immunol. 2017;18:716–724. doi: 10.1038/ni.3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see) Nat Rev Immunol. 2014;14:377–391. doi: 10.1038/nri3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kyewski B, Klein L. A CENTRAL ROLE FOR CENTRAL TOLERANCE. Annual Review of Immunology. 2006;24:571–606. doi: 10.1146/annurev.immunol.23.021704.115601. [DOI] [PubMed] [Google Scholar]
- 4.Takahama Y, Ohigashi I, Baik S, Anderson G. Generation of diversity in thymic epithelial cells. Nat Rev Immunol. 2017 doi: 10.1038/nri.2017.12. [DOI] [PubMed] [Google Scholar]
- 5.Perry JS, Hsieh CS. Development of T-cell tolerance utilizes both cell-autonomous and cooperative presentation of self-antigen. Immunol Rev. 2016;271:141–155. doi: 10.1111/imr.12403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson MS, Su MA. AIRE expands: new roles in immune tolerance and beyond. Nat Rev Immunol. 2016;16:247–258. doi: 10.1038/nri.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng MH, Anderson MS. Monogenic autoimmunity. Annu Rev Immunol. 2012;30:393–427. doi: 10.1146/annurev-immunol-020711-074953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298:1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 9.DeVoss J, Hou Y, Johannes K, Lu W, Liou GI, Rinn J, Chang H, Caspi RR, Fong L, Anderson MS. Spontaneous autoimmunity prevented by thymic expression of a single self-antigen. J Exp Med. 2006;203:2727–2735. doi: 10.1084/jem.20061864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shum AK, DeVoss J, Tan CL, Hou Y, Johannes K, O’Gorman CS, Jones KD, Sochett EB, Fong L, Anderson MS. Identification of an autoantigen demonstrates a link between interstitial lung disease and a defect in central tolerance. Sci Transl Med. 2009;1:9ra20. doi: 10.1126/scitranslmed.3000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taniguchi RT, DeVoss JJ, Moon JJ, Sidney J, Sette A, Jenkins MK, Anderson MS. Detection of an autoreactive T-cell population within the polyclonal repertoire that undergoes distinct autoimmune regulator (Aire)-mediated selection. Proc Natl Acad Sci U S A. 2012;109:7847–7852. doi: 10.1073/pnas.1120607109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hou Y, DeVoss J, Dao V, Kwek S, Simko JP, McNeel DG, Anderson MS, Fong L. An aberrant prostate antigen-specific immune response causes prostatitis in mice and is associated with chronic prostatitis in humans. J Clin Invest. 2009;119:2031–2041. doi: 10.1172/JCI38332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruserud Ø, Oftedal BE, Wolff AB, Husebye ES. AIRE-mutations and autoimmune disease. Current Opinion in Immunology. 2016;43:8–15. doi: 10.1016/j.coi.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 14.Kumar PG, Laloraya M, Wang C-Y, Ruan Q-G, Davoodi-Semiromi A, Kao K-J, She J-X. The Autoimmune Regulator (AIRE) Is a DNA-binding Protein. Journal of Biological Chemistry. 2001;276:41357–41364. doi: 10.1074/jbc.M104898200. [DOI] [PubMed] [Google Scholar]
- 15.Liston A, Gray DH, Lesage S, Fletcher AL, Wilson J, Webster KE, Scott HS, Boyd RL, Peltonen L, Goodnow CC. Gene dosage–limiting role of Aire in thymic expression, clonal deletion, and organ-specific autoimmunity. J Exp Med. 2004;200:1015–1026. doi: 10.1084/jem.20040581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oftedal BE, Hellesen A, Erichsen MM, Bratland E, Vardi A, Perheentupa J, Kemp EH, Fiskerstrand T, Viken MK, Weetman AP, et al. Dominant Mutations in the Autoimmune Regulator AIRE Are Associated with Common Organ-Specific Autoimmune Diseases. Immunity. 2015;42:1185–1196. doi: 10.1016/j.immuni.2015.04.021. This paper describes a new cohort of patients with dominant negative mutations in AIRE and non-classical APS-1, suggesting that AIRE mutations might be much more widespread than initally thought. [DOI] [PubMed] [Google Scholar]
- 17.Cetani F, Barbesino G, Borsari S, Pardi E, Cianferotti L, Pinchera A, Marcocci C. A novel mutation of the autoimmune regulator gene in an Italian kindred with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, acting in a dominant fashion and strongly cosegregating with hypothyroid autoimmune thyroiditis. J Clin Endocrinol Metab. 2001;86:4747–4752. doi: 10.1210/jcem.86.10.7884. [DOI] [PubMed] [Google Scholar]
- 18.Su MA, Giang K, Zumer K, Jiang H, Oven I, Rinn JL, Devoss JJ, Johannes KP, Lu W, Gardner J, et al. Mechanisms of an autoimmunity syndrome in mice caused by a dominant mutation in Aire. J Clin Invest. 2008;118:1712–1726. doi: 10.1172/JCI34523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Proekt I, Miller CN, Jeanne M, Fasano KJ, Moon JJ, Lowell CA, Gould DB, Anderson MS, DeFranco AL. LYN- and AIRE-mediated tolerance checkpoint defects synergize to trigger organ-specific autoimmunity. J Clin Invest. 2016;126:3758–3771. doi: 10.1172/JCI84440. This paper describes a novel digenic model of autoimmune susceptibility leading to uveitis in mice with a dominant negative mutation of Aire and deficiency in Lyn kinase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scapini P, Pereira S, Zhang H, Lowell CA. Multiple roles of Lyn kinase in myeloid cell signaling and function. Immunol Rev. 2009;228:23–40. doi: 10.1111/j.1600-065X.2008.00758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Selmi C. Diagnosis and classification of autoimmune uveitis. Autoimmun Rev. 2014;13:591–594. doi: 10.1016/j.autrev.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 22.Peluso R, Lupoli GA, Del Puente A, Iervolino S, Bruner V, Lupoli R, Di Minno MN, Foglia F, Scarpa R, Lupoli G. Prevalence of thyroid autoimmunity in patients with spondyloarthropathies. J Rheumatol. 2011;38:1371–1377. doi: 10.3899/jrheum.101012. [DOI] [PubMed] [Google Scholar]
- 23.Teh CE, Daley SR, Enders A, Goodnow CC. T-cell regulation by casitas B-lineage lymphoma (Cblb) is a critical failsafe against autoimmune disease due to autoimmune regulator (Aire) deficiency. Proc Natl Acad Sci U S A. 2010;107:14709–14714. doi: 10.1073/pnas.1009209107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodnow CC. Multistep pathogenesis of autoimmune disease. Cell. 2007;130:25–35. doi: 10.1016/j.cell.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 25.Bruserud O, Oftedal BE, Landegren N, Erichsen MM, Bratland E, Lima K, Jorgensen AP, Myhre AG, Svartberg J, Fougner KJ, et al. A Longitudinal Follow-up of Autoimmune Polyendocrine Syndrome Type 1. J Clin Endocrinol Metab. 2016;101:2975–2983. doi: 10.1210/jc.2016-1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahonen P, Myllarniemi S, Sipila I, Perheentupa J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322:1829–1836. doi: 10.1056/NEJM199006283222601. [DOI] [PubMed] [Google Scholar]
- 27.Ferre EM, Rose SR, Rosenzweig SD, Burbelo PD, Romito KR, Niemela JE, Rosen LB, Break TJ, Gu W, Hunsberger S, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight. 2016;1 doi: 10.1172/jci.insight.88782. This paper describes the first prospective observational natural history study of APS-1/APECED patients, who were enrolled consecutively and evaluated uniformly by a multidisciplinary team of specialists. It redefined the clinical and diagnostic features of the syndrome, which may allow for earlier recognition and better patient outcomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li D, Streeten EA, Chan A, Lwin W, Tian L, Pellegrino da Silva R, Kim CE, Anderson MS, Hakonarson H, Levine MA. Exome Sequencing Reveals Mutations in AIRE as a Cause of Isolated Hypoparathyroidism. J Clin Endocrinol Metab. 2017;102:1726–1733. doi: 10.1210/jc.2016-3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferre EMN, Rose SR, Rosenzweig SD, Burbelo PD, Romito KR, Niemela JE, Rosen LB, Break TJ, Gu W, Hunsberger S, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight. 2016;1 doi: 10.1172/jci.insight.88782. These papers describe the first cis-regulatory element of Aire. CNS1, a NF-kB responsive element, is responsible for Aire expression in the thymus and Cns1-deficient mice develop autoimmunity similar to Aire-deficient mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LaFlam TN, Seumois G, Miller CN, Lwin W, Fasano KJ, Waterfield M, Proekt I, Vijayanand P, Anderson MS. Identification of a novel cis-regulatory element essential for immune tolerance. J Exp Med. 2015;212:1993–2002. doi: 10.1084/jem.20151069. These papers describe the first cis-regulatory element of Aire. CNS1, a NF-kB responsive element, is responsible for Aire expression in the thymus and Cns1-deficient mice develop autoimmunity similar to Aire-deficient mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haljasorg U, Bichele R, Saare M, Guha M, Maslovskaja J, Kõnd K, Remm A, Pihlap M, Tomson L, Kisand K, et al. A highly conserved NF-κB-responsive B enhancer is critical for thymic expression of Aire in mice. European Journal of Immunology. 2015;45:3246–3256. doi: 10.1002/eji.201545928. [DOI] [PubMed] [Google Scholar]
- 32.Yanagihara T, Sanematsu F, Sato T, Uruno T, Duan X, Tomino T, Harada Y, Watanabe M, Wang Y, Tanaka Y, et al. Intronic regulation of Aire expression by Jmjd6 for self-tolerance induction in the thymus. Nat Commun. 2015;6:8820. doi: 10.1038/ncomms9820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rattay K, Claude J, Rezavandy E, Matt S, Hofmann TG, Kyewski B, Derbinski J. Homeodomain-interacting protein kinase 2, a novel autoimmune regulator interaction partner, modulates promiscuous gene expression in medullary thymic epithelial cells. J Immunol. 2015;194:921–928. doi: 10.4049/jimmunol.1402694. [DOI] [PubMed] [Google Scholar]
- 34.Chuprin A, Avin A, Goldfarb Y, Herzig Y, Levi B, Jacob A, Sela A, Katz S, Grossman M, Guyon C, et al. The deacetylase Sirt1 is an essential regulator of Aire-mediated induction of central immunological tolerance. Nat Immunol. 2015;16:737–745. doi: 10.1038/ni.3194. [DOI] [PubMed] [Google Scholar]
- 35.Khan IS, Taniguchi RT, Fasano KJ, Anderson MS, Jeker LT. Canonical microRNAs in thymic epithelial cells promote central tolerance. Eur J Immunol. 2014;44:1313–1319. doi: 10.1002/eji.201344079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shao W, Zumer K, Fujinaga K, Peterlin BM. FBXO3 Protein Promotes Ubiquitylation and Transcriptional Activity of AIRE (Autoimmune Regulator) J Biol Chem. 2016;291:17953–17963. doi: 10.1074/jbc.M116.724401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takaba H, Morishita Y, Tomofuji Y, Danks L, Nitta T, Komatsu N, Kodama T, Takayanagi H. Fezf2 Orchestrates a Thymic Program of Self-Antigen Expression for Immune Tolerance. Cell. 2015;163:975–987. doi: 10.1016/j.cell.2015.10.013. [DOI] [PubMed] [Google Scholar]
- 38.Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science. 2015;348:589–594. doi: 10.1126/science.aaa7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ochs HD, Gambineri E, Torgerson TR. IPEX, FOXP3 and regulatory T-cells: a model for autoimmunity. Immunol Res. 2007;38:112–121. doi: 10.1007/s12026-007-0022-2. [DOI] [PubMed] [Google Scholar]
- 40.Fujikado N, Mann AO, Bansal K, Romito KR, Ferre EM, Rosenzweig SD, Lionakis MS, Benoist C, Mathis D. Aire Inhibits the Generation of a Perinatal Population of Interleukin-17A-Producing gammadelta T Cells to Promote Immunologic Tolerance. Immunity. 2016;45:999–1012. doi: 10.1016/j.immuni.2016.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Devoss JJ, Shum AK, Johannes KP, Lu W, Krawisz AK, Wang P, Yang T, Leclair NP, Austin C, Strauss EC, et al. Effector mechanisms of the autoimmune syndrome in the murine model of autoimmune polyglandular syndrome type 1. J Immunol. 2008;181:4072–4079. doi: 10.4049/jimmunol.181.6.4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gavanescu I, Benoist C, Mathis D. B cells are required for Aire-deficient mice to develop multi-organ autoinflammation: A therapeutic approach for APECED patients. Proc Natl Acad Sci U S A. 2008;105:13009–13014. doi: 10.1073/pnas.0806874105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer S, Woodward M, Hertel C, Vlaicu P, Haque Y, Karner J, Macagno A, Onuoha SC, Fishman D, Peterson H, et al. AIRE-Deficient Patients Harbor Unique High-Affinity Disease-Ameliorating Autoantibodies. Cell. 2016;166:582–595. doi: 10.1016/j.cell.2016.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Landegren N, Sharon D, Freyhult E, Hallgren A, Eriksson D, Edqvist PH, Bensing S, Wahlberg J, Nelson LM, Gustafsson J, et al. Proteome-wide survey of the autoimmune target repertoire in autoimmune polyendocrine syndrome type 1. Sci Rep. 2016;6:20104. doi: 10.1038/srep20104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, Ujhazi B, Chen K, Lee YN, Tirosh I, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest. 2015;125:4135–4148. doi: 10.1172/JCI80477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolff AS, Karner J, Owe JF, Oftedal BE, Gilhus NE, Erichsen MM, Kampe O, Meager A, Peterson P, Kisand K, et al. Clinical and serologic parallels to APS-I in patients with thymomas and autoantigen transcripts in their tumors. J Immunol. 2014;193:3880–3890. doi: 10.4049/jimmunol.1401068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharma P, Allison James P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khan IS, Mouchess ML, Zhu ML, Conley B, Fasano KJ, Hou Y, Fong L, Su MA, Anderson MS. Enhancement of an anti-tumor immune response by transient blockade of central T cell tolerance. J Exp Med. 2014;211:761–768. doi: 10.1084/jem.20131889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen W, ten Dijke P. Immunoregulation by members of the TGF[beta] superfamily. Nat Rev Immunol. 2016;16:723–740. doi: 10.1038/nri.2016.112. [DOI] [PubMed] [Google Scholar]
- 50.Hauri-Hohl M, Zuklys S, Hollander GA, Ziegler SF. A regulatory role for TGF-[beta] signaling in the establishment and function of the thymic medulla. Nat Immunol. 2014;15:554–561. doi: 10.1038/ni.2869. [DOI] [PubMed] [Google Scholar]
- 51.Wolff ASB, Kärner J, Owe JF, Oftedal BEV, Gilhus NE, Erichsen MM, Kämpe O, Meager A, Peterson P, Kisand K, et al. Clinical and Serologic Parallels to APS-I in Patients with Thymomas and Autoantigen Transcripts in Their Tumors. The Journal of Immunology. 2014;193:3880–3890. doi: 10.4049/jimmunol.1401068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dertschnig S, Hauri-Hohl MM, Vollmer M, Holländer GA, Krenger W. Impaired thymic expression of tissue-restricted antigens licenses the de novo generation of autoreactive CD4<sup>+</sup> T cells in acute GVHD. Blood. 2015;125:2720–2723. doi: 10.1182/blood-2014-08-597245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dumont-Lagacé M, St-Pierre C, Perreault C. Sex hormones have pervasive effects on thymic epithelial cells. 2015;5:12895. doi: 10.1038/srep12895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dragin N, Bismuth J, Cizeron-Clairac G, Biferi MG, Berthault C, Serraf A, Nottin R, Klatzmann D, Cumano A, Barkats M, et al. Estrogen-mediated downregulation of AIRE influences sexual dimorphism in autoimmune diseases. The Journal of Clinical Investigation. 2016;126:1525–1537. doi: 10.1172/JCI81894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cavadini P, Vermi W, Facchetti F, Fontana S, Nagafuchi S, Mazzolari E, Sediva A, Marrella V, Villa A, Fischer A, et al. AIRE deficiency in thymus of 2 patients with Omenn syndrome. The Journal of Clinical Investigation. 2005;115:728–732. doi: 10.1172/JCI23087. [DOI] [PMC free article] [PubMed] [Google Scholar]